
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: PVT1 knockdown alleviates vancomycin-induced acute kidney injury by targeting miR-124 via inactivation of NF-κB signaling
Xiaoguang Zhu *,
Jun Shi,
Huicong li and
Fang Chen
*,
Jun Shi,
Huicong li and
Fang Chen
Department of Nephrology, Huaihe Hospital of Henan University, No. 115, Ximen Street, Gulou District, Kaifeng, Henan 475000, China. E-mail: zhuxiaoguangzxg@sina.com; Tel: +86-0371-23906586
First published on 12th September 2018
Abstract
Acute kidney injury (AKI) is a serious threat for human health and life. High dose of vancomycin (VAN) can give rise to AKI. The roles and molecular basis of long noncoding RNA plasmacytoma variant translocation 1 (PVT1) in VAN-induced AKI have been poorly defined till now. Protein levels of p65, phosphorylated p65 (p-p65), NF-κB inhibitor alpha (IκBα), phosphorylated IκBα (p-IκBα), Bcl-2 and Bax were measured by western blot assay. RNA levels of PVT1 and microRNA-124 (miR-124) were determined by RT-qPCR assay. HK-2 cell apoptosis was detected by an Annexin V-FITC apoptosis detection assay. Kidney functions were assessed by blood urea nitrogen (BUN) level, serum creatinine (Scr) level, histopathologic analysis, and TUNEL assay. Bioinformatical analysis, luciferase reporter assay, RIP and RNA pull down assays were performed to explore whether PVT1 could interact with miR-124. PVT1 was highly expressed in VAN-induced AKI models. Functional analysis revealed that PVT1 knockdown ameliorated VAN-induced AKI in vivo. Further exploration manifested that PVT1 directly interacted with miR-124. Moreover, the silencing of PVT1 abated VAN-induced HK-2 cell apoptosis in vitro, while this effect was reversed by the miR-124 inhibitor. Also, VAN treatment resulted in the reduction of miR-124 expression and the activation of NF-κB signaling in HK-2 cells. The inhibition of NF-κB alleviated VAN-induced HK-2 cell apoptosis. PVT1 activated NF-κB signaling by targeting miR-124 in VAN-induced HK-2 cells. PVT1 knockdown lessened VAN-induced AKI by targeting miR-124 via inactivating the NF-κB signaling, elucidating the critical roles and molecular basis of PVT1 in VAN-induced AKI and highlighting the diagnostic and therapeutic values of PVT1 in AKI.
Introduction
Acute kidney injury (AKI), a common clinical syndrome among hospitalized patients with major surgery and acute illness, is generally characterized by the acute reduction of kidney functions.1,2 AKI is a global public health concern with an estimated 13.3 million new cases and 1.7 million related deaths every year.3,4 Moreover, AKI is an independent risk factor for cardiovascular diseases, end stage renal disease (ESRD) and chronic kidney disease, bringing about massive healthcare costs.5–7 Despite remarkable advances in the understanding and management of AKI, it remains a serious threat for human health and life without other therapeutic interventions except fluid therapy and dialysis.8,9 Hence, it is imperative to investigate the pathophysiology of AKI so as to develop novel diagnostic and therapeutic strategies.Vancomycin (VAN), a commonly used glycopeptide antibiotic, is widely used for the treatment of staphylococcus aureus infection.10 However, the existence of VAN nephrotoxicity strikingly limited its usage in clinical trials.11,12 Also, VAN in higher doses has been reported to be correlated with increased AKI risk.13,14 Moreover, recent studies revealed that cellular apoptosis, mitochondrial dysfunction and proinflammatory oxidation played a vital role in the pathogenesis of VAN-induced AKI.15,16 In short, these data promoted us to further explore the nosetiology of AKI in VAN-induced AKI models.
Long noncoding RNAs (lncRNAs), a group of noncoding transcripts longer than 200 nucleotides, can regulate gene expression, maintain tissue homeostasis, and mediate disease progression.17 Moreover, recent studies indicated that lncRNAs might have potential diagnostic and therapeutic values in kidney diseases including AKI.18,19 For example, lncRNA TrAnscript Predicting Survival in AKI (TapSAKI) level was upregulated in plasma of patients with advanced AKI and TapSAKI could act as a predictor of mortality for patients with advanced AKI.20 LncRNA nuclear enriched abundant transcript 1 (NEAT1) overexpression aggravated lipopolysaccharide (LPS)-induced AKI by targeting microRNA-204 via activating NF-κB signaling in rat mesangial cells.21 PVT1, a long non-coding RNA without protein-coding potential, has been identified as a potential oncogenic factor in various cancers.22,23 Moreover, prior studies showed that PVT1 was closely associated with multiple kidney diseases such as end-stage renal disease,24,25 diabetic nephropathy,26,27 renal cancer28,29 and AKI.30 However, to our knowledge, the roles and molecular mechanisms of PVT1 in VAN-induced AKI have not been reported.
Mounting evidences show that lncRNAs can function as the molecular sponges of microRNAs (miRNAs) to exert their regulatory effects.31,32 MiRNAs, a class of small non-coding RNA molecules, play crucial roles in diverse biological processes such as cell apoptosis, proliferation and differentiation.33
Bioinformatical analysis indicated that PVT1 had a chance to interact with microRNA-124 (miR-124). MiR-124 has been widely reported to be a tumor suppressor in multiple cancers such as osteosarcoma,34 bladder cancer,35 and hepatocellular cancer.36 Also, miR-124 overexpression inhibited cell proliferation, invasion and migration in clear cell renal cell cancer.37 Moreover, the ectopic expression of miR-124 suppressed matrix metalloproteinase-2 (MMP2) protein translation and antagonized hypoxia-induced proliferation and invasion in renal proximal tubular epithelial cells (RPTEC).38 In short, these data indicated that miR-124 was closely associated with the pathogenesis of kidney diseases.
In the present research, we demonstrated that PVT1 knockdown alleviated VAN-induced AKI by targeting miR-124 via inactivating NF-κB signaling.
