
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrazine-solvothermal methods to synthesize polymeric thioarsenates from one-dimensional chains to a three-dimensional framework†
Jingyu Hana,
Shufen Lia,
Chunying Tangb,
Wei Zhenga,
Wenqing Jianga and
Dingxian Jia *a
*a
aCollege of Chemistry, Chemical Engineering and Materials Science, Soochow University, No. 199 Renai Road, Suzhou, 215123, P. R. China. E-mail: jiadingxian@suda.edu.cn
bCollege of Chemistry, Chemical Engineering and Materials Science, Shandong Normal University, No. 88 East Wenhua Road, Jinan, 250014, P. R. China
First published on 4th October 2018
Abstract
A series of polymeric Mn(II)-thioarsenates [Mn(en)3]n[(N2H4)2Mn6(μ6-S)(μ-N2H4)2(μ3-AsS3)4]n (1), [N2H5]n[{Mn(μ-N2H4)2(μ-AsS4)}·0.5en]n (2), [Mn(μ-trien){Mn(μ-N2H4)(μ-AsS3)}2]n (3), [{Mn(N2H4)}2(μ-N2H4)2{Mn(μ-N2H4)2(μ-AsS3)2}]n (4), [Mn3(μ-N2H4)6(μ3-AsS4)(μ2-AsS4)]n (5), and [Mn(NH3)6]n[{Mn(NH3)(μ-AsS4)}2]n (6) were synthesized using a hydrazine-solvothermal method. The thioarsenate units AsS3 and AsS4 coordinate to Mn(II) ions with variable coordination modes, forming a Mn–As–S ternary cluster (1), chains (2, 4–6), and layers (3), respectively. The hydrazine molecules act as inter-cluster, intra-chain and intra-layer bridging ligands to join the Mn(II) ions, resulting in hydrazine hybrid 1-D, 2-D, and 3-D Mn(II)-thioarsenate moieties in 1–5. Compounds 1–6 exhibit tunable semiconducting band gaps varying in the range of 2.19–2.47 eV. Compound 1 displays stronger antiferromagnetic coupling interactions than that of compound 2.
Introduction
Intense efforts have been continually devoted to the research on organic–inorganic hybrid materials, which not only have fundamental interest in their rich structural diversities but also pose important synthetic challenges and exhibit unique structure–property correlations.1 In the wide field of hybrids, chalcogenidometallates of main group metals are attractive for potential applications in a wide field of visible-light photocatalysts,2 photoluminescence,3 gas separators,4 nonlinear optical generators,5 photoconductors,6 semiconductors,4a,7 ion exchangers,8 and magnetism.9 Since Bedard et al. hydrothermally prepared microporous tin and germanium sulfides in aqueous amine solution in the late 1980s,10 the templating synthesis in liquids under mild hydro- or solvo-thermal conditions has been developed to be a versatile approach to the preparation of chalcogenidometallates. A large number of organic–inorganic chalcogenidometallates had been solvothermally prepared in organic solvents (such as organic amine or alkyl alcohol).11 In presence of both transition metals (TMs) and coordinative polyamines, the chalcogenidometallates which contain charge compensating cation [TM(polyamine)m]n+ had been obtained by the solvothermal methods.12 Recently, several methodologies have been developed in preparation of the crystalline chalcogenides. Thermal synthesis in ionic liquids (ionothermal method) has been employed in the synthesis of chalcogenidometallates which dominated by the works of Dehnen and Huang.13 The work of Zhang has shown that the surfactant-thermal method is a fruitful route to prepare chalcogenidoarsenates and chalcogenidoantimonates.14 Apparently, the templating synthesis in liquid make it possible to chose different templates or solvents to affect the thermal reactions, and further to tune the structures and properties of the final products.Hydrazine (N2H4) is an excellent candidate for the reaction media for the syntheses of chalcogenidometallates because of the unique properties of hydrazine: (1) strong reducing ability (standard reducing potential: φθ (N2/N2H4) = −1.16 V), (2) high polarity (dipole moment μ = 1.75 D),15,16a and (3) high coordination aptitude to metal ions with less steric hindrance. As a strong reducing agent, N2H4 can smoothly reduce elemental chalcogen to form chalcogenide Q2− anions or polychalcogenide Qn2− (Q = S, Se, Te) anions. This makes it possible to prepare chalcogenidometallates using elemental chalcogens instead of chalcogenides as starting materials under mild conditions. The high polarity and coordination aptitude of N2H4 are helpful