
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect conversion of C6 sugars to methyl glycerate and glycolate in methanol†
Lei Feng,
Gang Li,
Yueer Yan,
Wenrong Hou,
Yahong Zhang * and
Yi Tang*
* and
Yi Tang*
Department of Chemistry, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Laboratory of Advanced Materials, Collaborative Innovation Centre of Chemistry for Energy Materials (iChEM), Fudan University, Shanghai 200433, China. E-mail: zhangyh@fudan.edu.cn; yitang@fudan.edu.cn; Fax: +86-021-65643288; Tel: +86-021-65643288
First published on 28th August 2018
Abstract
The present work deals with the one-pot conversion of C6 sugars to methyl glycerate and glycolate via a cascade of retro-aldol condensation and oxidation processes catalyzed by using MoO3 as the Lewis acid catalyst and Au/TiO2 as the oxidation catalyst in methanol. Methyl glycerate (MGLY) is the product of C6 ketose (fructose), while methyl glycolate (MG) is produced from C6 aldose (mannose, glucose). It is found that a good one-pot match between two reactive processes is the key to the production of MGLY and MG with high yield (27.6% MGLY and 39.2% MG). A separated retro-aldol condensation and oxidation process greatly decreases their yields, and even no MGLY can be obtained in this separated process. We attribute this to high instability of glyceraldehyde/glycolaldehyde and their different reaction pathways which mainly depend on whether acetalization of retro-aldol products (glyceraldehyde and glycolaldehyde) occurs with methanol or not. This result opens a new prospect on the accumulation of C3 products other than lactate from biomass-derived carbohydrates.
1. Introduction
Much attention has been devoted to research on chemicals from renewable biomass in recent years.1–4 As the largest fraction of biomass, carbohydrates are acknowledged as an abundant and inexpensive source of carbon for the production of fuels and value-added chemicals.5–8 Nowadays, although a variety of strategies have been developed to convert carbohydrates into diverse platform chemicals containing C2 to C6 skeletons,9–15 a mild and highly efficient catalytic route is still desired, particularly for chemicals with high thermal instability. Methyl glycerate (MGLY) and methyl glycolate (MG) are industrially important chemicals, which can be regarded as solvents, extractants or as food additives. More importantly, they are also used as platform molecules in the synthesis of macromolecule polymer material.16–20 So far, many strategies, e.g., the coupling of formaldehyde and methyl formate, and the hydrogenation of dimethyl oxalate, have been developed to prepare MG from the different conventional substrates, and the corresponding catalysts containing heteropolyacids and homogenous organic metal or heterogeneous metal catalysts have been explored.21–25 Additionally, MG was also obtained from the oxidation of biomass-derived ethylene glycol in methanol by using nanoparticle Au catalyst under mild reaction conditions.26,27 Using the similar catalytic system, Heeres et al. studied the oxidative esterification of glycerol to MGLY.28 However, the direct production of MG and MGLY from sustainable biomass sugars is still desired. Recently, Zhang and co-workers reported the preparation of MG from cellulosic biomass by using tungsten-containing compounds as catalysts in methanol.29 Han et al. employed phosphomolybdic acid to prepare glycolic acid from cellulose in water.30 To obtain glycolic acid and its methyl ester, high temperature is an essential condition in their works. Nevertheless, this is not conducive to the production of MGLY because of the high instability of glyceraldehyde (GLA) produced in the retro-aldol process of hexose.31,32 Therefore, in the previous reports on C3 product of biomass sugar, only dehydration product of GLA, i.e., lactate, can be obtained in such catalytic system.33,34 There have been few reports on generating MGLY from C6 sugars or cellulose. Herein, we developed a one-pot catalytic system for the direct production of MGLY and MG from C6 ketose and aldose via a cascade of retro-aldol condensation and oxidation processes catalyzed by MoO3 as Lewis acid catalyst and nanoparticle Au supported on TiO2 (Au/TiO2) as oxidative catalyst, respectively. Moreover, a good one-pot match between two reaction processes is the key to obtain MG and MGLY with high yield. And it is found that a separated two-step process, i.e., separated retro-aldol condensation and oxidation processes, can decline their yields, and even no MGLY can be obtained by the two-step process. This can be attributed to the high instability of GLA and its different oxidative route from GA.2. Experimental section
Materials
D-(+)-Mannose (MAN, 99%), D-(+)-glucose (GLU, 99.5%), D-(−)-fructose (FRU, 99%), glycolaldehyde dimer (GA, 95%) and D-(−)-glyceraldehyde (GLA, 90%) were purchased from Sigma-Aldrich. Methyl glycolate (MG, 98%), 1,3-dihydroxyacetone (DHA, 99%), pyruvaldehyde (PYR, 40%), methyl lactate (ML, 98%), methanol (99.9%) were purchased from Macklin. Methyl glycerate (MGLY, 95%) was purchased from Molbase. Molybdenum(VI) oxide (MoO3) and potassium carbonate were purchased from Sinopharm Chemical Reagent Company. Gold nanoparticle supported on titanium oxides (Au/TiO2: gold 1% on titanium dioxide) was purchased from Aladdin. Deionized water was produced by a laboratory water purification system.Catalyst characterization
A Bruker D2 diffractometer, taken from 5° to 80° with Cu Kα radiation (λ = 0.154 nm), was employed for obtaining the X-ray diffraction (XRD) patterns of MoO3 and Au/TiO2 catalysts. The morphology of the Au/TiO2 was determined by transmission electron microscopy (TEM, Tecnai G2 F20 S-Twin, USA FEI Corp.). The scanning electron microscopy (SEM, Nova NanoSem 450, USA FEI Corp.) was employed to observe morphology of MoO3 catalyst.NH3-temperature programmed desorption (TPD) was used to determine the acid property of MoO3. Typically, 1.4 g of the sample was loaded in a U-shape quartz reactor, pretreated at 600 °C for 1 h and then cooled to 100 °C under He flow. The pretreated sample was saturated with 5% NH3/He for 1 h at 100 °C, with subsequent flushing at 100 °C for 1 h to remove the physisorbed ammonia. TPD analysis was carried out from 100 to 480 °C at a heating rate of 10 K min−1. Quantitative analysis of the desorbed ammonia was based on Thermal Conductivity Detector (TCD).