Materials and methods
Reagents and transfection
Pyrrolidinedithio-carbamate ammonium (PDTC) was purchased from Sigma-Aldrich, Inc. (St. Louis, MO, USA). Small interference RNA targeting PVT1 (si-PVT1) and its scramble control (si-NC), miR-124 mimic (miR-124) and its negative control (miR-NC), miR-124 inhibitor (anti-miR-124) and its negative control (anti-miR-NC) were purchased from GenePharma Co. Ltd. (Shanghai, China). Also, PVT1 overexpression plasmid was generated by subcloning PVT1 full-length fragments into pcDNA3.1 vector (Invitrogen, Carlsbad, CA, USA). All these substances were transfected into HK-2 cells using Lipofectamine 2000 reagent (Invitrogen).AKI mouse model
Male C57BL/6 mice (6–8 weeks) were obtained from Shanghai SLAC Laboratory Animal Co., Ltd (Shanghai, China). Mice were randomly divided into three groups: VAN-treated group (n = 8), VAN + si-NC group (n = 4), and VAN + si-PVT1 group (n = 4). VAN (Sigma-Aldrich) was intraperitoneally administered into mice at a single dose of 600 mg kg−1. Moreover, si-NC or si-PVT1 was injected into mice via tail vein 24 h prior to VAN treatment. Blood specimens and kidney tissues in VAN-treated group were collected at the indicated time points (0, 1, 3, 7 day) after VAN injection from two mice at a time. Blood samples and kidney tissues in VAN + si-NC and VAN + si-PVT1 groups were obtained on day 7 upon VAN administration. Then, serum was isolated from blood samples by centrifugation to measure the levels of serum creatinine (Scr) and blood urea nitrogen (BUN). Kidney tissues were harvested to perform following histological and biochemical analyses. All animal experiments were carried out with the approval of Experimental Animal Ethics Committee for Animal Research of Huaihe Hospital of Henan University following the national standard of care and use of laboratory animals and the guidelines of Institutional Committee for the Care and Use of Laboratory Animals in Huaihe Hospital of Henan University.Cell culture
Human proximal tubule epithelial cell line HK-2 was purchased from Cell Bank of Chinese Academy of Sciences (Shanghai, China) and was maintained in Keratinocyte Serum Free Medium (K-SFM, Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (Gibco), 5 ng ml−1 human recombinant epidermal growth factor (EGF, Gibco), 0.05 mg ml−1 bovine pituitary extract (BPE, Sigma-Aldrich, St. Louis, MO, USA) and 1× Gentamicin/Amphotericin solution (Gibco).The determination of Scr and BUN levels
Scr level was determined by Creatinine (serum) Colorimetric Assay Kit (Cayman Chemical, Ann Arbor, MI, USA) following the instructions by the manufacturer. BUN level was measured using a BUN detection kit (Solarbio Life Sciences, Beijing, China) according to the protocols of the manufacturer.Histological analysis
Resected kidney tissues were fixed with paraformaldehyde and then embedded using paraffin. Then, these kidney tissue sections were stained with haematoxylin and eosin (H&E) and photographed using an optical microscope to examine the morphological alterations such as tubular and epithelial changes. TUNEL assay was performed using the In Situ Cell Death Detection Kit (Sigma-Aldrich) to detect apoptosis pattern of kidney tissues.RT-qPCR
Total RNA was isolated from kidney tissues and HK-2 cells by Trizol reagent (Invitrogen) and reversely transcribed into cDNA using M-MLV Reverse Transcriptase (Invitrogen). Then, the relative expression of PVT1 (mouse or human) was measured using TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA, USA) for PVT1 (Hs01069023_m1 or Mm01203908_m1) and GAPDH (Hs00266705_g1 or Mm99999915_g1) with GAPDH as the endogenous control. MiR-124 relative level was determined by All-in-One™ miRNA Q-PCR Detection Kit (GeneCopoeia, San Diego, CA, USA) and primers for miR-124 and U6 snRNA (GeneCopoeia) with U6 snRNA as the internal control.Western blot assay
