
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of 4-isopropyl–thiotropolone as a novel anti-microbial: regioselective synthesis, NMR characterization, and biological evaluation†
Mohamed Elagawanyab,
Lamees Hegazya,
Feng Caoc,
Maureen J. Donlind,
Nigam Rathe,
John Tavisf and
Bahaa Elgendy *ag
*ag
aDepartments of Pharmacology and Physiology, Saint Louis University School of Medicine, St. Louis, MO, USA
bDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Damanhour University, Damanhour, Egypt
cJohn Cochran Division, Department of Veteran's Affairs Medical Center, 915 North Grand Blvd., St. Louis, MO 63106, USA
dEdward A. Doisy Department of Biochemistry and Molecular Biology, Saint Louis University School of Medicine, St. Louis, Missouri, USA
eDepartment of Chemistry and Biochemistry, Center for Nanoscience, University of Missouri – St. Louis, One University Boulevard, St. Louis, USA
fDepartment of Molecular Microbiology and Immunology, The Saint Louis University Liver Center, Saint Louis University School of Medicine, St. Louis, Missouri, USA
gChemistry Department, Faculty of Science, Benha University, Benha 13518, Egypt. E-mail: belgendy@fsc.bu.edu.eg
First published on 23rd August 2018
Abstract
We have synthesized and separated tosylated thujaplicin isomers for the first time, and elucidated their structures using 1D, 2D-NMR techniques and X-ray crystallography. The tosylated isomers were used to synthesize 4-isopropyl–thiotropolone and 6-isopropyl–thiotropolone in a regioselective manner. 1H and 13C Chemical shifts of synthesized isomers were fully assigned using several NMR experiments, and their isotropic magnetic shielding was calculated using the GIAO (Gauge Including Atomic Orbitals) method and the B3LYP def2-TZVPP level of theory. The calculated chemical shift values were in a good agreement with the experimental results. The biological activity of all synthesized compounds was evaluated against the fungal pathogen Cryptococcus neoformans and four different bacterial strains (Staphylococcus aureus (ATCC 29213), E. coli (ATCC 35218), Acinetobacter baumannii and Pseudomonas aeruginosa (ATCC 27853)). 4-Isopropyl–thiotropolone was found to inhibit Staphylococcus aureus in a low micro molar range and exhibit good therapeutic index and ADME properties. This compound can be used for future lead optimization to design inhibitors against Staphylococcus aureus (ATCC 29213).
Introduction
The troponoid core is well known in hundreds of natural products. The tropone ring has a unique structure and properties and despite their non-benzenoid nature, they are planar and possess aromatic character. Natural and synthetic compounds that have the troponoid core have profound biological activities and have been used as antitumor,1–3 antifungal,4 and antimicrobial agents.5 The biological activities of these compounds has been mainly attributed to their inhibitory effect on bimetallic enzymes.6,7 Colchicine (1) is the most recognized member of this family because it has been used for the treatment of gout8 and familial Mediterranean fever9 since the time of ancient Egyptians.10 Hinokitiol (β-thujaplicin) (2) is among the known tropolone derivatives that showed inhibitory effects against Chlamydia trachomatis and has clinical potential to be used as a topical drug to prevent sexually transmitted diseases.11Thiotropolones have not been widely explored compared to structurally related hydroxytropolones. Some examples exist in literature that shows anticancer activity such as thiocolchicin (3), which can act as tubulin12 or topoisomerase II inhibitor.13 Thiotropocin (4) is another structurally related antibiotic that was isolated from Pseudomonas sp. CB-104 in 1984.14,15 This antibiotic was found to exhibit antibacterial, antifungal, and antiprotozoal activities in vitro. Recently, Donlin and co-workers4 reported that thiotropolone 5 can inhibit the growth of the fungus Cryptococcus neoformans with a minimal 80% inhibitory concentration (MIC80) of 0.25 μM. In our effort to design and synthesize new thiotropolones to treat pathogenic fungi and multidrug resistant bacteria, we used the natural product β-thujaplicin (2) as starting point (Fig. 1).
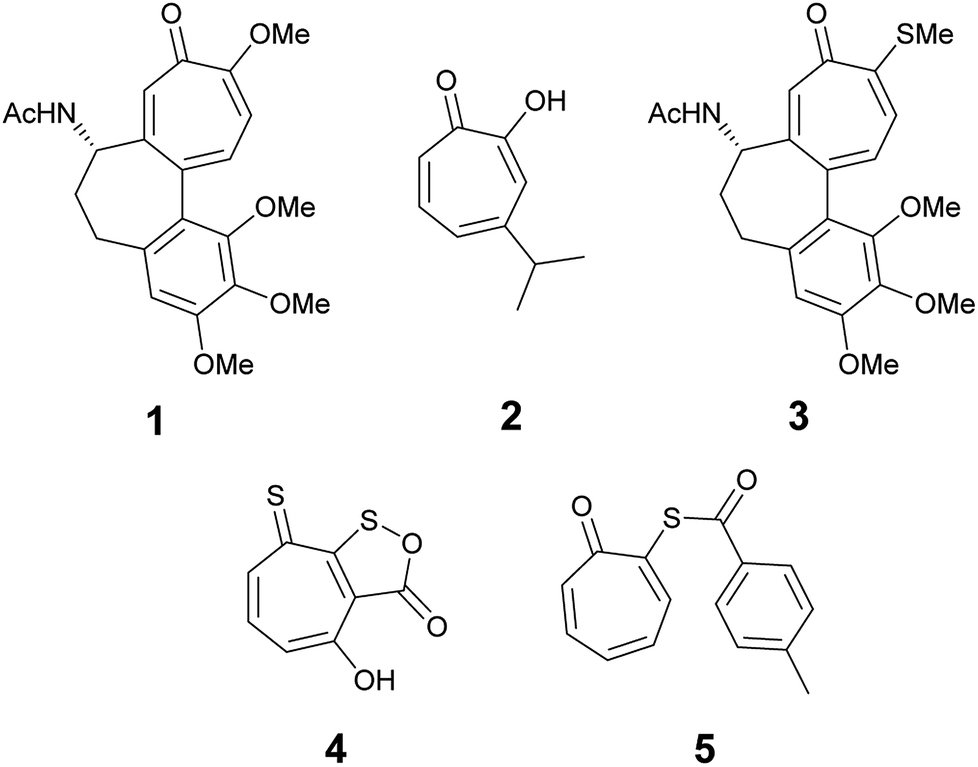 | ||
| Fig. 1 Examples of biologically active tropolones. | ||
Here, we report for the first time the isolation of the intractable mixture of tosylated thujaplicines, 8 and 9, their full NMR assignments and the X-ray structure of 8. Moreover, we report the synthesis, full NMR chemical shift assignments of 4-isopropyl and 6-isopropyl-2-mercaptocyclohepta-2,4,6-trien-1-one, 10 and 11, and their anti-Cryptococcus neoformans and antibacterial activity. Moreover, we have evaluated the ADME (adsorption, distribution, metabolism, excretion and toxicity) properties of tested compounds.
