
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of the cytochrome P450 enzymes involved in the oxidative metabolism of trantinterol using ultra high-performance liquid chromatography coupled with tandem mass spectrometry†
Kunjie Li,
Xingjie Guo ,
Feng Qin,
Zhili Xiong,
Longshan Zhao* and
Jia Yu*
,
Feng Qin,
Zhili Xiong,
Longshan Zhao* and
Jia Yu*
Department of Analytical Chemistry, School of Pharmacy, Shenyang Pharmaceutical University, 103 Wenhua Road, Shenyang 110016, China. E-mail: yujia_yc@163.com; longshanzhao@163.com; Fax: +86-24-2398-6285; Fax: +86-24-23986421; Tel: +86-24-2398-6285 Tel: +86-24-23986421
First published on 10th October 2018
Abstract
Trantinterol is a novel β2-adrenoceptor agonist used for the treatment of asthma. This study aimed to identify the cytochrome P450 enzymes responsible for the metabolism of trantinterol to form 4-hydroxylamine trantinterol (M1) and tert-butyl hydroxylated trantinterol (M2), which was achieved using the chemical inhibition study, followed by the metabolism study of trantinterol in a panel of recombinant CYPs, as well as the kinetic study with the appropriate cDNA-expressed P450 enzymes. A highly selective and sensitive ultra high-performance liquid chromatography tandem mass spectrometry method was developed and validated for the simultaneous determination of M1 and M2. The inhibition study suggested that CYP2C19 and CYP3A4/5 were involved in the formation of M1 and M2, and CYP2D6 only contributed to the formation of M1. Assays with cDNA-expressed CYP enzymes further showed that the relative contributions of P450 isoforms were 2C19 > 3A4 > 2D6 > 2E1 for the formation of M1, and 3A4 > 2C19 > 2D6 for the formation of M2. The enzyme kinetic analysis was then performed in CYP2C19, CYP2D6 and CYP3A4. The kinetic parameters were determined and normalized with respect to the human hepatic microsomal P450 isoform concentrations. All the results support the conclusion that CYP3A4 and CYP2C19 are the major enzymes responsible for formation of M1 and M2, while CYP2D6 and CYP2E1 also engaged to a lesser degree. The results imply that potential drug–drug interactions may be noticed when trantinterol is used with CYP2C19 and CYP3A4 inducers or inhibitors, and we should pay attention to this phenomenon in clinical study.
1. Introduction
Asthma is a chronic breathing disorder disease characterized by recurrent attacks of breathlessness and wheezing. β2-adrenoceptor agonists, such as salmeterol, tulobuterol and mabuterol, are currently used extensively for the treatment of asthma. Trantinterol, 2-(4-amino-3-chloro-5-trifluorome-thylphenyl)-2-tert-butylamino-ethanol hydrochloride (Fig. 1), exhibits both a selective β2-adrenoceptor agonist activity and potent trachea relaxing activity.1 It is currently being evaluated in phase III clinical trials in China for the treatment of asthma.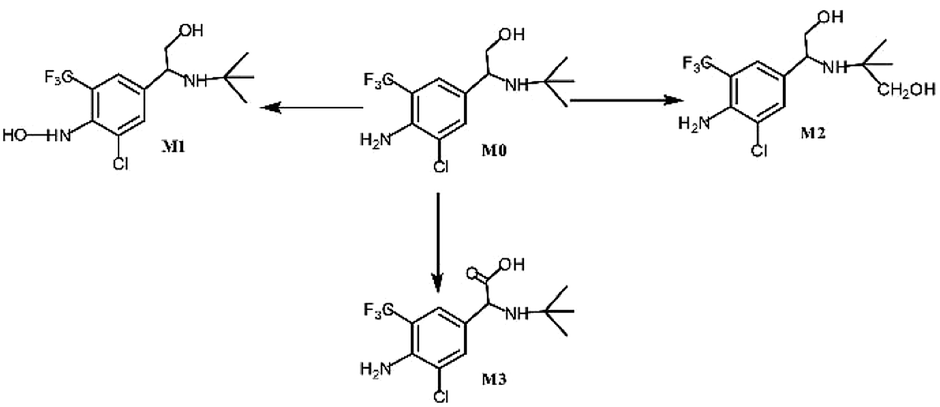 | ||
| Fig. 1 In vitro metabolic profiles of trantinterol. | ||
Our previous studies demonstrated that trantinterol was metabolized extensively in animals.2,3 The major metabolic pathways include oxidations of trantinterol to form 4-hydroxylamine trantinterol (M1), tert-butyl hydroxylated trantinterol (M2) and 1-carbonyl trantinterol (M3), as well as the glucuronide conjugations of the parent drug and phase I metabolites. Glutathione conjugation is another important in vivo metabolic pathway of trantinterol, which further undergoes catabolism and oxidation to form consecutive derivatives. Further in vitro metabolism study of trantinterol showed that incubation of trantinterol with human liver microsomes (HLMs) mainly yielded three phase I metabolites (M1, M2 and M3), and among these three metabolites, M1 and M2 were more abundant (Fig. 1). In a human excretion study, we observed that trantinterol was excreted mainly in the form of the glucuronide conjugate from human urine, followed by the total 4-hydroxylamine trantinterol (including free and glucuronide conjugate), total tert-butyl hydroxylated trantinterol (including free and glucuronide conjugate) and 1-carbonyl trantinterol.4
Cytochrome P450 enzymes (CYPs) play key roles in the clearance of many drugs, and it has been suggested that the activities and expression levels of CYPs can directly affect the bioavailability of many drugs.5 Moreover, CYPs can be inhibited or induced during concomitant medical treatment, which have been recognized as an important cause of many drug–drug interactions (DDIs), resulting in a lot of drug adverse events.6–10 In the past few years, a number of drugs have been withdraw from the market because of this reason.11,12 Thus, the in vitro identification of drug metabolizing CYPs will help develop better therapeutic strategies with regard to the prediction of possible drug–drug interactions during drug treatment, which can sufficiently improve the efficacy and reduce the toxicity.
Until now, the cytochrome P450 enzymes involved in the oxidative metabolism of trantinterol or of their relative contributions to the formation of trantinterol metabolites remain unclear. Therefore, the aim of this study was to identify the major CYP isoform(s) responsible for trantinterol hydroxylation metabolism, which was carried out using the selective chemical inhibition studies, followed by the metabolism study of trantinterol in a panel of recombinant CYPs, as well as the kinetic study with the appropriate cDNA-expressed P450 enzymes. The information obtained will help predict the potential drug–drug interactions during the clinical use of trantinterol.