Total proteins were extracted from HK-2 cells using RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific, Rockford, IL, USA) supplemented with Halt™ protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Protein concentrations were determined using a Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). Then, proteins were separated by SDS-PAGE and blotted onto nitrocellulose membranes (Millipore, Bedford, MA, USA). After blockade of nonspecific protein signals, the membranes were probed with primary antibodies against Bcl-2 (ab182858, 1:2000, Abcam, Cambridge, UK), Bax (ab32503, 1:2000, Abcam), NF-κB p65 (ab32536, 1:5000, Abcam), NF-κB p65 (phospho S536) (p-p65, ab76302, 1:10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Abcam), beta actin (β-actin, ab8227, 1:2000, Abcam), IκBα (#9242, 1:1000, Cell Signaling Technology, Danvers, MA, USA), or phospho-IκBα (p-IκBα, Ser32) (#2859, 1:1000, Cell Signaling Technology). Next, the membranes were incubated for 1 h at room temperature with Goat Anti-Rabbit IgG H&L (HRP) secondary antibody (ab97051, 1:20000, Abcam). Finally, protein signals were detected using Clarity™ and Clarity Max™ Western ECL Blotting Substrates (Bio-Rad Laboratories, Hercules, CA, USA). Relative intensity of protein bands was determined using Quantity One software (Bio-Rad Laboratories).
000, Abcam), beta actin (β-actin, ab8227, 1:2000, Abcam), IκBα (#9242, 1:1000, Cell Signaling Technology, Danvers, MA, USA), or phospho-IκBα (p-IκBα, Ser32) (#2859, 1:1000, Cell Signaling Technology). Next, the membranes were incubated for 1 h at room temperature with Goat Anti-Rabbit IgG H&L (HRP) secondary antibody (ab97051, 1:20000, Abcam). Finally, protein signals were detected using Clarity™ and Clarity Max™ Western ECL Blotting Substrates (Bio-Rad Laboratories, Hercules, CA, USA). Relative intensity of protein bands was determined using Quantity One software (Bio-Rad Laboratories).
Luciferase reporter assay
Partial sequences of wide type PVT1 containing conserved miR-124 binding sites were subcloned into psiCHECK-2 vector (Promega, Madison, WI, USA) to produce PVT1-WT reporter. PVT1-MUT reporter carrying mutant miR-124 binding sites was also constructed into psiCHECK-2 vector (Promega) by KOD-plus-mutagenesis kit (Toyobo, Osaka, Japan). Then, PVT1-WT or PVT1-MUT reporter was co-transfected into 293T cells along with miR-124 mimic or miR-NC. After 48 h of transfection, cells were collected and luciferase activities were measured through a dual luciferase reporter system (Promega).Annexin V-FITC apoptosis detection assay
Apoptosis index was determined by Annexin V-FITC and propidium iodide (PI) Apoptosis Detection Kit (Sigma-Aldrich) referring to the instructions of the manufacturers. Apoptosis cell percentage was analyzed using a FACSCaliburflow cytometry system (BD Biosciences, San Jose, CA, USA).RNA immunoprecipitation (RIP) assay
RNA immunoprecipitation assay was performed using Imprint® RNA Immunoprecipitation Kit (Sigma-Aldrich) and primary antibodies against IgG (Sigma-Aldrich) or Argonaute-2 (Ago 2, Sigma-Aldrich) according to the protocols of manufacturers. The levels of PVT1 and miR-124 in input, IgG and Ago2 immunoprecipitation complexes were detected by RT-qPCR assay.RNA pull down assay
RNA pull down assay was carried out to test possible interaction between PVT1 and miR-124 in HK-2 cells as described previously.39 Biotin-labeled wide type miR-124 (Bio-miR-124-WT), biotin-labeled mutant type miR-124 (Bio-miR-124-MUT) with mutant PVT binding sites, biotin-labeled wide type PVT1 probe (Bio-probe-PVT1-WT), biotin-labeled mutant type PVT1 probe containing mutant miR-124 binding sites (Bio-probe-PVT1-MUT) and a biotin-labeled control probe (Bio-NC) were synthesized by Sangon Inc. (Shanghai, China). Then, biotin-labeled probes or miRNAs were incubated with streptavidin-conjugated magnetic beads (Invitrogen) and added into HK-2 cell lysate. Finally, the enrichment degrees of PVT1 and miR-124 in eluted RNA complex were determined by RT-qPCR assay.Statistical analysis
