
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organic photovoltaics of diketopyrrolopyrrole copolymers with unsymmetric and regiorandom configuration of the side units†
Kenta Aoshima a,
Marina Idea and
Akinori Saeki*ab
a,
Marina Idea and
Akinori Saeki*ab
aDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: saeki@chem.eng.osaka-u.ac.jp
bPrecursory Research for Embryonic Science and Technology (PRESTO), Japan Science and Technology Agency (JST), 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 28th August 2018
Abstract
Diketopyrrolopyrrole (DPP) is a representative electron acceptor incorporated into narrow-bandgap polymers for organic photovoltaic cells (OPV). Commonly, identical aromatic units are attached to the sides of the DPP unit, forming symmetric DPP polymers. Herein we report the synthesis and characterization of DPP copolymers consisting of unsymmetric configurations of the side aromatics. The unsymmetric DPP copolymer with thienothiophene and benzene side moieties exhibits good solubility owing to the twisted dihedral angle at benzene and regiorandom configuration. A significant shallowing of the highest occupied molecular orbital level is observed in accordance with the electron-donating nature of the side units (benzene, thiophene, and thienothiophene). The overall power conversion efficiencies of the unsymmetric DPPs (2.3–2.4%) are greater than that of the centrosymmetric analogue (0.45%), which is discussed in view of bulk heterojunction morphology, polymer crystallinity, and space-charge-limited current mobilities. This comparative study highlights the effect of unsymmetric design on the molecular stacking and OPV performance of DPP copolymers.
Introduction
The need to meet a rising global demand for renewable energy sources has led to the exploration of materials for next-generation solar cells. As such, organic photovoltaic cells (OPV) based on a bulk heterojunction (BHJ) framework have been developed,1–4 in particular over the last two decades, wherein a bicontinuous network of binary5–7 or multiple8–10 components consisting of p-type and n-type conjugated materials is central to the photoelectric conversion process. The materials used include polymers and molecules that are mostly composed of covalently-bonded electron-donating and electron-accepting units to manipulate their electrochemical properties such as bandgap (Eg) and intermolecular interactions such as crystallinity11,12 and backbone orientation.13,14Diketopyrrolopyrrole (DPP)15–23 and isoindigo (IDG)24–34 are relatively strong electron acceptors and have been polymerized into narrow-bandgap polymers for OPV. Based on modification of IDG, Ashraf et al.,35 Pruissen et al.36 and Koizumi et al.37 have reported the synthesis of thienoisoindigo (TIDG) and its copolymers as organic field-effect transistors (OFETs), which is followed by applications of TIDG-based near-infrared (IR)-absorbing copolymers to OPV38–42 and OFET.43–47 IDG and TIDG have structural similarities to DPPs with symmetric side units of benzene (Ph) or thiophene (Th), respectively. However, TIDG exhibits a stronger electron-accepting tendency than Th-sandwiched DPP because of the direct attachment of Th to the electron-withdrawing ketopyrrole unit, which leads to a greatly narrowed Eg along with a low open-circuit voltage (Voc).38 Moreover, the most critical issue in the IR-absorbing polymers is that the low short-circuit current density (Jsc) resulting from the energy bandgap law48 causes shortening of the exciton lifetime38 through increased coupling with the low-energy vibrational mode. To balance the exciton lifetime and optical bandgap, Chen et al.49 and Ide et al.50,51 have synthesized a benzothienoisoindigo (BTIDG) unit that replaces one of the Th units of TIDG with Ph (Fig. 1). BTIDG includes an intramolecular attractive S–O interaction, and steric hindrance between the proton of Ph and oxygen of the keto unit, which constitutes a half-distorted π-plane. This unsymmetric molecular design elicited not only a moderate Eg but also improved film morphology (face-on orientation of polymer backbone and miscible BHJ network), resulting in increased power conversion efficiency (PCE).50 Thus, the unsymmetric structure and regiorandom configuration of BTIDG copolymers may contribute to the improved film characteristics associated with their solubilities, in analogy to the unsymmetric DPP polymers having Th and thienothiophene (TT) side units (PCE ∼ 6%).21
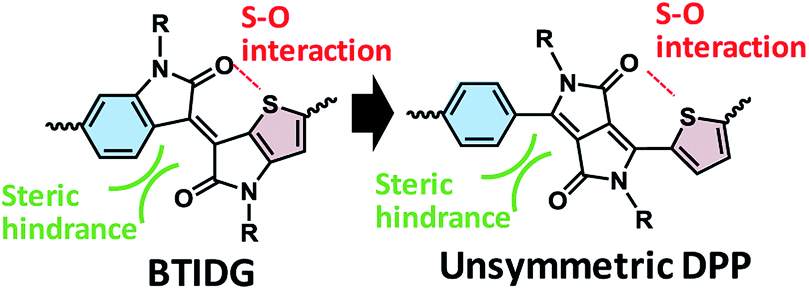 | ||
| Fig. 1 Molecular design of unsymmetric electron-accepting units. (Left) BTDIG, (right) unsymmetric DPP. | ||
Herein we report a comparative study on DPP-based copolymers with unsymmetric configurations of Ph and (Th or TT) units. Despite the structural similarity between BTIDG and Ph-DPP-Th (left and right in Fig. 1, respectively), they behave differently. The morphology, polymer orientation, and space-charge-limited current (SCLC) mobilities of DPP-based copolymer-[6,6]-phenyl-C61-butyric acid methyl ester (PCBM) blend films are evaluated and discussed, as are their PCEs in an OPV application.