for dissolving the metal chalcogenide species and prevent them from generating precipitation in the growing crystalline chalcogenides. In fact, hydrazine has been used as a reaction media in preparation of chalcogenides in the last decade and several hydrazine-adducts of metal chalcogenides were prepared in hydrazine solution at room temperature.16 However, the synthesis in hydrazine solvent under solvothermal conditions (hydrazine-solvothermal) remains less explored. The early works in this area afforded hydrazine-complexes of metal chalcogenides based on 15/16 and 14/16 chalcogenidometallates.17 Recently, we successfully prepared the heavier telluromercurates by the hydrazine-solvothermal method using powder Te as the sole tellurium source in the lower temperature range.18 In addition, several experiments have demonstrated that the reactive hydrazine molecule is also an important auxiliary reagent in the preparation of crystalline chalcogenidometallates, although it does not appear in the final products.13b,13h,19
Inspired by our previous work on fabrication of TM-containing chalcogenidoarsenates in different solvents,20 we are interested in understanding how the solvent and coordination effects of hydrazine to influence crystal growth of chalcogenidoarsenates under solvothermal conditions. Now, we chose Mn/As/S as a model system to systematically explore the hydrazine-solvothermal syntheses under different conditions. By changing the second solvent, six polymeric Mn(II)-thioarsenates [Mn(en)3]n[(N2H4)2Mn6(μ6-S)(μ-N2H4)2(μ3-AsS3)4]n (1), [N2H5]n[{Mn(μ-N2H4)2(μ-AsS4)}·0.5en]n (en = ethylenediamine) (2), [Mn(μ-trien){Mn(μ-N2H4)(μ-AsS3)}2]n (trien = triethylenetetramine) (3), [{Mn(N2H4)}2(μ-N2H4)2{Mn(μ-N2H4)2(μ-AsS3)2}]n (4), [Mn3(μ-N2H4)6(μ3-AsS4)(μ2-AsS4)]n (5), and [Mn(NH3)6]n[{Mn(NH3)(μ-AsS4)}2]n (6) were synthesized in N2H4 solvent under solvothermal conditions. The 2-D layer of 1 featuring a large [Mn6(μ6-S)(μ3-AsS3)4] cubic building block and 3-D framework of 3 show the unique synergistic coordination effect of N2H4 with organic amine. The terminal and bridging coordination modes of N2H4 ligands lead to various combinations between Mn2+ ion and [AsS3] or [AsS4] unit in compounds 1–5. This investigation could provide us a better understanding of the different effects of N2H4 solvent from the traditional organic amine solvent on preparation of Mn–As chalcogenides under solvothermal conditions.
Results and discussion
Syntheses
Title compounds were prepared using elements Mn, As, and S as starting materials in N2H4 solvent under solvothermal conditions in moderate temperature range (Scheme 1). Reaction of Mn, As, and S in the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) N2H4/en mixed solvent for 6 days produced a thioarsenate(III) [Mn(en)3]n[(N2H4)2Mn6(μ6-S)(μ-N2H4)2(μ3-AsS3)4]n (1). The reaction in the 2:1 (v/v) N2H4/en mixed solvent afforded a thioarsenate(V) [N2H5]n[{Mn(μ-N2H4)2(AsS4)}·0.5en]n (2). The same reactions with trien instead of en gave thioarsenates(III) [Mn(μ-trien){Mn(μ-N2H4)(μ3-AsS3)}2]n (3) and [{Mn(N2H4)}2(μ-N2H4)2{Mn(μ-N2H4)2(μ-AsS3)2}]n (4), respectively. Mn(II) ion does not bind the en or trien molecule in 2 and 4. The reaction in N2H4/DMF and N2H4/NH3 (aqueous ammonia, 25%) mixed solvents produced thioarsenates(V) [Mn3(μ-N2H4)6(μ3-AsS4)(μ-AsS4)]n (5) and [Mn(NH3)6]n[{Mn(NH3)(μ3-AsS4)}2]n (6), respectively. It is noteworthy that N2H4 molecule does not appear in compound 6. However, the attempt to synthesize 6 in ammonia solution without N2H4 had failed. The reactions without N2H4 in the temperature range 100–160 °C produced yellow solutions and small amount of grey precipitates. In compounds 1–5, N2H4 molecule acts as a ligand to the Mn2+ ion, and as a counterion in protonated form in compound 2. In the FT-IR spectra of compounds 1–5, the bands located between 986 and 996 cm−1 are attributed to the N–N vibrations of N2H4 molecules (Fig. S1–S5†).17a,17c The purity of bulk phases of title compounds were investigated using powder X-ray diffraction (PXRD). The PXRD patterns of title compounds are consistent with the simulated PXRD patterns based on single-crystal XRD data (Fig. S7†), respectively.