All data were obtained from at least three independent experiments, analyzed by SPSS21.0 software (SPSS Inc., Chicago, IL, USA) and displayed as mean ± standard deviation. Student's t-test was used to compare the difference between two groups, and one-way analysis of variance (ANOVA) was carried out for the comparison among multiple groups. The difference was statistically significant when P value was less than 0.05.Results
PVT1 was highly expressed in the kidneys of VAN-induced AKI mice
Firstly, biochemical parameters of BUN and Scr were used to assess the effect of VAN on kidney functions. Results showed that the injection of VAN (600 mg kg−1) caused an acute elevation of serum BUN and Scr levels in mice (Fig. 1A and B). Accordingly, histopathological analysis disclosed that VAN (600 mg kg−1) induced tubular distension and some epithelial degenerative changes such as vacuolization and coarse granulation of the cytoplasm in a time-dependent manner (Fig. 1C). Moreover, TUNEL assay revealed that VAN triggered more and more cell apoptosis with the prolongation of treatment time in kidney tissues (Fig. 1D). Also, pro-apoptosis protein Bax level was notably increased and anti-apoptosis protein Bcl-2 level was strikingly reduced in kidney tissues at the indicated time point (1, 3, 7 day) after VAN treatment, further confirming that VAN could induce cell apoptosis in kidney tissues (Fig. 1E). In a word, these data showed that VAN induced-mouse AKI models were successfully established. Also, we further demonstrated that VAN induced a time-dependent notable increase of PVT1 level in mice kidney tissues (Fig. 1F), hinting that PVT1 might be associated with the pathogenesis of AKI.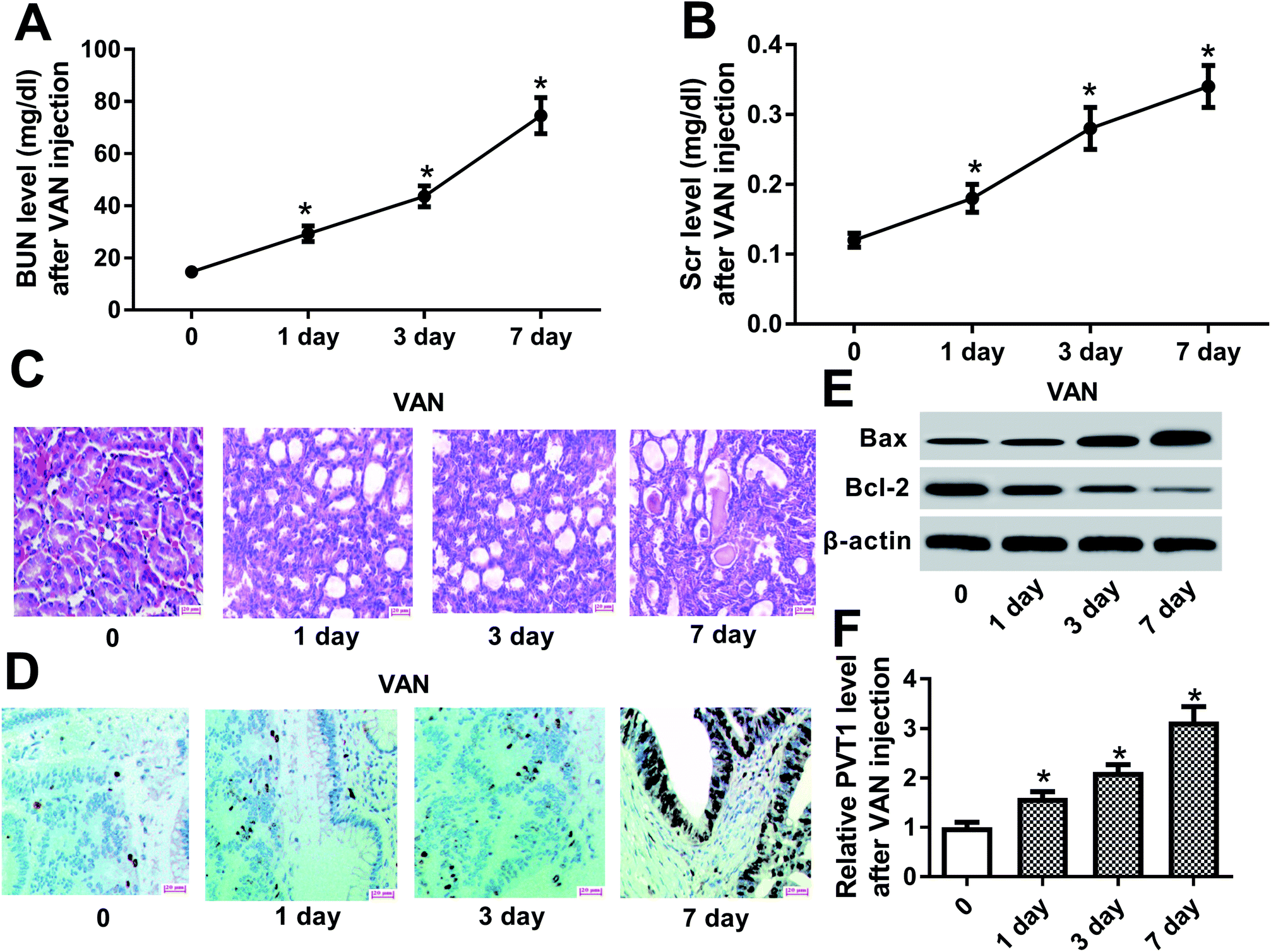 | ||
| Fig. 1 PVT1 was highly expressed in the kidneys of VAN-induced AKI mice. (A)–(E) Mice were intraperitoneally injected with VAN (600 mg kg−1) for the indicated time points (0, 1, 3, 7 day). (A) and (B) BUN and Scr levels were determined by respective commercial kits. (C) Histopathological alterations of kidney tissues were monitored by H&E staining. (D) TUNEL assay was performed to detect cell apoptosis pattern in kidney tissues. (E) Western blot assay was conducted to detect protein levels of Bax and Bcl-2 in kidney tissues. (F) PVT1 level in kidney tissues was measured by RT-qPCR assay. *P < 0.05. | ||
PVT1 knockdown ameliorated VAN-induced AKI in vivo
To further explore the roles and molecular mechanisms of PVT1 in VAN-induced AKI, siRNA specifically targeting PVT1 (si-PVT1) was injected into mice prior to VAN treatment. Following RT-qPCR assay confirmed that PVT1 level was markedly decreased in kidney tissues of mice with si-PVT1 injection (Fig. 2A). The depletion of PVT1 resulted in the reduction of BUN and Scr levels in VAN-induced AKI models (Fig. 2B and C). Also, the knockdown of PVT1 inhibited VAN-induced tubular distension and reduced VAN-induced epithelial degenerative changes in kidney tissues (Fig. 2D). Moreover, PVT1 silence suppressed VAN-triggered cell apoptosis in kidney tissues (Fig. 2E). Additionally, PVT1 knockdown resulted in the remarkable reduction of Bax level and the significant increase of Bcl-2 level in kidney tissues on day 7 after VAN stimulation (Fig. 2F). In summary, these results revealed that PVT1 knockdown ameliorated VAN-induced AKI in vivo.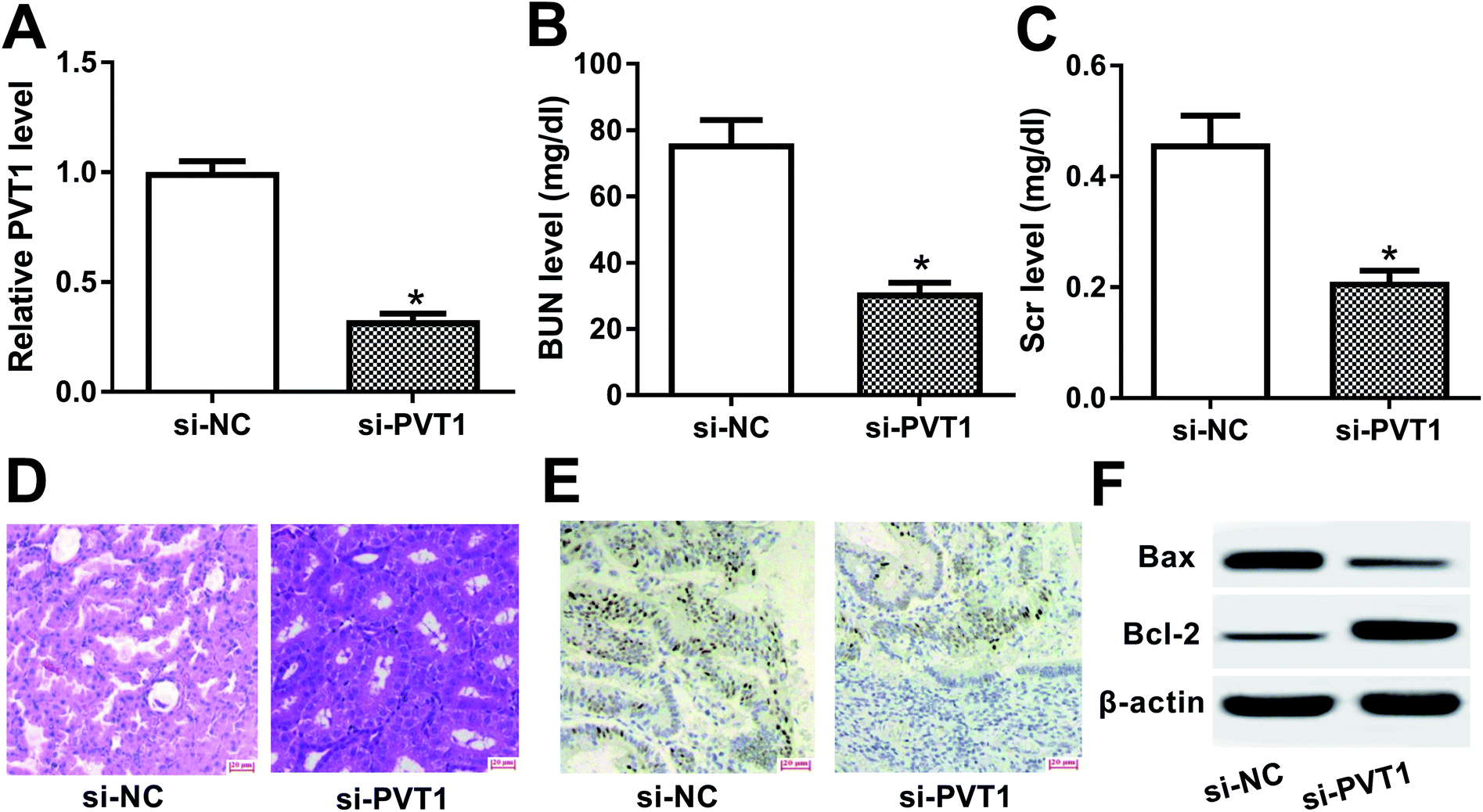 | ||
| Fig. 2 PVT1 knockdown ameliorated VAN-induced AKI in vivo. (A)–(E) Mice were injected with si-NC or si-PVT1 for 24 h and then intraperitoneally injected with VAN (600 mg kg−1) for 7 days. (A) PVT1 level in kidney tissues were measured by RT-qPCR assay. (B) and (C) BUN and Scr levels were determined by matching commercial kits. (D) Histopathological changes of kidney tissues were detected by H&E staining. (E) Cell apoptosis in kidney tissues was tested by TUNEL assay. (F) Western blot assay was conducted to detect protein levels of Bax and Bcl-2 in kidney tissues. *P < 0.05. | ||
PVT1 directly interacted with miR-124