Results and discussion
Tosylation is usually the first step toward functionalization of hydroxytropolones to prepare biologically active derivatives. Under standard conditions of tosylation, where base is used in excess, the formed anion of unsymmetrical hydroxytropolones (Fig. 2A) undergoes isomerization to give a mixture of isomers. During their attempt to synthesize O-alkyl derivatives of 4-isopropyl–tropolone as ribonucleotide reductase inhibitors, Crozet and his co-workers16 observed the formation of a mixture of 4- and 6-isopropyl derivatives (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture). They were only able to obtain sufficient amounts of the 4-isopropyl derivatives in a pure form suitable for testing. Similarly, tosylation of colchicine (1) gave the biologically inactive 9-tosyloxycolchicide isomer (6) as the major product (≈74%) while the biologically active 10-tosyloxycolchicide isomer (7) was the minor product (≈26%).17
:1 mixture). They were only able to obtain sufficient amounts of the 4-isopropyl derivatives in a pure form suitable for testing. Similarly, tosylation of colchicine (1) gave the biologically inactive 9-tosyloxycolchicide isomer (6) as the major product (≈74%) while the biologically active 10-tosyloxycolchicide isomer (7) was the minor product (≈26%).17
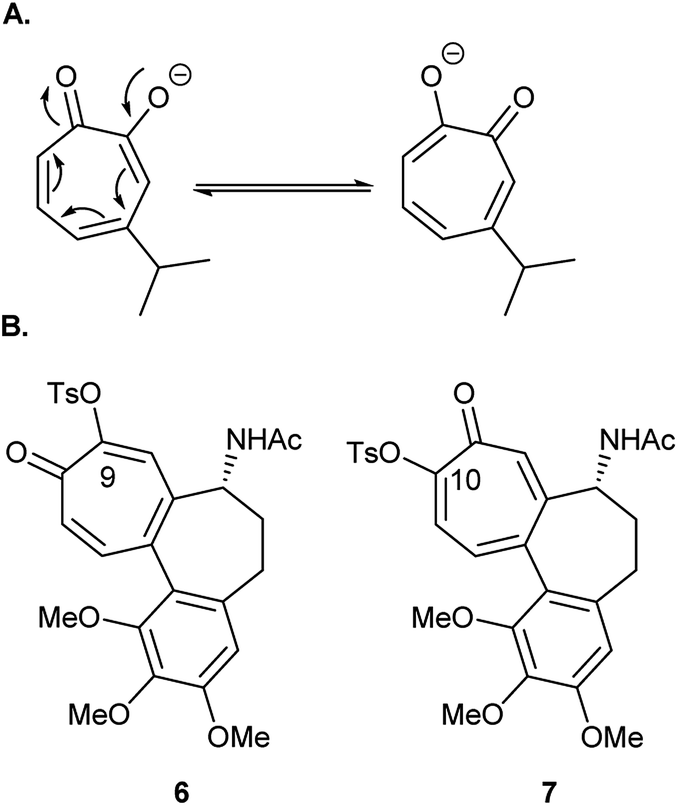 | ||
| Fig. 2 (A) β-Thujaplicin anions. (B) Tosyloxycolchicides 6 and 7. | ||
Tosylated thujaplicines, 8 and 9, are important synthetic intermediates in the synthesis of many biologically active tropolones. For example, they were used as an intermediate in the preparation of copper aminotropones, which were found to be more effective than a commercial toothpaste formulation in inhibiting plaque formation.18 The inability to separate 8 and 9 and their use as a mixture of isomers complicates the purification of the desired ligands. Obtaining such mixture of isomers is undesirable, and selective tosylation or facile separation of isomers is crucial from the synthetic point of view.18
Hinokitiol (β-thujaplicin) (2) was treated with p-toluenesulfonyl chloride in presence of pyridine in dichloromethane (DCM) at room temperature to give a mixture of 5-isopropyl-7-oxocyclohepta-1,3,5-trien-1-yl-4-methylbenzenesulfonate (8) and 3-isopropyl-7-oxocyclohepta-1,3,5-trien-1-yl-4-methylbenzenesulfonate (9) in ≈1:1 ratio. The isolation of this mixture was reported to be intractable.19 To obtain 9 (the isomer of the natural product) exclusively, it has to be through multistep synthesis. The first step involves iodination of 2 in position 7 followed by tosylation of hydroxyl group at position 2. The final step is to remove the iodine by hydrogenation using Pd/C (10%) in presence of anhydrous sodium acetate.19 Nevertheless, we were able to separate the two isomers using a Reveleris X2 flash chromatography system. The structures of 8 and 9 (Scheme 1) were elucidated using 1D and 2D NMR spectroscopy and single crystal X-ray diffraction.
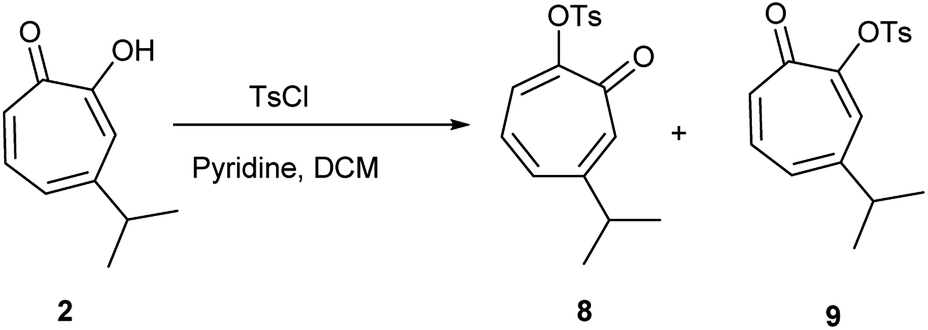 | ||
| Scheme 1 Synthesis of compounds 8 and 9. | ||
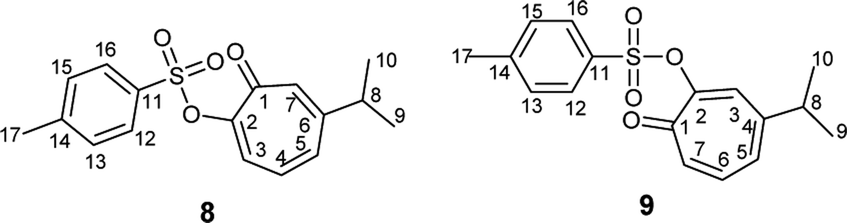
Compound 8 was obtained as colorless sharp needles. Its molecular formula C17H18O4S was determined by HRESIMS data (m/z 341.0816 [M + Na]+, calc. 341.0818). The analysis of 1H, 13C, COSY, HSQC, and HMBC spectra revealed the presence of a singlet proton H-7 (δC/δH 137.7/7.05, C-7). This proton shows two bond correlations with C-1 (δC 179.1) and C-6 (δC 157.3) and strong three bond correlations with C-2 (δC 154.5) and C-8 (δC 38.3). Furthermore, the one methine septet H-8 (δC/δH 38.3/2.73, C-8) shows two bond correlations to C-6 and three bond correlations to C-7. The two-methyl doublet H3-9 and H3-10 (δC/δH 22.6/1.17, C-9 and C-10) shows a strong three bond correlation to C-6. H-4 (δC/δH 129.1/6.88, C-4) shows strong three bond correlations to C-2 and C-6, which further confirms the location of tosyl and isopropyl groups (Table 1).
|
||||
|---|---|---|---|---|
| Atom | δExp 8 | δCalc 8 | δExp 9 | δCalc 9 |
| a 13C assignments were based on HSQC and HMBC spectra. 1H assignments were based on 1D-1H NMR and COSY spectra. | ||||
| C-1 | 179.1 | 188.9 | 178.9 | 189.1 |
| C-2 | 154.5 | 173.1 | 154.6 | 171.4 |
| C-3 | 129.2 | 140.9 | 131.7 | 145.6 |
| C-4 | 129.6 | 142.1 | 152.4 | 167.8 |
| C-5 | 136.6 | 143.4 | 130.5 | 138.2 |
| C-6 | 157.3 | 167.1 | 137.0 | 144.5 |
| C-7 | 137.7 | 155.5 | 138.9 | 152.4 |
| C-8 | 38.3 | 51.2 | 38.0 | 48.1 |
| C-9 and C-10 | 22.6 | 28.9 | 22.7 | 28.3 |
| C-11 | 133.4 | 151.9 | 133.4 | 151.7 |
| C-12 and C-16 | 128.4 | 141.4 | 128.5 | 141.4 |
| C-13 and C-15 | 129.2 | 138.5 | 129.6 | 138.7 |
| C-14 | 145.3 | 157.3 | 145.6 | 157.2 |
| C-17 | 21.7 | 27.3 | 21.8 | 27.4 |
| H-3 | 7.29 | 8.12 | 7.36 | 8.31 |
| H-4 | 6.88 | 7.69 | — | — |
| H-5 | 6.98 | 7.73 | 6.93 | 7.68 |
| H-6 | — | — | 7.14 | 7.94 |
| H-7 | 7.05 | 8.23 | 7.02 | 8.044 |
| H-8 | 2.73 | 3.47 | 2.80 | 3.64 |
| H-9 and H-10 | 1.17 | 2.01 | 1.21 | 2.05 |
| H-12 and H-16 | 7.89 | 10.15 | 7.90 | 10.07 |
| H-13 and H-15 | 7.31 | 8.27 | 7.33 | 8.28 |
| H-17 | 2.42 | 3.35 | 2.43 | 3.3445 |
We have calculated both 1H and 13C isotropic chemical shielding of 8 at the B3LYP/def2-TZVPP level of theory. Correlation plots between experimental and calculated chemical shifts shows very good correlations with correlation coefficients (R2) of 0.9946 and 0.9755 for 13C and 1H NMR chemical shifts, respectively (Fig. S50†).