:1 (v/v) N2H4/en mixed solvent for 6 days produced a thioarsenate(III) [Mn(en)3]n[(N2H4)2Mn6(μ6-S)(μ-N2H4)2(μ3-AsS3)4]n (1). The reaction in the 2:1 (v/v) N2H4/en mixed solvent afforded a thioarsenate(V) [N2H5]n[{Mn(μ-N2H4)2(AsS4)}·0.5en]n (2). The same reactions with trien instead of en gave thioarsenates(III) [Mn(μ-trien){Mn(μ-N2H4)(μ3-AsS3)}2]n (3) and [{Mn(N2H4)}2(μ-N2H4)2{Mn(μ-N2H4)2(μ-AsS3)2}]n (4), respectively. Mn(II) ion does not bind the en or trien molecule in 2 and 4. The reaction in N2H4/DMF and N2H4/NH3 (aqueous ammonia, 25%) mixed solvents produced thioarsenates(V) [Mn3(μ-N2H4)6(μ3-AsS4)(μ-AsS4)]n (5) and [Mn(NH3)6]n[{Mn(NH3)(μ3-AsS4)}2]n (6), respectively. It is noteworthy that N2H4 molecule does not appear in compound 6. However, the attempt to synthesize 6 in ammonia solution without N2H4 had failed. The reactions without N2H4 in the temperature range 100–160 °C produced yellow solutions and small amount of grey precipitates. In compounds 1–5, N2H4 molecule acts as a ligand to the Mn2+ ion, and as a counterion in protonated form in compound 2. In the FT-IR spectra of compounds 1–5, the bands located between 986 and 996 cm−1 are attributed to the N–N vibrations of N2H4 molecules (Fig. S1–S5†).17a,17c The purity of bulk phases of title compounds were investigated using powder X-ray diffraction (PXRD). The PXRD patterns of title compounds are consistent with the simulated PXRD patterns based on single-crystal XRD data (Fig. S7†), respectively.
 | ||
| Scheme 1 Solvothermal syntheses of compounds 1–6. | ||
Crystal structures
Compound 1 crystallizes in the monoclinic space group P21/c with four formulae in the unit cell (Table S1†). It consists of a [Mn(en)3]2+ complex cation, a 2-D [(N2H4)2Mn6(μ6-S)(μ-N2H4)2(μ3-AsS3)42−]n polymeric anion. The 2-D polymeric anion is constructed from a [Mn6(μ6-S)(μ3-AsS3)4] cluster and hydrazine molecules. There are four crystallographically independent As, seven Mn, and thirteen S atoms in 1. Each As3+ ion is coordinated by three S2− anions, forming a typical trigonal pyramid AsS3, which acts as the primary building unit (PBU). One AsS3 PBU chelates three Mn2+ ions as a μ3-AsS3 ligand, forming a Mn3AsS3 semi-cube (Fig. 1a). The three Mn2+ ions of Mn3AsS3 are capped by a S2− (S1) anion to