Catalytic reaction procedure
All the catalytic reactions were carried out in a 30 mL thick-walled glass reactor. Briefly, 50 mg of C6 sugar was dissolved in 5 g of methanol. And then, 12.5–50 mg of MoO3 and 40–100 mg of Au/TiO2 were added, respectively. Subsequently, the glass reactor containing reaction mixture was put into microwave instrument and pressurized with oxygen to 1 MPa and was heated to desired temperature under stirring with a magnetic stirring (600 rpm). For the conversion of FUR, a little amount of K2CO3 (3 mg) was added to form a weak alkaline environment for the purpose of easy oxidation process.35–37 After reacting for a certain time, the reactor was cooled to room temperature by compressed air. The mixture of liquid and solid was filtered to separate the solid catalysts from liquid fraction. The quantitative analysis of methanol-soluble products in the liquid fraction was conducted by high-performance liquid chromatography (Shimadzu, HPLC) and gas chromatography (Shimadzu, GC). And the solid fractions by filtration after the reaction were loaded in the reactor again to investigate the reusability of the catalysts. Additionally, the transformations of GA/GLA into MG/MGLY as well as the transformations of C6 sugars into GA/GLA were performed in the above same procedure, but either 100 mg of Au/TiO2 or 50 mg of MoO3 were added as single catalyst. Moreover, no O2 was needed during the catalytic process of C6 sugars into GA/GLA.Product analysis
Conversion of the substrate and yield of the product, except MGLY, were analysed on a Shimadzu HPLC (RI detector) equipped with a Shodex SC1011 (30 cm length) sugar column maintained at 80 °C using deionized water as the mobile phase with a flow rate of 1.0 mL min−1. The quantification analyses of samples were based on calibration curves obtained by injecting standard solutions of known concentrations (Fig. S1†). MGLY was analysed on GC (Shimadzu, FID detector) equipped with DC-FFAP capillary column (30 m × 0.25 mm × 0.25 μm). The injector and detector temperatures were set at 250 °C and 250 °C. Nitrogen was used as the carrier gas with a flow rate of 1.0 mL min−1. The split ratio was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100. GC-MS (Thermo Finnign Voyager, with a HP-Wax column of dimension 30 m × 0.25 mm × 0.25 μm and electron impact ionization) was used to confirm the products. Both the products of C6 ketose and aldose were further identified by the mass spectra. And 1H nuclear magnetic resonance (NMR) spectrum was performed to identify the reaction pathways of C6 ketose to MGLY and C6 aldose to MG. Conversion of substrate and carbon yield of product were calculated as follow.
:100. GC-MS (Thermo Finnign Voyager, with a HP-Wax column of dimension 30 m × 0.25 mm × 0.25 μm and electron impact ionization) was used to confirm the products. Both the products of C6 ketose and aldose were further identified by the mass spectra. And 1H nuclear magnetic resonance (NMR) spectrum was performed to identify the reaction pathways of C6 ketose to MGLY and C6 aldose to MG. Conversion of substrate and carbon yield of product were calculated as follow.