Then, bioinformatical analysis by miRcode online website was performed to seek for miRNAs that had a chance to interact with lncRNA PVT1. Among candidate targets of PVT1, miR-124 was selected by virtue of its vital roles in the development of kidney diseases (Fig. 3A). To further validate this prediction, the effect of miR-124 on the luciferase activities of PVT1-WT or PVT1-MUT reporter was measured by luciferase reporter assay. Results showed that the introduction of miR-124 mimic resulted in a conspicuous reduction of luciferase activity of PVT1-WT reporter in 293T cells, but had no effect on luciferase activity of PVT1-MUT reporter (Fig. 3B). It is well known that Ago2 is a core component of RNA-induced silencing complex (RISC), which plays vital roles in miRNA-mediated gene silencing. Hence, RIP assay was performed to find whether PVT1 and miR-124 co-existed in RISC. Results showed that PVT1 and miR-124 were substantially enriched in Ago2 immunoprecipitation complexes, further hinting the potential interaction between PVT1 and miR-124 in space (Fig. 3C). RNA pull down assay further demonstrated that endogenous PVT1 was considerably enriched by biotin-labeled wide type miR-124, but not by biotin-labeled mutant type miR-124 in HK-2 cells (Fig. 3D). Also, as expected, biotin-labeled wide type PVT1 probe rather than mutant type PVT1 probe could pull down remarkable endogenous miR-124 in HK-2 cells (Fig. 3D). In summary, these results disclosed that PVT1 could directly interact with miR-124 by putative binding sites.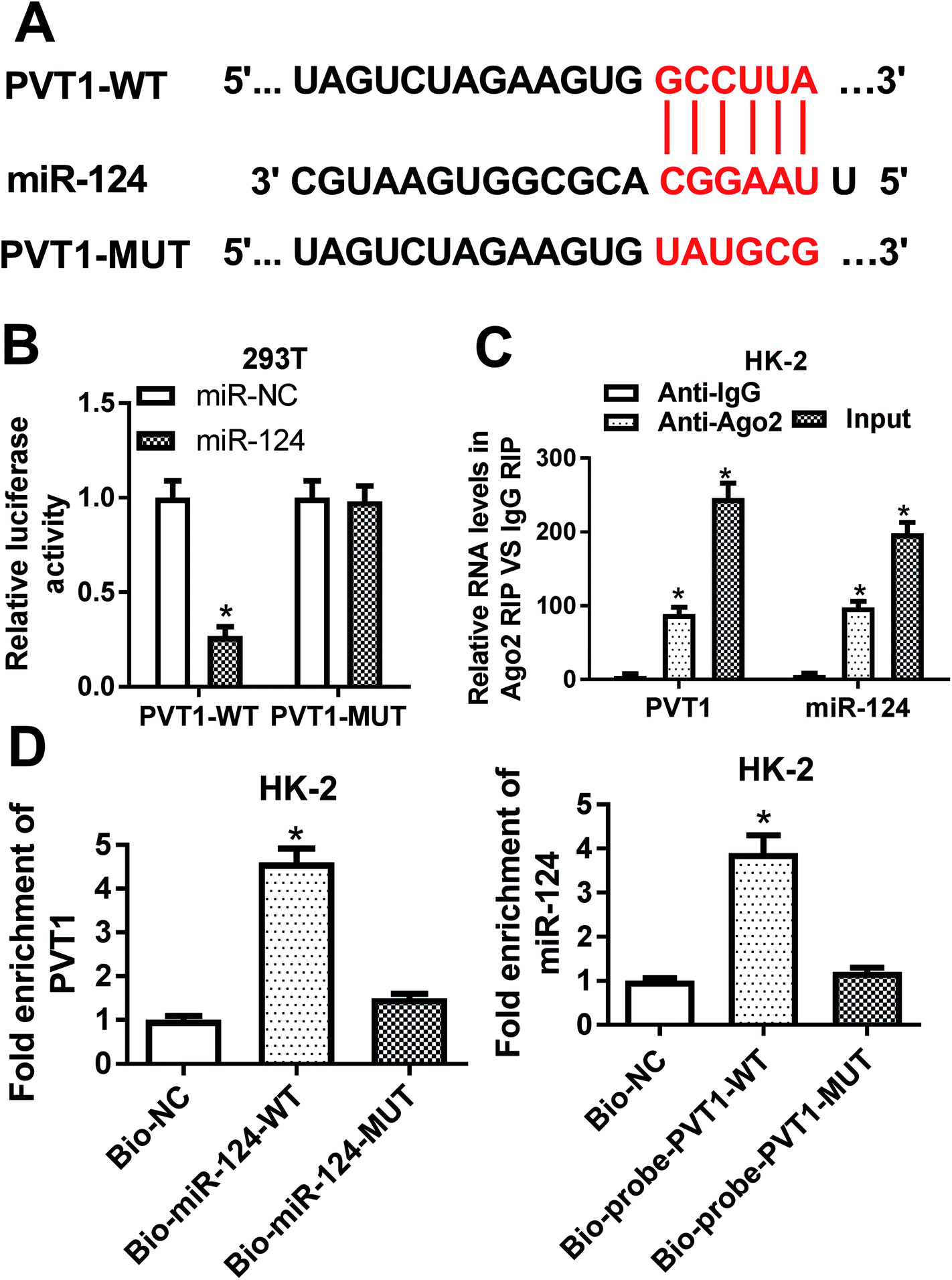 | ||
| Fig. 3 PVT1 directly interacted with miR-124. (A) Predicted binding sites between PVT1 and miR-124 by miRcode online website and mutant sites in PVT1-MUT reporter. (B) 293T cells were co-transfected with miR-NC or miR-124 and PVT1-WT reporter or PVT1-MUT reporter, followed by the determination of luciferase activities at 48 h after transfection. (C) RIP and RT-qPCR assays were carried out to measure the enrichment levels of PVT1 and miR-124 in IgG or Ago2 immunoprecipitation complex in HK-2 cells. (D) RNA pull down and RT-qPCR assays were performed to investigate whether miR-124 could interact with PVT1 by predicted binding sites in HK-2 cells. The experiments were repeated at least three times. *P < 0.05. | ||
The silencing of PVT1 abated VAN-induced HK-2 cell apoptosis in vitro
Then, expression pattern of PVT1 in VAN-treated HK-2 cells was further examined. As presented in Fig. 4A, PVT1 expression was markedly upregulated in VAN-treated HK-2 cells (Fig. 4A). And, the stimulation of VAN resulted in increased apoptosis index (Fig. 4B), reduced Bcl-2 (an anti-apoptosis protein) expression (Fig. 4C) and elevated Bax (a pro-apoptosis protein) expression (Fig. 4C) in HK-2 cells. In other words, VAN induced HK-2 cell apoptosis. Also, the transfection of si-PVT1 led to a notable reduction of PVT1 mRNA level in VAN-treated HK-2 cells (Fig. 4D). Moreover, the knockdown of PVT1 weakened VAN-induced apoptosis in HK-2 cells, as evidenced by the reduction of apoptosis index (Fig. 4E) and Bax expression (Fig. 4F) and the increase of Bcl-2 level (Fig. 4F) in VAN-stimulated cells following the transfection of si-PVT1.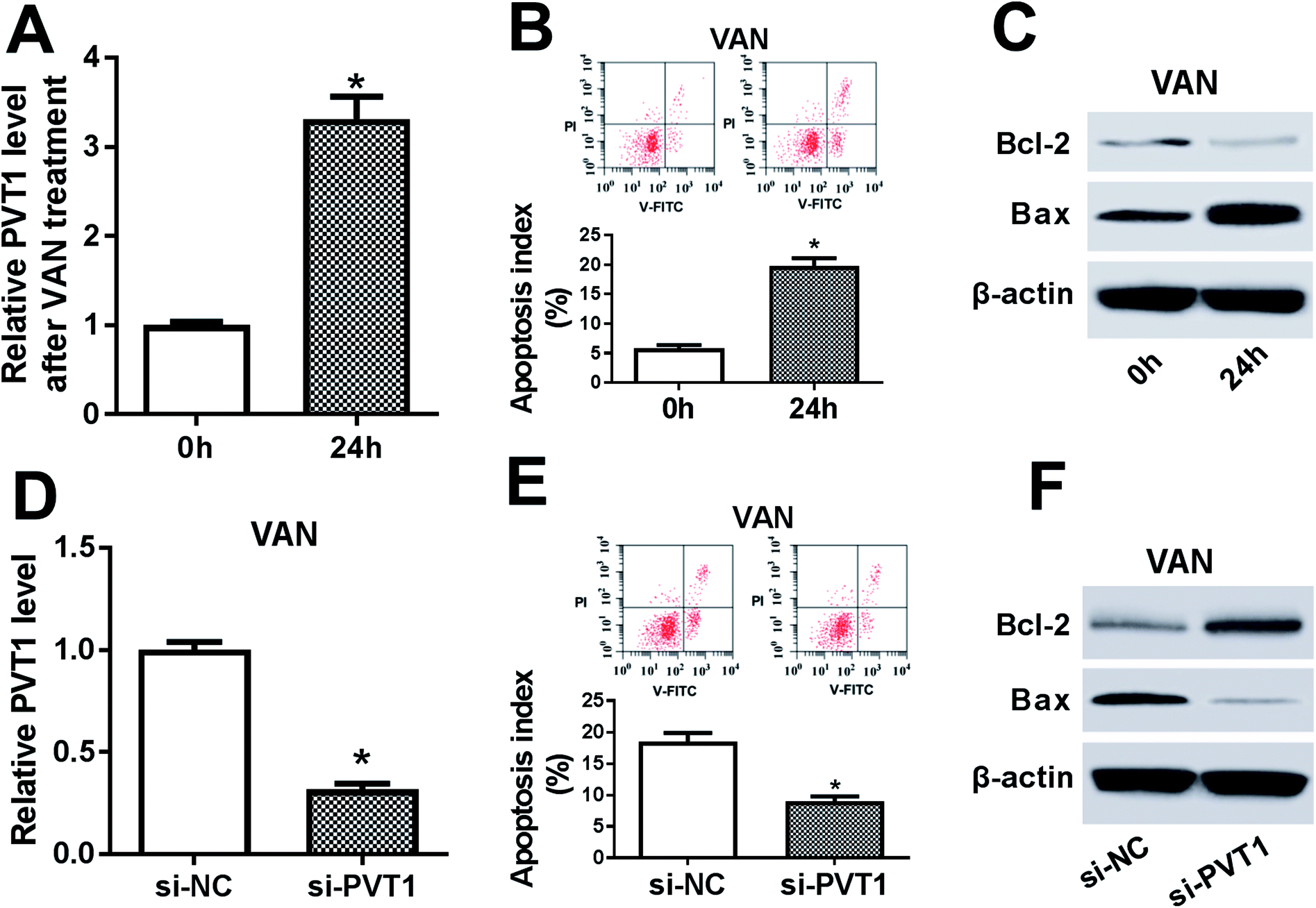 | ||
| Fig. 4 The silencing of PVT1 abated VAN-induced HK-2 cell apoptosis in vitro. (A)–(C) HK-2 cells were stimulated with 4 mM VAN for 24 h. (A) PVT1 level was measured by RT-qPCR assay. (B) Apoptosis index was determined by double-staining of Annexin V-FITC and PI using a flow cytometry. (C) Protein levels of Bcl-2 and Bax were detected by western blot assay. (D)–(F) HK-2 cells were transfected with si-NC or si-PVT1 for 24 h and then stimulated with 4 mM VAN for additional 24 h, followed by the determination of PVT1 (D), Bcl-2 (F) and Bax (F) levels, and apoptosis index (E). The experiments were repeated at least three times. *P < 0.05. | ||
The inhibition of miR-124 abrogated si-PVT1-mediated anti-apoptosis effect in VAN-treated HK-2 cells
