
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA green metal–organic framework to monitor water contaminants†
Priscilla Rocío-Bautistaa,
Verónica Pino *a,
Juan H. Ayalaa,
Catalina Ruiz-Pérezb,
Oriol Vallcorbac,
Ana M. Afonsoa and
Jorge Pasán*b
*a,
Juan H. Ayalaa,
Catalina Ruiz-Pérezb,
Oriol Vallcorbac,
Ana M. Afonsoa and
Jorge Pasán*b
aDepartment of Chemistry, Analytical Chemistry Division, Universidad de La Laguna, Apartado 456, 38200, La Laguna, Spain. E-mail: veropino@ull.edu.es
bLaboratorio de Rayos X y Materiales Moleculares (MATMOL), Department of Physics, Universidad de La Laguna, Apartado 456, 38200, La Laguna, Spain. E-mail: jpasang@ull.edu.es
cALBA Synchrotron Light Source, Cerdanyola del Vallés, Barcelona, 08920, Spain
First published on 5th September 2018
Abstract
The CIM-80 material (aluminum(III)-mesaconate) has been synthetized in high yield through a novel green procedure involving water and urea as co-reactants. The CIM-80 material exhibits good thermal stability with a working range from RT to 350 °C with a small contraction upon desolvation. Moreover, this material is stable in water at different pH values (1–10) for at least one week, and shows a LC50 value higher than 2 mg mL−1. The new material has been tested in a microextraction methodology for the monitoring of up to 22 water pollutants while presenting little environmental impact: only 20 mg of CIM-80 and 500 μL of acetonitrile are needed per analysis. The analytical performance of the CIM-80 in the microextraction strategy is similar to or even better for several pollutants than that of MIL-53(Al). The average extraction efficiencies range from ∼20% for heavy polycyclic aromatic hydrocarbons to ∼70–100% for the lighter ones. In the case of the emerging contaminants, the average extraction efficiency can reach values up to 70% for triclosan and carbamazepine.
1. Introduction
The monitoring of water quality undoubtedly provides empirical evidence to support decisions regarding health protection versus environmental issues.1,2 The frequent detection of pharmaceuticals, drugs, endocrine disrupting phenols, personal care products, and other contaminants, has become a global problem due to their potential to cause undesirable ecological and human health effects.3 Conventional environmental monitoring strategies are, paradoxically, also of environmental concern because they require the use of large amounts of halogenated organic solvents in the environmental sample preparation, thus generating enormous amounts of toxic waste.4 Alternatives have arisen to replace conventional approaches,5 mainly by utilizing microextraction techniques (that minimize and even eliminate the requirements of such solvents in the extraction step of the monitoring method),6 or by incorporating novel materials as successful extraction agents able to replace organic solvents or conventional sorbents.7,8Metal–organic frameworks (MOFs) clearly merit citation among novel sorbent materials in monitoring methods,9–11 particularly in microextraction methods such as solid-phase microextraction (SPME)12 or in miniaturized solid phase extraction in its dispersive mode (D-μSPE).13,14 D-μSPE is gaining a lot of attention nowadays from an environmental-friendly point of view, because its requirements of sorbents are really low (usually between 2–500 mg) and due to its simplicity. The method only requires proper dispersion of the sorbent in the aqueous sample (stirring, vortex…) to ensure the trapping of the contaminants by the sorbent material,15 followed by separation of the sorbent and further elution of the trapped analytes. Analytes are then ready to determination with the proper analytical technique. This way, D-μSPE can be utilized easily in laboratories worldwide without the need of expensive instrumentation to accomplish monitoring.
A number of properties of MOFs makes these materials almost ideal candidates for D-μSPE: good thermal and mechanical stability, uniform structured nanoscale cavities, uniform pore topologies, high adsorption affinity, and structural tunability. The possibility of performing in pore functionalization and outer-surface modifications have made MOFs attractive as highly versatile materials with task-specific properties.16 Recent studies have tried to correlate the structure of MOFs with that of pollutants intended to be monitored, with the goal of targeting MOFs for selective monitoring.9,17,18 Such studies pointed out the difficulties in predicting the adsorption of a target analyte (for analytes initially present in a water media) into a specific MOF material. In any case, the MOF's flexibility, the presence of unsaturated metal sites, large pore volumes, and a hydrophobic environment around the pores seem to be adequate characteristics when intending a MOF as generic sorbent for environmental monitoring.19
A step forward in the utilization of MOFs for water monitoring clearly requires the utilization of: (a) MOFs with high water stability and (b) greener MOFs in such microextraction methods. That is, the use of MOFs prepared following green chemistry routes: milder synthetic procedures and absence of toxic organic solvents.20 By incorporating MOFs prepared with greener strategies into D-μSPE monitoring methods, it is possible to avoid the paradoxical situation currently existing with environmental monitoring methods.
The MOF MIL-53(Al) has shown wide extraction capabilities for different contaminants, and thus it can be considered as a generic sorbent in D-μSPE.9,17 Green synthetic procedures have been developed to prepare MIL-53(Al), but they still need high temperatures and complex procedures to activate the material.21 To expand its extraction performance while improving its synthesis in terms of greenness, we have decided to use the mesaconate ligand, which includes a methyl functionalization in the fumaric acid. The aluminum(III)-mesaconate MOF (CIM-80) is a porous material with a crystal structure similar to that of MIL-68, good thermal and water stability and a synthetic procedure completely green without employing any organic solvent and short reaction times. At the time preparing this manuscript, this material has been reported with a similar green synthetic approach.20
To evaluate the performance of this greener CIM-80 as novel sorbent in D-μSPE to monitor waters, 22 different pollutants are selected for being representative analytes of different families. The D-μSPE method is completely optimized and validated with CIM-80; while comparing its analytical performance to that of MIL-53(Al) and other materials.