For the conversion of C6 sugars into GA/GLA and MG/MGLY:
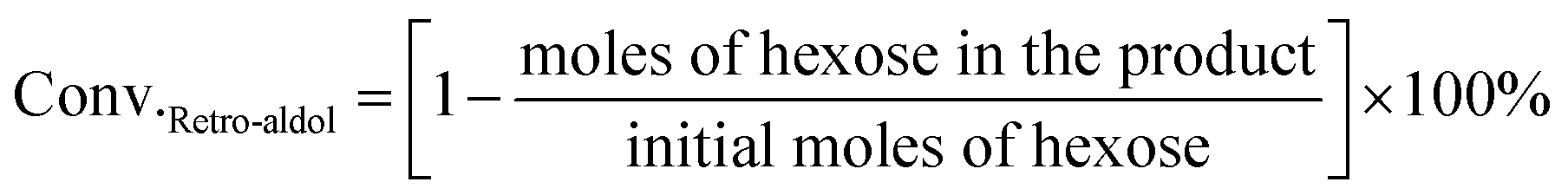
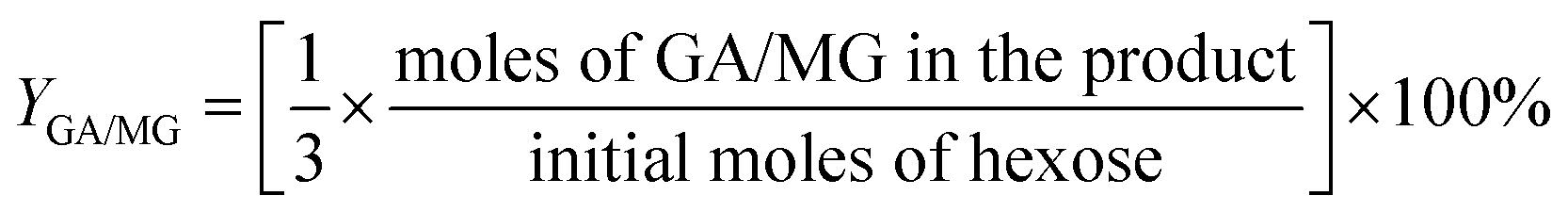
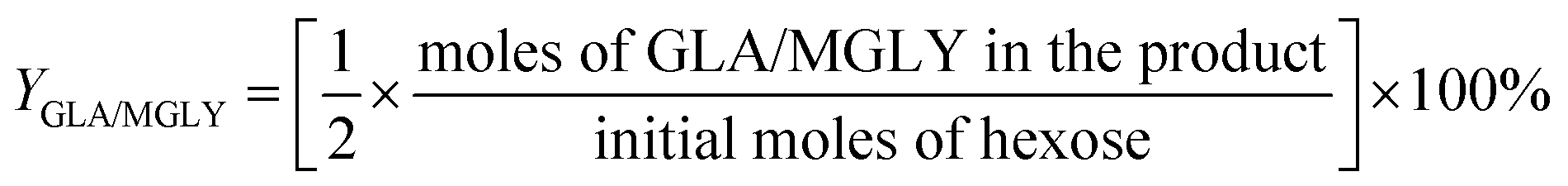

For the conversion of GA into MG:
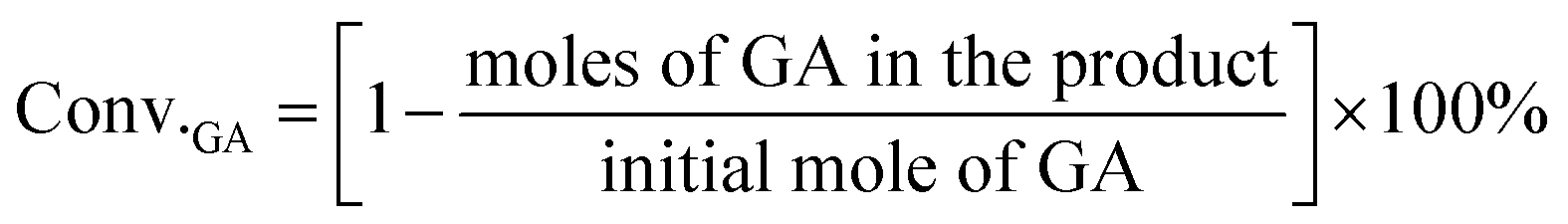
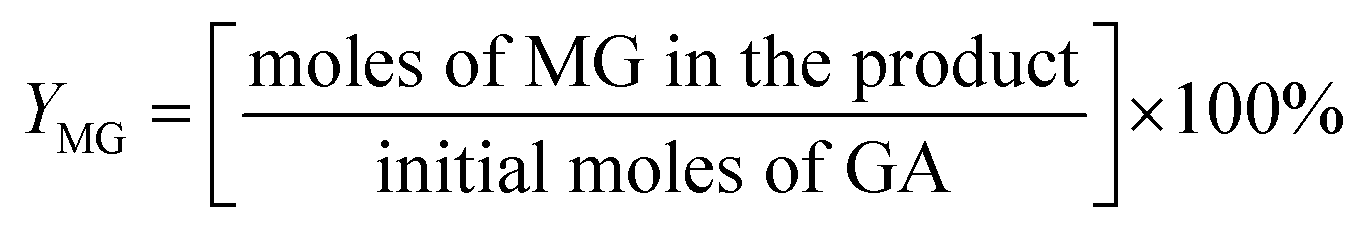
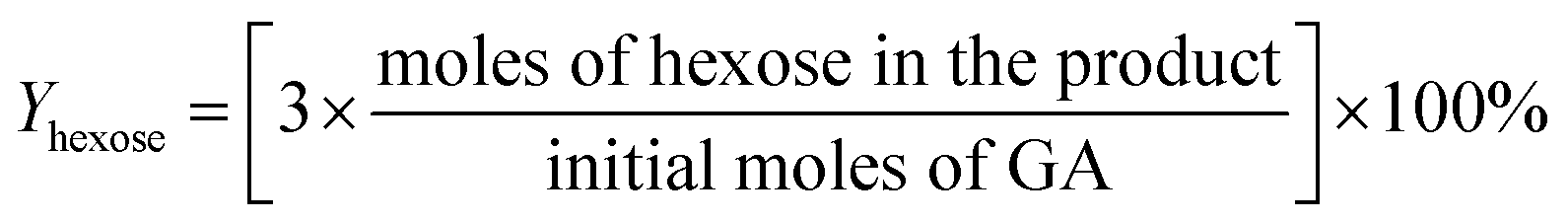

For the conversion of GLA into MGLY:
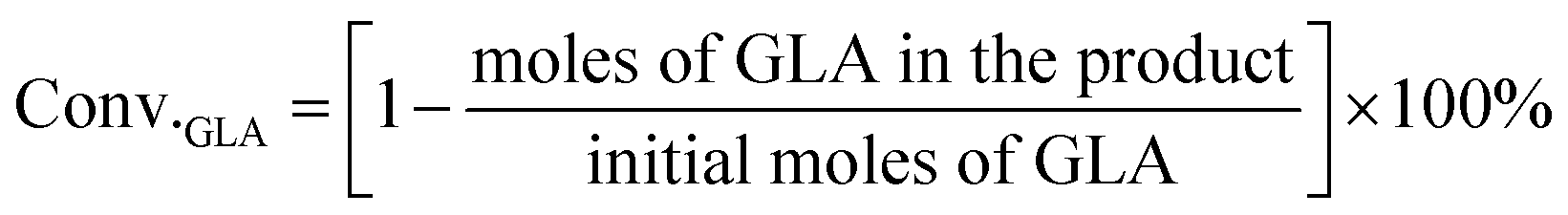

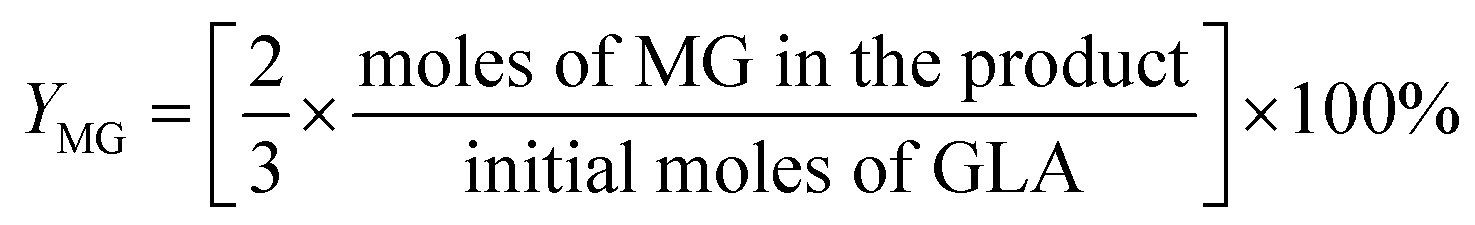
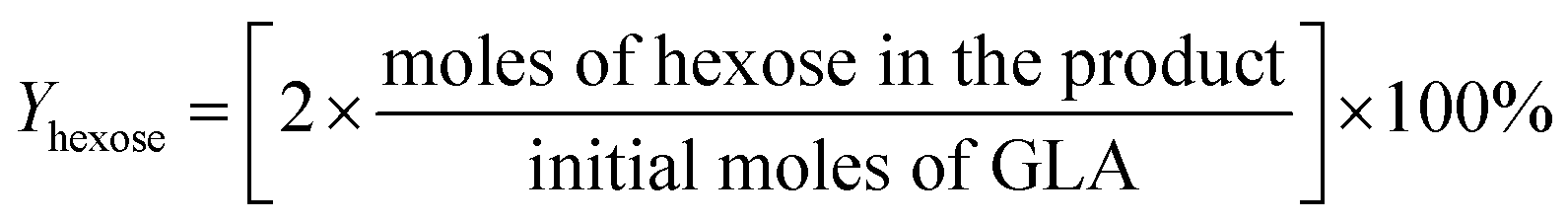
3. Results and discussion
One-pot conversion of various C6 sugars
To enable the productions of MGLY and MG from C6 sugars, a retro-aldol process and an oxidative esterification process are necessary (Scheme 1). As shown in Scheme 1, MGLY can be produced from C6 ketose (FRU) while MG is the product of C6 aldose (MAN, GLU) via a retro-aldol process and an oxidative esterification process. MoO3, as a suitable solid catalyst with good capacity to catalyze 1,2-intramolecular carbon shift, ensures that the retro-aldol process of C6 sugars can be achieved smoothly at moderate temperature.38–40 Such moderate retro-aldol reaction condition is very critical to its combination with the oxidative esterification process and the avoidance of the further side reactions such as dehydration, polymerization and over-oxidation of the retro-aldol products. Furthermore, for the oxidation catalyst, several conventional commercial noble metal catalysts are pre-screened by using GA as substrate. As shown in Table S1,† Au/TiO2 displays a compelling advantage on the oxidation of GA compared to other catalysts (Pt/C, Pd/C and Ru/C). Therefore, herein, MoO3 and Au/TiO2 catalysts are employed to jointly perform the conversion of C6 sugars toward MGLY and MG although a number of Lewis acid catalysts used in the retro-aldol process have been reported.41–44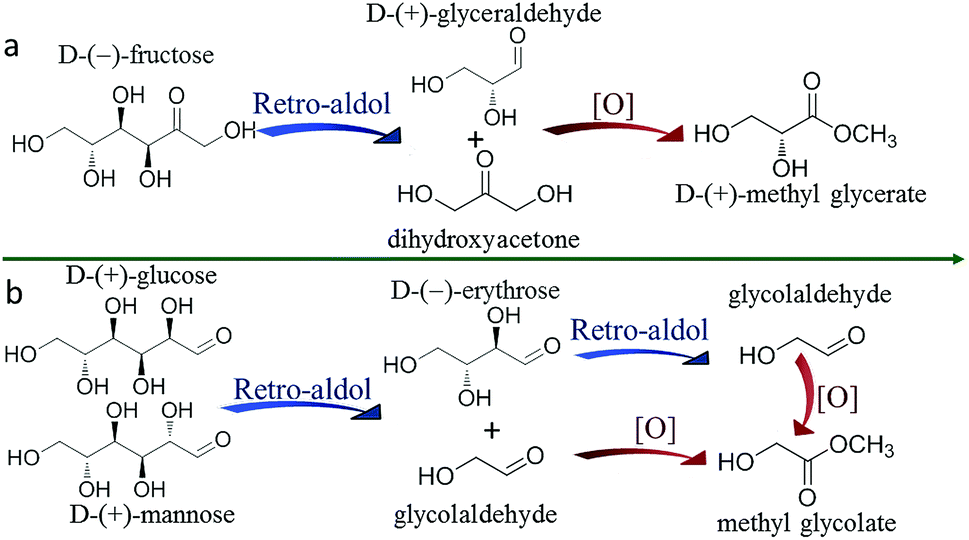 | ||
| Scheme 1 Direct conversion of C6 sugars to MGLY and MG via a retro-aldol process and an oxidative esterification process. | ||
XRD, SEM and NH3-TPD analyses were used to characterize the crystalline structure, size and acid activity of the used commercial MoO3 (Fig. 1a–c). The diffraction peaks in Fig. 1a at 2θ = 12.8°, 23.3°, 25.7°, 27.3°, 38.9° can be indexed to the reflection of (020), (110), (040), (021) and (060), which displays a typical layered α-MoO3 crystalline structure. The SEM image in Fig. 1b further confirms its laminate structure with a micro scale. Moreover, NH3-TPD result of MoO3 indicates that there is a maximum ammonia desorption at ∼150 °C in the range 100–480 °C of desorption profile (Fig. 1c), implying its Lewis acidity. The acid amount of MoO3 is 9.2 × 10−2 cm3 g−1 STP, which