2. Materials and methods
2.1 Chemicals and reagents
Trantinterol hydrochloride (99.3% purity) was generously provided by the Department of Pharmaceutical Chemistry, Shenyang Pharmaceutical University (Shenyang, China). 4-Hydroxylamine trantinterol (M1) was previously synthesized in our laboratory. tert-Butyl hydroxylated trantinterol was purified from microbial transformation culture in our laboratory. The purities of these two metabolites were above 99.0%, which were verified using high-performance liquid chromatography (HPLC). Clenbuterol (internal standard, IS, 99.4% purity) was purchased from the National Institute for Control of Pharmaceutical and Biological Products (Beijing, PR China). The chemical inhibitors for CYPs including α-Naphthoflavone (CYP1A2), quinidine (CYP2D6), sulfaphenazole (CYP2C9), ticlopidine (CYP2C19), ketoconazole (CYP3A4/5), clomethiazole (CYP2E1), and reduced nicotinamide adenine dinucleotide phosphate (NADPH) were all obtained from Sigma-Aldrich (St. Louis, MO, USA). Formic acid (HPLC grade) was purchased from Dikma (Richmond Hill, NY, USA). Methanol of HPLC grade was obtained from Tedia (Fairfield, OH, USA). All other reagents were of analytical reagent grade.Pooled human liver microsomes (HLMs) and cDNA-expressed human P450 enzymes (CYP1A2, CYP2D6, CYP2C9, CYP2C19, CYP3A4, CYP2E1 and control) prepared from baculovirus infected insect cells were obtained from BD Gentest Corporation (Woburn, MA, USA).
2.2 Biotransformation of trantinterol by human liver microsomes and cDNA-expressed P450 enzymes
Human liver microsomes and the following commercially available human cDNA-expressed P450 enzymes (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4 and CYP2E1) were incubated with trantinterol to identify the metabolites formed in vitro and to assess the involvement of each CYP isoform in trantinterol metabolites formation. The incubation system was set up in a 250 μL reaction tube, which contained human liver microsomes (1.0 mg mL−1) or cDNA-expressed P450 enzymes (50 pmol mL−1), trantinterol (50.0 μM) and NADPH (1 mM) in 100 mM Tris–HCl buffer (pH 7.4). Control incubates contained heat-inactivated human liver microsomes or control nonenzyme-expressing insect cell microsomes. After pre-incubation for 5 min at 37 °C, the reaction was initiated by adding NADPH. Reactions were kept at 37 °C for another 20 min, then terminated with 250 μL of ice-cold methanol containing 6.25 ng mL−1 clenbuterol (IS) and 0.5% ascorbic acid. All experiments were performed in duplicate. After centrifugation at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g for 10 min, a 10 μL aliquot of the supernatant was used for UHPLC-MS/MS analysis. In the following experiments, similar sample treatment procedures were used.
000 g for 10 min, a 10 μL aliquot of the supernatant was used for UHPLC-MS/MS analysis. In the following experiments, similar sample treatment procedures were used.
2.3 The effects of the selective chemical inhibitors on the formation of trantinterol metabolites
The incubation mixture (250 μL in duplicate) contained trantinterol (50.0 μM), HLM (0.5 mg mL−1), NADPH (1 mM) and a single P450 inhibitor in 100 mM Tris–HCl buffer (pH 7.4). Known selective inhibitors of CYP isoforms were used at concentrations between 1 and 10-fold of their reported Ki values.13–16 The following are the inhibitors selective for each P450 isoform and their concentrations used: α-naphthoflavone (CYP1A2 inhibitor, 0.01, 0.02, 0.05 and 0.1 μM), sulfaphenazole (CYP2C9 inhibitor, 0.3, 0.6, 1.5 and 3.0 μM), ticlopidin (CYP2C19 inhibitor, 1.2, 2.4, 6.0 and 12 μM), quinidine (CYP2D6 inhibitor, 0.4, 0.8, 2.0 and 4.0 μM), clomethiazole (CYP2E1 inhibitor, 12, 24, 60 and 120 μM), and ketoconazole (CYP3A4/5 inhibitor, 0.4, 0.8, 4.0 and 8.0 μM). The reactions were initiated by the addition of NADPH after a pre-incubation of HLMs with trantinterol and the inhibitors (except for ticlopidin) for 5 min at 37 °C. Metabolism-dependent inhibitor ticlopidin (CYP2C19 inhibitor) were pre-incubated with HLMs in the presence of NADPH for 30 min at 37 °C, then, trantinterol was added to initiate the reactions. The solutions were incubated at 37 °C for another 20 min. The other pretreatment procedures were same to those described above.Percentage of inhibition was calculated according to the following equation:17 I% = (R0 − Ri)/R0 × 100%, where I% represents the percentage of inhibition by each selective inhibitor on the formation of each metabolite, R0 represents the peak area ratio of the metabolite relative to the IS in the absence of the inhibitor, and Ri represents the peak area ratio of the metabolite relative to the IS in the presence of the inhibitor.
2.4 Kinetic study of trantinterol metabolites formation by human liver microsomes and cDNA-expressed P450 enzymes
Before assessment of the enzyme kinetics, the conditions for incubation time periods and protein concentrations both in HLMs and CYPs were optimized to achieve a linear metabolic rate. The formation rates of M1 and M2 were linear within 20 min at 0.5 mg mL−1 of human liver microsomal or 50 pmol mL−1 of CYP enzyme (data not shown). Therefore, for enzyme kinetic studies, the incubation mixture contained Tris–HCl buffer (100 mM, pH 7.4), NADPH (1 mM), HLM (0.5 mg mL−1) or 50 pmol mL−1 of cDNA-expressed P450 enzymes (CYP3A4, CYP2D6 or CYP2C19), and a series concentration of trantinterol. Twelve substrate concentrations (0, 10.0, 20.0, 40.0, 60.0, 80.0, 100, 120, 140, 160, 200, 400 mM) were evaluated in triplicate. The incubations were conducted at 37 °C for 20 min.2.5 Data analysis
The relative formation rates (peak area ratio/min pmol−1 CYP or mg HLM) of metabolites M1 and M2 were calculated and plotted against substrate concentration. The apparent kinetic parameters (Km and Vmax) for the formation of M1 and M2 were obtained by fitting the plot to the Michaelis–Menten equation [V = VmaxS/(Km + S)] using Graph Pad Prism (version 5.0, Graph-Pad Software Inc., San Diego, CA). In the equations above, S is the substrate concentration, V is the velocity of the reaction at each substrate concentration, Vmax is the maximum velocity of the reaction, Km is the substrate concentration at half of the Vmax. The intrinsic clearance (CLint) was further calculated as Vmax/Km.182.6 Quantitative determination of metabolites M1 and M2
:80, v/v) at a flow rate of 0.25 mL min−1 in an isocratic mode of elution.A Waters Micromass® Quattro micro™ API mass spectrometer ((Milford, MA, USA)) equipped with an electrospray ionization (ESI) source was used for detection and the data was acquired and processed using MassLynx™ NT 4.1 software (Waters, Milford, MA, USA). The ESI source was operated in positive ionization mode, and the MS parameters were optimized with the capillary voltage set at 1.0 kV and source temperature set at 110 °C. Nitrogen was used as desolvation and cone gas with the flow rate at 500 and 30 L h−1, respectively. Quantification was performed using multiple reaction monitoring (MRM) mode with the following transitions: m/z 327 → m/z 254 for 4-hydroxylamine trantinterol (M1), m/z 327 → m/z 238 for tert-butyl hydroxylated trantinterol (M2) and m/z 277 → m/z 203 for the internal standard clenbuterol. The optimized cone voltages and collision energies for M1, M2 and IS were 15, 12, 12 V and 15, 14, 12, 12 eV, respectively.