Compound 9 was obtained as a white powder. Its molecular formula C17H18O4S was determined by HRESIMS data (m/z 341.0818 [M + Na]+, calc. 341.0815). The analysis of 1H, 13C, COSY, HSQC, and HMBC spectra revealed the presence of a singlet proton (δC/δH 131.7/7.36, C-3). This proton shows two bond correlations with C-2 (δC 154.5) and C-4 (δC 152.4) and strong three bond correlations with C-1 (δC 178.9) and C-8 (δC 38.0). Furthermore, the one methine septet H-8 (δC/δH 38.0/2.80, C-8) shows two bond correlations to C-4 (δC 152.4) and three bond correlations to C-3. The two-methyl doublet H3-9 and H3-10 (δC/δH 22.7/1.21, C-9 and C-10) shows a strong three bond correlation to C-6. H-6 (δC/δH 137.0/7.14, C-6) shows strong three bond correlations to C-1 and C-4, which further confirms the location of carbonyl and isopropyl groups. Besides, H-7 (δC/δH 138.9/7.02, C-7) shows a strong three bond correlation to C-2, which confirms the position of tosyl group (Table 1).
We have calculated both 1H and 13C isotropic chemical shielding of 9 at the B3LYP/def2-TZVPP level of theory. Correlation plots between experimental and calculated chemical shifts shows very good correlations with R2 of 0.9962 and 0.9789 for 13C and 1H NMR chemical shifts, respectively (Fig. S51†).
Our NMR assignments were further confirmed by obtaining an X-ray crystal structure of compound 8 (Fig. 3). The X-ray shows that the tosyl group is in position 2 while isopropyl group is in position 6 of the tropolone ring. Compound 8 was crystallized from ethyl acetate (0.2 mL), ether (0.5 mL), and hexanes (10 mL) by solvent evaporation. Crystallographic data is given in Table S1.† The crystals of compound 8 crystallize in the monoclinic space group P21/c with four molecules in the unit cell (Z = 4) of dimensions a = 5.62, b = 28.31, c = 9.51 Å and β = 92.4°. V = 1512.31(11) Å3.
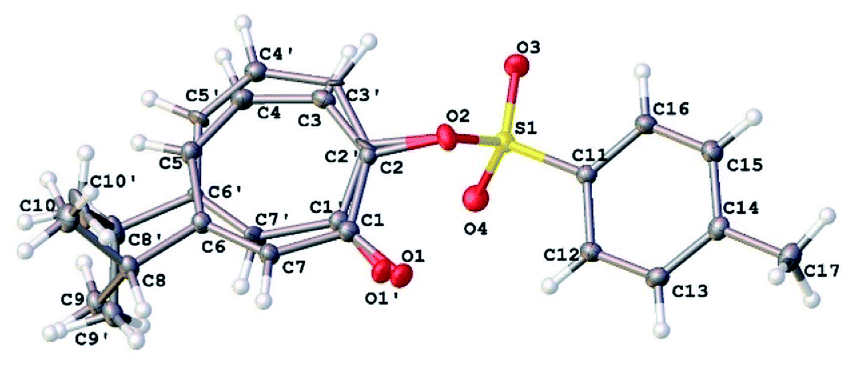 | ||
| Fig. 3 X-ray crystal structure of compound 8 showing the atom-numbering scheme. | ||
The 7-membered ring is disordered over 2 orientations (80:20%). The disorder was modelled with partial occupancy atoms and it indicates that the 7-membered ring was “breathing” by moving from less hindered to more hindered confirmation which facilitate the migration of tosyl group in substitution reactions. The tosyl group in 8 and 9 were shown to be fluxional and undergo thermally-induced shift between the two thujaplicin oxygen atoms. Molecular mechanics calculations by the MMFF94 force field suggests that this process takes place most likely through bipyramidal intermediate.20
6-Isopropyl–thiotropolone (10) and 4-isopropyl–thiotropolone (11) were readily accessible in good yields by reacting 8 and 9 with sodium hydrosulfide in ethanol at room temperature (Scheme 2). The structures of 10 and 11 were fully assigned using 1D and 2D NMR spectroscopy.
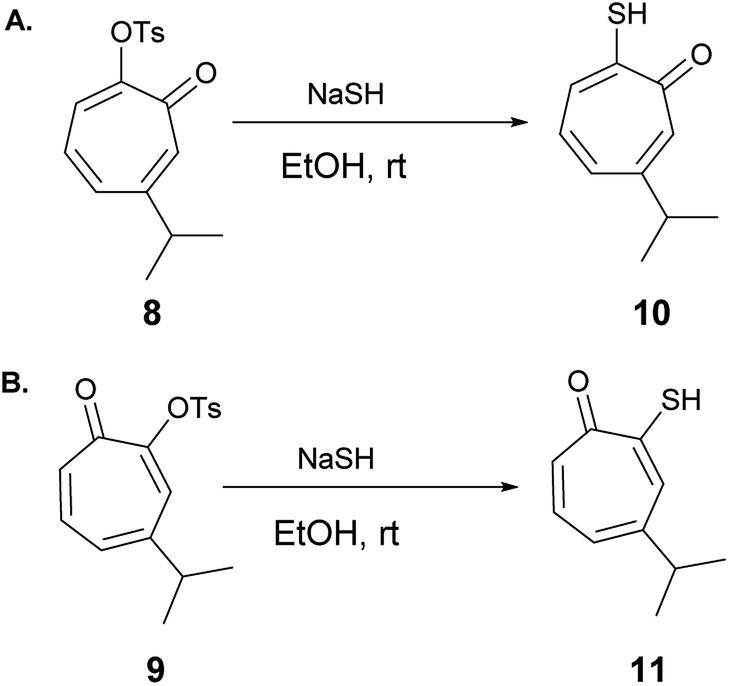 | ||
| Scheme 2 Synthesis of 6-isopropyl–thiotropolone (10) and 4-isopropyl–thiotropolone (11). | ||
2-Hydroxy-2,4,6-cycloheptatrien-1-one (tropolone 12) possesses hydroxyl group alpha to the carbonyl group and is expected to exhibit keto–enol tautomerism. Interestingly, this compound exists almost exclusively in the enol form (12A), which possess resonance stabilization because of its aromaticity. Density functional theory calculations of the keto–enol equilibrium constants of 2-hydroxy-2,4,6-cycloheptatrien-1-one (12A) and 3,5- and 3,6-cycloheptadiene-1,2-dione (12B and 12C) showed that the equilibrium is tilted in favour of the enol (12A) (Fig. 4).21
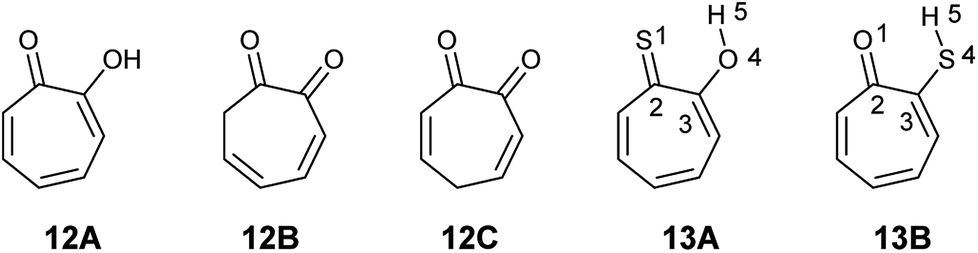 | ||
| Fig. 4 Hydroxytropolone tautomers (12A–C) and mercapto tropolone tautomers (13A and B). | ||