2. Experimental
2.1 Chemicals, reagents and materials
Aluminum nitrate nonahydrate (98%), mesaconic acid (99%), trimethylamine (>99%), urea (99%), and Na2CO3 (99%), were purchased from Sigma-Aldrich (Steinheim, Germany) and used for synthetizing the CIM-80. Dimethylformamide (99.9%) of high-performance liquid chromatography (HPLC) grade was acquired to Fluka-Sigma Aldrich (Steinheim, Germany). The following reagents were used to prepare the buffer: KCl, acquired to Panreac (Barcelona, Spain); and sodium acetate, acetic acid, and KH2PO4, with pro-analysis purity, acquired to Merck (Darmstadt, Germany).Carbamazepine (99.0%), methylparaben (95.5%), and atrazine (99.1%), were purchased to Sigma-Aldrich; progesterone (>99.99%) and triclosan (>99.99%) were obtained from US Pharmacopoeia Reference Standard (Basel, Switzerland); estrone (>99.99%) was purchased to European Pharmacopoeia Reference Standard (Strasburg, France); and benzophenone (99%) was supplied by Alfa Aesar (Karlsruhe, Germany). All these compounds were acquired as solid products. They were used to prepare individual standard solutions in acetonitrile (ACN). A standard solution with polycyclic aromatic hydrocarbon (PAHs) in ACN was supplied by Dr Ehrenstorfer GmbH (Augsburg, Germany), at a concentration of 10 mg L−1. The main characteristics of the studied contaminants are shown in Table S1 of the ESI.† Working standard solutions of all contaminants were prepared in ultrapure water.
Deionized water (Milli-Q, ultrapure grade) was obtained through a water purification system A10 MiliPore® (Watford, UK). ACN HiPerSolv Chromanorm liquid chromatography grade was purchased from VWR (Llinars del Vallés, Spain). Ultrapure water and ACN were used as mobile phases in ultra-high performance liquid chromatography (UHPLC). ACN was also required in the elution steps of D-μSPE. Methanol (MeOH) and isopropanol (>99%), supplied by Sigma-Aldrich, were used also as elution solvents. UHPLC mobile phases were always filtered with Durapore® membrane filters of 0.45 μm, supplied by Sigma-Aldrich.
Syringe filters Millex® Durapore® of polyvinylidene fluoride (PVDF) of 0.2 μm were purchased from Sartorius Stedim Biotech (Goettingen, Germany). They were used to filtrate all samples and standards before any UHPLC analysis.
The D-μSPE procedure required Pyrex® centrifuge tubes (Staffordshire, UK) with dimensions of 10 × 1.4 cm and volume of 15 mL. Solvothermal reactors of Teflon and stain steel autoclaves supplied by Parr Instrument Company (Moline, IL, USA) were used in the synthesis of the MOFs.
2.2 Instruments and equipment
A vortex from Reax-Control Heidolph GMBH (Schwabach, Germany), and an ultrasounds bath KM (Shenzhen Codyson Electrical Co., Ltd. Shenzhen, China) were utilized in the D-μSPE procedure.Phase identification of the synthetized CIM-80 was carried out by X-ray powder diffraction on a X'Pert Diffractometer (Panalytical, Netherlands) operating with Bragg–Brentano geometry. Data collection was carried out using Cu-Kα radiation (λ = 1.5418 Å) over the angular range from 5.01° to 80.00° with a total exposure time of 30 min. High resolution powder diffraction patterns were measured at the BL04-MSPD beamline22 of ALBA synchrotron (Barcelona, Spain), at 17.5 keV (λ = 0.70815 Å) equipped with the Mythen-II detector (Dectris) in the 0.37–43.2° angular range.
The microscopic morphology of the material was examined by a JSM6300 scanning electron microscope (SEM) from JEOL (Tokyo, Japan); supporting the material on a flat surface and silver cover. The surface area and pore volume of adsorbents were measured on a Gemini V 2365 Model (Micromeritics, Norcross, GA, USA) surface area analyzer at 77 K in the range 0.02 ≤ P/P0 ≤ 1.00. The Brunauer, Emmett and Teller (BET) method was used to calculate the surface area. Thermogravimetric analysis (TG/TDA) was carried out in a Perkin Elmer Pyris Diamond TGA/DTA equipment.
The UHPLC was the 1260 Infinity model from Agilent Technologies (Santa Clara, USA). Its quaternary pumps resist 600 bar. The instrument was equipped with a Rheodyne 7725i injection valve and an injection loop of 5 μL. The UV-Vis detector was a ProStar 325 LC Varian (Palo Alto, CA, USA), operating at 220 nm for the emerging pollutants studied. A multichannel fluorescence detector (FD) 1260 Infinity model from Agilent Technologies was also used. This detector operates with the program included in Table S2 of the ESI† for the group of PAHs studied. The UHPLC and the FD were controlled with the C.01.04 version of the OpenLab CDS ChemStation software (Agilent), whereas the UV-Vis data treatment was carried out with the 6.41 version LC Workstation software (Varian).
The separation of the PAHs was carried out using a Zorbax Eclipse PAHs column (1.8 μm, 50 × 4.6 mm) also from Agilent Technologies. The rest of target analytes were separated with a C18 Kinetex column (1.7 μm, 100 × 2.1 mm) supplied by Phenomenex (Torrance, CA, USA). The column thermostat was kept at 25 °C in both cases. Fig. S1 and S2 of the ESI† include a representative chromatogram obtained under optimum conditions with a standard solution, together with the specific group of conditions required for each separation.