The calibration standard samples and quality control (QC) samples (either in the pre-study validation or during the determination of actual samples) were prepared by spiking 50 μL aliquot of M1 and M2 at different concentrations with 125 μL of Tris–HCl buffer containing denatured microsomes (1.0 mg mL−1) or control nonenzyme-expressing insect cell microsomes (100 pmol mL−1), and 25 μL of NADPH (10 mol L−1) in a total volume of 250 μL. Afterwards, the samples were mixed with equal volume of ice-cold methanol containing 6.25 ng mL−1 of IS and 0.5% ascorbic acid, and then centrifuged at 15000g for 10 min at 4 °C. The standard and QC samples were conducted on each analysis run along with the actual samples following the same processing procedures.
The selectivity was investigated by comparing the chromatograms of blank samples conducted with denatured microsomes or control nonenzyme-expressing insect cell microsomes with those of corresponding standard samples spiked with two analytes and internal standard, as well as the incubation samples of trantinterol with HLMs or individual cDNA-expressed P450 enzymes. Calibration curves for M1 and M2 in HLMs and CYPs were constructed using standard samples by plotting the analyte-to-IS peak-area ratio (y) versus concentrations (x) of analytes using 1/x2 weighted least-square linear regression. Samples at low (23.13 ng mL−1), medium (241.0 ng mL−1) and high (2410 ng mL−1) concentrations of M1 and samples at low (3.072 ng mL−1), medium (32.77 ng mL−1) and high (384.0 ng mL−1) concentrations of M2 were chosen as quality control (QC) samples to determine the accuracy, precision and stability. The LLOQs for M1 and M2 were set at 9.640 and 1.843 ng mL−1, respectively. The intra- and inter-run precision as well as the accuracy of the analytes M1 and M2 were assessed by analyzing six replicates of LLOQ and QC samples on three consecutive days (one run per day), which consisted of two sets of calibration curves per day. The relative standard deviation (RSD) was used to report the precision. Accuracy was calculated from (measured − nominal)/nominal × 100%. The stability of metabolites M1 and M2 was assessed by analyzing three replicates of low and high QC samples under various conditions, including storage at 37 °C for 40 min and at 4 °C for 12 h.
3. Results
3.1 Determination of in vitro metabolites profile
HPLC/MS-MS analysis was performed on a Triple Quadrupole LC-MS System for trantinterol incubations conducted with human liver microsomes and cDNA-expressed human P450 enzymes. The metabolites formed in the reaction solution were separated using the method as described before2 and the structures of the analyzed metabolites were confirmed by comparison of their HPLC retention times and MS/MS fragments with the corresponding reference standards. Multiple reaction monitoring (MRM) mode was further used to quantify each metabolite, with clenbuterol as the internal standard.The results demonstrated that the major metabolites formed in human liver microsomes were M1 and M2, which represented approximately 17% and 6% of the total, respectively. Whereas, the amount of M3, identified as 1-carbonyl trantinterol, was less than 1%.