has been gotten by the Quantitative analysis of the desorbed ammonia based on Thermal Conductivity Detector (TCD). Furthermore, the XRD pattern of Au/TiO2 in Fig. 1a exhibits that only crystalline phase of TiO2 can be observed and no diffraction of Au (38.2°, 44.4°, 64.6°, 77.5°) can be found, which can be attributed to the small size and low amount of Au on the supported TiO2. And the size of Au nanoparticle is measured from TEM images and its size distribution is evaluated according to the images of hundreds of randomly selected particles, that is, its average size is calculated at 2 nm with statistic method (Fig. 1d).
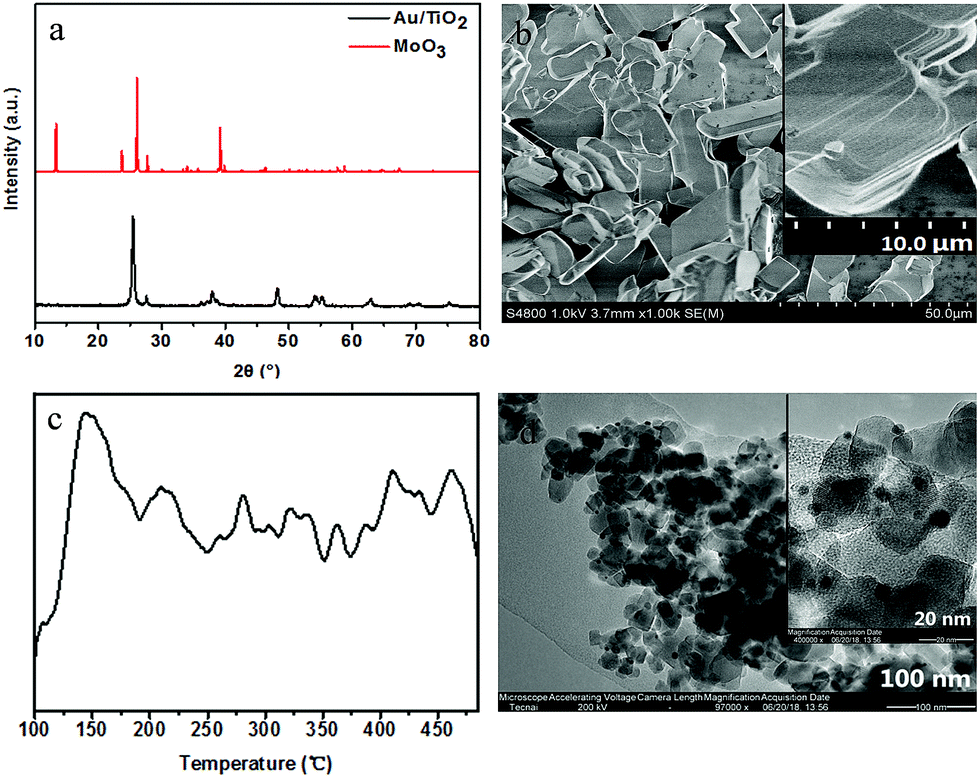 | ||
| Fig. 1 (a) XRD patterns of α-MoO3 and Au/TiO2; (b) SEM image of α-MoO3; (c) NH3-TPD profile of the α-MoO3 and (d) TEM of image of Au/TiO2. | ||
The conversion of various C6 sugars in methanol was carried out in the presence of the above MoO3 and Au/TiO2, and the results were listed in Table 1. Obviously, the data in Table 1 show that MGLY and MG can be smoothly obtained from C6 ketose and aldose, respectively. To be specific, the selectivity and yield of MGLY increase with the increasing reaction time, the highest yield of MGLY can reach 27.6% within 5 h at 363 K (Table 1, Fig. S2†). Meanwhile, the dehydration products of GLA, such as pyruvaldehyde (PYR) and methyl lactate (ML), increase with reaction time also (Table S2†). Moreover, low reaction temperature is more favourable to isomerization of hexoses rather than its retro-aldol splitting. However, as shown in Fig. S3† and Table 1, the main product is MG instead of MGLY when FRU is replaced by C6 aldose (MAN or GLU). The two aldoses display the similar product distributions in the presence of MoO3 and Au/TiO2 (Table S3†), and the highest selectivity and yield of MG are obtained within 4 h at 393 K. However, likely due to higher isomerization and epimerization tendency of GLU than MAN, GLU gives a lower yield of MG than MAN (Table 1). Besides, it can also be observed in Table 1 that high reaction temperature and long reaction time can result in a decreasing carbon balance. Because of the instability of substrates/intermediates, the increasing reaction temperature and time can induce the formation of undetectable humin/tar, and so decrease detectable carbon of the reaction system. Furthermore, the formations of MGLY and MG were further confirmed by GC-MS (Fig. S4 and S5†) and MS (Fig. S6†) results of their reaction solutions, respectively. It is worth noting that glyceric acid (Fig. S4a and S6a†) and glycolic acid (Fig. S5c and S6b†) are well observed in their reaction solutions except for MGLY and MG, respectively. Moreover, during the conversion of MAN, the acetal product of GA (1,1,2-trimethoxyethane, 2,2-dimethoxyethanol) and glycosidic MG are detected also (Fig. S5a, d, e and S6b†), indicating the acetalation/glycosidation reactions of GA/MG with methanol.