2.3 Synthesis of [Al(mesaconate)OH]·3H2O CIM-80
The synthesis of CIM-80 was optimized and the best procedure requires a mixture of mesaconic acid (1 mmol; 130 mg) and Al(NO3)3·9H2O (1 mmol; 375 mg) in 15 mL in deionized water containing 0.5 mmol of urea (30 mg), under constant stirring for 20 min. Then, the clear solution is transferred to a 23 mL Teflon lined stainless steel autoclave and kept at 150 °C for 3 h. Afterwards, the autoclave is cooled down to room temperature, and the obtained white product is isolated by filtration, washed with water, and air dried at 50 °C. Yield: 60% (based on Al). IR (cm−1): 3400 (broad), 1587s, 1408s, 1375m, 1305w, 993m, 916w, 812m, 658s, 619m, 481s. Elemental Analysis for C5H12O8Al (227.13 g mol−1): calcd: 26.4%, H 5.3%; found: C 26.8%, H 5.7%.2.4 D-μSPE-UHPLC procedure with CIM-80 as extraction sorbent of contaminants
The D-μSPE method using CIM-80, followed by UHPLC-UV-Vis or UHPLC-FD depending on the group of contaminants, was optimized properly. Under optimum conditions, 10 mL of water are put in contact with 20 mg of CIM-80 (previously activated at 150 °C overnight) in Pyrex® centrifuge tubes. The mixture is then subjected to vortex stirring during 3 min, followed by centrifugation during 5 min at 2739 × g. The supernatant aqueous phase is then carefully removed by decantation. Afterwards, 500 μL of elution solvent are added to the MOF remaining in the tube (which contains the trapped contaminants). Elution solvents tested were MeOH, ACN and isopropanol. Vortex is then applied for 3 min, followed again by centrifugation during 5 min at 2739 × g. The supernatant (eluate containing the contaminants initially trapped by the MOF) is sampled using a Pasteur pipette, filtered through 0.2 μm PVDF syringe filters, and directly injected in UHPLC-UV or UHPLC-FD.3. Results and discussion
3.1 Synthesis, structure and stability of CIM-80
The synthesis of [Al(mesaconate)OH] in water has been recently reported to require microwave heating at 90 °C of a mixture in water of aluminum(III) nitrate, mesaconic acid and sodium hydroxide.20 We have developed an alternative route using urea as base and conventional (and simple) heating, in an attempt to obtain single crystals to determine the crystal structure. Different reaction times (from 1.5 h to 32 h) were tested at a given temperature of 150 °C (the higher reaction temperature is needed to ensure the decomposition of urea gradually, thus releasing NH3 in the medium). At 1.5 h, there is no product in the reactor, but from 3 h to 32 h the CIM-80 is the product formed (Fig. S3†), and only an increment of 15% yield is achieved at the highest reaction time, but unfortunately no single crystals were obtained. Fig. S4† depicts a detailed view of the CIM-80 crystallites with maximum size of ca. 10 μm, but these ‘single’ crystals seem to be formed of various twin components, precluding their use in SCXRD. In order to keep the synthesis as green as possible, we established 3 h of reaction time as the optimum.The crystal structure of CIM-80 consists of a kagome-like MIL-68 framework with two different type of channels: large hexagonal and small triangular pores of 6 and 2 Å diameters, respectively, with the methyl groups of the mesaconic acid directed towards the large pores.20 After activation at 150 °C, and reduced pressure overnight, the N2 adsorption isotherm at 77 K leads to a BET surface area for this material of 891 m2 g−1. This value is close to the reported value of 1040 m2 g−1. The volume of micropore is 0.46 cm3 g−1, which is also in agreement with the value of 0.47 cm3 g−1 obtained from the poreblazer software.23