Fig. 2 shows the representative multiple reaction monitoring chromatograms of metabolites M1, M2 and IS in the incubation samples with HLMs. Similar ion chromatograms were observed in the CYP2C19, CYP2D6 and CYP3A4 incubation samples, except that the relative amount of these two metabolites were different.
 | ||
| Fig. 2 Representative MRM chromatograms of M1 (channel 2), M2 (channel 3) and IS (channel 1) in (A) Blank sample; (B) blank sample spiked with the analytes at the LLOQ and IS; (C) incubation sample of trantinterol with HLMs in the presence of NADPH for 20 min at 37 °C. | ||
3.2 Kinetic study of trantinterol in human liver microsomes
The formation rates of M1 and M2 from trantinterol by HLMs fit well to the Michaelis–Menten kinetic equation. The Michaelis–Menten enzyme kinetic plots are illustrated in Fig. 3. Table 1 summarizes the determined Michaelis–Menten kinetic parameters (Km, Vmax and CLint) for the formation of M1 and M2 from trantinterol by human liver microsomes. The Km values for the formation of M1 and M2 were 174.8 ± 19.15 and 136.9 ± 14.42 μM in HLMs, respectively. Based on the Michaelis–Menten parameters (Km and Vmax), the CLint value (3.618 mL per min per g protein) of M1 was higher than that of M2 (2.622 mL per min per g protein).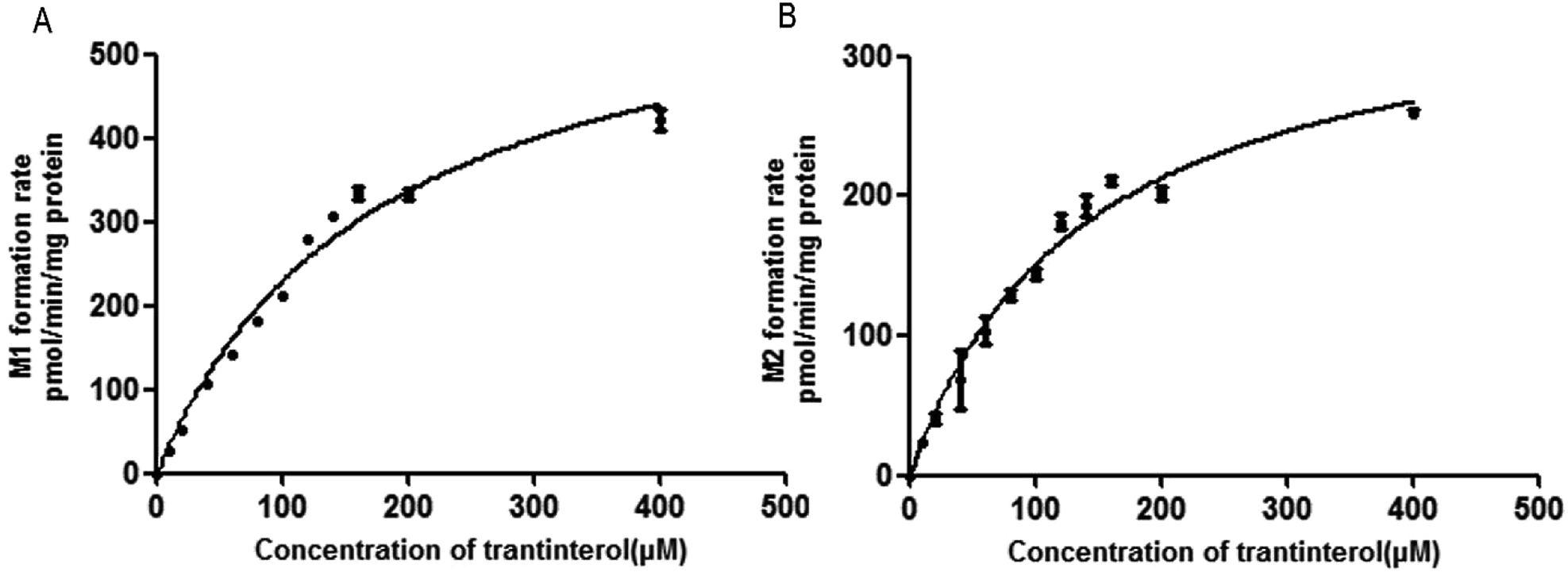 | ||
| Fig. 3 Michaelis–Menten enzyme kinetic plots for the formation of metabolites M1 (A) and M2 (B) in human liver microsomes. Each data point represents the mean ± SD of triplicate experiments. | ||
| Parameters | M1 | M2 |
|---|---|---|
| a The values are the mean ± SD of estimates from human liver microsomes in triplicate experiments. | ||
| Km (μM) | 174.8 ± 19.15 | 136.9 ± 14.42 |
| Vmax (pmol per min per mg protein) | 632.4 ± 35.91 | 358.9 ± 17.97 |
| CLint (mL per min per g protein) | 3.618 | 2.622 |
3.3 Effects of CYP isoform selective inhibitors on the biotransformation of trantinterol by human liver microsomes
Six inhibitors were used to determine which CYP isoform(s) predominate in the formation of M1 and M2. The effects of the selective inhibitors on the formation of the two metabolites are shown in Fig. 4. Our results revealed that the most pronounced decrease in the amounts of metabolites M1 and M2 formed was caused by ticlopidin (CYP2C19 inhibitor). In addition, ketoconazole (CYP3A4/5 inhibitor) was also found to inhibit the formation of the two metabolites to a lesser degree. Whereas, quinidine, which is the CYP2D6 selective inhibitor, could only inhibit the formation of M1. As is shown, the decrease in the amounts of metabolites fits well to the increase of concentration of the inhibitors, which further provides evidence for the effects of these inhibitors on the formation of trantinterol metabolites.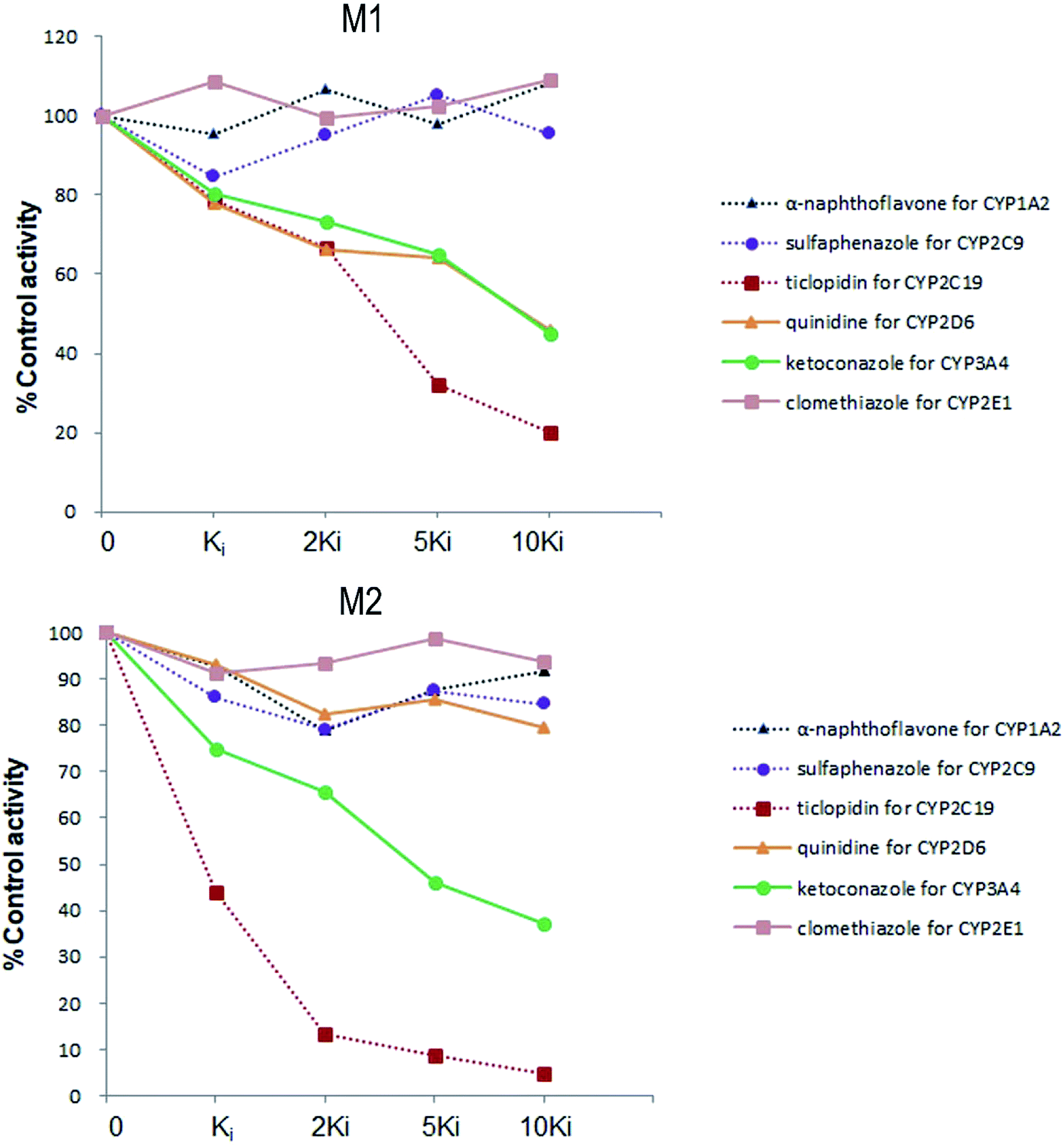 | ||
| Fig. 4 The chemical inhibition by CYP isoform-selective inhibitors on the formation of metabolites M1 and M2 in human liver microsomes. | ||
3.4 Metabolism of trantinterol in cDNA-expressed P450 enzymes