| Substrate | T (K) | t (h) | Conv.Retro-aldol (mol%) | Selec. (C mol%) | Y (C mol%) | Carbon balance (mol%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: 50 mg of FRU, 5 g of methanol, 50 mg of MoO3, 100 mg of Au/TiO2 and 3 mg of K2CO3, 1 MPa O2.b Reaction conditions: 50 mg of MAN, 5 g of methanol, 50 mg of MoO3, 100 mg of Au/TiO2, 1 MPa O2.c Reaction conditions: 50 mg of MAN, 5 g of methanol, 37.5 mg of MoO3, 80 mg of Au/TiO2, 1 MPa O2.d Reaction conditions: 50 mg of GLU, 5 g of methanol, 50 mg of MoO3, 100 mg of Au/TiO2, 1 MPa O2. Yields (Y) of all products are calculated by carbon atom yields from the original hexoses. Selectivity (Selec.) of MGLY is calculated by YMGLY/Conv.Retro-aldol × 100%, and the corresponding carbon balance is calculated by [1 − Conv.Retro-aldol + YMGLY + Yothers] × 100%, others: GLA, DHA, PYR, ML. Selectivity (Selec.) of MG is calculated by YMG/Conv.Retro-aldol × 100%, and the corresponding carbon balance is calculated by [1 − Conv.Retro-aldol + YMG + Yothers] × 100%, others: GA, DHA. | ||||||
| FRUa | MGLY | |||||
| 353 | 5 | 73.9 | 30.2 | 22.3 | 74.2 | |
| 363 | 1 | 45.7 | 21.4 | 9.8 | 82.7 | |
| 3 | 72.4 | 28.9 | 20.9 | 73.9 | ||
| 5 | 85.3 | 32.4 | 27.6 | 70.4 | ||
| 6 | 87.2 | 31.4 | 27.4 | 68.6 | ||
| MANb | MG | |||||
| 373 | 1 | 22.8 | 52.2 | 11.9 | 95.1 | |
| 2 | 41.9 | 51.0 | 21.4 | 91.5 | ||
| 3 | 53.8 | 50.5 | 27.2 | 89.4 | ||
| 4 | 55.2 | 56.5 | 31.2 | 88.7 | ||
| 5 | 55.8 | 57.9 | 32.3 | 87.5 | ||
| MANc | 393 | 1 | 72.5 | 33.4 | 24.2 | 78.0 |
| 2 | 77.8 | 42.0 | 32.7 | 74.8 | ||
| 3 | 79.7 | 45.4 | 36.2 | 73.9 | ||
| 4 | 80.2 | 48.9 | 39.2 | 71.8 | ||
| 5 | 80.3 | 47.8 | 38.4 | 70.7 | ||
| GLUd | 393 | 1 | 39.5 | 35.9 | 14.2 | 81.1 |
| 2 | 50.8 | 37.0 | 18.8 | 77.9 | ||
| 3 | 52.5 | 37.3 | 19.6 | 77.7 | ||
| 4 | 54.2 | 38.7 | 21.0 | 74.3 | ||
| 5 | 56.9 | 36.3 | 20.7 | 71.2 | ||
Two-step conversion of C6 sugars
In the experiment, we found that only trace of MGLY was obtained when the conversion of FRU toward MGLY was divided into two steps to carry out, i.e., MoO3-driven retro-aldol process first and then the oxidative esterification process catalyzed by Au/TiO2 later. This indicates that this one-pot integrated process is very favour of the formation of MGLY. To gain further insight into the conversion of FRU to MGLY (Scheme 1), the retro-aldol process of FRU catalyzed by MoO3 and oxidative esterification process of the corresponding triose (GLA) catalyzed by Au/TiO2 were investigated and optimized, respectively. According to the results shown in Table S4,† only about 5.0% of GLA can be achieved within the reaction of 3 h at 373 K when only MoO3 is added. Moreover, a certain amount of pyruvaldehyde (PYR) and methyl lactate (ML) can be obtained. This means that GLA is difficult to be stably accumulated in the single retro-aldol process of FRU, and it can further not only dehydrate into PYR and ML but also split into formaldehyde and GA. Therefore, the yield of GLA is very low in the conversion of FRU to GLA (Table S4†). This high instability of GLA also results in a low yield of MGLY even if GLA is directly used as substrate in its oxidative esterification process (Table S5†). Therefore, a real-time transformation of GLA, i.e., a one-pot process containing retro-aldol condensation and oxidative esterification processes together, pays to the formation of MGLY from FRU. Moreover, it is worth mentioning that the MGLY obtained from this one-pot process is a chiral compound because chiral center in α-carbon of GLA has not been influenced during the retro-aldol splitting of D-FRU toward GLA, which will be significant for its further transformation and practical application. Generally, this chiral MGLY can only be obtained by biological fermentation process.45Besides, the retro-aldol process of MAN and the oxidative esterification process of GA were also investigated and the reaction conditions were optimized. Unlike the case of GLA, GA can be stably obtained in methanol (Table S6†) and it can be also high selectively converted into MG (Table S7†). In more details, as shown in Table S6,† a short reaction time (1 h) and a high temperature (393 K) benefit the accumulation of GA, that is, a 45.4% of GA can be obtained from MAN in the presence of MoO3 within 1 h at 393 K. However, for the oxidative esterification of GA, the highest yield of MG can be obtained at 373 K (Table S7†). Therefore, we try to achieve the conversion of MAN toward MG via a two-step process under their optimum reaction conditions, that is, MAN is treated in the presence of MoO3 for 1 h at 393 K (Table 2, step 1), and then reaction mixture is further catalyzed by Au/TiO2 under the 1 MPa of O2 at 373 K for desired time after solid MoO3 is removed. A 30.4% of MG is ultimately obtained (Table 2, step 2), which is still lower than that (39.2%) achieved in the one-pot system. This result further demonstrates that the one-pot integrated process is conductive to the formation of methyl ester. In such one-pot catalytic system, the produced GLA/GA during the retro-aldol of C6 sugars can be timely converted by the oxidative esterification, which not only shifts the retro-aldol equilibrium toward right but also avoids the side reaction. Therefore, MG and MGLY are produced more efficiently from C6 sugars by the combination of MoO3 with Au/TiO2.