Next, RT-qPCR assay revealed that miR-124 was lower in VAN-treated HK-2 cells compared with untreated cells (Fig. 5A). Also, the knockdown of PVT1 resulted in a notable upregulation of miR-124 level, whereas this effect was markedly weakened by miR-124 inhibitor (Fig. 5B). Additionally, the downregulation of miR-124 abolished the inhibitory effect of PVT1 knockdown on cell apoptosis in VAN-treated HK-2 cells, as presented by the increase of apoptosis index and Bax expression, and the reduction of Bcl-2 level in si-PVT1-transfected and VAN-treated HK-2 cells upon the introduction of miR-124 inhibitor (Fig. 5C and D). In a word, these data revealed that the inhibition of miR-124 abrogated si-PVT1-mediated anti-apoptosis effect in VAN-treated HK-2 cells.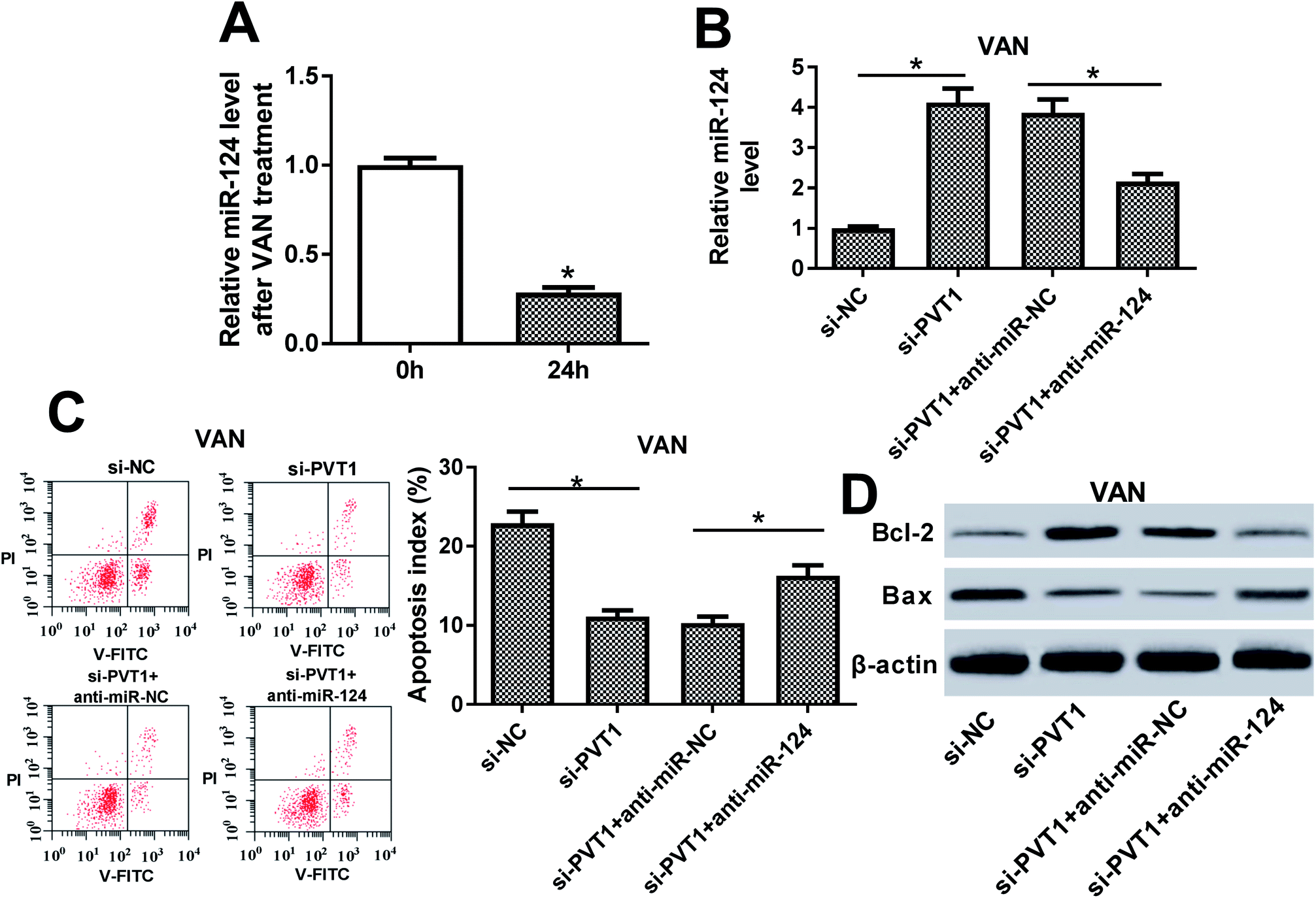 | ||
| Fig. 5 The inhibition of miR-124 abrogated si-PVT1-mediated anti-apoptosis effect in VAN-treated HK-2 cells. (A) HK-2 cells were stimulated with 4 mM VAN for 24 h, followed by the measurement of miR-124 level via RT-qPCR assay. (B)–(D) HK-2 cells were transfected with si-NC, si-PVT1, si-PVT1 + anti-miR-NC or si-PVT1 + anti-miR-124 for 24 h and then treated with 4 mM VAN for 24 h followed by the measurement of miR-124 (B), Bcl-2 (D) and Bax (D) levels, and apoptosis index (C). The experiments were repeated at least three times. *P < 0.05. | ||
The inhibition of NF-κB alleviated VAN-induced HK-2 cell apoptosis
We further demonstrated that VAN could induce a striking upregulation of p-p65 and p-IκBα levels in HK-2 cells (Fig. 6A), hinting that VAN could activate NF-κB signaling in HK-2 cells. Also, the inhibition of NF-κB by PDTC (a NF-κB inhibitor) notably weakened VAN-induced activation of NF-κB signaling in HK-2 cells (Fig. 6A). Moreover, the inactivation of NF-κB signaling by PDTC obviously suppressed VAN-induced HK-2 cell apoptosis, presented by reduced apoptosis index (Fig. 6B), increased Bcl-2 expression (Fig. 6C) and decreased Bax expression (Fig. 6C) in VAN-induced HK-2 cells upon the stimulation of PDTC. In summary, these data showed that the inhibition of NF-κB abated VAN-induced HK-2 cell apoptosis.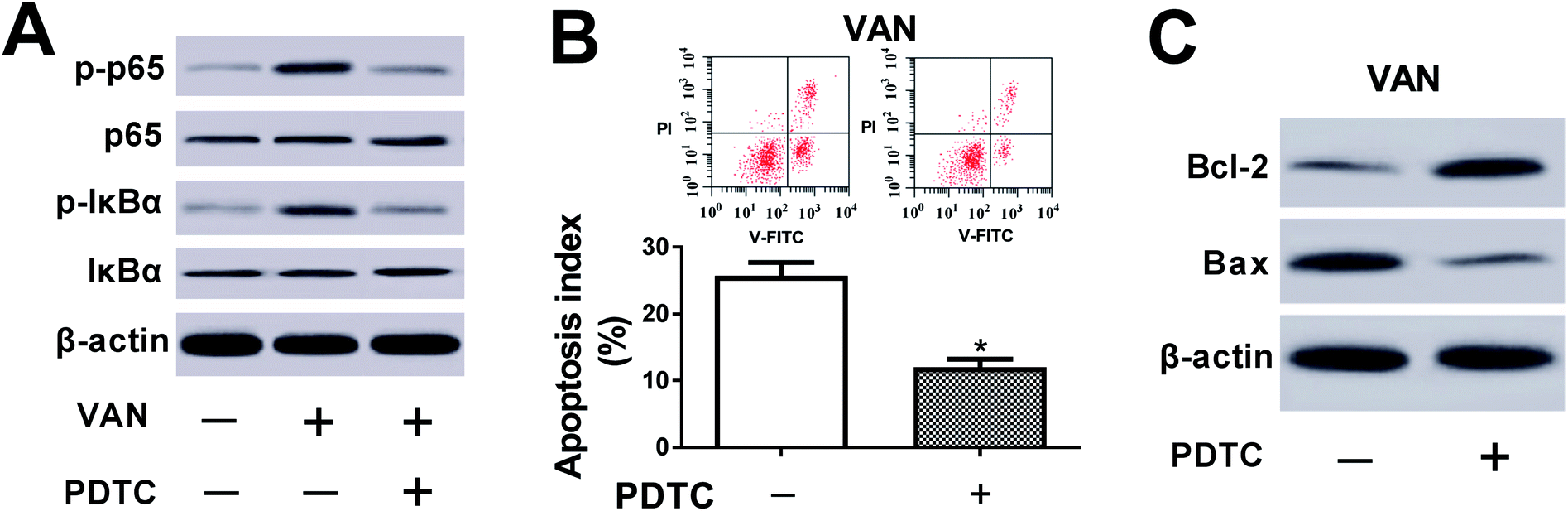 | ||
| Fig. 6 The inhibition of NF-κB alleviated VAN-induced HK-2 cell apoptosis. (A)–(C) HK-2 cells were treated with 4 mM VAN for 24 h or along with 50 μM PDTC for 1 h prior to VAN treatment. (A) and (C) Protein levels of p-p65, p65, p-IκBα, IκBα, Bcl-2 and Bax were measured by western blot assay. (B) Apoptosis index was determined using a flow cytometry. The experiments were repeated at least three times. *P < 0.05. | ||
PVT1 activated NF-κB signaling by targeting miR-124 in VAN-induced HK-2 cells
Next, western blot assay revealed that p-p65 and p-IκBα levels were remarkably increased in VAN-induced HK-2 cells upon the overexpression of PVT1 (Fig. 7A). That was to say, PVT1 activated NF-κB signaling in VAN-induced HK-2 cells. Additionally, miR-124 restoration strikingly weakened PVT1-induced NF-κB activation in VAN-induced HK-2 cells (Fig. 7A). Conversely, PVT knockdown resulted in a notable downregulation of p-p65 and p-IκBα levels in VAN-induced HK-2 cells, while this effect was reversed by miR-124 inhibitor (Fig. 7B). Taken together, these results showed that PVT1 activated NF-κB signaling by targeting miR-124 in VAN-induced HK-2 cells. Also, the schematic diagram of our hypothesis is presented in Fig. 8. In brief, VAN stimulation induced the upregulation of PVT1, which resulted in the sequential downregulation of miR-124 level. The reduction of miR-124 level promoted kidney cell apoptosis by activating NF-κB pathway.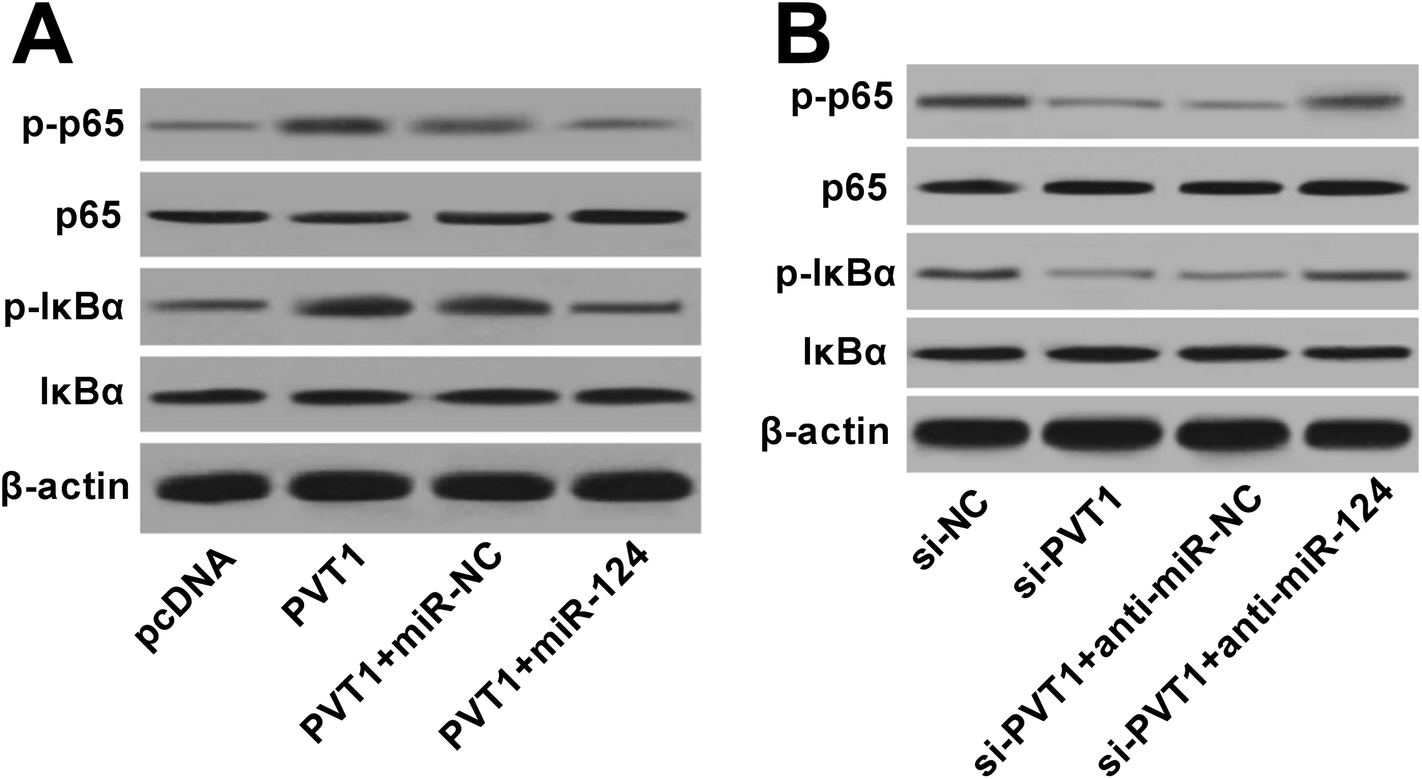 | ||
| Fig. 7 PVT1 activated NF-κB signaling by targeting miR-124 in VAN-induced HK-2 cells. (A) and (B) HK-2 cells were transfected with pcDNA, PVT1, PVT1 + miR-NC, PVT1 + miR-124, si-NC, si-PVT1, si-PVT1 + anti-miR-NC, or si-PVT1 + anti-miR-124 for 24 h and then treated with 4 mM VAN for 24 h. Then, protein levels of p-p65, p65, p-IκBα and IκBα were determined by western blot assay. The experiments were repeated at least three times.*P < 0.05. | ||