form a [Mn3(μ3-S)(μ3-AsS3)] cube (highlighted by purple bonds in Fig. 1a), which can be regarded as the secondary building unit (SBU). Four [Mn3(μ3-S)(μ3-AsS3)] cubic SBUs are joined via edge-sharing forming a cube-like heterometallic cluster [Mn6(μ6-S)(μ3-AsS3)4] (Fig. 1a). As a result, the S1 atom occupies the heart of the cubic cluster, and binds six Mn atoms as a μ6-S bridging ligand (Fig. 1b), whilst four As3+ cations occupy four vertexes and six Mn2+ cations are located at the face-centers of the cubic cluster. Each Mn2+ cation is further coordinated by a N atom from N2H4 molecule forming an octahedral coordination environment of MnS5N (Fig. 1a). The S2− anions have two different coordination modes in the [Mn6(μ6-S)(μ3-AsS3)4]2− cube. Except for the central S(1)2− ion with a μ6-S bridging mode (Fig. 1b), the remaining 12 sulfide anions (S2–S13) have the same μ3-S bridging modes binding two Mn2+ and one As3+ cations. The structure of the [Mn6(μ6-S)(μ3-AsS3)4]2− cubic cluster is characterized by an octahedral core Mn6(μ6-S) (Fig. 1b) which is constructed from the heart S2− anion and the six Mn2+ cations at the face-centers of the [Mn6(μ6-S)(μ3-AsS3)4]2− cube. The connectivity within the [Mn6(μ6-S)(μ3-AsS3)4]2− cluster is more easily understood when the Mn6(μ6-S) octahedron is capped by four μ3-AsS3 ligands at four opposite faces (Fig. S8†). The As–S, and Mn–S bond lengths (Table S2†) are in agreement with those observed in Mn-thioarsenates(III).9b,9c,20d The Mn(7)2+ ion is coordinated to six N atoms from three en ligands, forming a distorted octahedral [Mn(en)3]2+ complex cation (Fig. 1c).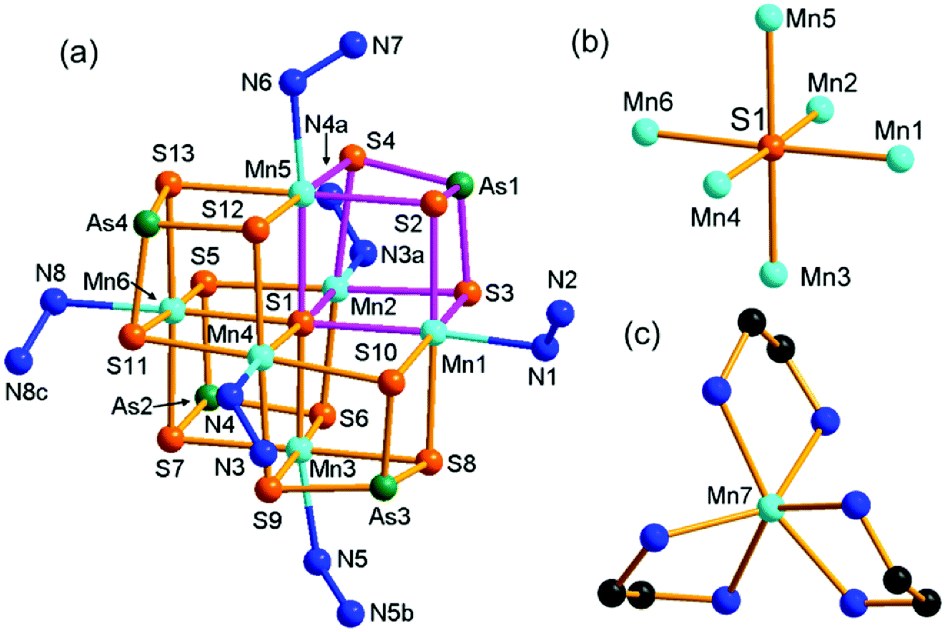 | ||
| Fig. 1 Structures for compound 1: [(N2H4)2Mn6(μ6-S)(μ-N2H4)2(μ3-AsS3)4]2− cluster showing the [Mn3(μ3-S)(μ3-AsS3)4] cube highlighted in purple bonds (a), Mn6(μ6-S) octahedron (b), and [Mn(en)3]2+ complex cation (c). Hydrogen atoms are omitted for clarity. Symmetry codes: (a) x − 1, y, z; (b) −x, −y + 1, −z; (c) −x + 1, −y + 1, −z + 1. | ||