A panel of six cDNA-expressed P450 enzymes was used to evaluate the contribution of the individual CYP isoform to the biotransformation of trantinterol. As shown in Fig. 5, CYP2C19, CYP2D6, CYP3A4 and CYP2E1 were able to generate M1, and CYP2C19 shows the strongest activity. The generation of M2 from trantinterol was more efficiently catalyzed by CYP3A4, followed by CYP2C19 and CYP2D6. In contrast to the observations in the chemical inhibition experiments, trace amount of M1 in the incubation samples of CYP2E1 and trace amount of M2 in CYP2D6 were also detected, which suggested that CYP2E1 and CYP2D6 might also contribute to the formation of M1 and M2, respectively.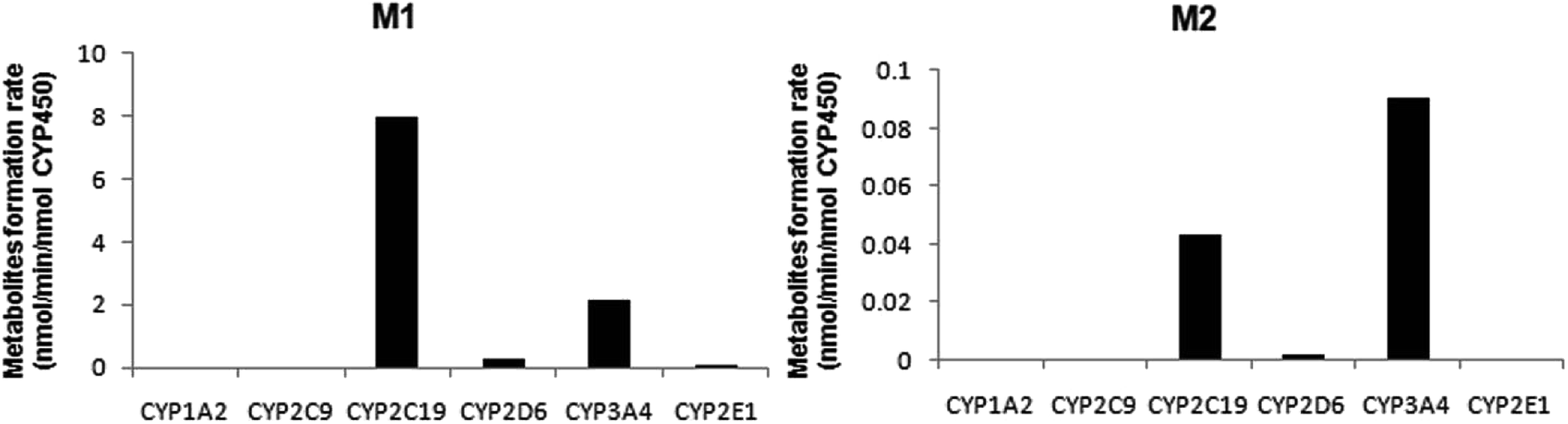 | ||
| Fig. 5 The formation rates of metabolites M1 and M2 after incubation of trantinterol with six cDNA-expressed P450 enzymes. Each CYP isoform was incubated with 50 μM trantinterol at 37 °C for 20 min. | ||
3.5 Kinetic study of trantinterol in cDNA-expressed P450 enzymes
Based on the initial screening analysis as described above, we also performed the enzyme kinetic studies for the formation of M1 and M2 from trantinterol (0–400 μM) after its incubation with recombinant CYP2C19, CYP2D6 and CYP3A4 (50 pmol mL−1). Enzyme kinetic plots of M1 formation from trantinterol by CYP2C19, CYP2D6 and CYP3A4 are shown in Fig. 6A, B and C, and plots of M2 formation by CYP2C19 and CYP 3A4 are shown in Fig. 6D and E. Estimated enzyme kinetic parameters for the formation of M1 and M2 by all the above CYP isoforms evaluated are given in Table 2. As shown in Table 2, the Km values suggest that trantinterol has a highest binding affinity to CYP2C19. Total intrinsic clearance estimates for M1 formation were 6.6-fold and 75.2-fold higher, respectively, for CYP2C19 compared with CYP3A4 and CYP2D6. However, the total intrinsic clearance estimate for M2 formation was a little bit lower for CYP2C19 compared with CYP3A4.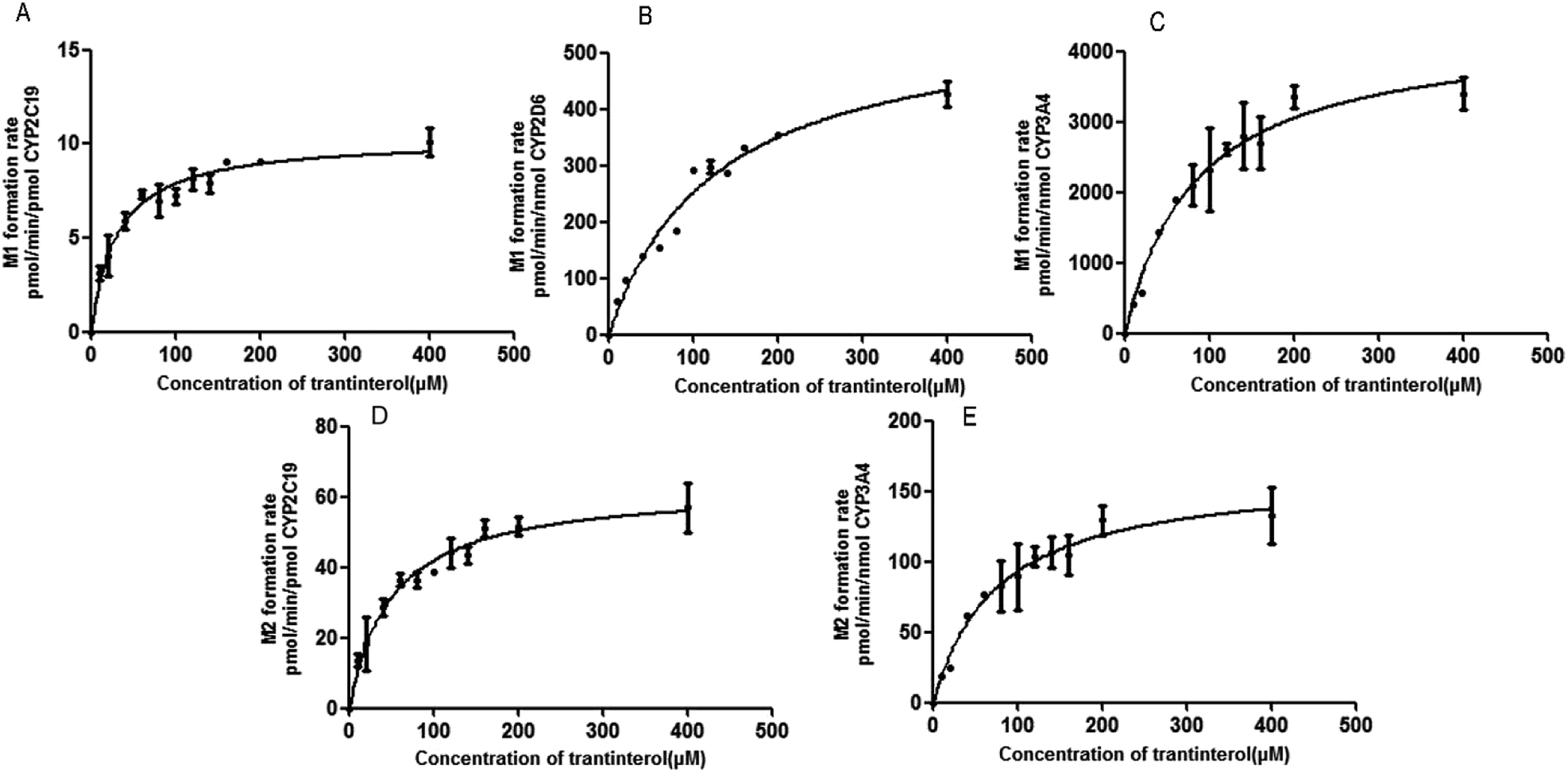 | ||
| Fig. 6 Michaelis–Menten enzyme kinetic plots of M1 formation by CYP2C19 (A), CYP2D6 (B) and CYP3A4 (C), and M2 formation by CYP2C19 (D) and CYP3A4 (E). Each data point represents the mean ± SD of triplicate experiments. | ||
| Parameters | M1 | M2 | |||
|---|---|---|---|---|---|
| CYP2C19 | CYP2D6 | CYP3A4 | CYP2C19 | CYP3A4 | |
| a The values are the mean ± SD of estimates from CYPs in triplicate experiments. | |||||
| Km (μM) | 29.88 ± 4.019 | 124.3 ± 15.58 | 81.75 ± 13.09 | 49.46 ± 6.729 | 74.56 ± 12.93 |
| Vmax (pmol per min per nmol enzyme) | 10270 ± 341.8 |
568.1 ± 32.65 | 4249 ± 266.1 | 62.83 ± 2.659 | 163.0 ± 10.54 |
| CLint (mL per min per μmol enzyme) | 343.7 | 4.570 | 51.98 | 1.270 | 2.186 |
| Relative contribution (%) | 45.32 | 0.34 | 54.34 | 6.83 | 93.2 |
3.6 Contributions of CYP isoforms to the formation of trantinterol metabolites