The corresponding thiotropolones have seven possible tautomers. The tautomeric equilibrium of all tautomers of thiotropolone was studied using Hartree Fock (HF) and density functional theory (B3LYP) at different levels of theory.22 Calculating electronic energy and Gibbs free energy showed that the enol forms were more stable than the keto forms because of its aromaticity. This observation was supported by calculating the nucleus independent chemical shifts of NMR chemical shifts of the studied thiotropolones.22 Compound 13A (2-hydroxy-2,4,6-cycloheptatriene-1-thione) was the most stable followed by compound 13B (2-mercapto-2,4,6-cycloheptatrien-1-one). The low energy of these two isomers is attributed to the favourable interactions between H5-S1 in 13A and H5-O1 in 13B.22
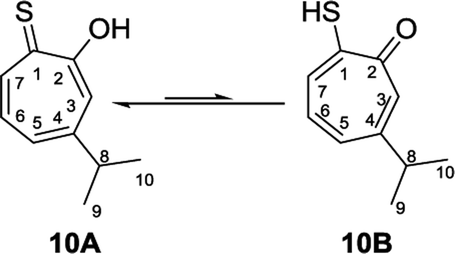
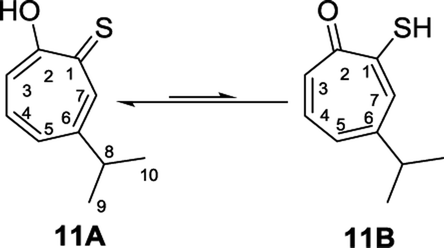
Nozoe et al.23 was the first to synthesize 2-mercaptotropone and it was suggested to exist in two tautomeric forms. Sulfur-substituted derivatives were obtained upon alkylation and acylation of this compound, which suggests that 2-mercaptotropone (13B) is the prevailing tautomer. However, studying the physicochemical properties of 2-mercaptotropone and its structural analogues 6-isopropyl-2-mercaptotropone (10) and 4-isopropyl-2-mercaptotropone (11) in depth suggested that the major tautomer is the 2-hydroxyl tropothione (13A).24
Compound 10 was obtained as a dark red oil. Its molecular formula (C10H11OS)2Na+was determined by HRESIMS data (m/z 381.0951 [M2 + Na]+, calc. 381.0953). The 1H NMR showed that this compound exist in CDCl3 as two tautomers 10A (≈87%) and 10B (≈13%). We were able to fully assign the chemical shifts of the major tautomer 10A (Table 2). The analysis of 1H, 13C, COSY, HSQC, and HMBC spectra revealed the presence of a singlet proton H-3 (δC/δH 119.6/7.45, C-3). This proton shows two bond correlations with C-2 (δC 172.5) and C-4 (δC 161.2) and strong three bond correlations with C-1 (δC 183.4), C-5 (δC 132.1) and C-8 (δC 38.8). Furthermore, the one methine septet H-8 (δC/δH 38.8/2.91, C-8) shows two bond correlations to C-4, C-9 and C-10 (δC 23.4) and three bond correlations to C-3 and C-5. The two-methyl doublet H3-9 and H3-10 (δC/δH 23.4/1.27, C-9 and C-10) shows two bond correlations to C-8 and a strong three bond correlation to C-4. H-5 (δC/δH 132.1/7.21, C-5) shows two bond correlations to C-6, and strong three bond correlations to C-3, C-7 and C-8. H-6 (δC/δH 133.8/7.14, C-3) shows strong three bond correlations to C-1 and C-4. H-7 (δC/δH 143.0/8.45, C-7) shows strong three bond correlations to C-2 and C-5 (Fig. 5).
|
|||||||
|---|---|---|---|---|---|---|---|
| Atom | δExp 10A | δCalc 10A | δCalc 10B | Atom | δExp 10A | δCalc 10A | δCalc 10B |
| C-1 | 183.4 | 210.3 | 180.0 | H-3 | 7.45 | 6.71 | 7.48 |
| C-2 | 172.5 | 179.0 | 185.9 | H-4 | — | — | — |
| C-3 | 119.6 | 118.2 | 141.2 | H-5 | 7.21 | 6.98 | 7.07 |
| C-4 | 161.2 | 164.9 | 164.1 | H-6 | 7.14 | 6.88 | 7.18 |
| C-5 | 132.1 | 132.6 | 133.9 | H-7 | 8.45 | 8.98 | 8.08 |
| C-6 | 133.8 | 130.9 | 136.8 | H-8 | 2.91 | 2.62 | 2.91 |
| C-7 | 143.0 | 159.8 | 130.8 | H3-9 | 1.27 | 1.46 | 1.42 |
| C-8 | 38.8 | 47.8 | 47.9 | H3-10 | 1.27 | 1.45 | 1.47 |
| C-9 | 23.4 | 26.9 | 25.2 | OH or SH | 9.50 | 8.98 | 4.04 |
| C-10 | 23.4 | 24.3 | 24.0 | ||||
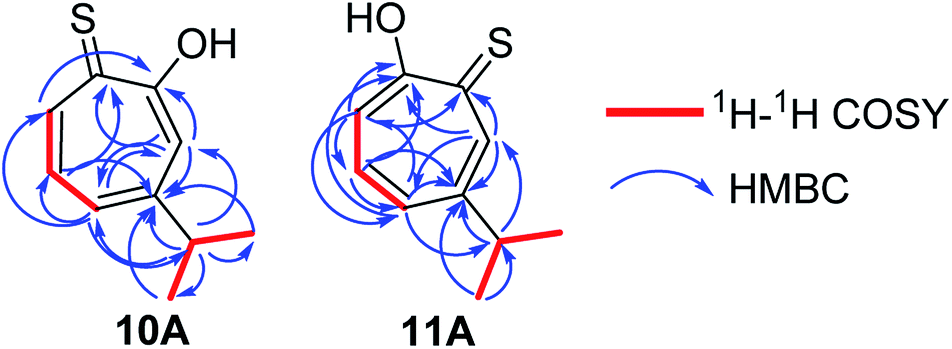 | ||
| Fig. 5 Key 1H–1H COSY and HMBC correlations of 10A and 11A. | ||
We have calculated both 1H and 13C isotropic chemical shielding of 10A and 10B at the B3LYP/def2-TZVPP level of theory. Correlation plots between experimental and calculated chemical shifts shows very good correlations in case of 10A with R2 of 0.9828 and 0.99 for 13C and 1H NMR chemical shifts, respectively (Fig. 6). The calculated chemical shift values of isomer 10B correlated to less extent with the experimental values (see Fig. S52†).