In compound 1, the [Mn6(μ6-S)(μ3-AsS3)4]2− clusters are connected via hydrazine molecules into a layered structure. There are two coordination types for the N2H4 molecule binding the Mn(II) centers. Four Mn(II) ions (Mn2, Mn3, Mn4, Mn6) of the [Mn6(μ6-S)(μ3-AsS3)4]2− cluster are respectively linked to four neighbor [Mn6(μ6-S)(μ3-AsS3)4]2− clusters via four bidentate μ-N2H4 ligands, while the two remaining Mn(II) ions (Mn1, Mn5) at the neighbor face are terminated by a monodentate N2H4 ligand (Fig. 2a). As a result, the [Mn6(μ6-S)(μ3-AsS3)4]2− clusters are connected into a 2-D [(N2H4)2Mn6(μ6-S)(μ-N2H4)2(μ3-AsS3)42−]n waved layer perpendicular to the b axis (Fig. 2b and S9a†). Parallel stacking of the waved layers generates square-shaped channels running along the a axis of the unit cell, and the [Mn(en)3]2+ counter cations are wrapped in the channels between the layers (Fig. S9b†). Weak N–H⋯S hydrogen bonds are observed between the layers with the donor of NH2 groups from N2H4 molecules, and acceptor of S atom from the [Mn6(μ6-S)(μ3-AsS3)4]2− cluster (Table S8†). The inter-layer hydrogen bonds connected [(N2H4)2Mn6(μ6-S)(μ-N2H4)2(μ3-AsS3)42−]n layers into a 3D network.
 | ||
| Fig. 2 Structures of the [Mn6(μ6-S)(μ3-AsS3)4] cluster showing the N2H4 linkers between the Mn(II) ions (a), and the 2D [(N2H4)2Mn6(μ6-S)(μ-N2H4)2(μ3-AsS3)42−]n network (b) in 1. Symmetry codes: (a) x − 1, y, z; (b) −x, −y + 1, −z; (c) −x + 1, −y + 1, −z + 1. | ||
In our previous work, we prepared Mn(II)-thioarsenates [Mn(dien)2][Mn6(μ6-S)(μ-N2H4)3(μ3-AsS3)4]·H2O (dien = diethylenetriamine) [7, monoclinic, Cc (no. 9)] and [Mn(1,2-dap)3][(N2H4)2Mn6(μ6-S)(μ-N2H4)2(μ3-AsS3)4] (1,2-dap = 1,2-diaminopropane) [8, monoclinic, P21 (no. 4)] in the dien/N2H4 or 1,2-dap/N2H4 mixed solvents, respectively.20d The [Mn6(μ6-S)(μ3-AsS3)4]2− cluster is connected into a 3-D {[Mn6(μ6-S)(μ-N2H4)3(μ3-AsS3)4]2−}n anionic framework via three μ-N2H4 bridging ligands in 7, while it is connected into a 2-D {[(N2H4)2Mn6(μ6-S)(μ-N2H4)2(μ3-AsS3)4]2−}n anionic layer via two μ-N2H4 bridging ligands in 8. Compound 1 contains the same 2-D {[(N2H4)2Mn6(μ6-S)(μ-N2H4)2(μ3-AsS3)4]2−}n anionic layer as the anionic layer of compound 8. But it crystallizes in a different space group of P21/c (no. 14). Contrarily, the [Mn6(μ6-S)(μ3-AsS3)4] clusters observed in 1, 7, and 8 had been never obtained for the syntheses of Mn/As/S in en, dien or 1,2-dap without addition of N2H4,21 indicating the unique templating effect of N2H4 in the crystal growth of thioarsenates.