In order to determine the contributions of CYP isoforms to the formation of trantinterol metabolites M1 and M2, the average protein contents of the main P450s in human hepatic microsomes [CYP1A2, 52; CYP2C19, 14; CYP2C9, 73; CYP2D6, 8; CYP3A4, 111; and CYP2E1, 61 pmol per mg microsomal protein]21,22 as well as the actual intrinsic clearance values for various P450s were considered and calculated together. The relative contribution of each CYP isoform was shown in Table 2, which suggest that CYP3A4 and CYP2C19 are the predominant CYP isoforms responsible for the formation of M1, which contributed 54.34% and 45.32%, respectively. For the formation of M2, CYP3A4 and CYP2C19 contributed 93.2% and 6.83%, respectively.3.7 Method validation
The selectivity of the method was firstly determined. As is shown in Fig. 2, M1, M2 and IS were well separated, and no interference was observed at the elution times of the analytes and internal standard. The calibration curves showed good linearity (γ2 > 0.99) over the concentration range of 9.640–3212 ng mL−1 for M1 and 1.843–512.0 ng mL−1 for M2 in the HLMs and CYPs systems. The data of intra and inter-day precision and accuracy from QC samples in the incubation samples of HLMs and CYPs are summarized in Table S1,† which demonstrated that the method was accurate and precise for M1 and M2 analysis. The results from all stability tests are listed in Table S2,† which indicated that the two analyzed metabolites were stable over all steps of the procedures. All these results demonstrated that the developed UPHLC-MS-MS method is selective, sensitive, and reliable for quantification of M1 and M2 in the in vitro system.4. Discussion
Identification of the CYP enzymes involved in the drug metabolism helps to predict the potential drug interactions in vivo as well as the effect of polymorphic enzyme activity on drug disposal. Although there are more than 50 CYP isoforms, CYP1A2, CYP2C9, CYP2D6, CYP2C19, CYP3A4 and CYP2E1 are the major isoforms responsible for the metabolism of most of market drug.14,15,23,24 Therefore, the present study identified the major CYP enzymes involved in trantinterol biotransformation and assessed the contribution of the above six CYP450 isoforms to the formation of metabolites M1 and M2.Some analytical methods have been used for the study of enzyme identification and the determination of enzyme kinetic parameters. Among these methods, HPLC-MS/MS method has been adopted extensively in the recent years due to the high-selectivity, high-sensitivity, and high-speed separations, but reference standards of the analyzed metabolites are required for the quantification.24–28 In our study, reference standards of the two metabolites were synthesized or purified in our laboratory, and the structures were confirmed using nuclear magnetic resonance (NMR) as described previously.2 The purities of these two metabolites were determined by HPLC method to be above 99.0%. An UHPLC-MS/MS method, which uses UHPLC columns packed with sub-2 μm fully porous particles, was further developed and validated, which demonstrated to be rapid, selective, sensitive, reproducible and accurate. The eluent in the first 1 min was discarded using a switch valve, which could avoid the damages of the high polar constituents (mainly inorganic salts and proteins) to the ion source.