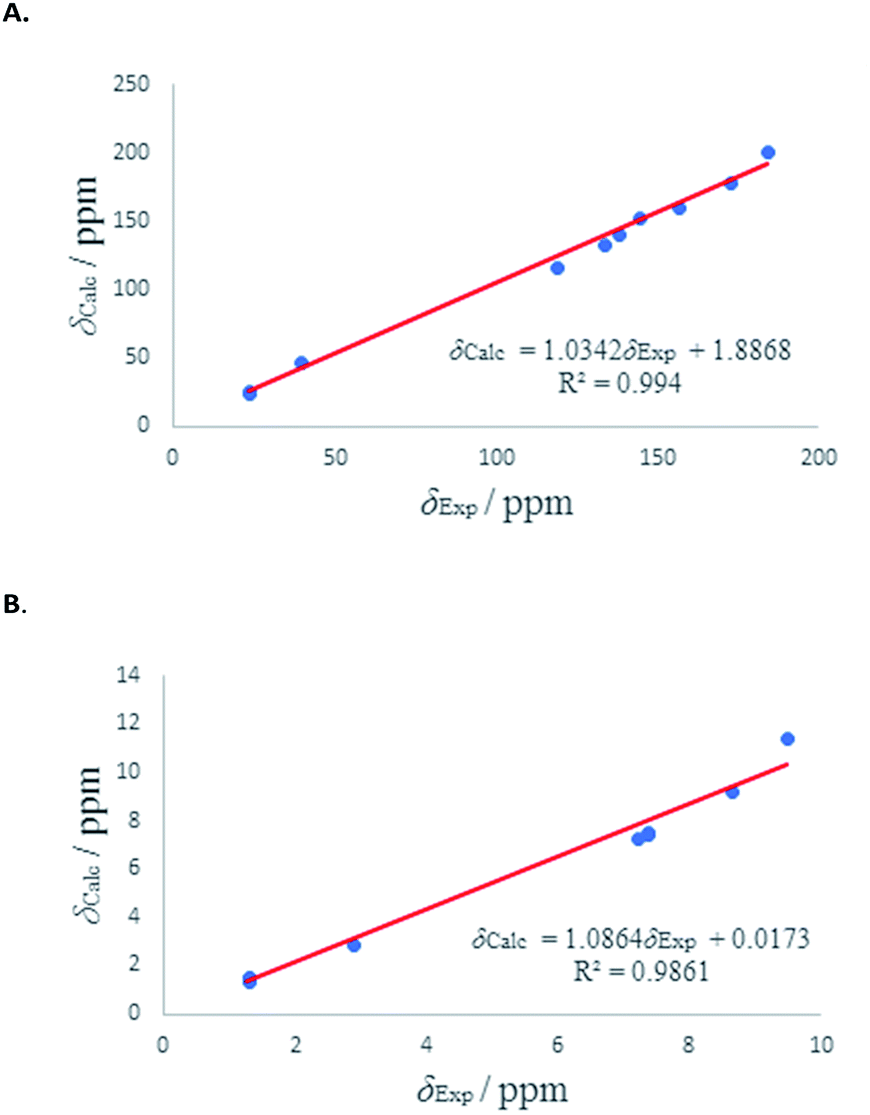 | ||
| Fig. 6 (A) Experimental 13C chemical shifts (δ) of 10A (vs.) calculated. (B) Experimental 1H chemical shifts (δ) of 10A (vs.) calculated. | ||
Compound 11 was obtained as orange microcrystals. Its molecular formula (C10H11OS)2Na+ was determined by HRESIMS data (m/z 381.0950 [M2 + Na]+, calc. 381.0953). This compound exists in CDCl3 as a mixture of two tautomers 11A (≈96%) and 11B (≈4%). We were able to fully assign the chemical shifts of the major tautomer 11A (Table 3). The analysis of 1H, 13C, COSY, HSQC, and HMBC spectra revealed the presence of a singlet proton H-7 (δC/δH 144.0/8.62, C-7). This proton shows two bond correlations with C-1 (δC 184.1) and C-6 (δC 156.5) and strong three bond correlations with C-2 (δC 172.2) and C-5 (δC 133.0). Furthermore, the one methine septet H-8 (δC/δH 39.0/2.88, C-8) shows two bond correlations to C-6 (δC 156.5) and three bond correlations to C-7. The two-methyl doublet H3-9 and H3-10 (δC/δH 23.1/1.27, C-9 and C-10) shows two bond correlations to C-8 and a strong three bond correlation to C-6. H-5 (δC/δH 133.0/7.19, C-5) shows two bond correlations to C-6, strong three bond correlations to C-3 and C-8, and four bond correlations to C-2. H-3 (δC/δH 118.2/7.34, C-3) shows two bond correlations to C-2 and C-4 and strong three bond correlations to C-1 and C-5. H-4 (δC/δH 137.9/7.35, C-4) shows two bond correlations to C-5 and strong three bond correlations to C-2 and C-6 which further confirms the location of the isopropyl groups (Fig. 5).
|
|||||||
|---|---|---|---|---|---|---|---|
| Atom | δExp 11A | δCalc 11A | δCalc 11B | Atom | δExp 11A | δCalc 11A | δCalc 11B |
| C-1 | 184.1 | 202.0 | 181.5 | H-3 | 7.34 | 7.50 | 7.29 |
| C-2 | 172.2 | 179.8 | 185.5 | H-4 | 7.35 | 7.54 | 7.51 |
| C-3 | 118.2 | 117.1 | 138.3 | H-5 | 7.19 | 7.34 | 7.07 |
| C-4 | 137.9 | 142.0 | 140.4 | H-6 | — | — | — |
| C-5 | 133.0 | 134.3 | 131.1 | H-7 | 8.62 | 9.32 | 8.27 |
| C-6 | 156.5 | 161.1 | 163.3 | H-8 | 2.88 | 2.94 | 2.91 |
| C-7 | 144.0 | 152.8 | 135.2 | H3-9 | 1.27 | 1.47 | 1.51 |
| C-8 | 39.0 | 47.8 | 47.7 | H3-10 | 1.27 | 1.51 | 1.44 |
| C-9 | 23.1 | 26.4 | 24.0 | OH or SH | 9.47 | 11.46 | 4.10 |
| C-10 | 23.1 | 25.4 | 28.1 | ||||
1H and 13C isotropic chemical shielding of 11A and 11B were calculated at the B3LYP/def2-TZVPP level of theory (Table 3). Correlation plots between experimental and calculated chemical shifts shows very good correlations in the case of 11A, with R2 of 0.994 and 0.9861 for 13C and 1H NMR chemical shifts, respectively (Fig. 7). The calculated chemical shift values of isomer 11B correlated to less extent with the experimental values (see Fig. S53†).
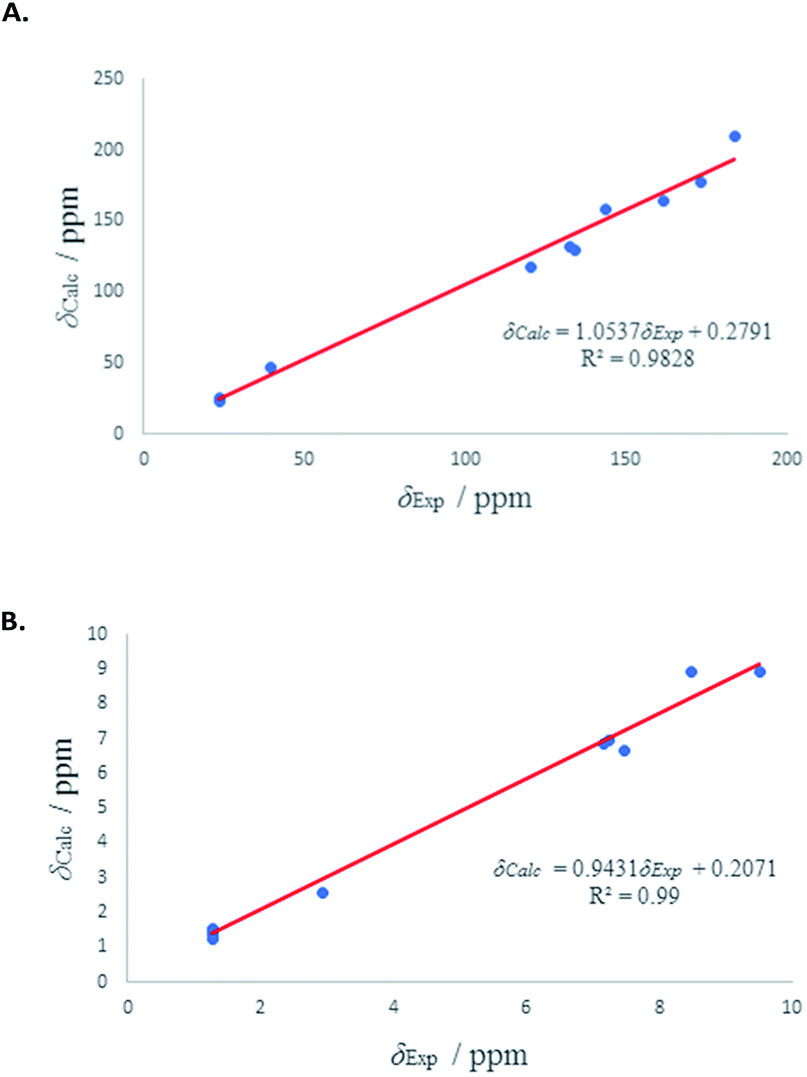 | ||
| Fig. 7 (A) Experimental 13C chemical shifts (δ) of 11A (vs.) calculated. (B) Experimental 1H chemical shifts (δ) of 11A (vs.) calculated. | ||
Computational details
Electronic structure calculations were performed using ORCA.25,26 Crystal structures of compounds 8 and 9 were optimized at the DFT theory BLYP with basis sets deft2-SVP and RI approximation. Geometries of compounds 10 and 11, and their isomers were modelled using the Avogadro program.27 Their geometries were optimized at the B3LYP functional28,29 and def2-TZVPP basis set.30 The RijCosX approximation and deft2/J auxiliary basis set were used during optimization. The absolute 1H NMR chemical shifts (δH), 13C NMR chemical shifts (δC) for all compounds were calculated with the GIAO (Gauge Including Atomic Orbitals) method31–33 at B3LYP def2-TZVPP level of theory. The TMS shielding was calculated at the same level of theory and relative chemical shifts were then estimated by subtracting the calculated chemical shift from TMS. The calculated 1H and 13C isotropic chemical shielding of TMS are 31.942/183.923.Biological evaluation
The broad anti-bacterial activity of β-thujaplicin (Hinokitiol, 2) was reported several decades ago.34,35 Compound 2 could modestly inhibit S. aureus, E. coli, and A. baumannii in our experimental conditions, with MIC80 values of 66.7, 44.4 and 51.2 μM, respectively. Compound 8, which is the rearranged tosyl isomer of the natural product 2, has no activity against all tested bacteria (MIC80 > 100 μM). However, this compound showed similar activity to 2 against C. neoformans (MIC80 = 24 μM for 8 vs. MIC80 = 21 μM for 2). Interestingly, compound 9, which is the tosylated derivative of the natural product 2 showed 8-fold increase in the antibacterial activity against S. aureus, but diminished activity against the Gram-negative bacteria E. coli, A. baumannii, and P. aeruginosa. Compound 9 has similar activity to 2 and 8 against C. neoformans (MIC80 = 24 μM) (Table 4).| Comp. | Bacteria MIC80 (μM) | C. neoformans MIC80 | CC50 | ||||
|---|---|---|---|---|---|---|---|
| S. aureus (29213) | E. coli (35218) | A. baumannii (from a patient) | P. aeruginosa (27853) | CC50 (neutral red) | CC50 (MTS) | ||
| 24 | 66.7 | 44.4 | 51.2 | >100 | 21 (ref. 4) | na | 66.4 |
| 8 | >100 | >100 | >100 | >100 | 24 | 31.8 | 33.6 |
| 9 | 8.8 | >100 | >100 | >100 | 24 | 19.5 | 34.2 |
| 10 | >100 | >100 | >100 | >100 | 50 | 39.7 | 95 |
| 11 | 16 | >100 | >100 | >100 | 24 | 30.4 | 64.2 |