| Step | T (K) | t (h) | Conv.Retro-aldol (mol%) | YGA | YMG | Carbon balance (mol%) |
|---|---|---|---|---|---|---|
| (C mol%) | ||||||
| a Reaction conditions: 50 mg of MAN, 5 g of methanol, 50 mg of MoO3 (step 1); 5 mL reaction solution of step 1 without MoO3, 100 mg of Au/TiO2, 1 MPa O2 (step 2). Yields (Y) of products are calculated by carbon atom yield from the original hexose. Carbon balance is calculated by [1 − Conv.Retro-aldol + YGA + YMG + YDHA] × 100%. | ||||||
| 1 | 393 | 1 | 71.1 | 45.6 | 0 | 74.5 |
| 2 | 373 | 1 | 76.4 | 3.5 | 29.5 | 59.3 |
| 2 | 78.8 | 3.2 | 30.4 | 56.8 | ||
In addition, it is worth mentioning that MoO3 can also give an oxidation activity by the reduction of Mo(VI) to Mo(IV),38 which means that oxidative products could be formed in the presence of only MoO3. However, in our experiments, when only MoO3 is used as catalyst, only retro-aldol process of FRU/MAN can be observed and no MGLY or MG can be detected. However, similar to that of literature,38 a long reaction time (more than 5 h) could partly lead to the colour change of MoO3 from white to dark blue although no MGLY or MG can be observed, indicating the existence of other complex oxidized by-products. Moreover, when MoO3 is used in the presence of O2, the colour change of MoO3 becomes negligible because of the oxidizing environment. Furthermore, if MoO3 is used in the presence of O2, as shown in Table S8,† only trace amount of MG can be observed with the reaction of 3 h, which can be attributed to the oxidation of O2 with a low activity. Even if the reaction time is prolonged to 9 h, only 10.8% of MG is achieved (Table S8†). Therefore, we can confirm that the MoO3 and Au/TiO2 in the presence of O2 are responsible for the retro-aldol activity and oxidative activity during the conversion of hexoses toward MGLY and MG, respectively.
Optimization of reaction conditions and recycling of catalysts
In further maximizing the yields of MG and MGLY, the amount and the ratio of two catalysts were optimized also. As shown in Fig. 2a (left) and Table S9,† high or low mass ratio of MoO3 to Au/TiO2 results in a low selectivity and yield of MGLY, and the highest yield of MGLY is obtained within 5 h when their mass ratio is set to 1:2 (50 mg MoO3 and 100 mg Au/TiO2). Similar to that of MGLY formation, the highest yield of MG can be obtained when the mass ration of MoO3 to Au/TiO2 is about 1:2 (Table S10†). These results indicate that an appropriate match between the amounts of two catalysts is the key to obtain the product with high yield. Under these optimal conditions, the influences of hexose concentration in the MGLY/MG yields were studied (Fig. 2, Table S11 and S12†). It is found that changes in the FRU concentration from 1.0 to 10.0 wt% do not significantly affect the yield of MGLY, and nearly 20% of MGLY was obtained even at a high FRU concentration of 10 wt% (Fig. 2a, right). However, as shown in Fig. 2b (left), although there is not obvious change in the yield of MG when the concentration of MAN is increased from 1.0 to 2.0 wt%, MG yield decreased significantly with the increase of MAN concentration from 2.0 to 10.0 wt%. In a word, a low hexose concentration is beneficial to their direct conversion toward MGLY/MG. Subsequently, we also tested the prospect of reusing solid MoO3 and Au/TiO2 in the reaction of producing MG from MAN (Fig. 2b, right, Table S13†). After a reaction cycle was finished, the used MoO3 and Au/TiO2 catalysts were filtered out directly from the reaction system and then were added into the next cycle experiment of the conversion of MAN to MG as catalysts. It is found that the MG yield is slightly dropped after four cycles, which could be caused by the part aggregation of Au nanoparticles loaded on the TiO2 from 2 nm to 5 nm during the recyclability test experiment (Fig. 1d vs. Fig. S7†). The slight activity decrease of Au nanoparticle can be also confirmed by the slight accumulation of GA in the fourth cycle in Table S13.† Because the conversion of in situ produced GA from retro-aldol process of MAN to MG is converted into MG under the catalysis of Au/TiO2, the activity decrease of Au/TiO2 catalyst results in the accumulation of GA in the reaction system.
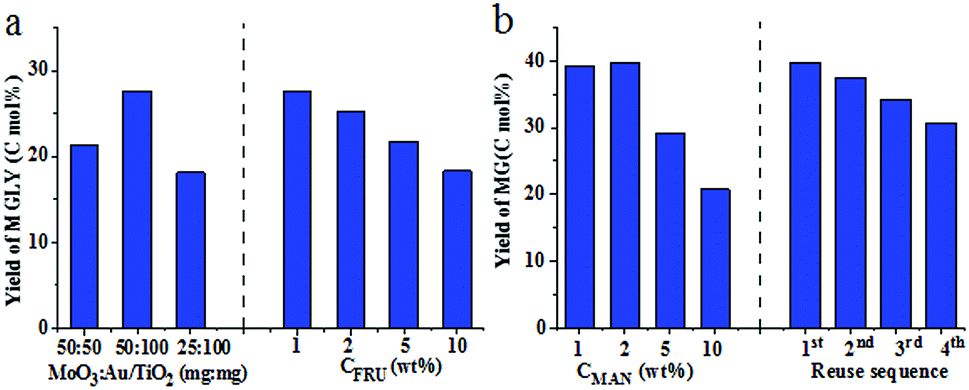 | ||
| Fig. 2 (a) Carbon yield of MGLY for FRU conversion in methanol containing different mass ratios of MoO3 to Au/TiO2 (left), and different FRU concentrations (right); (b) carbon yield of MG for MAN conversion in methanol with different MAN concentrations (left), and reuse of MoO3 and Au/TiO2 catalysts. | ||
Reaction pathway