As steps preceding the enzyme identification, metabolism of trantinterol in human liver microsomes was investigated. The in vitro study showed that the biotransformation of trantinterol to metabolites M1 and M2 were NADPH dependent, indicating the involvement of cytochrome P450s in trantinterol metabolism. The results also demonstrated that the metabolites formed in vitro were structurally identical to the phase I metabolites formed in vivo, but the relative abundances of these metabolites were different. The major metabolites formed in HLMs were M1 and M2, with only slight amount of M3 detected. Whereas in the rat and dog urine as well as in human excretion and pharmacokinetic study, M3 was the one of the predominant metabolites.2–4,19,29 The results indicated that CYPs may play important roles in the metabolism of trantinterol to form 4-hydroxylamine trantinterol (M1) and tert-butyl hydroxylated trantinterol (M2). While due to the relative low abundance of metabolite M3 in HLMs and the high abundance in vivo, we may come up with the hypotheses that CYP450 enzymes may play minor roles in the metabolism of trantinterol to 1-carbonyl trantinterol (M3), which deserves further investigation. This study mainly focuses on the identification of CYPs involved in the formation of M1 and M2.
The kinetic study of trantinterol in HLMs was first studied, which suggested that the clearance value (CLint) of M1 was higher than that of M2, demonstrating the much higher catalytic efficiency of human liver microsomes for the formation of M1 relative to M2. This is consistent with the in vivo observation that the cumulative excretion amount of M1 was more than M2.4,19
The CYP isoforms involved in the formation of metabolites M1 and M2 were further carried out with the initial evaluation of the effects of selective chemical inhibitors on metabolites formation in HLMs, followed by the screening of metabolic turnover by cDNA-expressed P450 enzymes.
An ideal chemical inhibitor should inhibit only one single CYP isoform. However, some chemical inhibitors can inhibit multiple CYP isoforms at the same time, or high concentrations of inhibitors may inhibit several CYP isoforms.6 Besides, the effectiveness of a competitive inhibitor depends on the concentration of both drugs and inhibitors. Thus, a series of increased concentrations of chemical specific inhibitors in a low concentration range were finally used in our study to avoid the cross-reactivity. Moreover, selective chemical inhibition studies designed for the identification of the major CYP enzymes involved in a drug's metabolism should use drug concentration at approximately its Km value.16 Therefore, a substrate concentration of 50.0 μM was used because of the linearity, the apparent Km value of trantinterol determined in our previous kinetic experiments, the percent of conversion, and the detection sensitivity for the two metabolites. These inhibition studies suggested that CYP2C19 and CYP3A4/5 were involved in the formation of both of the two metabolites, and CYP2D6 only contributed to the formation of M1.
The results from further screening studies with cDNA-expressed P450 enzymes were more or less consistent with the conclusions from the chemical inhibition experiments, while some tiny differences were also observed. The results from the inhibition studies showed that specific chemical inhibitors of CYP1A2, CYP2C9 and CYP2E1 displayed no inhibition effect on the formation of M1 and M2. In addition, quinidine, the CYP2D6 inhibitor, showed no inhibition on the formation of M2, although it could inhibit the formation of M1 to a similar degree compared with ketoconazole (CYP3A4/5 inhibitor). In contrast to these observations, trace amount of M1 was also found in the incubation of CYP2E1, and trace amount of M2 was found when trantinterol was incubated with CYP2D6, suggesting the tiny inhibition effects of CYP2E1 and CYP2D6 on the formation of M1 and M2, respectively. One reasonable explanation for this discrepancy is that a CYP isoform may show catalytic activity in the absence of other isoenzymes, while due to the competitive inhibition, it may show little or no metabolism activity in the presence of other P450 isoenzymes.30 Overall, the consistency of the results from the chemical inhibition study and the cDNA-expressed P450 enzymes proved the reliability of our conclusion, which suggested that CYP2C19 and CYP3A4 contributed the most to the hydroxylation of trantinterol.
IF a drug is metabolized by only one CYP isoform, explanation of the results would be relatively simple and straightforward. However, if more than one CYP isoform is involved in the metabolism of a drug, measurement of enzyme activity alone does not provide information on the relative importance of an individual pathway. Thus, it is necessary to perform the kinetic studies in order to assess the contribution of each P450 isoform to the hepatic clearance of the drugs. Therefore, the enzyme kinetic analysis was then performed using the appropriate cDNA-expressed P450 enzymes according to the above screening study, which demonstrated that CYP2C19 and CYP3A4 have the highest catalytic efficiency towards M1 and M2 formation, respectively, corresponding to the highest CLint values. In addition, CYP2D6 has the lowest catalytic efficiency towards M1 formation, and the highest Km value also indicated its lowest affinity for trantinterol. This is in accordance with the observations in the screening study that CYP2D6 only provides slight contribute for the formation of M1. Due to the low concentrations of M1 in CYP2E1 and M2 in CYP2D6, their kinetic parameters were not determined.
As the relative contributions of individual CYP isoform to the clearance of one drug depend on both the catalytic activity and the relative content of each isoform in the liver, and this may result in the contrary results from studies with cDNA-expressed P450 enzymes and human liver microsomes.31 Thus, intrinsic clearance values measured with individual cDNA-expressed enzymes have to be normalized with the average protein content of each CYP isoform in human hepatic microsomes. The relative contribution of CYP3A4 to M2 formation was the highest because of both the high intrinsic clearance and average amount. Although CYP2C19 reveals the highest intrinsic clearance in vitro, it is presumably not the most relevant pathway in vivo due to its relatively low content in the liver. In addition, CYP2C19 had the highest affinity for the formation of M1 and M2 compared with other CYP isoforms, corresponding to the lowest Km. However, the calculation of relative contributions of CYPs confirmed the role of CYP3A4 in trantinterol metabolism. The important role of CYP3A4 in trantinterol metabolism was also confirmed by the observation that the metabolism of trantinterol in HLMs was inhibited to a large degree by ketoconazole, a selective inhibitor for CYP3A4/5.
Trantinterol is a selective β2-adrenergic receptor agonist with long-lasting effect, and long-time use of trantinterol is proposed for patients suffered with asthma. Hence, combination of trantinterol with other drugs will be common. As a newly developed drug, it is necessary to use the in vitro data to predict the possible drug–drug interaction, which will help to guide the clinical use of trantinterol. Identification of the main enzymes involved the trantinterol metabolism as well as the characterization of its inhibition and induction effects on various enzymes are two important aspects for the prediction of metabolism-based DDI. At present there are few investigations about the possible DDIs of trantinterol with other drugs. Kun Jiang et al.32 reported the effects of trantinterol on the activities of various CYP isoforms, which indicated that trantinterol showed no significant CYP450 inhibition or induction. Our results of this study further predict that potential drug–drug interactions may occur when trantinterol is co-administered with CYP3A4 and CYP2C19 inhibitors or inducers. In addition, we should notice that CYP2C19 is genetically polymorphic,33,34 and the hydroxylation of trantinterol may be affected to a certain degree by the wide interindividual variation of CYP2C19 activity caused by polymorphisms. However, whether these predicted interactions of trantinterol have significant clinical relevance must be evaluated by clinical studies.