The mercapto derivative of 8, compound 10, has no antibacterial activity against all four tested bacteria. The activity of 10 against C. neoformans (MIC80 = 50 μM) is >2-fold less than the corresponding hydroxy tropolone 2. The mercapto derivative of 9, compound 11, which is the direct isomer of 2, showed promising antibacterial activity against S. aureus (MIC80 = 16 μM), but no activity against the other tested bacteria. Compound 11 showed similar activity as 2, 8, and 9 against C. neoformans (MIC80 = 24 μM) (Table 4).
We tested the cytotoxicity of the synthesized compounds in hepatoblastoma cells using an MTS cytotoxicity assay that measures mitochondrial function and a neutral red assay that measures lysosomal function and calculated the CC50, which is the concentration of inhibitor required to reduce cell viability 50% relative to untreated cells (Table 4). Compound 10 have the lowest toxicity among all synthesized compounds in both assays. The toxicity of compound 11 is relatively low, and comparable to the natural product β-thujaplicin (2).
In silico ADME evaluation
We calculated a set of physical descriptors and pharmaceutical properties of all tested compounds using Qikprop to predict their absorption, distribution, metabolism, and excretion (ADME) properties (Table 5). All compounds are in compliance with Lipinski's rule of five36 and Jorgensen's rule of three37 and are predicted to have good oral bioavailability and excellent absorption through cell membranes.| Comp. | amol_MW | bQPlogPo/w |
cHBD | dHBA | e% Human oral absorption | fPSA | gQplogS |
hQPPCaco | i#Metab | jQplogBB |
kQPPMDCK |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Molar weight in Daltons (130–725).b Logarithm of partitioning coefficient between n-octanol and water phases (range for 95% of drugs: −2 to 6).c Number of hydrogen bonds donors (0–6).d Number of hydrogen bond acceptors (2–20).e Predicted human oral absorption on a 0–100% scale, based on a multiple linear regression model (<25% low, >80% high).f Polar surface area (7–200).g Predicted aqueous solubility, logS. S in mol dm−3 (−6.5 to 0.5).h Predicted apparent Caco-2 cell permeability in nm s−1 as a model for the gut-blood barrier (<25 poor, >500 excellent).i Number of possible metabolic reactions (2–8).j Predicted brain/blood partition coefficient (−3 to 1.2).k Predicted apparent MDCK cell permeability in nm s−1 as a mimic for blood/brain barrier (<25 poor, >500 excellent). Qikrop predictions are for non-active transport. |
|||||||||||
| 2 | 164.204 | 1.443 | 1 | 2.75 | 89.394 | 50.125 | −2.193 | 1040.346 | 2 | −0.396 | 516.318 |
| 8 | 318.387 | 2.1 | 0 | 6.5 | 92.748 | 68.778 | −2.742 | 975.826 | 2 | −0.615 | 490.424 |
| 9 | 318.387 | 2.216 | 0 | 6.5 | 94.321 | 68.77 | −2.952 | 1095.291 | 2 | −0.591 | 555.56 |
| 10A | 180.264 | 2.351 | 0.8 | 2.5 | 100 | 28.102 | −2.794 | 3286.638 | 2 | 0.207 | 4437.612 |
| 10B | 180.264 | 2.4 | 1 | 2.25 | 100 | 29.939 | −2.881 | 3010.296 | 2 | 0.168 | 3962.203 |
| 11A | 180.264 | 2.36 | 0.8 | 2.5 | 100 | 28.166 | −2.843 | 3222.618 | 2 | 0.195 | 4334.35 |
| 11B | 180.264 | 2.398 | 1 | 2.25 | 100 | 30.01 | −2.884 | 2998.094 | 2 | 0.166 | 3935.7 |
Conclusions
Tosylated thujaplicin isomers 8 and 9 were separated for the first time and their structures were fully characterized. Isomers 8 and 9 were used to synthesize the two mercapto isomers of β-thujaplicin, 10 and 11, in a regioselective manner. 1H and 13C Chemical shifts of synthesized isomers were fully assigned using several NMR experiments, and their isotropic magnetic shielding was calculated computationally. The calculated chemical shift values were in a good agreement with the experimental results. When the biological activity of the synthesized compounds was evaluated against C. neoformans and four different bacterial strains, 4-isopropyl–thiotropolone (11) displayed good potency against Staphylococcus aureus (ATCC 29213). The relatively non-toxic nature of this compound and excellent predicted ADME properties make it a potential lead compound to develop potent inhibitors against Staphylococcus aureus infection.Experimental section
All materials were purchased from commercial suppliers and used without further purification. Purification was done by Reveleris X2 flash chromatography. The purities of the final compounds were characterized by high-performance liquid chromatography (HPLC) using a gradient elution program (Ascentis Express Peptide C18 column, acetonitrile/water 5/95 → 95/5, 5 min, 0.05% trifluoracetic acid) and UV detection (254 nM). The purities of final compounds were 95% or greater. NMR spectra was recorded on a Bruker NMR 400 MHz Avance III spectrometer operating at 400 MHz for 1H NMR and 100 MHz for 13C NMR. Chemical shifts are given in part per million (ppm) relative to tetramethylsilane (TMS), coupling constants J are given in Hertz. High-resolution mass spectra were obtained using electrospray ionization on a Bruker 12 T APEX-Qe FTICR-MS with an Apollo II ion source at the COSMIC Laboratory facility at Old Dominion University, VA.Synthesis of compounds 8 and 9
p-Toluenesulfonyl chloride (2.3 g, 12.2 mmol) was added to a solution of tropolone (2 g, 12.2 mmol) and DIPEA (4.3 mL, 24.4 mmol) in DCM (50 mL), and the reaction mixture was stirred overnight. The resulting mixture was successively washed with 1 M HCl (30 mL), water (30 mL) and brine (30 mL). The organic layer was dried over Na2SO4, filtered and the solvent was evaporated in vacuo yielding a mixture of the two isomers, which were separated by flash chromatography using (EtOAc/hexanes 3:7) with a flow rate 10 mL min−1 to obtain each isomer in a pure form.