Compound 2 crystallizes in the orthorhombic space group Pbca with eight formulae in the unit cell. It consists of a protonated hydrazine [N2H5]+ cation, a 1-D polymeric [{Mn(μ-N2H4)2(μ-AsS4)}−]n anion, and half an en molecule. There are one crystallographically independent Mn, one As, and four S atoms in 2. The As5+ ion is coordinated with four S2− ions to form an [AsS4]3− unit. As shown in Fig. 3, the Mn2+ ion is repeatedly linked by two μ-N2H4 bridging ligands, forming an infinite [Mn(μ-N2H4)22+]n chain. The Mn(II) centers of the [Mn(μ-N2H4)22+]n are further joined by S1 of the [AsS4]3− unit, giving the chain-like [{Mn(μ-N2H4)2(μ-AsS4)}−]n polymeric anion (Fig. 3). The third hydrazine molecule exists as monoprotonated [N2H5]+ cation to act as the counter ion of the polymeric anion. The [AsS4]3− unit exhibits tetrahedral geometry with S–As–S bond angles in the range of 104.24(15)–112.67(18)° (Table S3†). The As–S bond lengths are shorter than those of [AsS3]3− observed in compound 1 (Tables S2 and S3†). The Mn2+ ion is coordinated by four N and two S atoms, forming a distorted octahedron MnN4S2 with axial angles varying in the range of 165.8(4)–173.0(4)°. The Mn–S and Mn–N bond lengths are consistent with the corresponding bond lengths found in compound 1 (Tables S2 and S3†). The [{Mn(μ-N2H4)2(μ-AsS4)}−]n anionic chains run parallel along the a axis and stack a layer via N–H⋯S hydrogen bonds (Fig. 3). [N2H5]+ cation and en molecules are located between the anionic layers (Fig. S10†).
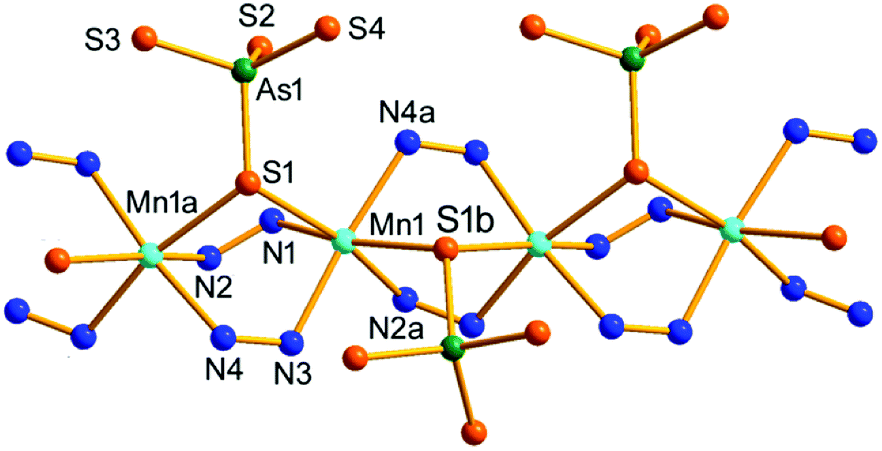 | ||
| Fig. 3 Structure of the [{Mn(μ-N2H4)2(μ-AsS4)}−]n anionic chain in 2 with the labeling scheme. Symmetry codes: (a) x − 1/2, y, −z + 1/2; (b) x + 1/2, y, −z + 1/2. | ||
Compound 3 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one formula in the unit cell. It contains one crystallographically independent As, three half Mn, and four S atoms. As shown in Fig. 4, the As3+ ion is coordinated with three S2− anions to form a typical trigonal pyramid AsS3 with As–S bond lengths ranging 2.337(5)–2.350(4) Å (Table S4†). Mn(1)2+ is coordinated by two S atoms from two AsS3 trigonal pyramids, and four N atoms from two trien molecules, forming a distorted octahedron MnS2N4 with axial angles in the range of 79.4(5)–100.6(5)°. Mn(2)2+ and Mn(3)2+ ions are joined by a μ-N2H4 bridging ligand, and are capped by the AsS3 unit with S1 binding Mn2, S3 binding Mn3, and S2 binding both Mn2 and Mn3. Meanwhile, the S1 atom coordinates to Mn1, and the asymmetric structural unit [Mn(μ-trien){Mn(μ-N2H4)(μ3-AsS3)}2] of 3 is formed (Fig. 4). Both Mn(2)2+ and Mn(3)2+ ions are octahedrally coordinated by four S atoms from two AsS3 units, and two N atoms from two μ-N2H4 ligands, forming MnS4N2 octahedra. The Mn–S and Mn–N bond lengths are comparable to the corresponding bond lengths found in compound 1 and 2 (Tables S2–S4†).