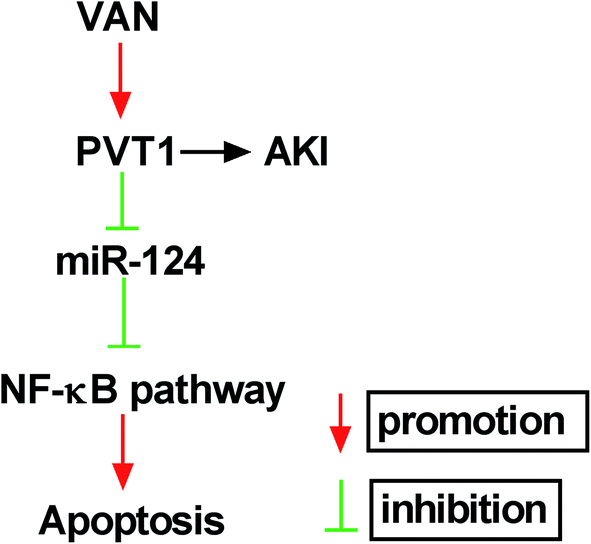 | ||
| Fig. 8 The schematic diagram of PVT1 knockdown alleviates vancomycin-induced acute kidney injury by targeting miR-124 via inactivating NF-κB signaling. | ||
Discussion
Abovementioned evidence manifests that non-coding RNAs including lncRNAs and microRNAs are closely associated with development and progression of AKI.40 Prior studies also showed that PVT1 contributed to the development and progression of diabetic nephropathy by inducing extracellular matrix accumulation in the kidney.26,27 Additionally, Huang et al. pointed out that the downregulation of PVT1 relieved LPS-induced AKI by reducing expressions of p-JNK, p-c-Jun, p-IκBα and p-p65 in HK-2 cells.30 Our study further demonstrated that PVT1 expression was markedly upregulated in VAN-induced AKI models. Also, PVT1 knockdown alleviated VAN-induced AKI in vivo, as evidenced by subtle serum BUN and Scr levels, slight histopathological changes and mild kidney cell apoptosis in VAN-treated mice following the depletion of PVT1. Additionally, PVT1 silence inhibited VAN-induced apoptosis in HK-2 cells.Moreover, miRNAs have been reported to be implicated in the development and the pathogenesis of renal disorders such as AKI, lupus nephritis and diabetic nephropathy.41,42 For instance, the inhibition of miR-301a-5p impeded VAN-induced kidney cell apoptosis in vitro and ameliorated VAN-induced AKI in vivo.43 In the present study, we demonstrated that PVT1 could directly interact with miR-124. MiR-124 has been demonstrated to be downregulated in unilateral ureteral occlusion (UUO) rats and TGF-β1-induced HK-2 cells, and the inhibition of miR-124 aggravated EMT and renal interstitial fibrosis.44 Also, miR-124 overexpression resulted in the alleviation of candidiasis-induced AKI and the reduction of kidney lesion and excessive inflammatory responses in septic mice.45 Our study further demonstrated that the inhibition of miR-124 could reverse si-PVT1-mediated anti-apoptosis effect in VAN-treated HK-2 cells, hinting that PVT1 knockdown suppressed VAN-induced AKI by targeting miR-124 in vitro.
As mentioned above, a previous finding indicated that PVT1 aggravated LPS-induced AKI by the activation of NF-κB signaling in HK-2 cells.30 NF-κB signaling was activated in AKI models and the inhibition of NF-κB signaling ameliorated AKI.46–48 Our study showed that VAN induced the activation of NF-κB signaling and the inhibition of NF-κB by PDTC lessened VAN-induced apoptosis in HK-2 cells. Moreover, PVT1 further activated NF-κB signaling in VAN-treated HK-2 cells, while this effect was weakened by increased miR-124.
Collectively, our data showed that PVT1 knockdown weakened VAN-induced AKI by targeting miR-124 via inactivating NF-κB signaling, deepening our understanding on etiology of VAN-induced AKI and providing some potential targets and strategies for the alleviation of VAN nephrotoxicity.
Author's contributions
This work was designed and conceived by Xiaoguang Zhu and Jun Shi. The experiment procedures and data analysis were carried out by Huicong li, Fang Chen, Jun Shi and Xiaoguang Zhu. The manuscript was prepared by Xiaoguang Zhu and Jun Shi.Conflicts of interest
There is no conflict of interest regarding the publication of this paper.Acknowledgements
None.References
- K. Singbartl and J. A. Kellum, Kidney Int., 2012, 81, 819–825 CrossRef PubMed.
- O. Rewa and S. M. Bagshaw, Nat. Rev. Nephrol., 2014, 10, 193–207 CrossRef PubMed.
- R. L. Mehta, J. Cerda, E. A. Burdmann, M. Tonelli, G. Garcia-Garcia, V. Jha, P. Susantitaphong, M. Rocco, R. Vanholder, M. S. Sever, D. Cruz, B. Jaber, N. H. Lameire, R. Lombardi, A. Lewington, J. Feehally, F. Finkelstein, N. Levin, N. Pannu, B. Thomas, E. Aronoff-Spencer and G. Remuzzi, Lancet, 2015, 385, 2616–2643 CrossRef.