According to the above results, we can conclude that MG can be obtained through both one-pot and two-step processes, but MGLY can only be accumulated in the one-pot process. Moreover, it is found that the acetal product of GA (1,1,2-trimethoxyethane, 2,2-dimethoxyethanol) and glycosidic MG can be observed in the products during the conversion of MAN (Fig. S5 and S6b†), however, no corresponding derivative species of GLA in the conversion of FUR can be detected. Based on these facts, we propose that the formations of MGLY and MG from hexoses experience different routes (Scheme 2), which could be responsible for their different reaction results in the above two reaction processes. In our proposed reaction routes, after C6 aldose and ketose is split into GA and GLA via retro-aldol process, respectively, GA can react with methanol at once and convert into dimethoxyethanol and 1,1,2-trimethoxyethane via acetalization process, and then the acetal products can be further oxidized into MG (Scheme 2). However, the produced GLA is inclined to directly convert into glyceric acid and MGLY in the presence of Au/TiO2 and O2 (Scheme 2). Obviously, taking the instability of aldehyde group into account, its acetalization with methanol can make it stable within a short time. Therefore, the acetalization of GA in methanol assures its stability and accumulation during the retro-aldol process (Table S6†), and so MG can be obtained in the two-step process due to the good stability of GA acetal (Table 2). On the contrary, GLA tends to further dehydrate into PYR and ML or split into GA and formaldehyde because of its instability (Tables S4 and S5†). To confirm this, 1H-NMR analyses of GA and GLA in methanol before/after thermal treatment were studied. Moreover, to avoid the other side reactions, no catalyst was added during the thermal treatment. As shown in Fig. 3a, a simple thermal treatment (393 K for 30 min) results in significant change of 1H chemical shifts of GA. For instance, the aldehyde H-1 resonated at 9.61 ppm disappears whereas two new chemical shifts appear at 4.48 and 4.37 ppm, which can be assigned to those H of acetal and hemiacetal of aldehyde in GA (Fig. S8†). However, the 1H chemical shifts of GLA before/after thermal treatment display few change, indicating that no other product of GLA is formed in methanol (Fig. 3b). Additionally, the stability effect of acetalation process on GA can be further confirmed by the conversion of GA in methanol. As shown in Table S14,† when GA is directly added into methanol in the absence of oxidation catalyst, it displays a very low conversion in HPLC analysis because of deacetalization of acetal product during the elution process even if the thermal treatment temperature is increased to 190 °C. However, its GC result shows that the acetal product is observed and its yield can be more than 90%. This demonstrates not only the dominant acetalation process of GA in methanol but also its stabilization effect on GA. On the contrary, the difficult acetalation of GLA facilitates the formation of ML, and so leads to the poor accumulation of GLA. These results further support our proposed formation routes of MGLY and MG (Scheme 2). This finding of solvent effect also provides the possibility to stabilize the active products and so achieve a high product yield and even to change the product distribution.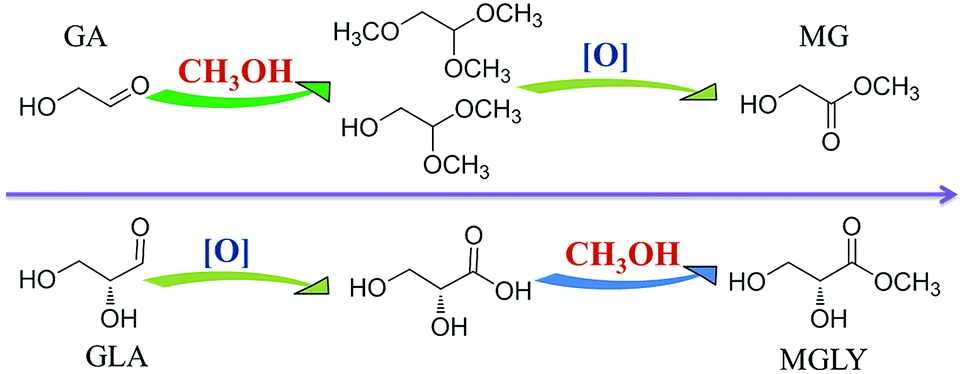 | ||
| Scheme 2 The proposed different reaction pathways of GA to MG and GLA to MLGY. | ||
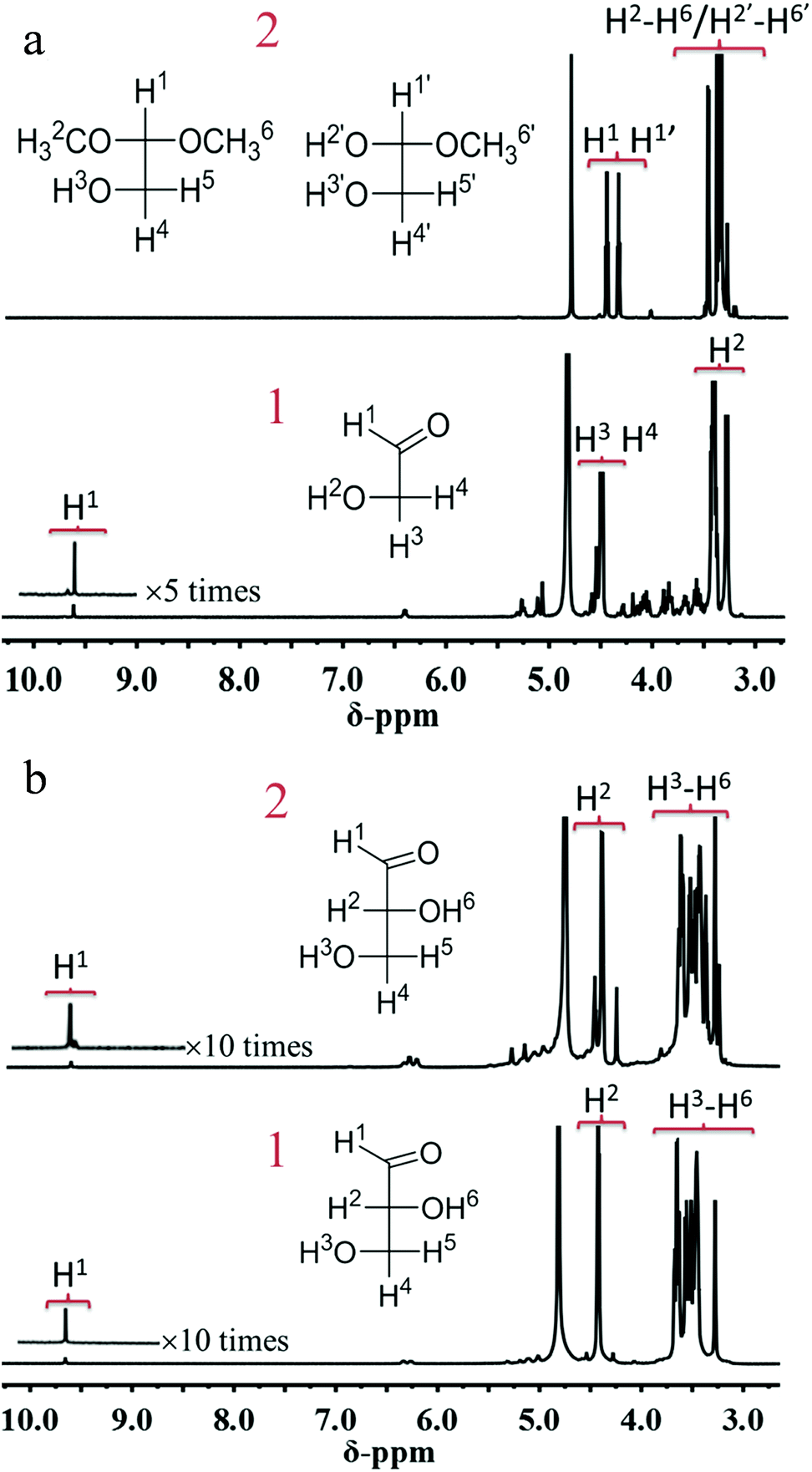 | ||
| Fig. 3 (a) 1H-NMR spectra of GA in methanol before (1)/after (2) thermal treatment, (b) 1H-NMR spectra of GLA in methanol before (1)/after (2) thermal treatment. | ||
4. Conclusions
We have proposed a one-pot strategy to prepare MGLY and MG from biomass-derived hexoses in the presence of MoO3 and Au/TiO2. It is found that a good match between two cascade reactions is the key to obtain MG and MGLY with high yield, and a separated two-step process, i.e., separated retro-aldol condensation and oxidation processes, can decline their yields because of the high instability of GLA and GA. We also proposed that the formations of MGLY and MG from hexoses experience different reaction routes, and whether acetalation of instable aldehyde group with methanol plays an important role on the yields of MGLY and MG. The results provide a new glimpse on the accumulation of the instable product via a solvent effect. Future studies will focus on further achieving a higher reaction efficiency as well as carbon atom utilization through the use of more efficient catalysts and solvents.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Science Foundation of China (21673045, 21473037, U1463206, 21433002).Notes and references
- A. Yamaguchi, O. Sato, N. Mimura and M. Shirai, Catal. Commun., 2015, 67, 59–63 CrossRef.