with one formula in the unit cell. It contains one crystallographically independent As, three half Mn, and four S atoms. As shown in Fig. 4, the As3+ ion is coordinated with three S2− anions to form a typical trigonal pyramid AsS3 with As–S bond lengths ranging 2.337(5)–2.350(4) Å (Table S4†). Mn(1)2+ is coordinated by two S atoms from two AsS3 trigonal pyramids, and four N atoms from two trien molecules, forming a distorted octahedron MnS2N4 with axial angles in the range of 79.4(5)–100.6(5)°. Mn(2)2+ and Mn(3)2+ ions are joined by a μ-N2H4 bridging ligand, and are capped by the AsS3 unit with S1 binding Mn2, S3 binding Mn3, and S2 binding both Mn2 and Mn3. Meanwhile, the S1 atom coordinates to Mn1, and the asymmetric structural unit [Mn(μ-trien){Mn(μ-N2H4)(μ3-AsS3)}2] of 3 is formed (Fig. 4). Both Mn(2)2+ and Mn(3)2+ ions are octahedrally coordinated by four S atoms from two AsS3 units, and two N atoms from two μ-N2H4 ligands, forming MnS4N2 octahedra. The Mn–S and Mn–N bond lengths are comparable to the corresponding bond lengths found in compound 1 and 2 (Tables S2–S4†).
 | ||
| Fig. 4 The asymmetric structural unit of 3 with the labeling scheme, showing coordination environments of each Mn2+ ion. Hydrogen atoms are omitted for clarity. Symmetry codes: (a) −x + 1, −y + 1, −z + 1; (b) −x, −y + 1, −z + 2; (c) −x, −y + 2, −z + 2; (d) x, y − 1, z. | ||
In 3, the trien molecule acts as a tetradentate μ-trien bridging ligand. It chelates a Mn(1)2+ ion with two N atoms, and chelates another Mn(1)2+ ion with the remaining two N atoms. As a result, the Mn(1)2+ ions are repeatedly joined by the μ-trien bridging ligands, forming an infinite planar [{Mn(1)(μ-trien)}2+]n chain (Fig. 5a). It is notable that the tetradentate amine trien usually chelates the same TM2+ ion with four N atoms in the syntheses of TM-containing chalcogenidometallates.22 The μ-trien bridging ligand in 3 is seldom observed, and only few examples were reported.23 The Mn(2)2+ and Mn(3)2+ ions are interconnected by a μ3-AsS3 and a μ-N2H4 bridging ligands, generating an infinite [Mn2{(μ-N2H4)(μ3-AsS3)}22−]n chain (Fig. 5b). The S1 atoms of the [Mn2{(μ-N2H4)(μ3-AsS3)}22−]n chain coordinate to the Mn(1)2+ ions on both sides of the planar [{Mn(1)(μ-trien)}2+]n chain (Fig. 5a and b), to complete coordination number of six for the Mn(1)2+ ion. As a result, a neutral 3-D framework of [Mn(μ-trien)(Mn2{(μ-N2H4)(μ-AsS3)}2)]n is obtained (Fig. 5c). In the framework, the [{Mn(μ-trien)}2+]n cationic chains run along the a axis, while the [Mn2{(μ-N2H4)(μ3-AsS3)}22−]n anionic chains run along the b axis of the unit cell (Fig. 5c and d). Intermolecular N–H⋯S hydrogen bonds are observed between the cationic and anionic chains (Fig. S11, Table S8†).