Results and discussion
Three DPP copolymers were prepared, including a symmetric Ph-DPP-Ph copolymer (P1) as control, an unsymmetric Ph-DPP-Th copolymer (P2), and an unsymmetric Ph-DPP-TT copolymer (P3), as shown in Fig. 2. Ph-DPP-Ph rather than more planar and common Th-DPP-Th unit15,17–23,52,53 was chosen as control in order to examine the change in the backbone planarization from twist (Ph-DPP-Ph) to half-distortion (Ph-DPP-Th (or TT)) (vide infra). DPP units were synthesized according to previously described procedures (see Experimental) and polymerized with an electron-donor unit of 2-dimensional benzobisthiophene-appending alkylthiophene side units (BDT-Th) via Stille coupling reaction. The resultant unsymmetric DPP polymers are regiorandom, similarly to the pyridylthiadiazole-based molecules and polymers reported by Bazan et al.54,55 The weight-averaged molecular weights (Mws) and polydispersity indices (PDIs) of the polymers, as characterized by size exclusion chromatography, were 98.4 kg mol−1 (2.7) for P1, 22.2 kg mol−1 (2.4) for P2, and 31.6 kg mol−1 (2.5) for P3. These polymers showed glass transition at ∼147 °C and the identical profiles during two cycles of differential scanning calorimetry (up to 300 °C), exhibiting no decomposition below this temperature (Fig. S1†). Solubility of P3 was as large as ∼100 mg mL−1 even in toluene at room temperature, which is presumably derived from the twisted backbone at the benzene-DPP connection (vide infra) and unsymmetric, regiorandom configuration. When comparing solubilities of P1 and P3 in toluene–hexane mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vol%), they are well-soluble (∼18 and ∼13 mg mL−1, respectively).
:1 vol%), they are well-soluble (∼18 and ∼13 mg mL−1, respectively).
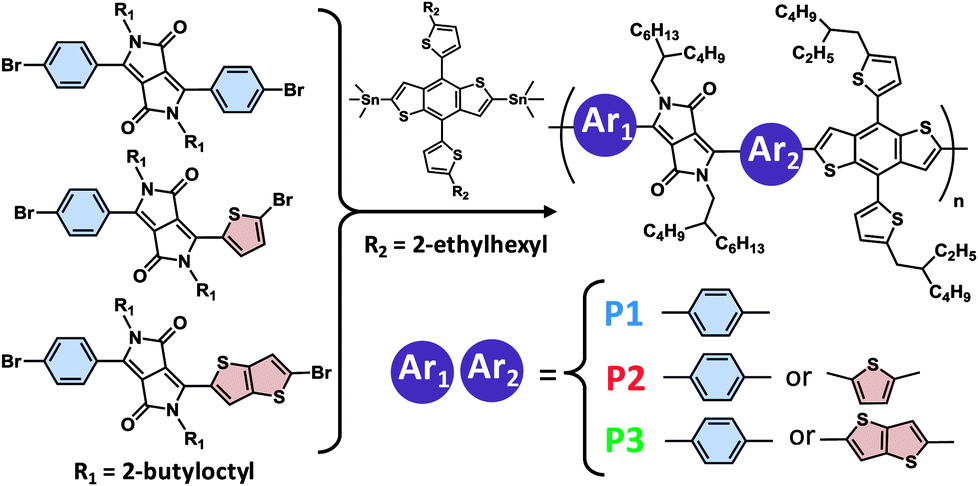 | ||
| Fig. 2 Chemical structures of the unsymmetric, regiorandom DPP-based copolymers. Polymerization was carried out by Stille coupling method using Pd(PPh3)4 catalyst. | ||
Fig. 3a shows the photoabsorption spectra of P1–P3 in diluted chlorobenzene solutions and as films. All the polymers exhibited mostly unchanged maxima attributed to an intramolecular charge transfer band in both solution and film phases (545 nm for P1, 629 nm for P2, and 622 nm for P3). Meanwhile, P2 and P3 exhibited accompanying shoulder peaks in the longer-wavelength region, suggestive of their more extensive conjugation systems and higher crystallinities as compared to P1. Fig. 3b displays the electrochemical properties of the polymers, including the highest occupied molecular orbital (HOMO) levels as evaluated using photoelectron yield spectroscopy (PYS) and the Egs estimated from the photoabsorption spectra of the films (Fig. S2†). The HOMO levels drastically shifted upward, from −5.62 eV for P1, −5.37 eV for P2, and −5.13 eV for P3. The Egs of P2 and P3 were very similar (1.67 and 1.66 eV, respectively), and are up to 0.4 eV narrower than that of P1 (2.04 eV). The narrowing of the Egs and shallowing of HOMO levels are rationalized by the order of the electron-donating strengths of Ph, Th, and TT. Density functional theory (DFT) calculations of donor–acceptor–donor units shown in Fig. 3c revealed a contrasting change in the dihedral angle between the BDT-Th donor unit and DPP, where Ph-side angles were almost double (20–25°) those of the Th (or TT)-side angles (11°). The structural planarization of P2 and P3 is likely to partly affect their narrowed Egs and the appearance of shoulder peaks (improved crystallinity in a film).
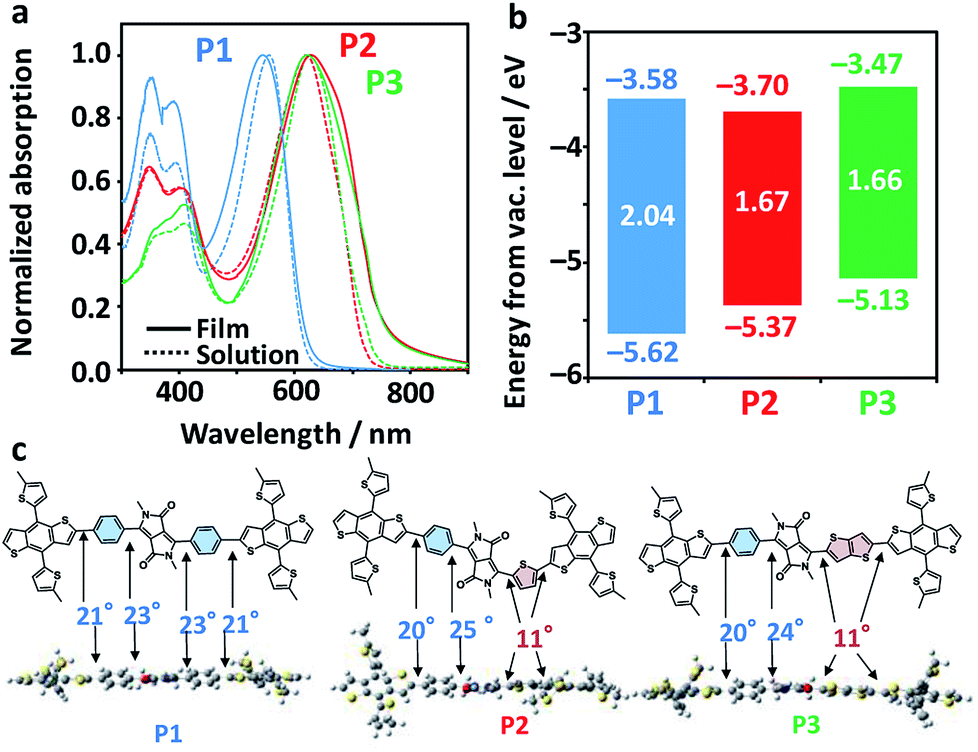 | ||
| Fig. 3 (a) Photoabsorption spectra of P1–P3 in chlorobenzene solutions (dotted lines) and as films (solid lines). (b) Energy diagram of the copolymers. HOMO (the bottom of each bar) and Eg (the centre of each bar) were evaluated by PYS and the photoabsorption onset in the film, respectively. LUMO (the top of each bar) was calculated by adding Eg and HOMO. (c) Horizontal view, along with dihedral angles at respective bonds. The donor–acceptor–donor compounds were geometry-optimized using DFT with B3LYP/6-31G*. Alkyl chains were replaced by methyl groups to simplify the calculation. | ||
Inverted-type OPV devices (ITO/ZnO/active layer/MoOx/Ag) were fabricated from chlorobenzene solutions with 3 vol% 1,8-diiodooctane (DIO) as the solvent additive. We have surveyed the processing conditions including thermal annealing (TA, 120 °C for 10 min) and copolymer:PCBM blend ratios (1:1, 1:2, and 1:3) (Fig. S3 and Table S1†). The highest PCE obtained for P1 was 0.45% (average 0.38 ± 0.04%), while P2 and P3 achieved higher PCEs of 2.30% (average 1.83 ± 0.25%) and 2.40% (average 1.97 ± 0.32%), respectively (Fig. 4a and Table 1). The low PCE of control P1 is linked to the coarse BHJ morphology and low crystallinity (low electron mobility, vide infra), while the symmetric copolymer composed of BDT-Th and Th-DPP-Th with the identical alkyl chains have reportedly showed high PCE (1% (ref. 52) and 6.5% (ref. 53)). Despite the low PCE of P1, it exhibited the highest Voc (0.99 V) among the polymers (P2: 0.875 V and P3: 0.813 V), which is in accordance with the HOMO levels of the respective polymers. The improvement in PCE for P2 and P3 is mainly caused by the Jsc (1.25, 6.50, and 7.19 mA cm−2 for P1, P2, and P3, respectively), as evident from the large difference in the external quantum efficiency (EQE) spectra (Fig. 4b). The EQE spectra display the same shape as the corresponding photoabsorption spectra of the polymers, and the integrated Jsc over the EQE spectra are consistent with those under pseudo-sunlight exposure (100 mW cm−2). Note that the small Jsc of P1 is not limited by the narrow Eg, but rather is governed by BHJ morphology (vide infra) that simultaneously affects the exciton migration, charge separation, and charge transport. The SCLC hole mobilities (μh) of pristine polymers monotonically increased from 2.5 × 10−6 cm2 V−1 s−1 for P1 to 1.0 × 10−5 cm2 V−1 s−1 for P2, and further to 8.5 × 10−5 cm2 V−1 s−1 for P3 (Table S1 and Fig. S4†). In contrast, μhs of the optimized blend films were low and mostly comparable (2.8 × 10−6 cm2 V−1 s−1 for P1:PCBM, 2.1 × 10−6 cm2 V−1 s−1 for P2:PCBM, and 2.2 × 10−6 cm2 V−1 s−1 for P3:PCBM), while the SCLC electron mobilities (μe) of the blend films increased between P1 (1.4 × 10−5 cm2 V−1 s−1) and P2 (1.9 × 10−4 cm2 V−1 s−1), and further to P3 (7.9 × 10−4 cm2 V−1 s−1) (Table 1 and Fig. S4†). The μe values of P2:PCBM and P3:PCBM, which were an order of magnitude larger than that of P1:PCBM, agrees with the higher crystallinity of the former polymers (vide infra). Therefore, μe, which is closely related to the purity and percolation network of PCBM domains facilitated by the crystallization process of the polymer in the blends, is likely a dominant factor influencing overall PCE.
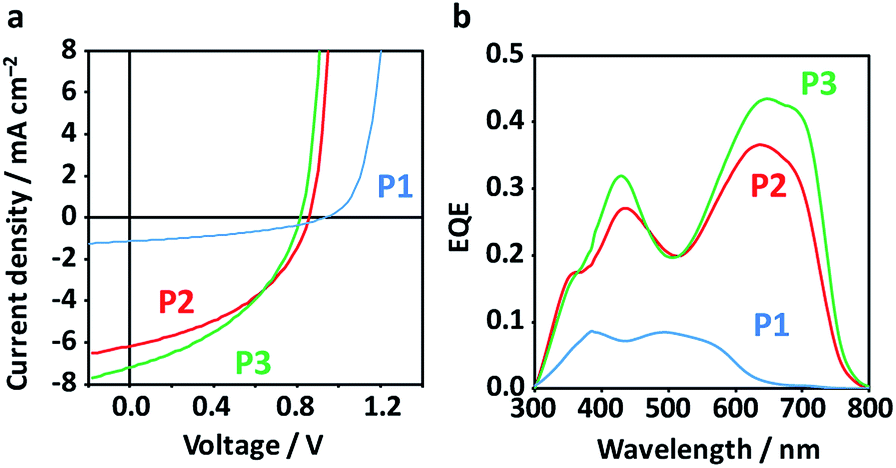 | ||
| Fig. 4 (a) Current density–voltage curves of the best-performing OPV devices under pseudo-sunlight (100 mW cm−2). (b) EQE spectra of the corresponding devices. | ||
:PCBM OPV performancesa and SCLC mobilities
| Polymer (p:n) |
L/nm | Jsc (JEQEsc)c/mA cm−2 | Voc/V | FFd | PCE/% | PCEavee/% | μhf/cm2 V−1 s−1 | μef/cm2 V−1 s−1 |
|---|---|---|---|---|---|---|---|---|
| a Inverted cell (ITO/ZnO/active layer/MoOx/Ag) under simulated sunlight (100 mW cm−2).b Thermal annealing at 120 °C for 10 min.c JEQEsc is the integrated Jsc over the EQE spectrum.d Fill factor.e Average over at least six devices. The error is a standard deviation.f SCLC mobility of a hole-only device (μh) and an electron-only device (μe). | ||||||||
| P1 (1:2)b |
80 | 1.25 (1.05) | 0.992 | 0.380 | 0.45 | 0.38 ± 0.04 | 2.8 × 10−6 | 1.4 × 10−5 |
| P2 (1:1) |
70 | 6.50 (6.35) | 0.875 | 0.435 | 2.30 | 1.83 ± 0.25 | 2.1 × 10−6 | 1.9 × 10−4 |
| P3 (1:1)b |
80 | 7.19 (7.48) | 0.813 | 0.411 | 2.40 | 1.97 ± 0.32 | 2.2 × 10−6 | 7.9 × 10−4 |
Surface morphologies of the blend films were observed by atomic force microscopy (AFM), which revealed large circular grains (∼170 nm diameter) in P1:PCBM (Fig. 5a). This contrasted the miscible, small grains observed in both P2:PCBM and P3:PCBM (∼50 nm diameter). The root mean square (rms) roughness of these height images were mostly comparable: 3.9 nm for P1:PCBM, 2.0 nm for P2:PCBM, and 3.1 nm for P3:PCBM. The 2-dimensional grazing-incidence X-ray diffraction (2D-GIXD) images exhibited weak, less-oriented patterns for all of the blend films (Fig. 5b). The diffraction profile of P3:PCBM in the out-of-plane (OOP) direction comprises the two peaks attributed to the interlamellar distance (dIL) of 1.87 nm and π–π stacking distance (dππ) of 0.364 nm. The dILs of P1:PCBM and P2:PCBM were 1.35 and 1.77 nm, respectively, while their π–π stacking peaks in the outer region of the hallow due to the alkyl chains (scattering vector q ∼ 14 nm−1) were not observed. Regardless of having identical side alkyl chains, the dILs of P2 and P3 were larger than that of P1. This is probably due to perturbation of the interdigitation of the side alkyl chains by the winding, regiorandom configuration of the backbone, as seen in the DFT calculations (Fig. 3c). The dIL of P1:PCBM was approximately half the end-to-end distance of the expanded 2-ethylhexyl chains of BDT-Th (∼2.7 nm), indicating that the side chains were well-interdigitated. However, the peak was relatively broad, and the crystallite size calculated using Scherrer's relation56 was small (8.4 nm). In contrast, the crystallite sizes of interlamellar and π–π stacking of P3:PCBM were 13.5 and 3.1 nm, respectively. The former is obviously larger than those of P1:PCBM (8.4 nm) and P2:PCBM (11.5 nm). The same dILs were obtained in the pristine P1–P3 polymers without PCBM (Fig. S5†). Notably, blending with PCBM increased the interlamellar crystallite sizes of P1–P3 from 4.0 to 8.4 nm, 4.3 to 11.5 nm, and 4.1 to 13.5 nm, respectively (Table S2†). In particular, P3 demonstrated the largest increase, which accompanies the appearance of a π–π stacking peak in the 2D-GIXD profile. Despite the increased crystallinity in the blend films, hole mobilities were decreased and became comparable among the polymers. This may be due to the decreased connectivity of polymer crystallites, suffered from the presence of PCBM domains in BHJ films.
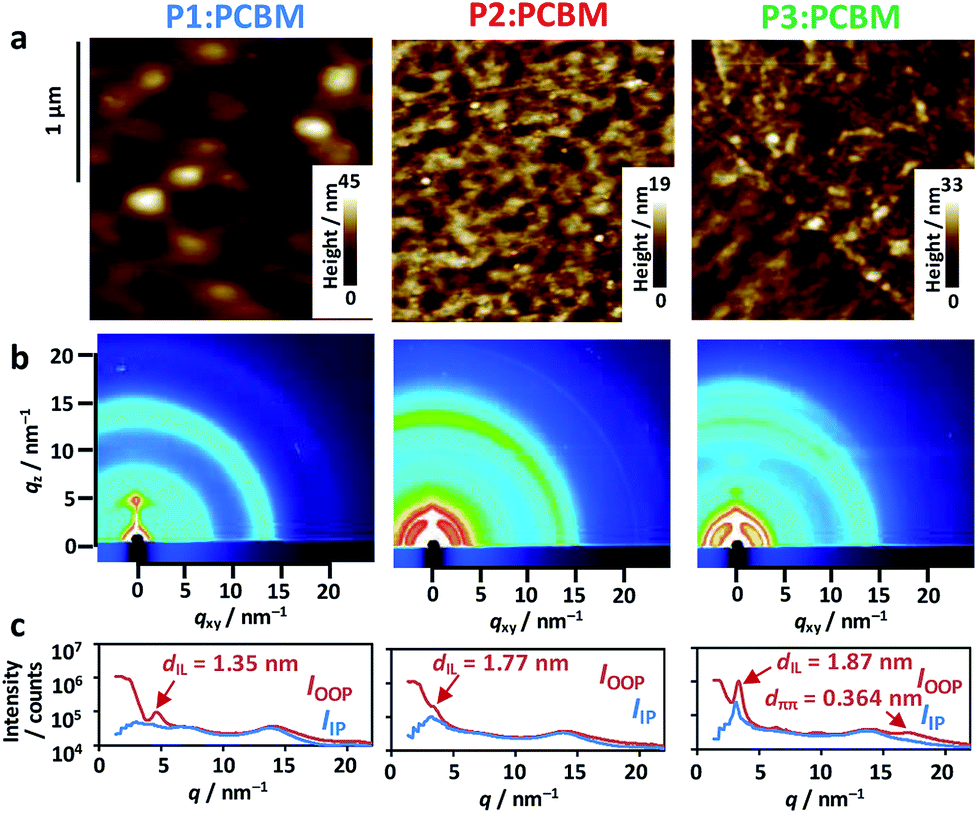 | ||
| Fig. 5 (a) AFM height images of the P1–P3 copolymer:PCBM blend films. (b) 2D-GIXD images of the blend films. (c) Out-of-plane (OOP, red line) and in-plane (IP, blue line) diffraction profiles. The interlamellar (dIL) and π–π stacking distances (dππ) are appended. | ||
Conclusions
We synthesized and characterized DPP-BDT-Th copolymers having unsymmetric and regiorandom configurations of the side aromatic rings. The unsymmetric DPP copolymers (P2 and P3) showed greater PCEs (2.3–2.4%) than the symmetric analogue P1 (0.45%), which is mainly attributed to the improved BHJ morphology associated with increased solubility of the unsymmetric polymers. The crystallinities of P2 and P3 in their respective PCBM blends were increased as compared to the corresponding pristine films, leading to enlarged crystallite sizes and relatively good μes. Nonetheless, the insufficient PCEs of P2 and P3 result from the low polymer crystallinities and low μhs (10−6 cm2 V−1 s−1) in the PCBM blends as compared to the reported symmetric Th-DPP-Th copolymers (PCE = 6–7%).19,21,53 The comparative study shows how an unsymmetric molecular design increases the solubility and affects the film morphology and OPV performance of DPP copolymers, broadening the library of conjugated polymers for applications in organic electronics.Experimental
General measurement
Steady-state photoabsorption spectroscopy was performed using a Jasco V-570 UV-vis spectrophotometer. Molecular weights (weight-averaged: Mw) and polydispersity index (PDI) of polymers were determined using the size exclusion chromatography (SEC) (gel permeation chromatography: GPC) method with polystyrene standards. SEC-GPC analysis was performed with chloroform as an eluent at a flow rate of 1 cm3 min−1 at 40 °C, on a SHIMADZU LC-20AT, CBM-20A, CTO-20A chromatography instrument connected to a SHIMADZU SPD-M20A UV-vis detector. Photoelectron yield spectroscopy (PYS) of the polymer films on indium-tin-oxide (ITO) glass was performed on a Bunko Keiki BIP-KV2016K instrument. 2D-GIXD experiments were conducted at the SPring-8 on the beam line BL46XU using 12.39 keV (λ = 1 Å) X-ray. The GIXD patterns were recorded with a 2-D image detector (Pilatus 300K). Atomic force microscopy (AFM) was carried out on a Bruker Innova AFM microscope. Differential scanning calorimetry (DSC) was performed using a Netzsch model DSC204F1 Phoenix under N2 at 10 °C min−1 (sample weight = 2.1–2.9 mg). Film thicknesses were measured using a Bruker Dektak XT surface profiler. Solubility of the polymers were measured by dissolving weighted copolymers in chlorobenzene at 80 °C, cooling down to room temperature, filtrating (0.20 μm) the solution, weighting the remained copolymer in a filter, and measuring the volume of the solution.Synthesis of polymers
Symmetric and unsymmetric DPP monomers were synthesized according to the references (Fig. 6).21,57–61 The details of the synthesis are provided in ESI.† The polymers (P1, P2, and P3) were synthesized via Stille coupling using (PPh3)4Pd catalyst from the relevant Br-DPP-Br monomer and (Me3Sn)-(BDT-2Th)-(SnMe3) monomer (Fig. 2). The yields were 36% for P1, 18% for P2, and 75% for P3. The 1H NMR spectra of monomers (Fig. S6–S8) and polymers (Fig. S9) are provided in ESI.†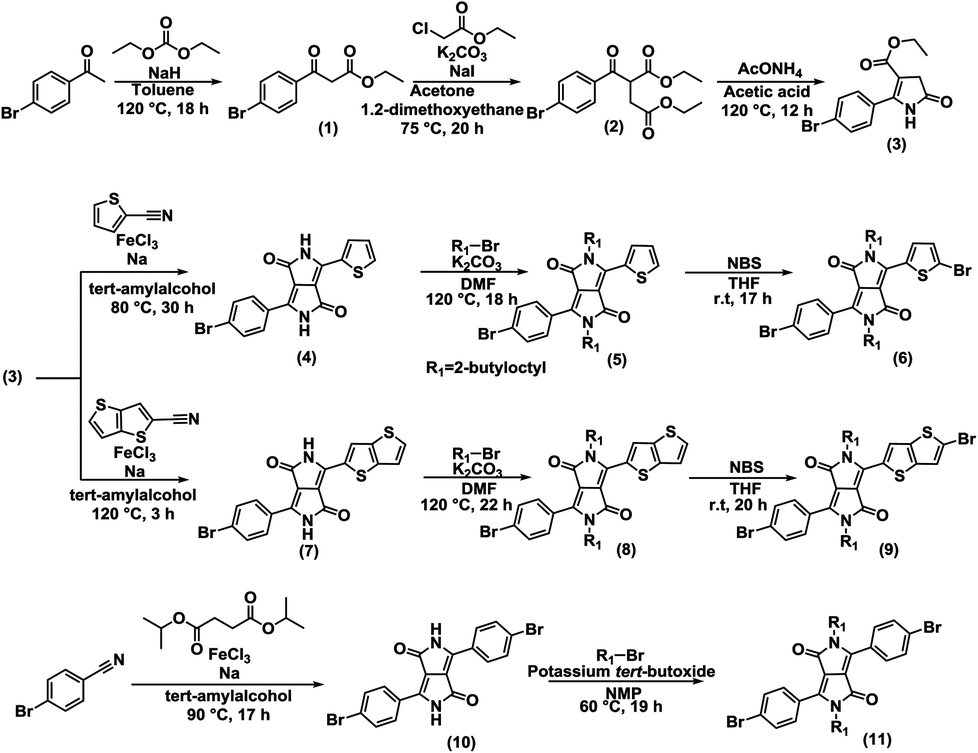 | ||
| Fig. 6 Synthesis of symmetric and unsymmetric DPP monomers. The enlarged figure and details of the synthesis are provided in ESI.† | ||
Organic photovoltaic cell (OPV)
A ZnO layer was fabricated onto a cleaned ITO layer by spin-coating with a ZnO precursor solution (0.1 g mL−1 zinc acetate dihydrate and 0.028 g mL−1 ethanolamine in 2-methoxyethanol). The substrate was annealed on a hot plate at 200 °C for 30 min. An active layer was cast on top of the ZnO layer in a nitrogen glove box by spin-coating. An anode consisting of 10 nm MoOx and 100 nm Ag layers was sequentially deposited on top of the active layers, through a shadow mask, by thermal evaporation in a vacuum chamber. The resulting device configuration was an ITO (120–160 nm)/ZnO (30 nm)/active layer/MoOx (10 nm)/Ag (100 nm) with an active area of 7.1 mm2. Current density–voltage curves were measured using a source-measure unit (ADCMT Corp., 6241A) under AM 1.5 G solar illumination at 100 mW cm−2 (1 sun, monitored by a calibrated standard cell, Bunko Keiki SM-250KD) from a 300 W solar simulator (SAN-EI Corp., XES-301S). The EQE spectra were measured by a Bunko Keiki model BS-520BK equipped with a Keithley model 2401 source meter. The monochromated light power was calibrated by a silicon photovoltaic cell, Bunko Keiki model S1337-1010BQ.Space-charge-limited current (SCLC)
The SCLC device structures consisted of an ITO/PEDOT:PSS/active layer (100–200 nm)/Au for the hole, and Al/active layer (∼200 nm)/LiF/Al for the electron. The other procedures are similar to those of the OPV device. The mobility was determined by fitting a current density–voltage curve into the Mott–Gurney law, J = 9ε0εrμV2(8L3)−1, where ε0 is the permittivity of free space, εr is the dielectric constant of the material, μ is the mobility, V is the voltage drop across the device, and L is the thickness of the active layer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant-in-Aid for Scientific Research (A) (Grant No. JP16H02285) and the PRESTO program (Grant No. JPMJPR15N6) from the JST of Japan.References
- A. J. Heeger, Adv. Mater., 2014, 26, 10 CrossRef PubMed.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633 CrossRef PubMed.
- Y. Ma, Z. Kang and Q. Zheng, J. Mater. Chem. A, 2017, 5, 1860 RSC.
- Y. Yan, X. Liu and T. Wang, Adv. Mater., 2017, 29, 1601674 CrossRef PubMed.
- M. Pfannmöller, W. Kowalsky and R. R. Schröder, Energy Environ. Sci., 2013, 6, 2871 RSC.
- A. J. Moulé and K. Meerholz, Adv. Funct. Mater., 2009, 19, 3028 CrossRef.
- H. Utzat, S. D. Dimitrov, S. Wheeler, E. Collado-Fregoso, P. Shakya Tuladhar, B. C. Schroeder, I. McCulloch and J. R. Durrant, J. Phys. Chem. C, 2017, 121, 9790 CrossRef.
- H. Benten, D. Mori, H. Ohkita and S. Ito, J. Mater. Chem. A, 2016, 4, 5340 RSC.
- L. Lu, M. A. Kelly, W. You and L. Yu, Nat. Photonics, 2015, 9, 491 CrossRef.
- T. Kumari, S. M. Lee, S.-H. Kang, S. Chen and C. Yang, Energy Environ. Sci., 2017, 10, 258 RSC.
- N. E. Jackson, K. L. Kohlstedt, B. M. Savoie, M. O. de la Cruz, G. C. Schatz, L. X. Chen and M. A. Ratner, J. Am. Chem. Soc., 2015, 137, 6254 CrossRef PubMed.
- S. Yoshikawa, A. Saeki, M. Saito, I. Osaka and S. Seki, Phys. Chem. Chem. Phys., 2015, 17, 17778 RSC.
- C. J. Mueller, E. Gann, C. R. Singh, M. Thelakkat and C. R. McNeill, Chem. Mater., 2016, 28, 7088 CrossRef.
- I. Osaka and K. Takimiya, Adv. Mater., 2017, 29, 1605218 CrossRef PubMed.
- J. C. Bijleveld, A. P. Zoombelt, S. G. J. Mathijssen, M. M. Wienk, M. Turbiez, D. M. de Leeuw and R. A. J. Janssen, J. Am. Chem. Soc., 2009, 131, 16616 CrossRef PubMed.
- A. T. Yiu, P. M. Beaujuge, O. P. Lee, C. H. Woo, M. F. Toney and J. M. J. Fréchet, J. Am. Chem. Soc., 2012, 134, 2180 CrossRef PubMed.
- J. W. Jung, F. Liu, T. P. Russell and W. H. Jo, Energy Environ. Sci., 2012, 5, 6857 RSC.
- C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859 CrossRef PubMed.
- W. Li, K. H. Hendriks, A. Furlan, W. S. C. Roelofs, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2013, 135, 18942 CrossRef PubMed.
- J.-H. Kim, M. Lee, H. Yang and D.-H. Hwang, J. Mater. Chem. A, 2014, 2, 6348 RSC.
- Y. Ji, C. Xiao, Q. Wang, J. Zhang, C. Li, Y. Wu, Z. Wei, X. Zhan, W. Hu, Z. Wang, R. A. J. Janssen and W. Li, Adv. Mater., 2016, 28, 943 CrossRef PubMed.
- W. Li, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, Acc. Chem. Res., 2016, 49, 78 CrossRef PubMed.
- G. B. Yoon, H.-Y. Kwon, S.-H. Jung, J.-K. Lee and J. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 39502 CrossRef PubMed.
- P. Sonar, H.-S. Tan, S. Sun, Y. M. Lam and A. Dodabalapur, Polym. Chem., 2013, 4, 1983 RSC.
- T. Lei, J.-Y. Wang and J. Pei, Acc. Chem. Res., 2014, 47, 1117 CrossRef PubMed.
- R. Stalder, J. Mei, K. R. Graham, L. A. Estrada and J. R. Reynolds, Chem. Mater., 2014, 26, 664 CrossRef.
- C.-C. Ho, C.-A. Chen, C.-Y. Chang, S. B. Darling and W.-F. Su, J. Mater. Chem. A, 2014, 2, 8026 RSC.
- Z. Ma, D. Dang, Z. Tang, D. Gedefaw, J. Bergqvist, W. Zhu, W. Mammo, M. R. Andersson, O. Inganäs, F. Zhang and E. Wang, Adv. Energy Mater., 2014, 4, 1301455 CrossRef.
- C.-C. Ho, C.-A. Chen, C.-Y. Chang, S. B. Darling and W.-F. Su, J. Mater. Chem. A, 2014, 2, 8026 RSC.
- W. Yue, R. S. Ashraf, C. B. Nielsen, E. Collado-Fregoso, M. R. Niazi, S. A. Yousaf, M. Kirkus, H.-Y. Chen, A. Amassian, J. R. Durrant and I. McCulloch, Adv. Mater., 2015, 27, 4702 CrossRef PubMed.
- M. Tomassetti, F. Ouhib, A. Wislez, A.-S. Duwez, H. Penxten, W. Dierckx, I. Cardinaletti, R. A. A. Bovee, G. W. P. van Pruissen, C. Jérôme, J. Manca, W. Maes and C. Detrembleur, Polym. Chem., 2015, 6, 6040 RSC.
- Y. Deng, W. Li, L. Liu, H. Tian, Z. Xie, Y. Geng and F. Wang, Energy Environ. Sci., 2015, 8, 585 RSC.
- S. Nishinaga, H. Mori and Y. Nishihara, Macromolecules, 2015, 48, 2875 CrossRef.
- X. Chen, Z. Zhang, J. Liu and L. Wang, Polym. Chem., 2017, 8, 5496 RSC.