6-(1-Methylethyl)-2-[[(4-methylphenyl)sulfonyl]oxy]-2,4,6-cycloheptatrien-1-one (8)
White needles (46%), Rf = 0.655 (EtOAc/hexanes 1:1). 1H NMR (400 MHz, chloroform-d) δ 7.92 (d, J = 7.3 Hz, 2H), 7.38–7.26 (m, 3H), 7.08 (s, 1H), 7.01 (d, J = 11.4 Hz, 1H), 6.91 (t, J = 10.3 Hz, 1H), 2.82–2.67 (m, 1H), 2.44 (s, 3H), 1.20 (d, J = 6.7 Hz, 6H); 13C NMR (100 MHz, chloroform-d) δ 179.2, 157.5, 154.6, 145.4, 137.8, 136.7, 133.5, 129.7, 129.6, 129.3, 128.6, 38.4, 22.7, 21.8; LC/MS m/z: 319.1 [M + H]+, 341 [M + Na]+. HRESIMS m/z 341.0816 [M + Na]+ (calcd for C17H18O4SNa+, 341.0818).
4-(1-Methylethyl)-2-[[(4-methylphenyl)sulfonyl]oxy]-2,4,6-cycloheptatrien-1-one (9)
White powder (43%), Rf = 0.483 (EtOAc/hexanes 1:1). 1H NMR (400 MHz, chloroform-d) δ 7.93 (d, J = 7.6 Hz, 2H), 7.41–7.32 (m, 3H), 7.17 (dd, J = 12.1, 8.7 Hz, 1H), 7.04 (d, J = 12.3 Hz, 1H), 6.96 (d, J = 8.6 Hz, 1H), 2.89–2.75 (m, 1H), 2.46 (s, 3H), 1.24 (d, J = 6.8 Hz, 6H); 13C NMR (100 MHz, chloroform-d) δ 178.9, 154.5, 152.4, 145.5, 139.0, 136.9, 133.4, 131.8, 130.5, 129.6, 128.6, 38.1, 22.8, 21.8; LC/MS m/z: 319.1 [M + H]+, 341 [M + Na]+. HRESIMS m/z 341.0815 [M + Na]+ (calcd for C17H18O4SNa+, 341.0818).
General procedure for the synthesis of 10 and 11
NaSH (0.122 g, 2.17 mmol) was added portion wise to a solution of 8 or 9 (0.150 g, 0.54 mmol) in ethanol (10 mL). The reaction mixture was stirred for 2 h at room temperature and was monitored by LCMS and TLC till completion. Ethanol was evaporated, and the resulting solid was dissolved in water and acidified with HCl (2 M) then extracted with DCM (3 × 20 mL) to yield the desired product. Purification was performed by flash chromatography using a mixture of EtOAc/hexanes to obtain the desired compound.2-Hydroxy-4-isopropylcyclohepta-2,4,6-triene-1-thione (10)
Red dark oil, Rf = 0.876 (EtOAc/hexanes 1:1). 1H NMR (400 MHz, chloroform-d) δ 9.71 (s, 1H), 8.46 (d, J = 10.6 Hz, 1H), 7.47 (s, 1H), 7.23 (d, J = 9.7 Hz, 1H), 7.16 (t, J = 10.2 Hz, 1H), 2.99–2.87 (m, 1H), 1.29 (d, J = 6.9 Hz, 7H); 13C NMR (101 MHz, CDCl3) δ 183.5, 172.5, 161.4, 143.1, 134.0, 132.2, 119.7, 38.9, 23.5; LC/MS m/z: 181 [M + H]+. HRESIMS m/z 381.0951 [M2 + Na]+ (calcd for (C10H11OS)2Na+, 381.0953).
7-Hydroxy-3-isopropylcyclohepta-2,4,5-triene-1-thione (11)
Orange crystal, Rf = 0.793, (EtOAc/hexanes 1:1). 1H NMR (400 MHz, chloroform-d) δ 9.86 (s, 1H), 8.63 (s, 1H), 7.39–7.26 (m, 2H), 7.26–7.11 (m, 1H), 2.94–2.83 (m, 1H), 1.27 (d, J = 6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 184.3, 172.2, 156.6, 144.1, 138.0, 133.1, 118.3, 39.1, 23.5; LC/MS m/z: 181 [M + H] +. HRESIMS m/z 381.0950 [M2 + Na]+ (calcd for (C10H11OS)2Na+, 381.0953).
X-ray structure determination of 8
A crystals of approximate dimensions 0.544 × 0.254 × 0.132 mm3 was mounted on a MiTeGen cryoloop in a random orientation. Preliminary examination and data collection were performed using a Bruker X8 Kappa Apex II Charge Coupled Device (CCD) Detector system single crystal X-ray diffractometer equipped with an Oxford Cryostream LT device. All data were collected using graphite monochromated Mo K radiation (=0.71073 Å) from a fine focus sealed tube X-ray source. Preliminary unit cell constants were determined with a set of 36 narrow frame scans. Typical data sets consist of combinations of and ϕ scan frames with typical scan width of 0.5 and counting time of 10 seconds per frame at a crystal to detector distance of 4.0 cm. The collected frames were integrated using an orientation matrix determined from the narrow frame scans. Apex II and SAINT software packages (Bruker Analytical X-ray, Madison, WI, 2016) were used for data collection and data integration. Analysis of the integrated data did not show any decay. Final cell constants were determined by global refinement of 7781 reflections harvested from the complete data set. Collected data were corrected for systematic errors using SADABS (Bruker Analytical X-ray, Madison, WI, 2016) based on the Laue symmetry using equivalent reflections.Crystal data and intensity data collection parameters are listed in Table 1S.† Structure solution and refinement were carried out using the SHELXTL-PLUS software package.40 The structure was solved and refined successfully in the monoclinic space group P21/c. Full matrix least-squares refinements were carried out by minimizing w(Fo2 − Fc2)2. The non-hydrogen atoms were refined anisotropically to convergence. The 7-membered ring is disordered, and the two orientations were refined with geometrical restraints to 82:18%. All hydrogen atoms were treated using appropriate riding model (AFIX m3). The final residual values and structure refinement parameters are listed in Table 1S.†
Complete listings of positional and isotropic displacement coefficients for hydrogen atoms, anisotropic displacement coefficients for the non-hydrogen atoms and other geometrical parameters are listed as ESI (Tables 2S to 7S†). Table of calculated and observed structure factors are available in electronic format.