The thermal stability of the material has been studied through TG/DTA analysis and temperature dependent X-ray diffraction. The TG shows a 25% weight loss below 100 °C that has been assigned to three crystallization water molecules. After that, the material does not show any process up to 400 °C, when the decomposition starts (see Fig. S5 of the ESI†). The crystallinity of CIM-80 was studied as a function of temperature (see Fig. 1 and S6†), observing well defined peaks in all the powder diffraction patterns up to 200 °C (the maximum temperature reached in the experiment), although some variations in the peak intensities were found. The evolution of the cell volume with the temperature was studied from the fitting of the powder patterns at different temperatures (see Fig. 2 and S7 of the ESI†). It is observed a reduction in the cell volume up to 100 °C, indicating that the release of the crystallization water molecules produces a small contraction of the pores (0.4% of the cell volume). Above 100 °C, the cell volume increases as expected, and when the sample is cooled down again to room temperature, the cell volume is smaller than the hydrated one since the water molecules are not recovered in the process. The thermal expansion parameter calculated is 13.1 MK−1, which is within the range of other metal–organic materials.24
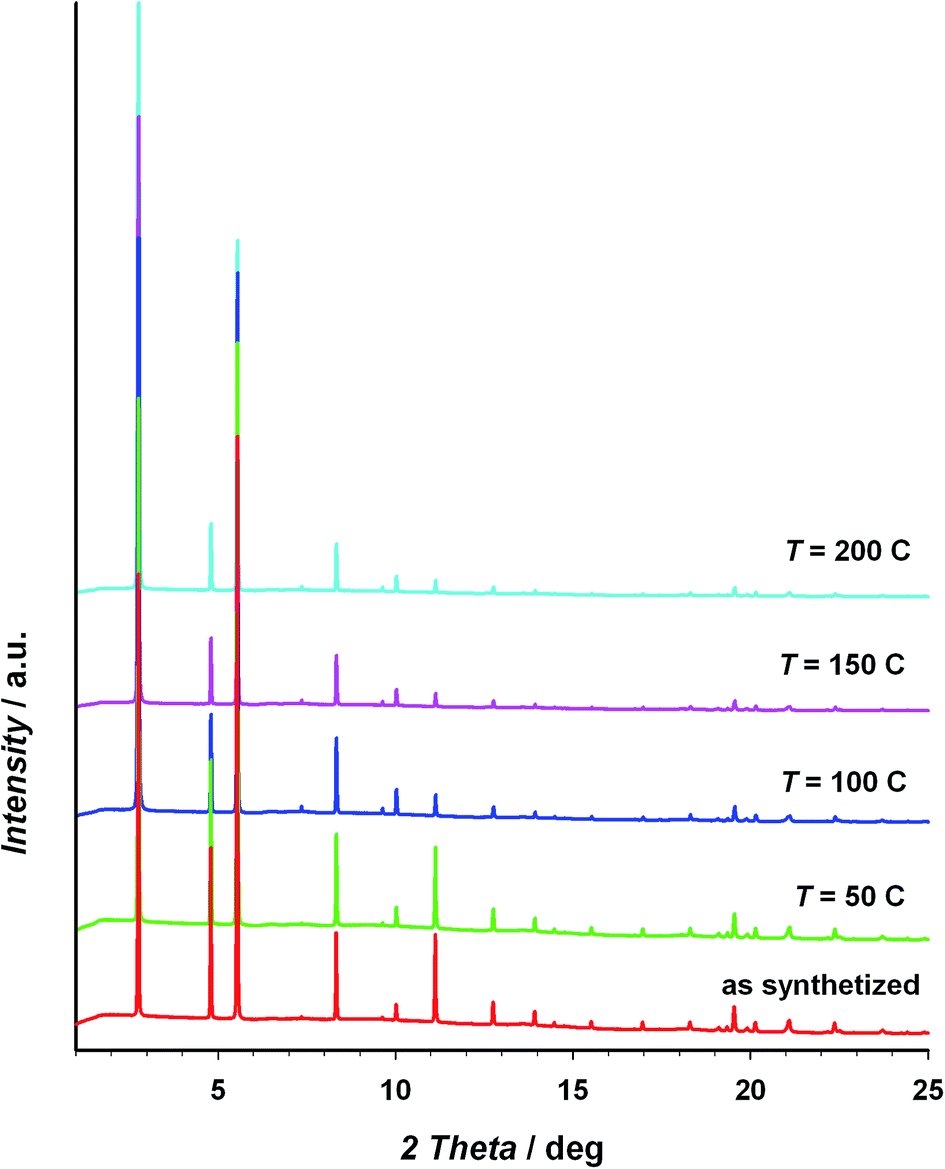 | ||
| Fig. 1 Powder X-ray diffraction patterns at different temperatures of the CIM-80 compound. | ||
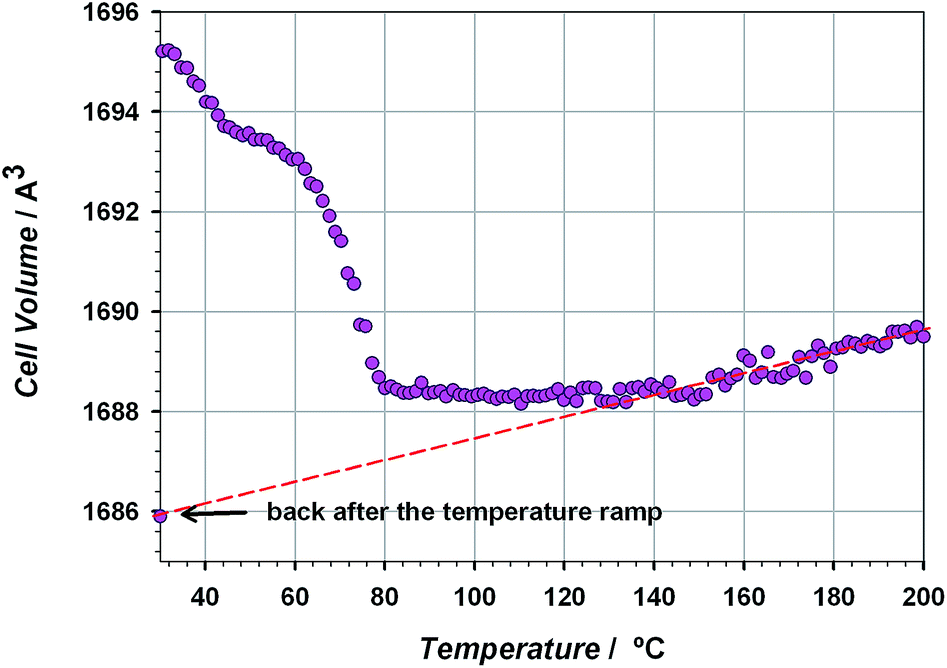 | ||
| Fig. 2 Cell volume variation with the temperature upon heating. The contraction is associated with the solvent loss. The red dashed line indicates the fitting of the data to get the expansion coefficient. | ||
We are interested in the application of this material as an extractant for different water samples, and thus we have tested the stability of the material at working conditions. The CIM-80 material is stable in water and after two weeks immersed in water, the material does not show signs of degradation or loss of crystallinity. CIM-80 was also tested in waters at different pH values (1, 5, 10) keeping the material for one week immersed in the solution. No significate changes are observed in the powder patterns, supporting the robustness of the framework (see Fig. S8 of the ESI†). Since the analytical methodology includes a vortex agitation step, the CIM-80 was subjected to 8 min of vortex in water keeping its integrity (see Fig. S8 of the ESI†).
The cytotoxicity of the MOF CIM-80 was evaluated following a strategy previously reported for other MOFs,25 with details of the procedure included in Fig. S9 of the ESI.† The LC50 value obtained was higher than 2 mg mL−1, which was the maximum value tested in the study. Therefore, it is possible to work with quite high values of CIM-80 without provoking cytotoxicity issues. Other LC50 values reported for MOFs are, for example, 0.70–1.10 mg mL−1 for MIL-100 or 0.02–0.10 mg mL−1 for ZIF-8,26 clearly supporting the greenness of this MOF: green synthesis and non-cytotoxicity issues from its utilization.
3.2 Evaluation of the analytical extraction performance of CIM-80 in D-μSPE-UHPLC
Up to 22 analytes, including conventional contaminants such as PAHs10,27,28 and emerging contaminants such as preservatives, UV filters or drugs19 have been selected as target compounds (Table S1 of the ESI†) to evaluate the ability of the designed MOF to trap them from environmental waters through monitoring strategies. Target analytes have been selected trying to have a representative group of contaminants (persistent and emergent) with different structures and properties, thus showing the ability of the MOF to extract them by D-μSPE-UHPLC.To evaluate the performance of CIM-80 as adequate extraction sorbent for these water contaminants, aqueous standards of the target analytes (at low concentration values to mimic environmental levels) are subjected to the entire D-μSPE-UHPLC method. Thus, the extraction efficiency (ER, in %) is estimated by comparison of the obtained chromatographic signal with the expected signal if the analytes are efficiently extracted and eluted by the MOF in D-μSPE. Another efficient tool to evaluate the performance of the D-μSPE method is the enrichment factor (EF), that is, the preconcentration achieved by the procedure if comparing the isolated UHPLC determination method with the D-μSPE-UHPLC method. Tables S3 and S4 of the ESI† include the calibrations obtained with only UHPLC-UV (for the 7 emerging pollutants monitored) and UHPLC-FD (for the 15 PAHs monitored), without the D-μSPE approach, together with a number of analytical features.
Clearly, a number of parameters need optimization in the D-μSPE procedure. To simplify such optimization, several parameters are fixed to common values in the initial stage. This way, the water sample (or aqueous standard) volume is fixed to 10 mL (to simplify the further centrifugation steps while being easy to sample in environmental fields); the vortex time is limited to 3 min (to ensure a quick method); and the elution solvent volume is 500 μL (to ensure a preconcentration procedure, and to minimize environmental wastes). Fig. 3 shows a scheme of the entire D-μSPE procedure. Under these conditions, the main variables optimized were the amount of MOF required and the type of elution solvent. It has been pointed that these two variables are the key points to ensure proper efficiency in D-μSPE.13
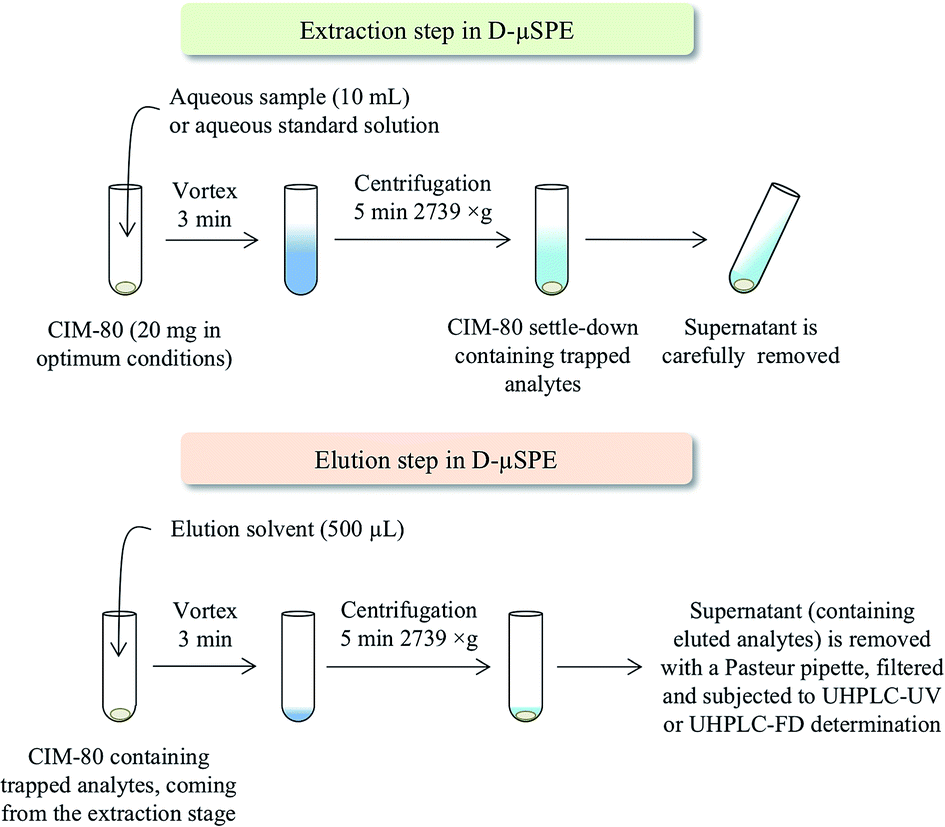 | ||
| Fig. 3 Scheme of the dispersive μ-SPE procedure. | ||
Values between 5 and 50 mg of CIM-80 were tested with the fixed conditions abovementioned to select the best amount to work with, while ensuring a microextraction procedure. The elution solvent in these experiments was MeOH. Experiments were carried out in triplicate with the group of emerging pollutants, for being those more difficult to extract in conventional methods if compared to more hydrophobic analytes such as PAHs.19 From Fig. S10 of the ESI,† it can be observed that 20 mg is the best amount to utilize. Lower amounts appear to be insufficient whereas higher amounts probably make the elution step more difficult, thus provoking decreases in the overall efficiency. The exception was estrone, for which the best amount was the lowest value tested: 5 mg. In order to have the best amount for the majority of compounds, 20 mg of CIM-80 was fixed for further experiments, and the method was further optimized by evaluating the influence of the elution solvent.
Three different solvents were tested in the elution step: ACN, MeOH and isopropanol, for being miscible with the UHPLC mobile phases (and thus avoiding solvent evaporation and solvent exchange steps). Fig. 4 shows the extraction efficiencies (ER in %) obtained, specifically for PAHs using UHPLC-FD (Fig. 4A) and for the emerging contaminants using UHPLC-UV (Fig. 4B), while using the fixed conditions above mentioned (and 20 mg of CIM-80).
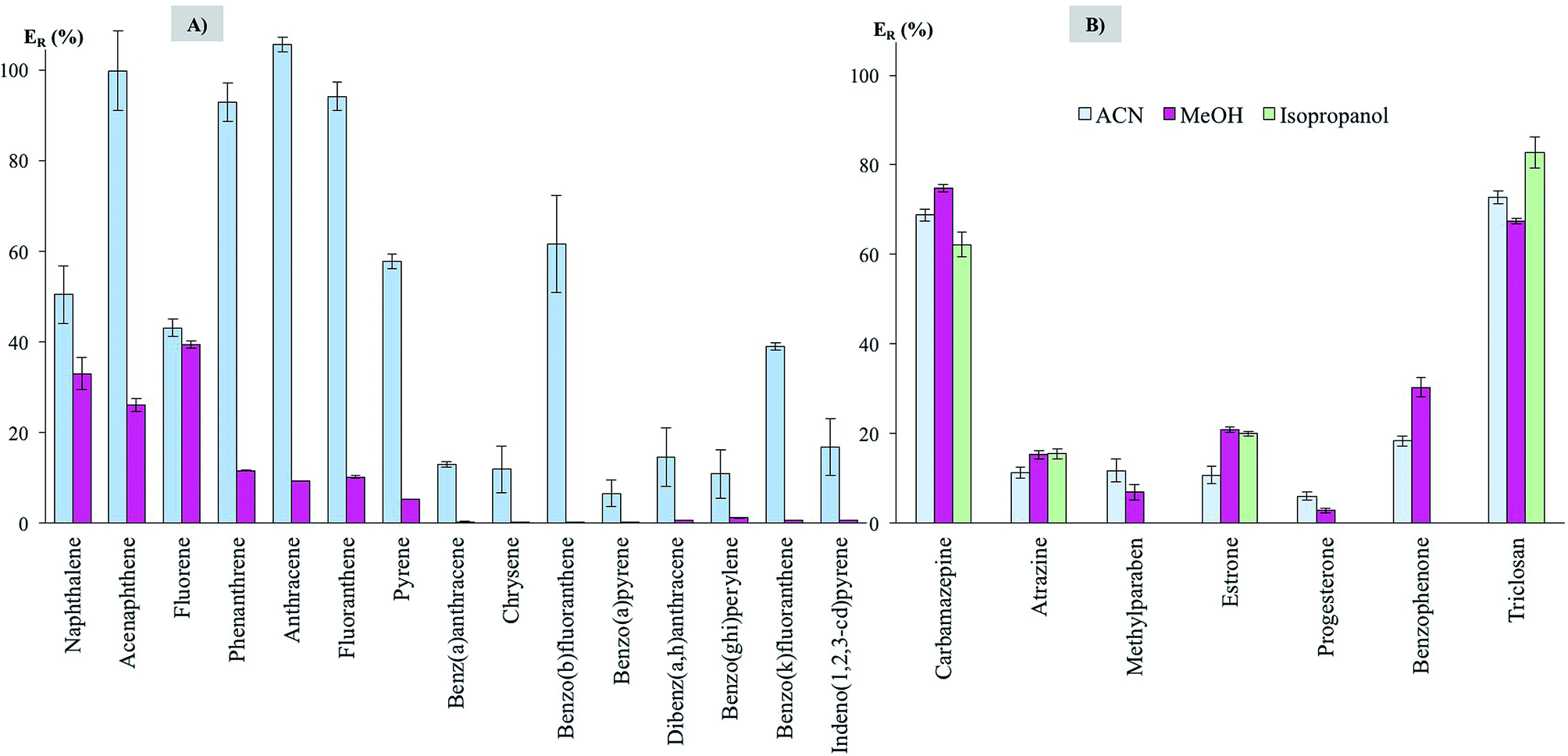 | ||
| Fig. 4 Extraction efficiencies obtained for the group of contaminants studied, using CIM-80 as sorbent of the D-μSPE method and HPLC-FD (A) or HPLC-UV (B), when using different elution solvents. Remaining experimental conditions as described in the text. | ||
ACN is, in general, the elution solvent that generates the higher efficiencies. This is particularly clear for PAHs. On the other hand, isopropanol, given its polarity, was unable to elute the PAHs trapped by the MOF. For the emerging contaminants, MeOH behaves nicely, with ACN showing lower performance. Isopropanol would be a better choice for estrone and triclosan. In any case, differences in efficiencies when using ACN, MeOH or isopropanol, with few exceptions, are lower than 10% for emerging contaminants. Therefore, with the purpose of setting up a generic procedure for different contaminants, ACN is the elution solvent selected for PAHs whereas MeOH is the one selected for the emerging contaminants.
The higher efficiencies observed for lighter PAHs can be explained for a better adsorption of the analytes by the MOF material and a better elution when ACN is used. However, for bulkier PAHs the efficiencies are systematically lower, and even negligible when MeOH is the eluent, maybe pointing out the difficulties in the extraction of these bulkier analytes. In the case of the emerging contaminants, the selection of the eluent is not a key point, since the differences are low. Therefore, dissimilarities in the efficiencies should come from the extraction process. The methyl groups of the mesaconate ligand pointing toward the channels would favour the interaction with the more hydrophobic analytes, and this is supported with the good efficiencies obtained for lighter PAHs. This may be the cause for better extraction in the case of carbamazepine, benzophenone or triclosan. However, other factors should play an important role in the case of, for example, methylparaben. For this analyte a better extraction was expected regarding its similarities with the other ones (aromatic rings, hydrogen bond donor and acceptor groups, smaller size). This points out for the difficulties in establishing general rules of affinity MOF-analyte when using MOFs in D-μSPE.13
Regarding efficiencies, they range from ∼50 to ∼100% for lighter PAHs, which can be highlighted if considering that efficiencies in microextraction methods are hardly 100%.29,30 For heavy PAHs, average efficiencies are ∼20%. For the majority of emerging contaminants, average efficiencies are ∼15%, being ∼70% for carbamazepine and triclosan. Independently on the efficiency, precision values (as relative standard deviation, RSD, in %) are always lower than 10.9%, thus supporting the adequate reproducibility of the microextraction strategy. It must be considered that efficiencies are acceptable in a microextraction method as long as the precision values are adequate (RSD < 20%) and the enrichment factor attained by the method fulfills environmental policies of sensitivity.
Under the optimum conditions described, the quality analytical parameters obtained for the D-μSPE-UHPLC monitoring method are shown in Table 1. Maximum EF values were obtained for light PAHs such as acenaphthene, phenanthrene, anthracene and fluoranthene, and for emerging pollutants such as carbamazepine (a drug) and triclosan (a disinfectant); which is totally in agreement with the efficiencies obtained for these compounds with the entire method.
| Analyte | EFa | RSDb (%) | LODc (ng L−1) | LOQd (ng L−1) |
|---|---|---|---|---|
| a Enrichment factor, being EFmax = 20.b Relative standard deviation (n = 3).c Estimated limit of detection for the entire D-μSPE-UHPLC method.d Estimated limit of quantification for the entire D-μSPE-UHPLC method. | ||||
| Carbamazepine | 15 | 1.5 | 1.1 × 102 | 3.5 × 102 |
| Atrazine | 3.1 | 1.8 | 1.1 × 103 | 3.6 × 103 |
| Methylparaben | 1.4 | 17 | 8.3 × 103 | 2.8 × 104 |
| Estrone | 4.2 | 6.6 | 3.5 × 103 | 1.2 × 104 |
| Progesterone | 0.6 | 5.3 | 2.1 × 104 | 7.1 × 104 |
| Benzophenone | 6.1 | 19 | 3.7 × 102 | 1.2 × 103 |
| Triclosan | 13 | 6.2 | 3.3 × 102 | 1.1 × 103 |
| Naphthalene | 10 | 5.0 | 3.6 | 12 |
| Acenaphthene | 18 | 3.5 | 4.2 | 14 |
| Fluorene | 8.6 | 1.8 | 0.93 | 3.1 |
| Phenanthrene | 19 | 1.9 | 1.6 | 5.3 |
| Anthracene | 21 | 0.59 | 0.75 | 2.5 |
| Fluoranthene | 19 | 1.3 | 0.81 | 2.7 |
| Pyrene | 11 | 1.1 | 0.78 | 2.6 |
| Benz(a)anthracene | 2.6 | 1.9 | 7.8 | 26 |
| Chrysene | 2.4 | 9.5 | 1.8 | 6.0 |
| Benzo(b)fluoranthene | 12 | 7.0 | 1.7 | 5.6 |
| Benzo(a)pyrene | 1.3 | 11 | 2.6 | 8.5 |
| Dibenz(a,h)anthracene | 2.9 | 10 | 2.9 | 9.8 |
| Benzo(ghi)perylene | 2.2 | 12 | 4.5 | 15 |
| Benzo(k)fluoranthene | 7.8 | 0.85 | 2.7 | 8.9 |
| Indeno(1,2,3-cd)pyrene | 3.4 | 15 | 9.3 | 31 |
One of the main important features of environmental monitoring methods is the sensitivity achieved, in order to reach the levels imposed by regulations. Thus, the estimated limits of quantification obtained with the proposed microextraction method using CIM-80 range from 2.5 to 31 ng L−1 for PAHs, and from 0.35 to 71 μg L−1 for emerging contaminants. The United States Environmental Protection Agency (US-EPA) has established that the benzo(a)pyrene content in drinking waters should not be higher than 0.2 μ L−1.31 The European Union (EU) legislation terms as priority PAHs: benzo(a)pyrene, benzo(b)fluoranthene, benzo(k)fluoranthene, benzo(ghi)perylene and indeno(1,2,3-cd)pyrene, while establishing a maximum value for benzo(a)pyrene of 0.1 μg L−1 in superficial and drinking waters.32 The sum of the content of the rest of priority PAHs in superficial waters must not exceed 0.1 μg L−1. The EU also sets maximum concentration values for anthracene and fluorene: 0.4 μg L−1 and 1 μg L−1, respectively. Clearly, the sensitivity achieved using CIM-80 in the D-μSPE-UHPLC-FD method fits perfectly within the imposed limits. Regarding emerging contaminants, there is still a lack of regulations despite growing concerns of their presence in environmental reservoirs. It is also important mentioning that the use of MS detection over UV or DAD clearly improves the sensitivity. If the current D-μSPE method with CIM-80 is used in combination with LC-MS rather than with UHPLC-UV, much lower limits of detection will be obtained.
If comparing the performance of CIM-80 for the group of analytes studied with other materials (including conventional ones and also other MOFs) in SPE methods from literature (Table S5 of the ESI†), it is important to mention that the sensitivity obtained is comparable to those reports.10,19 This also proves the efficiency of the D-μSPE using the proposed CIM-80.
Regarding emerging pollutants, a previous study of our group on the use of the MOF MIL-53(Al) and HPLC-DAD19 presents comparable sensitivity for carbamazepine, atrazine, and estrone. Thus, the novel material CIM-80 is able to operate as sorbent material in a monitoring method with comparable (and even better) performance of other materials, including the similar MOF MIL-53(Al), but clearly compiling with green chemistry requirements, because its synthesis can be considered as environmental-friendly, and also because its use ensures low cytotoxicity.
Regarding the water monitoring of PAHs (Table S5 of the ESI†), previous studies of our group using a magnetic composite based on MOF and UHPLC-FD27 shows quite similar sensitivity, and even better when using CIM-80. In this case, lower amount of material and simpler (and greener) preparation routes are also involved.
4. Conclusions
We have synthesized a new promising material through a green procedure for the determination of pollutants in water and using an optimized environmental-friendly methodology that minimizes wastes and maximizes efficiency. The CIM-80 material obtained is stable in water at different pH values, it has a working temperature range from RT to 350 °C, and it also presents low cytotoxicity, with an impressive LC50 value higher than 2 mg L−1. The efficiencies shown in the water analysis for 22 pollutants are similar or even better than those reported for MIL-53(Al) and other materials. Ongoing work is aimed to incorporate this material in other miniaturized extraction techniques, particularly considering its analytical performance and partial selectivity with triclosan and carbamazepine, and to give more insights into the extraction mechanism of emerging contaminants through crystallography of inclusion compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
V. P. and C. R.-P. thank the MINECO for the Projects Ref. MAT2014-57465-R and MAT2017-89207-R. P. R.-B. thanks her FPI PhD research contract associated to the Project Ref. MAT2014-57465-R. We acknowledge the beamtime awarded at BL04 – MSPD beamline at ALBA Synchrotron and the collaboration of the ALBA staff. The research group of Jacob Lorenzo-Morales from the ULL-University Institute of Tropical Diseases and Public Health of the Canary Islands, and quite particularly the help of Ines Sifaoui, are greatly acknowledged. We thank the SEGAI services of the University of La Laguna, specially the technicians, for the help provided.Notes and references
- P. Bauerová, J. Vinklerová, J. Hraníček, V. Čorba, L. Vojtek, J. Svobodová and M. Vinkler, Sci. Total Environ., 2017, 601–602, 1556–1565 CrossRef PubMed.
- Z. Tousova, P. Oswald, J. Slobodnik, L. Blah, M. Muz, M. Hu, W. Brack, M. Kraus, C. Di Paolo, Z. Tarcai, T.-B. Seiler, H. Hollert, S. Koprivica, M. Ahel, J. E. Schollée, J. Hollender, M. J.-F. Suter, A. O. Hidasi, K. Schirmer, M. Sonavane, S. Ait-Aissa, N. Creusot, Fr. Brion, J. Froment, A. C. Almeida, K. Thomas, K. E. Tollefsen, S. Tufi, X. Ouyang, P. Leonards, M. Lamoree, V. O. Torrens, A. Kolkman, M. Schriks, P. Spirhanzlova, A. Tindall and T. Schulze, Sci. Total Environ., 2017, 601–602, 1849–1868 CrossRef PubMed.