- A. J. Lewington, J. Cerdá and R. L. Mehta, Kidney Int., 2013, 84, 457–467 CrossRef PubMed.
- L. S. Chawla, P. W. Eggers, R. A. Star and P. L. Kimmel, N. Engl. J. Med., 2014, 371, 58–66 CrossRef PubMed.
- S. A. Silver, J. Long, Y. Zheng and G. M. Chertow, J. Hosp. Med., 2017, 12, 70 CrossRef PubMed.
- S. G. Coca, S. Singanamala and C. R. Parikh, Kidney Int., 2012, 81, 442–448 CrossRef PubMed.
- J. Vanmassenhove, J. Kielstein, A. Jorres and W. V. Biesen, Lancet, 2017, 389, 2139–2151 CrossRef.
- A. S. Levey and M. T. James, Ann. Intern. Med., 2017, 167, Itc66–Itc80 CrossRef PubMed.
- R. S. Griffith, J. Antimicrob. Chemother., 1984, 14(Suppl. D), 1–5 CrossRef PubMed.
- K. A. Mergenhagen and A. R. Borton, J. Pharm. Pract., 2014, 27, 545–553 CrossRef PubMed.
- O. Bamgbola, Ther. Adv. Endocrinol. Metab., 2016, 7, 136–147 CrossRef PubMed.
- E. A. Sinclair, G. Yenokyan, A. McMunn, J. J. Fadrowski, A. M. Milstone and C. K. Lee, Ann. Pharmacother., 2014, 48, 1555–1562 CrossRef PubMed.
- C. A. Knoderer, K. R. Nichols, K. C. Lyon, M. M. Veverka and A. C. Wilson, J. Pediatric Infect. Dis. Soc., 2014, 3, 127–131 CrossRef PubMed.
- Y. Arimura, T. Yano, M. Hirano, Y. Sakamoto, N. Egashira and R. Oishi, Free Radicals Biol. Med., 2012, 52, 1865–1873 CrossRef PubMed.
- Y. Sakamoto, T. Yano, Y. Hanada, A. Takeshita, F. Inagaki, S. Masuda, N. Matsunaga, S. Koyanagi and S. Ohdo, Eur. J. Pharmacol., 2017, 800, 48–56 CrossRef PubMed.
- G. Hu, F. Niu, B. A. Humburg, K. Liao, S. Bendi, S. Callen, H. S. Fox and S. Buch, Oncotarget, 2018, 9, 18648–18663 Search PubMed.
- J. M. Lorenzen and T. Thum, Nat. Rev. Nephrol., 2016, 12, 360 CrossRef PubMed.
- J. Zhou, H. Chen and Y. Fan, Oncotarget, 2017, 8, 100196 Search PubMed.
- J. M. Lorenzen, C. Schauerte, J. T. Kielstein, A. Hübner, F. Martino, J. Fiedler, S. K. Gupta, R. Faulhaberwalter, R. Kumarswamy and C. Hafer, Clin. Chem., 2015, 61, 191–201 CrossRef PubMed.
- Y. Chen, J. Qiu, B. Chen, Y. Lin, Y. Chen, G. Xie, J. Qiu, H. Tong and D. Jiang, Int. Immunopharmacol., 2018, 59, 252–260 CrossRef PubMed.
- M. Cui, L. You, X. Ren, W. Zhao, Q. Liao and Y. Zhao, Biochem. Biophys. Res. Commun., 2016, 471, 10–14 CrossRef PubMed.
- X. Pan, G. Zheng and C. Gao, Clin. Lab., 2018, 655–662 Search PubMed.
- R. L. Hanson, D. W. Craig, M. P. Millis, K. A. Yeatts, S. Kobes, J. V. Pearson, A. M. Lee, W. C. Knowler, R. G. Nelson and J. K. Wolford, Diabetes, 2007, 56, 975–983 CrossRef PubMed.
- M. P. Millis, D. Bowen, C. Kingsley, R. M. Watanabe and J. K. Wolford, Diabetes, 2007, 56, 3027–3032 CrossRef PubMed.
- M. L. Alvarez and J. K. DiStefano, PLoS One, 2011, 6, e18671 CrossRef PubMed.
- M. L. Alvarez, M. Khosroheidari, E. Eddy and J. Kiefer, PLoS One, 2013, 8, e77468 CrossRef PubMed.
- Q. Wu, F. Yang, Z. Yang, Z. Fang, W. Fu, W. Chen, X. Liu, J. Zhao, Q. Wang and X. Hu, Oncotarget, 2017, 8, 101865–101875 Search PubMed.
- T. Yang, H. Zhou, P. Liu, L. Yan, W. Yao, K. Chen, J. Zeng, H. Li, J. Hu and H. Xu, Oncotarget, 2017, 8, 85353–85367 Search PubMed.
- W. Huang, X. Lan, X. Li, D. Wang, Y. Sun, Q. Wang, H. Gao and K. Yu, Int. Immunopharmacol., 2017, 47, 134–140 CrossRef PubMed.
- M. D. Paraskevopoulou and A. G. Hatzigeorgiou, Methods Mol. Biol., 2016, 1402, 271–286 CrossRef PubMed.
- J. H. Yoon, K. Abdelmohsen and M. Gorospe, Semin. Cell Dev. Biol., 2014, 34, 9–14 CrossRef PubMed.
- Y. Cai, X. Yu, S. Hu and J. Yu, Genomics, Proteomics Bioinf., 2009, 7, 147–154 CrossRef.
- S. Geng, X. Zhang, J. Chen, X. Liu, H. Zhang, X. Xu, Y. Ma, B. Li, Y. Zhang and Z. Bi, PLoS One, 2014, 9, e91566 CrossRef PubMed.
- X. Wang, Q. Wu, B. Xu, P. Wang, W. Fan, Y. Cai, X. Gu and F. Meng, FEBS J., 2016, 282, 4376–4388 CrossRef PubMed.
- Q. Q. Cai, Y. W. Dong, R. Wang, B. Qi, J. X. Guo, J. Pan, Y. Y. Liu, C. Y. Zhang and X. Z. Wu, Sci. Rep., 2017, 7, 40733 CrossRef PubMed.
- H. Butz, P. M. Szabó, H. W. Z. Khella, R. Nofechmozes, A. Patocs and G. M. Yousef, Oncotarget, 2015, 6, 12543–12557 CrossRef PubMed.
- S. Zell, R. Schmitt, S. Witting, H. H. Kreipe, K. Hussein and J. U. Becker, Nephron Extra, 2013, 3, 50–58 CrossRef PubMed.
- W. Liu, C. Ma, B. Yang, C. Yin, B. Zhang and Y. Xiao, Biochem. Biophys. Res. Commun., 2017, 493, 1168–1175 CrossRef PubMed.
- P. Zhou, Z. Chen, Y. Zou and X. Wan, Kidney Blood Pressure Res., 2016, 41, 757–769 CrossRef PubMed.
- K. Bhatt, M. Kato and R. Natarajan, Am. J. Physiol. Renal Physiol., 2016, 310, F109–F118 CrossRef PubMed.
- Q. Wei, Q. S. Mi and Z. Dong, IUBMB Life, 2013, 65, 602–614 CrossRef PubMed.
- J. Wang, H. Li, S. Qiu, Z. Dong, X. Xiang and D. Zhang, Cell Death Dis., 2017, 8, e3120 CrossRef PubMed.
- H. Zhou, L. Gao, Z. H. Yu, S. J. Hong, Z. W. Zhang and Z. Z. Qiu, Nephrology, 2018 DOI:10.1111/nep.13394.
- X. Y. Li, Y. Q. Zhang, G. Xu, S. H. Li and H. Li, Int. J. Mol. Med., 2018, 41, 3468–3476 Search PubMed.
- D. Kumar, S. K. Singla, V. Puri and S. Puri, PLoS One, 2015, 10, e115947 CrossRef PubMed.
- R. Yan, Y. Li, L. Zhang, N. Xia, Q. Liu, H. Sun and H. Guo, Int. Urol. Nephrol., 2015, 47, 861 CrossRef PubMed.
- C. C. Cao, X. Q. Ding, Z. L. Ou, C. F. Liu, P. Li, L. Wang and C. F. Zhu, Kidney Int., 2004, 65, 834–845 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |