
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: FOXC1 silencing promotes A549 cell apoptosis through inhibiting the PI3K/AKT/hedgehog/Gli2 signaling pathway†
Pei Wang *,
Hongbing Ma,
Yong Li,
Dong Chen,
Xiaohui Li and
Xiang Gao
*,
Hongbing Ma,
Yong Li,
Dong Chen,
Xiaohui Li and
Xiang Gao
Department of Cardiothoracic Surgery, Huaihe Hospital of Henan University, Baobei Road No. 8, Kaifeng 475000, China. E-mail: peiwangtx@126.com; Fax: +86-0371-23906599; Tel: +86-0371-23906599
First published on 2nd October 2018
Abstract
Lung cancer begins in the lung and is a leading cause of premature death. Forkhead box C1 (FOXC1) has been reported to play an important role in different types of cancer, and evidence suggests that FOXC1 is highly expressed in non-small cell lung cancer (NSCLC) patients. However, the function and molecular mechanism of FOXC1 in the NSCLC cell line A549 is still unclear. In the present study, we indicate that FOXC1 is expressed in the NSCLC cell lines A549, H460, and SK-MES-1 at a high level compared with control human bronchial epithelial (HBE) cells. FOXC1 silencing promotes A549 cell apoptosis, whereas it inhibits cell survival. The levels of anti-apoptosis protein Bcl-2 decreased and the expression of pro-apoptosis protein Bax increased in FOXC1 silenced cells. Further studies show that FOXC1 knockdown inhibits the PI3K/AKT/hedgehog/Gli2 pathway. Overexpressed AKT or Gli2 reversed the effects of FOXC1 silencing on A549 cell survival and apoptosis. Taken together, our results conclude that FOXC1 silencing reduced the survival of cancer cells and promoted their apoptosis, and that the PI3K/AKT/hedgehog/Gli2 pathway plays an important role in the functioning of FOXC1 silencing.
1. Introduction
Lung cancer is the most common type of malignant tumor worldwide and the leading cause of death from cancer in people.1,2 Non-small cell lung cancer (NSCLC) is the main type of lung cancer, and its proportion of cases is as high as 85%.3 The other type of lung cancer is small cell lung cancer, which accounts for 15% of cases.4 Although NSCLC can be divided into three sub-classifications, squamous cell carcinoma, large cell carcinoma and adenocarcinoma, the different histological subtypes of NSCLC do not affect chemotherapeutic or surgical decisions.5 Evidence indicates that the development of lung cancer is associated with tobacco use and cigarette smoking.6 However, about 25% of lung cancer patients all over the world have never smoked, according to global statistics from 2002.7 Recent studies have confirmed that many molecules are implicated in the regulation of lung cancer growth and apoptosis, and these genes act as oncogenes or tumor suppressor genes.8,9 Thus, studying the function and mechanism of these effective molecules would greatly facilitate the management of lung cancer.Members of the forkhead box (FOX) family of transcription factors have been divided into 17 subclasses (FoxA–FoxR), containing more than 41 members.10 FOX proteins have a three-dimensional structure, including three helices and two W1 and W2 loops.11 Evidence suggests that FOX family members play roles in malignant cell growth, tumorigenesis, and carcinogenesis.12,13 Members of the FOX family also affect the maintenance, initiation, drug resistance and progression of cancers, and they are involved in cancer cell proliferation, invasion, metastasis, apoptosis and differentiation.13 Forkhead box C1 (FOXC1), also named Fkh-1, Mf1, or FREAC3, is located at chromosome 6p25, encoding a 533 amino acid protein, and is characterized by a distinct fork head domain that can bind DNA.14–17 FOXC1, a member of the FOX family, is an important transcriptional regulator, which plays roles in a wide variety of cancers.18 However, the biological functions and specific mechanisms of FOXC1 in NSCLC have not yet been fully understood.
The hedgehog (HH) pathway, one of the most important signal transduction pathways, has been reported to play an important role in homeostasis, and tissue development and repair.19 Intercellular signaling proteins from the HH family were isolated in the early 1990s and act as pivotal mediators in embryogenesis processes.20 There is convincing evidence that the HH pathway is involved in tumorigenesis, and cancer development and treatment.21–24 Evidence has demonstrated that HH signaling contributes to glioma stem-like cell proliferation and tumorigenesis; moreover, the HH pathway may be an effective target for glioblastoma multiforme treatment.25,26 F. Wang et al. indicated that sulforaphane plays an impactful role on the gefitinib tolerance of human lung cancer cells via regulating sonic hedgehog signaling.27
In the current study, we investigated the function and mechanism of FOXC1 in NSCLC. We demonstrated that FOXC1 is highly expressed in NSCLC cells in vitro, and knockdown in the level of FOXC1, using FOXC1 siRNA, abolished A549 cell growth and induced cell apoptosis. Finally, we further corroborated that FOXC1 depletion inhibits the PI3K/AKT/hedgehog/Gli2 pathway, and that the signal pathway plays a vital role in FOXC1 siRNA induced cell apoptosis.
2. Methods
2.1. Antibodies
The antibodies used in this study were all obtained from Abcam (Abcam, Cambridge, MA, USA). The following antibodies were used: rabbit anti-human FOXC1 monoclonal antibody; rabbit anti-human Bcl-2 monoclonal antibody; rabbit anti-human Bax monoclonal antibody; rabbit anti-human pAKT monoclonal antibody; rabbit anti-human AKT polyclonal antibody; and rabbit anti-human Gli2 polyclonal antibody.2.2. Cell culture and transfection
Human bronchial epithelial cells (HBE) and the human NSCLC cell lines, A549, H460, and SK-MES-1, were purchased from American Type Cell Collection (ATCC, Manassas, VA, USA). Cells were grown in RPMI-1640 medium containing 10% fetal bovine serum, supplemented with 10% FBS, 100 μg ml−1 streptomycin and 100 U ml−1 penicillin. All NSCLC cells were cultured under 5% CO2 humidified conditions at 37 °C. Cells were transfected with FOXC1 siRNA (si-FOXC1; siRNA sequence: 5′-AACgggAATAgTAgCTgTCAA-3′) or non-specific control siRNA (si-NC), or co-transfected with si-FOXC1 and pcDNA3.1-AKT, si-FOXC1 and pcDNA3.1-Gli2, or si-FOXC1 and pcDNA3.1 empty vector, using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions.2.3. Cell survival assay
A549 cells were cultured and transfected as above. Cell survival was quantified using a commercially available kit, the CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) (Promega, Madison, WI, USA), according to the manufacturer's instructions.2.4. MTT assay
Cell survival was also determined via MTT assays. Briefly, A549 was cultured and transfected as above. Cells were plated into 96-well plates in quadruplicate at a density of 1 × 104 cells per well. 10 μL (1 mg mL−1) of MTT per well was added to the cells for another 4 h at 37 °C. The formed formazan was solubilized with DMSO and the absorbance was then measured at 570 nm.2.5. Apoptosis assay
The Cell Death Detection ELISA Plus kit (Roche Diagnostics GmbH, Mannheim, Germany) was used to measure cell apoptosis through detecting mono- and oligo-nucleosome cytoplasmic histone-associated DNA fragments in vitro.28,292.6. RT-PCR
The total RNA from A549 cells was isolated and purified using TRIzol reagent (Invitrogen) and RNeasy mini columns (Qiagen, Valencia, CA), according to the manufacturer's instructions, respectively. cDNA was synthesized via reverse transcription, using the First-Strand Synthesis System (Invitrogen). RT-PCR was performed using SYBR Green Real-Time PCR (TaKaRa), following the manufacturer's instructions. The primer sequences used in this study are as follows: FOXC1 forward: 5′-CAGCATCCGCCACAACCTCT-3′; reverse: 5′-GCAGCCTGTCCTTCTCCTCCT-3′.2.7. Western blot
The total protein was isolated from A549 cells using RIPA lysis buffer. Protein concentrations were determined using a BCA kit and equal quantities of protein were electrophoresed with 10% SDS-PAGE. The protein was then transferred onto a PVDF membrane and blocked with 5% skim milk. The primary proteins were added to the membranes overnight at 4 °C. Subsequently, horseradish peroxidase conjugated goat anti-rabbit IgG antibody (Abcam) was used as a secondary antibody.2.8. Statistical analysis
The data in this article are expressed as means ± standard deviations (SD) from triplicates of three independent experiments. GraphPad Prism 5.0 (GraphPad Software Inc., San Diego, CA, USA) and SPSS 22.0 software (SPSS, Chicago, IL) were used for statistical analyses. The significances of differences between groups were analyzed using one-way ANOVA, followed by the Bonferroni t test, with * indicating p values < 0.05.3. Results
3.1. FOXC1 increases in A549 cells
FOXC1 has been shown to be involved in the hypoxia-enhanced progression of lung cancer,33 however, the specific functions and mechanisms of FOXC1 in lung cancer are still unclear. Our study first determined the expression of FOXC1 in human bronchial epithelial (HBE) cells and the human NSCLC cell lines A549, H460 and SK-MES-1 cells. As shown in Fig. 1, compared with normal HBE cells, RT-PCR and western blot analysis displayed a significant increase in the expression of FOXC1 in A549, H460 and SK-MES-1 cells at both mRNA and protein levels.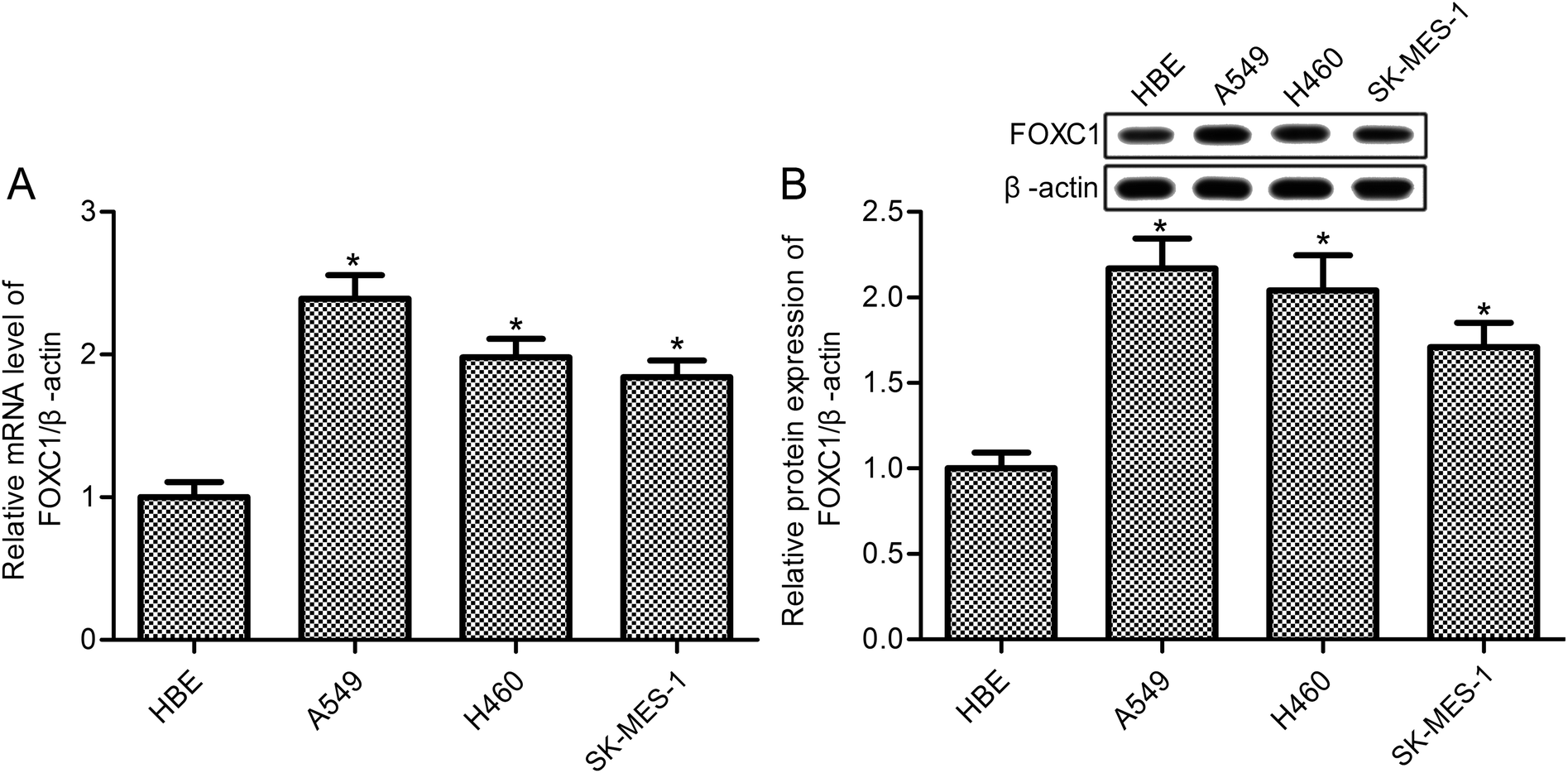 | ||
| Fig. 1 FOXC1 is increased in NSCLC cells. (A) Relative FOXC1 expression levels are examined via RT-PCR studies; and (B) relative FOXC1 protein expression is examined via western blot studies. NSCLC: non-small cell lung cancer; and HBE: human bronchial epithelial. The data in this article are expressed as means ± standard deviations (SD) of triplicates of three independent experiments. *: p values < 0.05 versus the HBE group. | ||
3.2. FOXC1 silencing inhibits A549 cell survival
To investigate the role of FOXC1, we developed a FOXC1 silencing cell model of A549 cells; the results indicate that FOXC1 expression was markedly reduced in A549 cells transfected with FOXC1 siRNA (Fig. 2A and B). To validate the functional role of FOXC1 silencing on non-small cell lung cancer cell growth, cell survival rates were assayed. FOXC1 depletion impeded A549 cell survival compared with the control group (Fig. 2C; p < 0.05). Moreover, the decrease in cell survival was confirmed in A549 cells transfected with FOXC1 siRNA (Fig. 2D; p < 0.05) via an MTT assay. However, cell survival was did not change obviously between the siNC and control groups (Fig. 2C and D; p > 0.05). To validate the effects of FOXC1 siRNA on NSCLC cell lines, SK-MES-1 cells were transfected with FOXC1 siRNA and the control siNC (ESI Fig. 1A and B†). As shown in ESI Fig. 1C,† FOXC1 depletion suppressed SK-MES-1 cell survival compared with the control group (p < 0.05).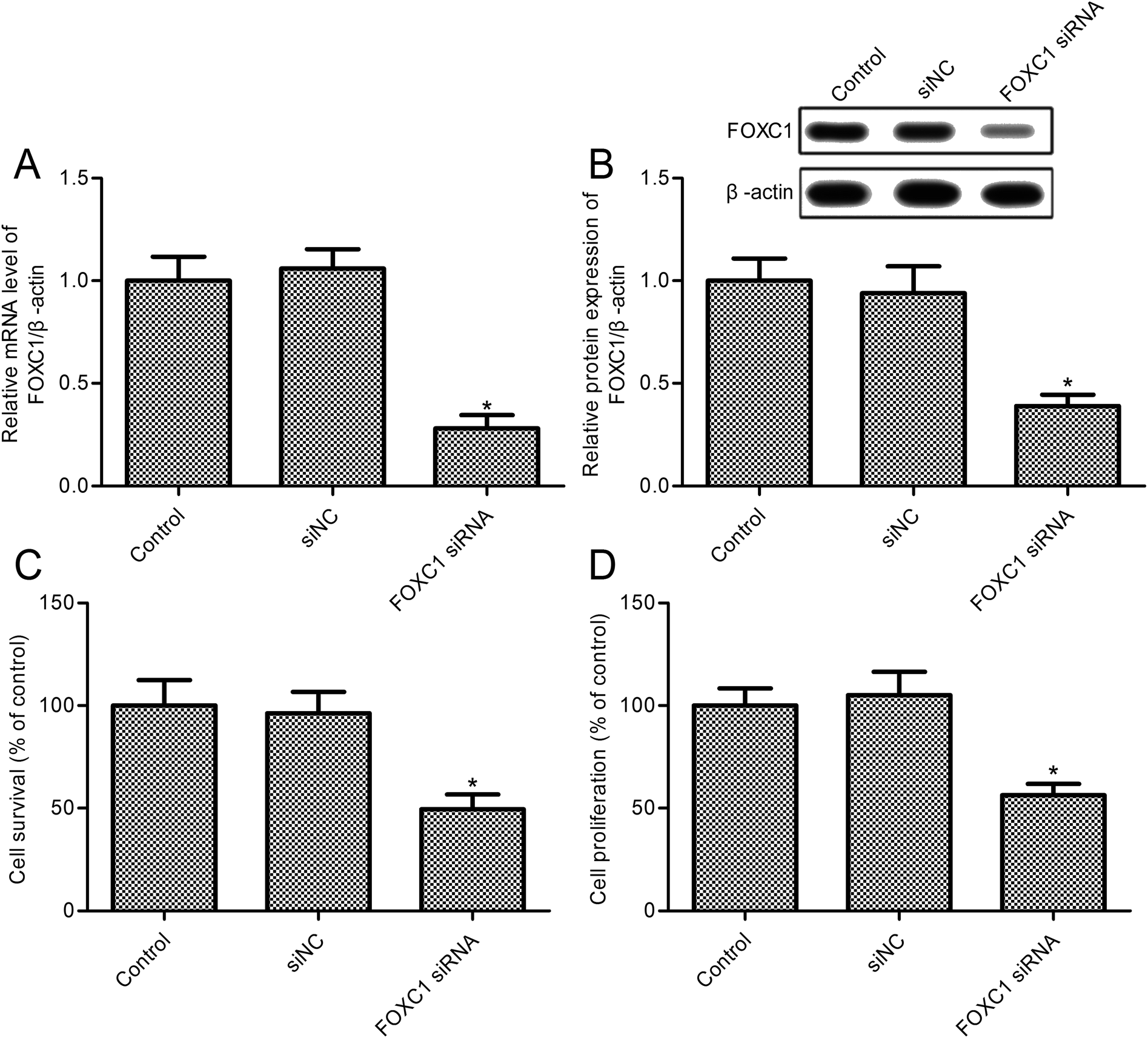 | ||
| Fig. 2 FOXC1 depletion reduces A549 cell survival. (A) Relative FOXC1 expression levels are examined via RT-PCR studies; (B) relative FOXC1 protein expression is examined via western blot studies; (C) CellTiter 96® AQueous One Solution Cell Proliferation Assays (MTS) are used to determine cell survival; and (D) relative cell survival is examined via MTT assays. A549 cells are transfected with FOXC1 siRNA or non-specific control siRNA (siNC) for 48 h. The data in this article are expressed as means ± standard deviations (SD) of triplicates of three independent experiments. *: p values < 0.05 versus the control group. | ||
3.3. FOXC1 silence promotes A549 cell apoptosis
Further studies suggest that FOXC1 knockdown in A549 cells dramatically elevated cell apoptosis (Fig. 3A). Also, FOXC1 silencing increased SK-MES-1 cell apoptosis compared with the control group (ESI Fig. 1B;† p < 0.05). To confirm the function of FOXC1 in cell apoptosis, the expressions of the apoptosis related proteins Bcl-2 and Bax were also determined. As shown in Fig. 3B, FOXC1 silencing decreased the expression of the anti-apoptosis protein Bcl-2, whereas it increased the expression of the pro-apoptosis protein Bax. These results suggest that FOXC1 may be involved in lung cancer cell apoptosis.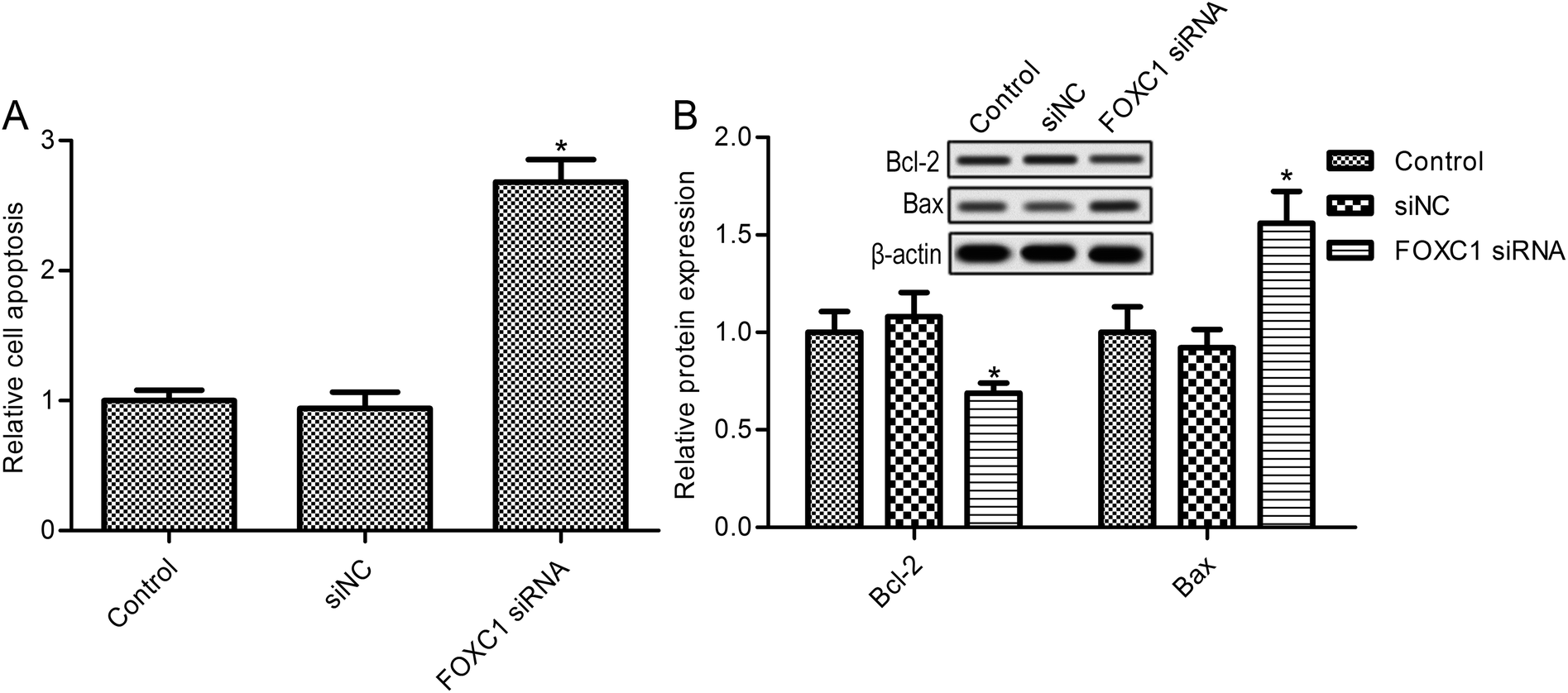 | ||
| Fig. 3 Knockdown of FOXC1 increases NSCLC cell apoptosis. (A) A Cell Death Detection ELISA Plus kit is used to detect cell apoptosis; and (B) the relative protein expressions of Bcl-2 and Bax are examined via western blot assays. A549 cells are transfected with FOXC1 siRNA or non-specific control siRNA (siNC) for 48 h. The data in this article are expressed as means ± standard deviations (SD) of triplicates of three independent experiments. *: p values < 0.05 versus the control group. | ||
3.4. The regulation of the PI3K/AKT and hedgehog/Gli2 pathways is related to FOXC1
We next looked for the signaling pathway that was involved in the function of FOXC1; evidence suggests that FOXC1 was associated with the regulation of the PI3K/AKT and hedgehog pathways.30,31 We next determined the effects of FOXC1 silencing on the expression of p-AKT and Gli2. As shown in Fig. 4, the depletion of FOXC1 markedly suppressed p-AKT and Gli2 expression compared with the control group. Additionally, overexpression of AKT reversed the inhibitory effects of FOXC1 silencing on Gli2 expression. These results indicated that FOXC1 mediated the PI3K/AKT/hedgehog/Gli2 pathway in A549 cells.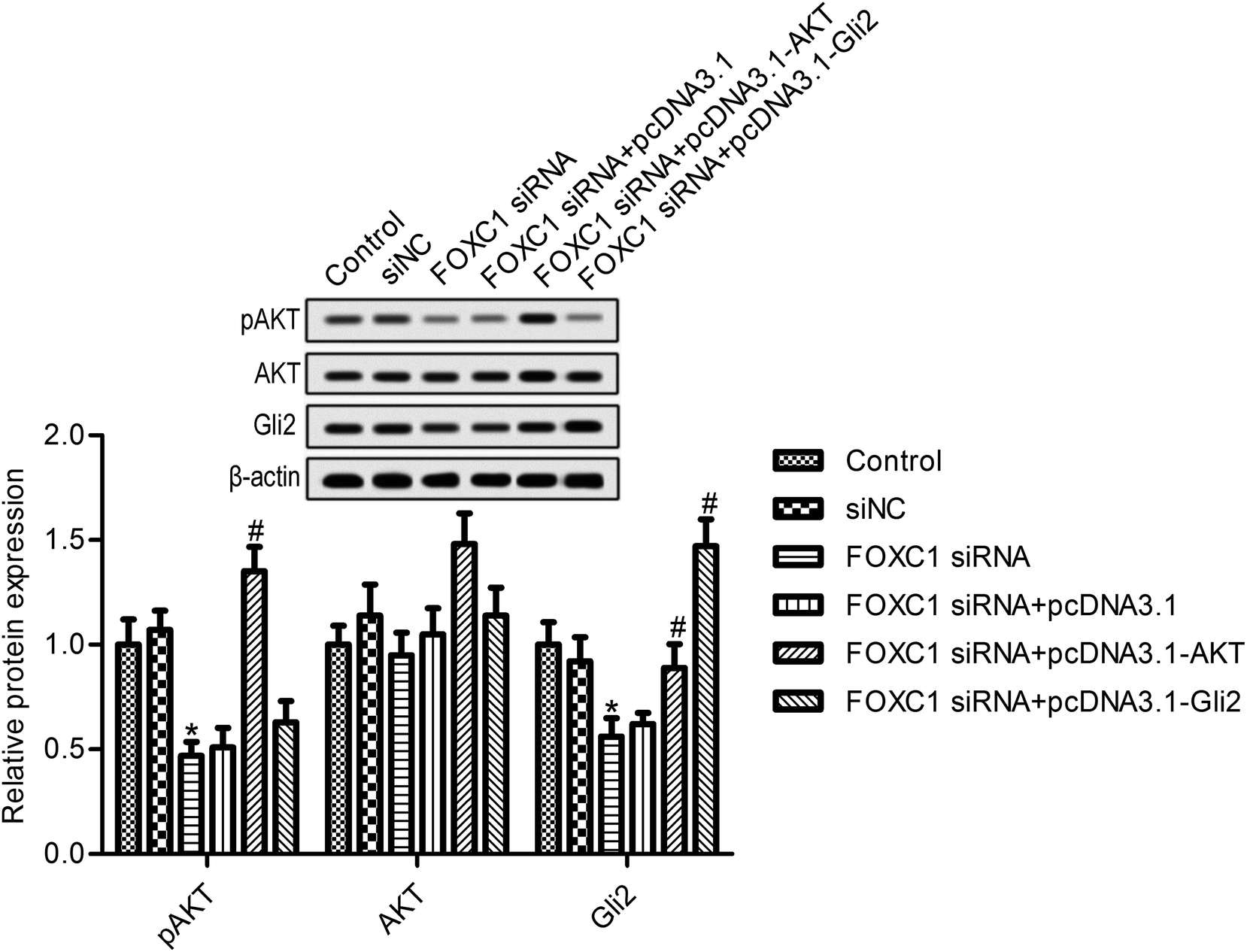 | ||
| Fig. 4 Knockdown of FOXC1 inhibits the PI3K/AKT/hedgehog/Gli2 pathway. The relative protein expressions of pAKT, AKT and Gli2 are examined via western blot assays. A549 cells are transfected with FOXC1 siRNA or non-specific control siRNA (siNC) for 48 h, or co-transfected with FOXC1 siRNA and pcDNA3.1 vector or pcDNA3.1-AKT vector for 48 h. The data in this article are expressed as means ± standard deviations (SD) of triplicates of three independent experiments. *: p values < 0.05 versus the control group; #: p values < 0.05 versus the FOXC1 siRNA group. | ||
3.5. AKT or Gli2 overexpression reverses the effects of FOXC1 silencing on A549 cell biology
To further assess the relationship between the function of FOXC1 and the PI3K/AKT/hedgehog/Gli2 pathway, we overexpressed AKT or Gli2 in A549 cells transfected with FOXC1 siRNA. As shown in Fig. 4, AKT or Gli2 overexpression reversed the FOXC1 silencing induced inhibition of p-AKT or Gli2 expression. Cell survival and apoptosis were determined. Results suggest that AKT or Gli2 overexpression increased A549 cell survival compared with the FOXC1 siRNA group (Fig. 5A and B; p < 0.05); moreover, cell apoptosis was decreased in A549 cells co-transfected with FOXC1 siRNA and pcDNA3.1-AKT or pcDNA3.1-Gli2, compared with the FOXC1 siRNA group (Fig. 5C; p < 0.05).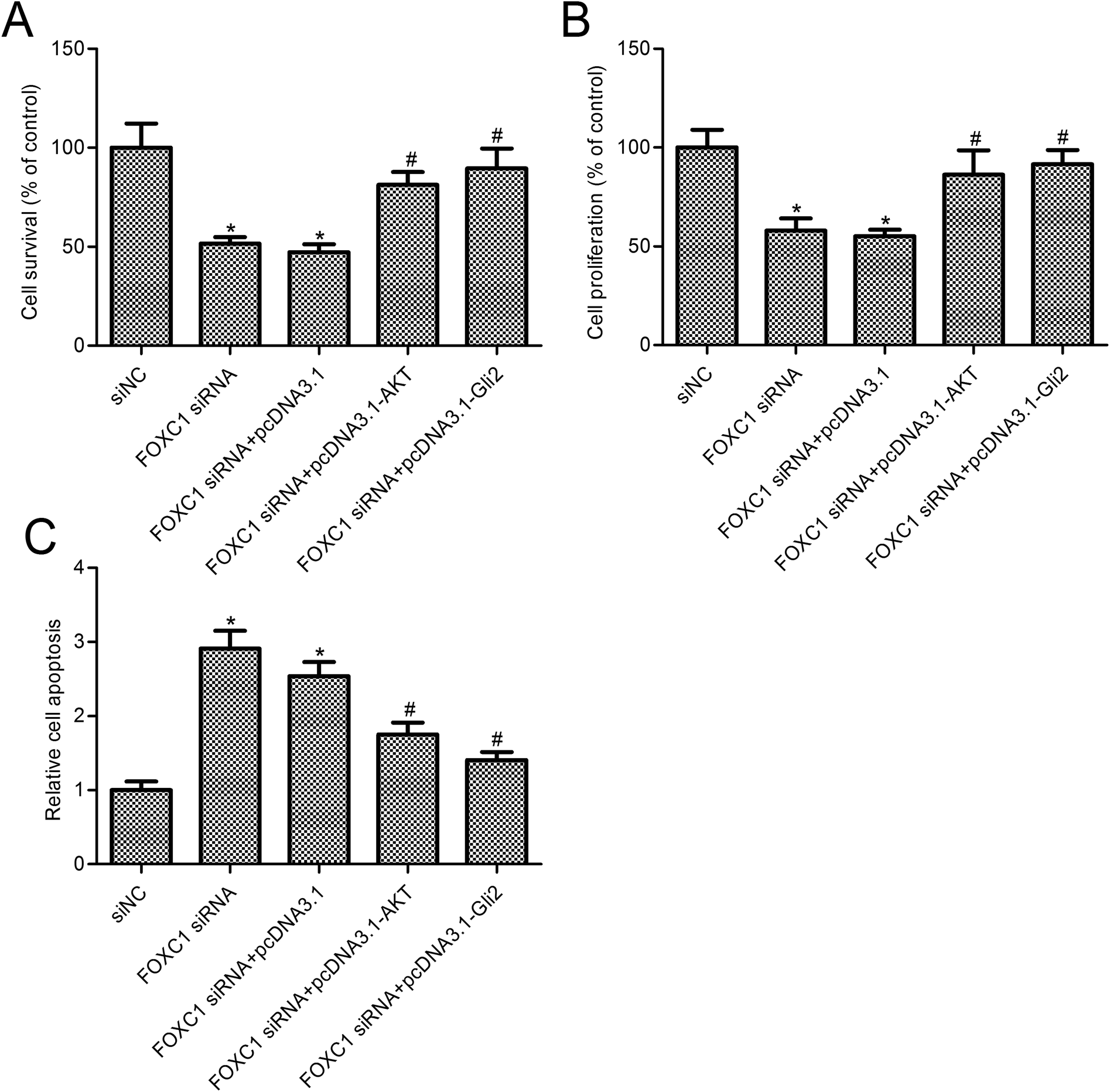 | ||
| Fig. 5 The PI3K/AKT/hedgehog/Gli2 pathway is involved in the function of FOXC1 knockdown in A549 cells. (A) CellTiter 96® AQueous One Solution Cell Proliferation Assays (MTS) are used to determine cell survival; (B) relative cell survival is examined via MTT assays; and (C) a Cell Death Detection ELISA Plus kit is used to detect cell apoptosis. A549 cells are transfected with FOXC1 siRNA or non-specific control siRNA (siNC) for 48 h, or co-transfected with FOXC1 siRNA and pcDNA3.1 vector or pcDNA3.1-AKT vector or pcDNA3.1-Gli2 vector for 48 h. The data in this article are expressed as means ± standard deviations (SD) of triplicates of three independent experiments. *: p values < 0.05 versus the control group; #: p values < 0.05 versus the FOXC1 siRNA group. | ||
4. Discussion
According to published estimates, during embryogenesis, the transcription factor FOXC1 plays a role in the development of the brain, eyes and mesoderm,32,33 and evidence indicates that FOXC1 may act as an important diagnostic biomarker for basal-like breast cancer.34,35 Evidence indicates that FOXC1 is expressed in nasopharyngeal carcinomas at a high level, and mediates N-cadherin, fibronectin and vimentin expression, affecting the epithelial-to-mesenchymal transition process of cancer.36 Data presented by Aravalli et al. confirmed that FOXC1 protein activation plays important roles in the inflammation and subsequent metastasis of hepatocellular carcinomas.17 Yu-Jung Lin et al. suggested that FOXC1, which acts as a hypoxia-responsive gene, affects the tumor microenvironment and is involved in the hypoxia-enhanced progression of lung cancer.37 These reports indicate that FOXC1 plays a role in lung cancer progression under hypoxia conditions, however, the function and mechanism of FOXC1 in NSCLC cell survival and apoptosis are still unclear. Our study firstly demonstrates that FOXC1 is highly expressed in NSCLC cell lines when compared with control cells. Further studies suggest that the depletion of FOXC1 significantly inhibits cancer cell survival and promotes cell apoptosis.The HH pathway, a highly conserved developmental pathway, shows low activity in differentiated adult tissue and has been regarded as an important mediator in cancer pathogenesis and progression.26 Bo Ram Kim et al. have demonstrated that a HH pathway associated with genipin induces colorectal cancer cell apoptosis.38 Moreover, a recent study found that sonic HH and PI3K pathways were implicated in fibroblast activation protein-α induced growth and the migration of lung cancer cells.39 The zinc-finger protein glioma-associated oncogene family members 1, 2, and 3 (Gli1, Gli2, and Gli3) are members of the HH pathway.40 A growing body of evidence suggests that the transcriptional activation of Gli genes and Gli protein posttranslational modification related signaling pathways contribute to different types of cancer formation.41 Moreover, evidence indicates that FOXC1 activates HH signaling in basal-like breast cancer cells and the knocked down expression of Gli2 in MDA-MB-231 cells reversed the activation of HH signaling by FOXC1.42 In agreement with the above results, the present study found that the HH/Gli2 pathway contributes to FOXC1 silencing induced NSCLC cell apoptosis; our study showed that FOXC1 silencing suppressed Gli2 expression, and Gli2 overexpression reversed the effects of FOXC1 depletion on NSCLC cell apoptosis.
Emerging studies have revealed that many signaling pathways exhibit crosstalk with the HH signaling pathway, such as TGF-β, rat sarcoma viral oncogene homolog GTPase (RAS)-extracellular signal-regulated kinase (ERK),43 Notch,44 Wnt,45 TGF-β,46 epidermal growth factor receptor (EGFR)47 and phosphatidylinositol 3-kinase (PI3K)-AKT-mechanistic target of rapamycin (mTOR).48 Liu Huang et al. confirmed that FOXC1 mediated cervical carcinoma cell proliferation, migration, invasion and epithelial–mesenchymal transition are dependent on the PI3K/AKT signaling pathway.31 Consistent with these reports, our results found that the AKT pathway was involved in the regulation of the HH signaling pathway. AKT overexpression reversed the inhibition of Gli2 induced by FOXC1 silencing.
5. Conclusions
In conclusion, our studies suggest that FOXC1 suppression enhanced NSCLC cell apoptosis and decreased cell survival. We confirmed that FOXC1 inhibition reduced the expression of AKT and HH/Gli2 protein expression. Finally, we found that the overexpression of AKT or Gli2 both reversed the effects of FOXC1 silencing on NSCLC cell biology. Overall, our data highlight the function and molecular mechanism of FOXC1 in NSCLC cell biology, and provide evidence that FOXC1 may be an effective target in relation to lung cancer cell growth.Conflicts of interest
The authors have no competing interests to disclose.Acknowledgements
None.References
- C. Dela Cruz, L. Tanoue and R. Matthay, Clin. Chest Med., 2011, 32, 605–644 CrossRef PubMed.
- A. Jemal, R. Siegel, E. Ward, Y. Hao, J. Xu and M. Thun, Ca-Cancer J. Clin., 2009, 59, 225–249 CrossRef PubMed.
- S. Peters, A. Adjei, C. Gridelli, M. Reck, K. Kerr and E. Felip, Ann. Oncol., 2012, 23, vii56–64 CrossRef PubMed.
- M. Reck and K. Rabe, N. Engl. J. Med., 2017, 377, 849–861 CrossRef CAS PubMed.
- A. Thomas, S. V. Liu, D. S. Subramaniam and G. Giaccone, Nat. Rev. Clin. Oncol., 2015, 12, 511–526 CrossRef CAS PubMed.
- P. Middha, S. Weinstein, S. Männistö, D. Albanes and A. Mondul, Nicotine Tob. Res., 2018 DOI:10.1093/ntr/nty115.
- D. Parkin, F. Bray, J. Ferlay and P. Pisani, Ca-Cancer J. Clin., 2005, 55, 74–108 CrossRef PubMed.
- S. Zohre, N. Kazem, A. Abolfazl, R. Mohammad, M. Aliakbar, A. Effat, D. Zahra, D. Hassan and Z. Nosratollah, Asian Pac. J. Cancer Prev., 2014, 15, 6581–6586 CrossRef PubMed.
- W. Zhang, X. Wu, L. Hu, Y. Ma, Z. Xiu, B. Huang, Y. Feng and X. Tang, Oncol. Res., 2017, 25, 843–852 CrossRef PubMed.
- S. Myatt and E. Lam, Nat. Rev. Cancer, 2007, 7, 847–859 CrossRef CAS PubMed.
- J. Wang, W. Li, Y. Zhao, n. Kang, W. Fu, X. Zheng, X. Pang and G. Du, Pharmacol. Ther., 2018, 181, 183–196 CrossRef CAS PubMed.
- O. Lehmann, J. Sowden, P. Carlsson, T. Jordan and S. Bhattacharya, Trends Genet., 2003, 19, 339–344 CrossRef CAS PubMed.
- E. Lam, J. Brosens, A. Gomes and C. Koo, Nat. Rev. Cancer, 2013, 13, 482–495 CrossRef CAS PubMed.
- H. Zhu, Life Sci., 2016, 144, 194–201 CrossRef CAS PubMed.
- S. Pierrou, M. Hellqvist, L. Samuelsson, S. Enerbäck and P. Carlsson, EMBO J., 1994, 13, 5002–5012 CrossRef CAS PubMed.
- F. Berry, R. Saleem and M. Walter, J. Biol. Chem., 2002, 277, 10292–10297 CrossRef CAS PubMed.
- R. Aravalli and T. Greten, Gastroenterology, 2015, 149, 861–863 CrossRef PubMed.
- Z. Yang, S. Jiang, Y. Cheng, T. Li, W. Hu, Z. Ma, F. Chen and Y. Yang, Ther. Adv. Med. Oncol., 2017, 9, 797–816 CrossRef CAS PubMed.
- Y. Abe and N. Tanaka, J. Dev. Biol., 2017, 5, 12–30 CrossRef PubMed.
- P. Ingham and A. McMahon, Genes Dev., 2001, 15, 3059–3087 CrossRef CAS PubMed.
- Z. Du, F. Zhou, Z. Jia, B. Zheng, S. Han, J. Cheng, G. Zhu and P. Huang, Iran. J. Basic Med. Sci., 2016, 19, 1171–1176 Search PubMed.
- Y. Wang, G. Jin, Q. Li, Z. Wang, W. Hu, P. Li, S. Li, H. Wu, X. Kong, J. Gao and Z. Li, J. Cancer, 2016, 7, 2067–2076 CrossRef CAS PubMed.
- M. Amarante, G. Vitiello, M. Rosa, I. Mancilla and M. Watanabe, Acta Oncol., 2018, 1–9 Search PubMed.
- Z. Yao, L. Han, Y. Chen, F. He, B. Sun, S. Kamar, Y. Zhang, Y. Yang, C. Wang and Z. Yang, Cell Death Dis., 2018, 9, 701 CrossRef PubMed.
- T. Takezaki, T. Hide, H. Takanaga, H. Nakamura, J. Kuratsu and T. Kondo, Cancer Sci., 2011, 102, 1306–1312 CrossRef CAS PubMed.
- J. Melamed, J. Morgan, S. Ioele, J. Gleghorn, J. Sims-Mourtada and E. Day, Oncotarget, 2018, 9, 27000–27015 CrossRef PubMed.
- F. Wang, W. Wang, J. Li, J. Zhang, X. Wang and M. Wang, Oncol. Lett., 2018, 15, 109–114 Search PubMed.
- T. Tominaga, T. Tsuchiya, K. Mochinaga, J. Arai, N. Yamasaki, K. Matsumoto, T. Miyazaki, T. Nagasaki, A. Nanashima, K. Tsukamoto and T. Nagayasu, BMC Cancer, 2016, 16, 354 CrossRef PubMed.
- P. Geiger, B. Mayer, I. Wiest, S. Schulze, U. Jeschke and T. Weissenbacher, BMC Cancer, 2016, 16, 870 CrossRef PubMed.
- B. Han, Y. Qu, Y. Yu-Rice, J. Johnson and X. Cui, Mol. Cell. Oncol., 2016, 3, e1131668 CrossRef PubMed.
- L. Huang, Z. Huang, Y. Fan, L. He, M. Ye, K. Shi, B. Ji, J. Huang, Y. Wang and Q. Li, Am. J. Transl. Res., 2017, 9, 1297–1306 CAS.
- T. Kume, K. Deng, V. Winfrey, D. Gould, M. Walter and B. Hogan, Cell, 1998, 93, 985–996 CrossRef CAS PubMed.
- K. Maclean, J. Smith, L. St Heaps, N. Chia, R. Williams, G. Peters, E. Onikul, T. McCrossin, O. Lehmann and L. Adès, Am. J. Med. Genet., Part A, 2005, 132, 381–385 CrossRef PubMed.
- T. Jensen, T. Ray, J. Wang, X. Li, W. Naritoku, B. Han, F. Bellafiore, S. Bagaria, A. Qu, X. Cui, C. Taylor and P. Ray, J. Natl. Cancer Inst., 2015, 107, 148–156 CrossRef PubMed.
- P. Ray, J. Wang, Y. Qu, M. Sim, J. Shamonki, S. Bagaria, X. Ye, B. Liu, D. Elashoff, D. Hoon, M. Walter, J. Martens, A. Richardson, A. Giuliano and X. Cui, Cancer Res., 2010, 70, 3870–3876 CrossRef CAS PubMed.
- L. Ou-Yang, S. Xiao, P. Liu, S. Yi, X. Zhang, S. Ou-Yang, S. Tan and X. Lei, Mol. Med. Rep., 2015, 12, 8003–8009 CrossRef PubMed.
- Y. Lin, W. Shyu, C. Chang, C. Wang, C. Wu, H. Lee, L. Chen and C. Hsieh, Theranostics, 2017, 7, 1177–1191 CrossRef CAS PubMed.
- B. Kim, Y. Jeong, Y. Na, S. Park, M. Jo, J. Kim, S. Jeong, S. Lee, H. Kim, S. Oh and D. Lee, Oncotarget, 2017, 8, 101952–101964 Search PubMed.
- J. Jia, T. Martin, L. Ye, L. Meng, N. Xia, W. Jiang and X. Zhang, Int. J. Mol. Med., 2018, 41, 275–283 Search PubMed.
- J. Jiang and C. Hui, Dev. Cell, 2008, 15, 801–812 CrossRef CAS PubMed.
- C. Hui and S. Angers, Annu. Rev. Cell Dev. Biol., 2011, 27, 513–537 CrossRef CAS PubMed.
- B. Han, Y. Qu, Y. Jin, Y. Yu, N. Deng, K. Wawrowsky and X. Cui, Cell Rep., 2015, 13, 1046–1058 CrossRef CAS PubMed.
- T. Mazumdar, J. DeVecchio, A. Agyeman, T. Shi and J. Houghton, Oncotarget, 2011, 2, 638–645 CrossRef PubMed.
- A. Androutsellis-Theotokis, R. Leker, F. Soldner, D. Hoeppner, R. Ravin, S. Poser, M. Rueger, S. Bae, R. Kittappa and R. McKay, Nature, 2006, 442, 823–826 CrossRef CAS PubMed.
- K. Suzuki, Y. Yamaguchi, M. Villacorte, K. Mihara, M. Akiyama, H. Shimizu, M. Taketo, N. Nakagata, T. Tsukiyama, T. Yamaguchi, W. Birchmeier, S. Kato and G. Yamada, Development, 2009, 136, 367–372 CrossRef CAS PubMed.
- D. Javelaud, M. Pierrat and A. Mauviel, FEBS Lett., 2012, 586, 2016–2025 CrossRef CAS PubMed.
- M. Eberl, S. Klingler, D. Mangelberger, A. Loipetzberger, H. Damhofer, K. Zoidl, H. Schnidar, H. Hache, H. Bauer, F. Solca, C. Hauser-Kronberger, A. Ermilov, M. Verhaegen, C. Bichakjian, A. Dlugosz, W. Nietfeld, M. Sibilia, H. Lehrach, C. Wierling and F. Aberger, EMBO Mol. Med., 2012, 4, 218–233 CrossRef CAS PubMed.
- Y. Wang, Q. Ding, C. Yen, W. Xia, J. Izzo, J. Lang, C. Li, J. Hsu, S. Miller, X. Wang, D. Lee, J. Hsu, L. Huo, A. Labaff, D. Liu, T. Huang, C. Lai, F. Tsai, W. Chang, C. Chen, T. Wu, N. Buttar, K. Wang, Y. Wu, H. Wang, J. Ajani and M. Hung, Cancer Cell, 2012, 21, 374–387 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06041j |
| This journal is © The Royal Society of Chemistry 2018 |