Bacterial strains
Staphylococcus aureus (ATCC 29213), Escherichia coli (ATCC 35218), and Pseudomonas aeruginosa (ATCC 27853) were obtained from the American Type Culture Collection (ATCC). Acinetobacter baumannii was collected from the microbiology laboratory at the John Cochran division of the St. Louis VA Medical Center (STLVAMC) under a Subcommittee on Research Safety (SRS)-approved protocol.Determination of the minimum inhibitory concentration
MIC80s were determined by the broth microdilution method recommended by the Clinical and Laboratory Standards Institute (CLSI) in cation-adjusted Mueller–Hinton II broth (CAMHB). A 1.5-fold dilution series of the compounds was prepared in CAMHB. Overnight bacterial culture was added to the diluted compounds in a 96 well plate after adjusting the bacterial concentration to 5 × 105 CFU mL−1 in final concentration. After 16–24 hours incubation at 35 °C ± 2 °C, the plates were read at 600 nM in microplate reader. The MIC80 was defined as the concentration of an antibacterial agent that inhibited bacteria growth ≥80% compared to vehicle-treated control cultures. All values were determined at least twice independently, and the average number is reported.Inhibition of C. neoformans growth
Compounds were tested C. neoformans var grubii, KN99 (serotype A, MATα) in a limiting dilution assay with a starting optical density (650 nM) of 0.001 in YNB-02 (0.67% yeast nitrogen base, 0.2% dextrose, pH 7.0 with 50 mM MOPs) + 1% DMSO. Cells were incubated without shaking for 48 hours at 35 °C and cell density was measured at 650 nM. The minimal inhibitory concentration was determined using compound concentrations from 0.19 to 50 μM of the compound in YNB-02 + 1% DMSO. Each assay was done in triplicate and all values are the average of two or more independent assays. The data are presented as the average cell density as a percent of DMSO-only treated cells. MICs are reported as the minimal concentration needed to inhibit 80% of C. neoformans growth relative to vehicle-treated controls.Cytotoxicity in hepatoma cells
HepDES19 cells38 (1.0 × 104 cells per well) were seeded in 96-well plates and incubated in Dulbecco's Modified Eagle Medium (DMEM) with 10% FBS plus 1% penicillin/streptomycin solution, 1% nonessential amino acids, and 1% glutamine. The compounds were diluted in the medium to the desired concentrations at a final concentration of 1% DMSO and added to the cells 48 h after plating, with each concentration tested in triplicate.MTS cytotoxicity assay (CC50)
Soluble MTS reagent [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, Promega] was added 72 h after incubation, the cultures were incubated for 90 min, and absorbance was read at 490 nm. The CC50 was calculated as the concentration of inhibitor required to reduce cell viability 50% relative to untreated cells. The data are plotted as log[inhibitor] versus response and fit to a variable slope model using GraphPad Prism (v6, www.graphpad.com).4Neutral red assay (CC50)
Cells were incubated with the compound for 72 hours and cytotoxicity was measured using a neutral red assay.39 The data were transformed to log[inhibitor] and fit to a 4-variable slope curve using GraphPad Prism. The concentration at which 50% of cells were inhibited relative to vehicle-treated control is reported as the CC50 value.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Saint Louis University Start up fund (No. 203437). Funding from the National Science Foundation (MRI, CHE-0420497) for the purchase of the Apex-II diffractometer is acknowledged. We would like to thank Ms Ashley Anderson for help with acquiring NMR.Notes and references
- M. Yamato, K. Hashigaki, Y. Yasumoto, J. Sakai, R. F. Luduena, A. Banerjee, S. Tsukagoshi, T. Tashiro and T. Tsuruo, J. Med. Chem., 1987, 30, 1897–1900 CrossRef PubMed.
- K. Sugawara, M. Ohbayashi, K. Shimizu, M. Hatori, H. Kamei, M. Konishi, T. Oki and H. Kawaguchi, J. Antibiot., 1988, 41, 862–868 CrossRef PubMed.
- M. Yamato, J. Ando, K. Sakaki, K. Hashigaki, Y. Wataya, S. Tsukagoshi, T. Tashiro and T. Tsuruo, J. Med. Chem., 1992, 35, 267–273 CrossRef PubMed.
- M. J. Donlin, A. Zunica, A. Lipnicky, A. K. Garimallaprabhakaran, A. J. Berkowitz, A. Grigoryan, M. J. Meyers, J. E. Tavis and R. P. Murelli, Antimicrob. Agents Chemother., 2017, 61, e02574-16 CrossRef PubMed.
- W. H. Johnston, J. J. Karchesy, G. H. Constantine and A. M. Craig, Phytother. Res., 2001, 15, 586–588 CrossRef PubMed.
- S. R. Piettre, A. Ganzhorn, J. Hoflack, K. Islam and J. M. Hornsperger, J. Am. Chem. Soc., 1997, 119, 3201–3204 CrossRef.
- D. M. Himmel, K. A. Maegley, T. A. Pauly, J. D. Bauman, K. Das, C. Dharia, A. D. Clark, K. Ryan, M. J. Hickey, R. A. Love, S. H. Hughes, S. Bergqvist and E. Arnold, Structure, 2009, 17, 1625–1635 CrossRef PubMed.
- S. J. Newberry, J. D. FitzGerald, A. Motala, M. Booth, M. A. Maglione, D. Han, A. Tariq, C. E. O'Hanlon, R. Shanman, W. Dudley and P. G. Shekelle, Ann. Intern. Med., 2017, 166, 27–36 CrossRef PubMed.
- S. Özen, E. D. Batu and S. Demir, Front. Immunol., 2017, 8, 253 Search PubMed.
- A. Bayes-Genis, Y. Adler, A. B. De Luna and M. Imazio, Eur. Heart J., 2017, 38, 1706–1709 CrossRef PubMed.
- H. Yamano, T. Yamazaki, K. Sato, S. Shiga, T. Hagiwara, K. Ouchi and T. Kishimoto, Antimicrob. Agents Chemother., 2005, 49, 2519–2521 CrossRef PubMed.
- A. Muzaffar, A. Brossi, C. M. Lin and E. Hamel, J. Med. Chem., 1990, 33, 567–571 CrossRef PubMed.
- J. Guan, X. K. Zhu, Y. Tachibana, K. F. Bastow, A. Brossi, E. Hamel and K. H. Lee, J. Med. Chem., 1998, 41, 1956–1961 CrossRef PubMed.
- S. Tsubotani, Y. Wada, K. Kamiya, H. Okazaki and S. Harada, Tetrahedron Lett., 1984, 25, 419–422 CrossRef.
- K. Kintaka, H. Ono, S. Tsubotani, S. Harada and H. Okazaki, J. Antibiot., 1984, 37, 1294–1300 CrossRef PubMed.
- I. Tamburlin-Thumin, M. P. Crozet, J. C. Barriere, M. Barreau, J. F. Riou and F. Lavelle, Eur. J. Med. Chem., 2001, 36, 561–568 CrossRef PubMed.
- M. Cavazza and F. Pietra, Org. Biomol. Chem., 2003, 1, 3002–3003 RSC.
- M. C. Barret, P. H. Bhatia, G. Kociok-Köhn and K. C. Molloy, Transition Met. Chem., 2014, 39, 543–551 CrossRef.
- T. Yanagisawa, K. Kosakai, T. Tomiyama, M. Yasunami and K. Takase, Chem. Pharm. Bull., 1990, 38, 3355–3358 CrossRef PubMed.
- M. Cavazza and F. Pietra, Tetrahedron Lett., 2003, 44, 1895–1897 CrossRef.
- S. W. Paine and A. Salam, Chem. Phys., 2006, 331, 61–66 CrossRef.
- S. W. Paine and A. Salam, Int. J. Quantum Chem., 2012, 113, 1245–1252 CrossRef.
- T. Nozoe, M. Satô and K. Matsui, Proc. Jpn. Acad., 1953, 29, 22–26 CrossRef.
- T. Nozoe and K. Matsui, Bull. Chem. Soc. Jpn., 1961, 34, 616–618 CrossRef.
- N. Frank, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 2, 73–78 Search PubMed.
- N. Frank, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, e1327 Search PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1986, 84, 4524–4529 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. London, J. Phys. Radium, 1937, 8, 397–409 CrossRef.
- R. Ditchfield, J. Chem. Phys., 1972, 56, 5688–5691 CrossRef.
- T. Helgaker, M. Jaszuński and K. Ruud, Chem. Rev., 1999, 99, 293–352 CrossRef PubMed.
- Y. Arima, Y. Nakai, R. Hayakawa and T. Nishino, J. Antimicrob. Chemother., 2003, 51, 113–122 CrossRef PubMed.
- Y.-H. Shih, D.-J. Lin, K.-W. Chang, S.-M. Hsia, S.-Y. Ko, S.-Y. Lee, S.-S. Hsue, T.-H. Wang, Y.-L. Chen and T.-M. Shieh, PLoS One, 2014, 9, e94941 CrossRef PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef PubMed.
- W. L. Jorgensen and E. M. Duffy, Adv. Drug Delivery Rev., 2002, 54, 355–366 CrossRef PubMed.
- H. Guo, D. Jiang, T. Zhou, A. Cuconati, T. M. Block and J.-T. Guo, J. Virol., 2007, 81, 12472–12484 CrossRef PubMed.
- G. Repetto, A. del Peso and J. L. Zurita, Nat. Protoc., 2008, 3, 1125 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1856450. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra06297h |
| This journal is © The Royal Society of Chemistry 2018 |