- Y. Ma, W. Tan, K. Wang, J. Wang, J. Jiang and J. Xu, ACS Sustainable Chem. Eng., 2017, 5, 5880–5886 CrossRef.
- L. Dehabadi, M. H. Mahaninia, M. Soleimani and L. D. Wilson, ACS Sustainable Chem. Eng., 2017, 5, 2970–2980 CrossRef.
- S. Liu, Appl. Energy, 2015, 144, 114–128 CrossRef.
- L. Yang, X. Yang, E. Tian, V. Vattipalli, W. Fan and H. Lin, J. Catal., 2016, 333, 207–216 CrossRef.
- Z. Liu, W. Li, C. Pan, P. Chen, H. Lou and X. Zheng, Catal. Commun., 2011, 15, 82–87 CrossRef.
- K. Zhang, S. Wu, H. Yang, H. Yin and G. Li, RSC Adv., 2016, 6, 77499–77506 RSC.
- B. O. de Beeck, M. Dusselier, J. Geboers, J. Holsbeek, E. Morre, S. Oswald, L. Giebeler and B. F. Sels, Energy Environ. Sci., 2015, 8, 230–240 RSC.
- S. Saravanamurugan and A. Riisager, Catal. Sci. Technol., 2014, 4, 3186–3190 RSC.
- Y. Wang, W. Deng, B. Wang, Q. Zhang, X. Wan, Z. Tang, Y. Wang, C. Zhu, Z. Cao, G. Wang and H. Wan, Nat. Commun., 2013, 4, 2141–2147 CrossRef PubMed.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC.
- F. Cao, T. J. Schwartz, D. J. Mcclelland, S. H. Krishna, J. A. Dumesic and G. W. Huber, Energy Environ. Sci., 2015, 8, 1808–1815 RSC.
- T. Wang, M. W. Nolte and B. H. Shanks, Green Chem., 2014, 16, 548–572 RSC.
- H. Chen, J. Tan, Y. Zhu and Y. Li, Catal. Commun., 2016, 73, 46–49 CrossRef.
- Y. Z. Wang, S. De and N. Yan, Chem. Commun., 2016, 52, 6210–6224 RSC.
- Y. Cui, B. Wang, C. Wen, X. Chen and W. L. Dai, ChemCatChem, 2016, 8, 527–531 CrossRef.
- C. Wen, Y. Cui, X. Chen, B. Zong and W. L. Dai, Applied Catalysis, B: Environmental, 2015, 162, 483–493 CrossRef.
- Y. Sun, H. Wang, J. Shen, H. Liu and Z. Liu, Catal. Commun., 2009, 10, 678–681 CrossRef.
- J. Zheng, H. Lin, X. Zheng, X. Duan and Y. Yuan, Catal. Commun., 2013, 40, 129–133 CrossRef.
- H. Fan, J. Tan, Y. Zhu, H. Zheng and Y. Li, J. Mol. Catal. A: Chem., 2016, 425, 68–75 CrossRef.
- D. He, W. Huang, J. Liu and Q. Zhu, Catal. Today, 1999, 51, 127–134 CrossRef.
- D. H. He, W. G. Huang, J. Y. Liu and Q. M. Zhu, J. Mol. Catal. A: Chem., 1999, 145, 335–338 CrossRef.
- Y. Sun, H. Wang, J. Shen, H. Liu and Z. Liu, Catal. Commun., 2009, 10, 678–681 CrossRef.
- B. Wang, Q. Xu, H. Song and G. Xu, J. Energy Chem., 2016, 16, 78–80 Search PubMed.
- A. Y. Yin, C. Wen, W. L. Dai and K. N. Fan, Applied Catalysis, B: Environmental, 2011, 108, 90–99 CrossRef.
- T. Hayashi, T. Inagaki, N. Itayama and H. Baba, Catal. Today, 2006, 117, 210–213 CrossRef.
- Y. H. Ke, X. X. Qin, C. L. Liu, R. Z. Yang and W. S. Dong, Catal. Sci. Technol., 2014, 4, 3141–3150 RSC.
- R. K. Purushothaman, J. V. Haveren, D. S. Es, I. Melian-Cabrera and H. J. Heeres, Green Chem., 2012, 14, 2031–2037 RSC.
- G. Xu, A. Wang, J. Pang, X. Zhao, J. Xu, N. Lei, J. Wang, M. Zheng, J. Yin and T. Zhang, ChemSusChem, 2017, 10, 1390–1394 CrossRef PubMed.
- J. Zhang, X. Liu, M. Sun, X. Ma and Y. Han, ACS Catal., 2012, 2, 1698–1702 CrossRef.
- F. D. Bobbink, J. Q. Zhng, Y. Pierson, X. Chen and N. Yan, Green Chem., 2015, 17, 1024–1031 RSC.
- J. Y. Zhang, B. L. Hou, A. Q. Wang, Z. L. LI, H. Wang and T. Zhang, AIChE J., 2014, 60, 3804–3813 CrossRef.
- K. Isobe, M. Kataoka, J. Ogawa, J. Hasegawa and S. Shimizu, New Biotechnol., 2012, 29, 177–182 CrossRef PubMed.
- H. A. Rass, N. Essayem and M. Besson, Green Chem., 2013, 15, 2240–2251 RSC.
- C. P. Ferraz, M. A. Garcia, É. Teixeira-Neto and L. M. Rossi, RSC Adv., 2016, 6, 25279–25285 RSC.
- N. Mei, B. Liu, J. D. Zheng, K. L. Lv, D. G. Tang and Z. H. Zhang, Catal. Sci. Technol., 2015, 5, 3194–3202 RSC.
- X. L. Tong, Z. H. Liu, L. H. Yu and Y. D. Li, Chem. Commun., 2015, 51, 3674–3677 RSC.
- M. Orazov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11777–11782 CrossRef PubMed.
- Y. E. Yan, L. Feng, G. Li, S. Y. Lin, Z. Sun, Y. H. Zhang and Y. Tang, ACS Catal., 2017, 7, 4473–4478 CrossRef.
- E. Miliordos, S. Caratzoulas and D. G. Vlachos, Appl. Catal., A, 2017, 530, 75–82 CrossRef.
- N. Ji, T. Zhang, M. Zheng, A. Wang, H. Wang, X. Wang and J. G. Chen, Angew. Chem., Int. Ed., 2008, 47, 8510–8513 CrossRef PubMed.
- Z. Tai, J. Zhang, A. Wang, M. Zheng and T. Zhang, Chem. Commun., 2012, 48, 7052–7054 RSC.
- T. Deng and H. Liu, J. Mol. Catal. A: Chem., 2014, 388, 66–73 CrossRef.
- T. Ennaert, A. J. Van, J. Dijkmans, C. R. De, W. Schutyser, M. Dusselier, D. Verboekend and B. F. Sels, Chem. Soc. Rev., 2016, 45, 584–611 RSC.
- H. Habe, Y. Shimada, T. Yakushi, H. Hattori, Y. Ano and T. Fukuoka, Appl. Environ. Microbiol., 2009, 75, 7760–7766 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05612a |
| This journal is © The Royal Society of Chemistry 2018 |