- M. I. Vasquez, A. Lambrianides, M. Schneider, K. Kümmerer and D. Fatta-Kassinos, J. Hazard. Mater., 2014, 279, 169–189 CrossRef PubMed.
- Z. Huang and H. K. Lee, Trends Anal. Chem., 2012, 39, 228–244 CrossRef.
- J. K. Ludwig, Green Chem. Lett. Rev., ed. M. Lancaster, The Royal Society of Chemistry, Cambridge, UK, 3rd edn, 2017, ISBN: 978-1-78262-294-9 Search PubMed.
- É. A. Souza-Silva, R. Jiang, A. Rodríguez-Lafuente, E. Gionfriddo and J. Pawliszyn, Trends Anal. Chem., 2015, 71, 224–235 CrossRef.
- T. D. Ho, A. J. Canestraro and J. L. Anderson, Trends Anal. Chem., 2011, 695, 18–43 Search PubMed.
- J. Tian, J. Xu, F. Zhu, T. Lu, C. Su and G. Ouyang, J. Chromatogr. A, 2013, 1300, 2–16 CrossRef PubMed.
- P. Rocío-Bautista, C. Martínez-Benito, V. Pino, J. Pasán, J. H. Ayala, C. Ruiz-Pérez and A. M. Afonso, Talanta, 2015, 139, 13–20 CrossRef PubMed.
- P. Rocío-Bautista, V. Pino, J. H. Ayala, J. Pasán, C. Ruiz-Pérez and A. M. Afonso, J. Chromatogr. A, 2016, 1436, 42–50 CrossRef PubMed.
- E. Tahmasebi, M. Y. Masoomi, Y. Yamini and A. Morsali, Inorg. Chem., 2015, 54, 425–433 CrossRef PubMed.
- P. Rocío-Bautista, I. Pacheco-Fernández, J. Pasán and V. Pino, Anal. Chim. Acta, 2016, 939, 26–41 CrossRef PubMed.
- P. Rocío-Bautista, P. González-Hernández, V. Pino, J. Pasán and A. M. Afonso, Trends Anal. Chem., 2017, 90, 114–134 CrossRef.
- F. Maya, C. Palomino-Cabello, R. M. Frizzarin, J. M. Estela, G. Turnes-Palomino and V. Cerdà, Trends Anal. Chem., 2017, 90, 142–152 CrossRef.
- J. Płotka-Wasylka, N. Szczepaśnka, M. Guardia and J. Namieśnik, Trends Anal. Chem., 2015, 73, 19–38 CrossRef.
- M. R. Sohrabi, Microchim. Acta, 2014, 181, 435–444 CrossRef.
- G. Gao, S. Li, S. Li, Y. Wang, P. Zhao, X. Zhang and X. Hou, Talanta, 2018, 180, 358–367 CrossRef PubMed.
- T. Wang, J. Wang, C. Zhang, Z. Yang, X. Dai, M. Cheng and X. Hou, Analyst, 2015, 140, 5308–5316 RSC.
- P. Rocío-Bautista, V. Pino, J. Pasán, I. López-Hernández, J. H. Ayala, C. Ruiz-Pérez and A. M. Afonso, Talanta, 2018, 179, 775–783 CrossRef PubMed.
- H. Reinsch, T. Homburg, N. Heidenreich, D. Frçhlich, S. Hennninger, M. Wark and N. Stock, Chem.–Eur. J., 2018, 24, 2173–2181 CrossRef PubMed.
- P. A. Bayliss, I. A. Ibarra, E. Pérez, S. Yang, C. C. Tang, M. Poliakoff and M. Schröder, Green Chem., 2014, 16, 3796–3802 RSC.
- F. Fauth, I. Peral, C. Popescu and M. Knapp, Powder Diffr., 2013, 28, S360–S370 CrossRef.
- L. Sarkisov and A. Harrison, Mol. Simul., 2011, 15, 1248–1257 CrossRef.
- Y.-S. Wei, K.-J. Chen, P.-Q. Liao, B.-Y. Zhu, R.-B. Lin, H.-L. Zhou, B.-Y. Wang, W. Xue, J.-P. Zhang and X.-M. Chen, Chem. Sci., 2013, 4, 1539–1546 RSC.
- K. A. Mocniak, I. Kubajewska, D. E. M. Spillane, G. R. Williams and R. E. Morris, RSC Adv., 2015, 5, 83648–83656 RSC.
- C. Tamames-Tabar, D. Cunha, E. Imbuluzqueta, F. Ragon, C. Serre, M. J. Blanco-Prieto and P. Horcajada, J. Mater. Chem. B, 2014, 2, 262–271 RSC.
- P. Rocío-Bautista, V. Pino, J. Pasán, J. H. Ayala, C. Ruiz-Pérez and A. M. Afonso, LC-GC, 2018, 36, 464–471 Search PubMed.
- M. J. Trujillo-Rodríguez, O. Nacham, K. D. Clark, V. Pino, J. L. Anderson, J. H. Ayala and A. M. Afonso, Anal. Chim. Acta, 2016, 934, 106–113 CrossRef PubMed.
- J. López-Darias, M. Germán-Hernández, V. Pino and A. M. Afonso, Talanta, 2010, 80, 1611–1618 CrossRef PubMed.
- M. J. Trujillo-Rodríguez, P. Rocío-Bautista, V. Pino and A. M. Afonso, Trends Anal. Chem., 2013, 51, 87–106 CrossRef.
- D. Lerda, Polycyclic aromatic hydrocarbons (PAHs) factsheet, European Commission, Institute for reference materials and measurements, 2011 Search PubMed.
- Decision No 2455/2001/EC of the European parliament and of the council of 20 November 2001, Establishing the list of priority substances in the field of water policy and amending Directive 2000/60/EC, Official Journal of the European Union, L331/1.
Footnote |
| † Electronic supplementary information (ESI) available: Diffraction patterns, thermodiffractograms, TG plot, chromatograms, SEM picture, tables. See DOI: 10.1039/c8ra05862h |
| This journal is © The Royal Society of Chemistry 2018 |