- R. S. Ashraf, A. J. Kronemeijer, D. Ian James, H. Sirringhaus and I. McCulloch, Chem. Commun., 2012, 48, 3939 RSC.
- G. W. P. Van Pruissen, F. Gholamrezaie, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem., 2012, 22, 20387 RSC.
- Y. Koizumi, M. Ide, A. Saeki, C. Vijayakumar, B. Balan, M. Kawamoto and S. Seki, Polym. Chem., 2013, 4, 484 RSC.
- M. Ide, Y. Koizumi, A. Saeki, Y. Izumiya, H. Ohkita, S. Ito and S. Seki, J. Phys. Chem. C, 2013, 117, 26859 CrossRef.
- M. Karakawa and Y. Aso, Macromol. Chem. Phys., 2013, 214, 2388 CrossRef.
- C. Lu, H.-C. Chen, W.-T. Chuang, Y.-H. Hsu, W.-C. Chen and P.-T. Chou, Chem. Mater., 2015, 27, 6837 CrossRef.
- K. Wang, W. y. Su, B. Guo, X. Guo, M. Zhang and Y. Li, Synth. Met., 2016, 220, 134 CrossRef.
- M. Ide, Y. Koizumi and A. Saeki, J. Photopolym. Sci. Technol., 2016, 29, 565 CrossRef.
- G. K. Dutta, A.-R. Han, J. Lee, Y. Kim, J. H. Oh and C. Yang, Adv. Funct. Mater., 2013, 23, 5317 CrossRef.
- I. Meager, M. Nikolka, B. C. Schroeder, C. B. Nielsen, M. Planells, H. Bronstein, J. W. Rumer, D. I. James, R. S. Ashraf, A. Sadhanala, P. Hayoz, J.-C. Flores, H. Sirringhaus and I. McCulloch, Adv. Funct. Mater., 2014, 24, 7109 Search PubMed.
- G. Kim, S.-J. Kang, G. K. Dutta, Y.-K. Han, T. J. Shin, Y.-Y. Noh and C. Yang, J. Am. Chem. Soc., 2014, 136, 9477 CrossRef PubMed.
- G. Kim, H. Kim, M. Jang, Y. K. Jung, J. H. Oh and C. Yang, J. Mater. Chem. C, 2016, 4, 9554 RSC.
- H. J. Cho, S.-J. Kang, S. M. Lee, M. Jeong, G. Kim, Y.-Y. Noh and C. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 30755 CrossRef PubMed.
- J. S. Wilson, N. Chawdhury, M. R. A. Al-Mandhary, M. Younus, M. S. Khan, P. R. Raithby, A. Köhler and R. H. Friend, J. Am. Chem. Soc., 2001, 123, 9412 CrossRef PubMed.
- M. S. Chen, J. R. Niskala, D. A. Unruh, C. K. Chu, O. P. Lee and J. M. J. Fréchet, Chem. Mater., 2013, 25, 4088 CrossRef.
- M. Ide, A. Saeki, Y. Koizumi, T. Koganezawa and S. Seki, J. Mater. Chem. A, 2015, 3, 21578 RSC.
- M. Ide and A. Saeki, Chem. Lett., 2017, 46, 1133 CrossRef.
- Y. Wang, F. Yang, Y. Liu, R. Peng, S. Chen and Z. Ge, Macromolecules, 2013, 46, 1368 CrossRef.
- L. Dou, W.-H. Chang, J. Gao, C.-C. Chen, J. You and Y. Yang, Adv. Mater., 2013, 25, 825 CrossRef PubMed.
- L. Ying, F. Huang and G. C. Bazan, Nat. Commun., 2017, 8, 14047 CrossRef PubMed.
- X. Liu, Y. Sun, L. A. Perez, W. Wen, M. F. Toney, A. J. Heeger and G. C. Bazan, J. Am. Chem. Soc., 2012, 134, 20609 CrossRef PubMed.
- T. Erb, U. Zhokhavets, G. Gobsch, S. Raleva, B. Stühn, P. Schilinsky, C. Waldauf and C. J. Brabec, Adv. Funct. Mater., 2005, 15, 1193 CrossRef.
- M. A. Evans, D. C. Smith, J. M. Holub, A. Argenti, M. Hoff, G. A. Dalglish, D. L. Wilson, B. M. Taylor, J. D. Berkowitz, B. S. Burnham, K. Krumpe, J. T. Gupton, T. C. Scarlett, R. W. Durham Jr and I. H. Hall, Arch. Pharm. Pharm. Med. Chem., 2003, 336, 181 CrossRef PubMed.
- Y. Xu, Y. Jin, W. Lina, J. Peng, H. Jiang and D. Cao, Synth. Met., 2010, 160, 2135–2142 CrossRef.
- X. Guo, R. P. Ortiz, Y. Zheng, M.-G. Kim, S. Zhang, Y. Hu, G. Lu, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2011, 133, 13685 CrossRef PubMed.
- K. Chung, M. S. Kwon, B. M. Leung, A. G. Wong-Foy, M. S. Kim, J. Kim, S. Takayama, J. Gierschner, A. J. Matzger and J. Kim, ACS Cent. Sci., 2015, 1, 94 CrossRef PubMed.
- M. Sasikumar, D. Bharath, G. S. Kumar, N. R. Chereddya, S. Chithiravel, K. Krishnamoorthy, B. Shanigaram, K. Bhanuprakash and V. J. Rao, Synth. Met., 2016, 220, 236 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, Tables S1–S3, and Fig. S1–S9. See DOI: 10.1039/c8ra05903a |
| This journal is © The Royal Society of Chemistry 2018 |