5. Conclusions
Taken together, CYP3A4 and CYP2C19 are the major enzyme responsible for formation of M1 and M2, with CYP2D6 and CYP2E1 also engaged to a lesser degree. Our findings suggested that potential drug–drug interactions may be noticed when trantinterol is used combined with CYP2C19 and CYP3A4 inducers or inhibitors, and we should concern about this phenomenon in clinical study.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 81102505).Notes and references
- L. L. Gan, M. W. Wang, M. S. Cheng and L. Pan, Biol. Pharm. Bull., 2003, 26, 323 CrossRef CAS.
- K. J. Li, F. Qin, L. J. Jing, F. M. Li and X. J. Guo, Anal. Bioanal. Chem., 2013, 405, 2619 CrossRef CAS PubMed.
- Y. J. Wang, X. M. Lu, K. Jiang, Z. L. Xiong, M. S. Cheng and F. M. Li, Biomed. Chromatogr., 2010, 24, 274 CAS.
- F. Qin, L. J. Wang, K. J. Li, Z. L. Xiong and F. M. Li, J. Chromatogr. B, 2015, 997, 64 CrossRef CAS PubMed.
- S. Rendic, Drug Metab. Rev., 2002, 34, 83 CrossRef CAS PubMed.
- W. Guo, X. W. Shi, W. Wang, W. L. Zhang and J. X. Li, Environ. Toxicol. Pharmacol., 2014, 38, 901 CrossRef CAS PubMed.
- H. H. Tsai, H. W. Lin, Y. H. Lu, Y. L. Chen and G. B. Mahady, PLoS One, 2013, 8, e64255 CrossRef CAS PubMed.
- W. Qu and X. Z. Liu, Biomed. Chromatogr., 2018, 32, e4149 CrossRef PubMed.
- C. E. Leonard, C. M. Brensinger, W. B. Bilker, S. E. Kimmel, X. Han, Y. H. Nam, J. J. Gagne, M. J. Mangaali and S. Hennessy, Int. J. Cardiol., 2017, 228, 761 CrossRef PubMed.
- A. Yamada, N. Shimizu, T. Hikima, M. Takata, T. Kobayashi and H. Takahashi, Biochemistry, 2016, 55, 388838 Search PubMed.
- C. Lecefel, P. Eloy, B. Chauvin, B. Wyplosz, V. Amilien, L. Massias, A.-M. Taburet, H. Francois and V. Furlan, J. Clin. Pharm. Ther., 2015, 40, 119 CrossRef CAS PubMed.
- C. Lu, A. Suri, W. C. Shyu and S. Prakash, Biopharm. Drug Dispos., 2014, 35, 543 CrossRef CAS PubMed.
- M. Bourrie, V. Meunier, Y. Berger and G. Fabre, J. Pharmacol. Exp. Ther., 1996, 277, 321 CAS.
- Y. Sai, R. Dai, T. J. Yang, K. W. Krausz, F. J. Gonzalez, H. V. Gelboin and M. Shou, Xenobiotica, 2000, 30, 327 CrossRef CAS PubMed.
- X. Q. Li, T. B. Andersson, M. Ahlström and L. Weidolf, Drug Metab. Dispos., 2004, 32, 821 CrossRef CAS.
- U. S. Food and Drug Administration, Drug Interactions & Labeling-Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers, https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm093664.htm, Accessed November, 2017.
- W. H. Jing, Y. L. Song, R. Yan and Y. T. Wang, J. Pharm. Biomed. Anal., 2013, 77, 175 CrossRef CAS PubMed.
- A. J. Sadeque, S. Palamar, K. A. Usmani, C. Chen, M. A. Cerny and W. G. Chen, Drug Metab. Dispos., 2016, 44, 570 CrossRef PubMed.
- K. J. Li, Y. J. Wang, L. L. Zhang, F. Qin, X. J. Guo and F. M. Li, J. Chromatogr. B, 2013, 934, 89 CrossRef CAS PubMed.
- U. S. Food and Drug Administration. Guidance for Industry: Bioanalytical Method Validation, https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070107.pdf, Accessed May 2001.
- A. Rostami-Hodjegan, A. Rostami-Hodjegan and G. T. Tucker, Nat. Rev. Drug Discovery, 2007, 6, 140 CrossRef PubMed.
- K. Abass, P. Reponen, S. Mattila and O. Pelkonen, Chem.-Biol. Interact., 2010, 185, 163 CrossRef CAS PubMed.
- D. W. Nebert and D. W. Russell, Lancet, 2002, 360, 1152 CrossRef.
- X. M. Zhuang, L. Chen, Y. Tan, H. Y. Yang, C. Lu, Y. Gao and H. Li, Chin. J. Nat. Med., 2017, 15, 695 Search PubMed.
- W. H. Jing, Y. L. Song, R. Yan and Y. T. Wang, J. Pharm. Biomed. Anal., 2013, 77, 175 CrossRef CAS PubMed.
- L. M. Nielsen, N. B. Holm, S. Leth-Petersen, J. L. Kristensen, L. Olsen and K. Linnet, Drug Test. Anal., 2016, 107, 2411 Search PubMed.
- T. E. Perez, K. L. Mealey, T. L. Grubb, S. A. Greene and M. H. Court, Drug Metab. Dispos., 2016, 44, 1963 CrossRef CAS PubMed.
- S. Nicole and S. Gisela, Anal. Bioanal. Chem., 2014, 406, 2325 CrossRef PubMed.
- F. Qin, B. C. Yin, L. J. Wang, K. J. Li, F. M. Li and Z. L. Xiong, J. Pharm. Biomed. Anal., 2016, 117, 413 CrossRef CAS PubMed.
- G. T. Tucker, J. B. Houston and S. M. Huang, Biochem. Pharmacol., 2001, 70, 103 CAS.
- X. A. He, X. Luo, Z. Liu, G. Y. Hu and Z. N. Chen, Xenobiotica, 2011, 41, 844 CrossRef CAS PubMed.
- K. Jiang, K. J. Li, F. Qin, X. M. Lu and F. M. Li, Toxicol. In Vitro, 2011, 25, 1033 CrossRef CAS PubMed.
- K. Probst-Schendzielorz, R. Viviani and J. C. Stingl, Expert Opin. Drug Metab. Toxicol., 2015, 11, 1219 CrossRef CAS PubMed.
- Y. Yao, J. P. Lewis, J. Hulot and A. S. Stuart, Expert Opin. Drug Metab. Toxicol., 2015, 11, 1599 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06219f |
| This journal is © The Royal Society of Chemistry 2018 |