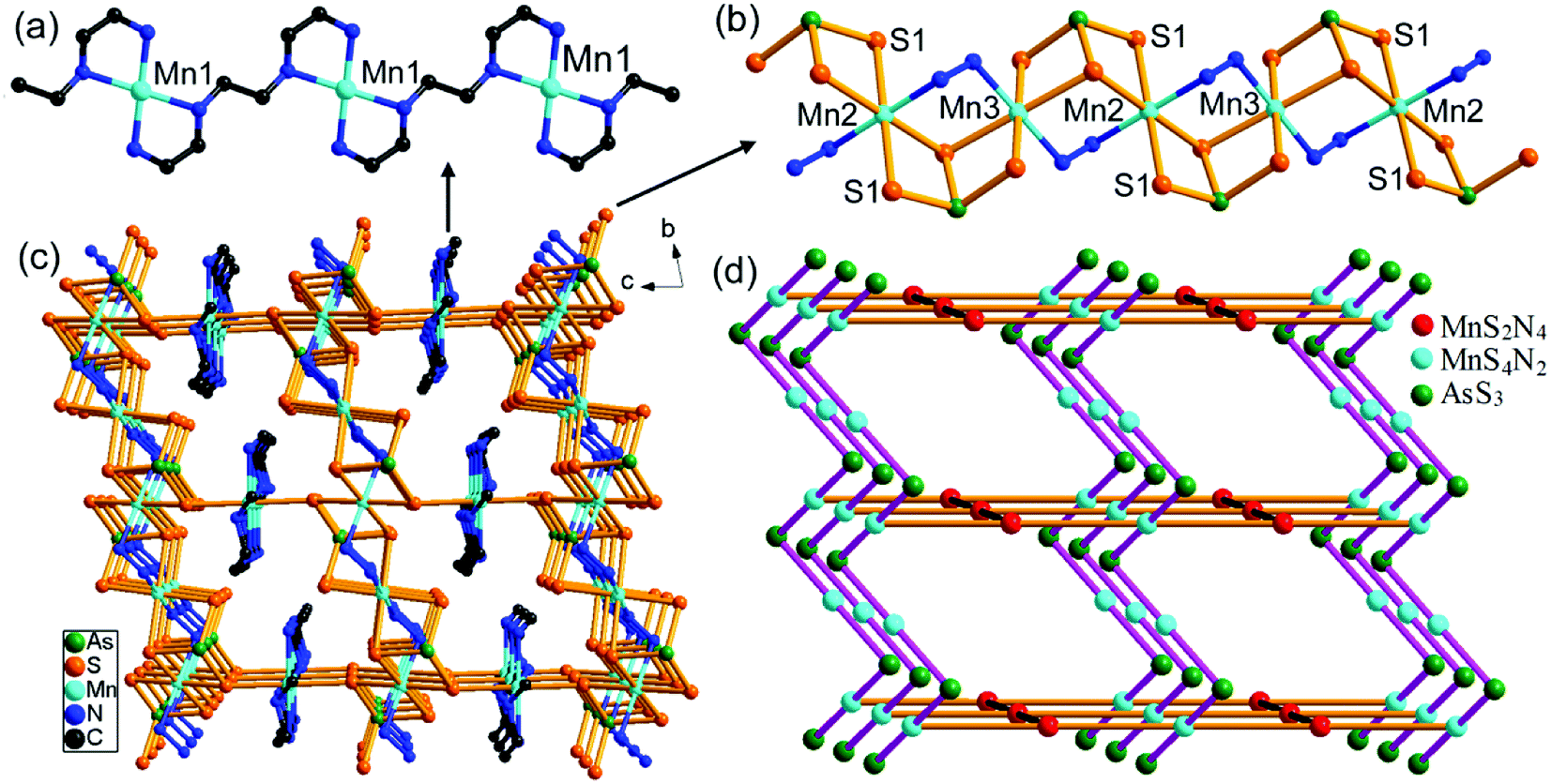 | ||
| Fig. 5 Crystal structures for compound 3: (a) planar [{Mn(μ-trien)}2+]n cationic chain. (b) [Mn2{(μ-N2H4)(μ3-AsS3)}22−]n anionic chain (hydrogen atoms are omitted for clarity). (c) Packing diagram of 3-D [Mn(μ-trien)(Mn2{(μ-N2H4)(μ-AsS3)}2)]n framework. (d) Schematic depiction of the 3-D framework in 3. | ||
Compound 4 crystallizes in the triclinic space group P. It consists of a neutral [{Mn(N2H4)}2{Mn(μ-N2H4)2(μ-AsS3)2}(μ-N2H4)2]n chain. The chain is composed of two and two halves crystallographically independent Mn, two As, and six S atoms. As shown in Fig. 6, As(1)3+ and As(2)3+ ions are both coordinated with three S2− anions to form trigonal pyramids As(1)S3 and As(2)S3, respectively. Mn(1)2+ and Mn(2)2+ ions are joined by a μ-N2H4 bridging ligand and by the As(1)S3 trigonal pyramid in μ-AsS3 coordination mode, meanwhile the Mn(2)2+ ion binds a terminal N2H4 molecule. As a result, a binuclear [Mn2(N2H4)(μ-N2H4)(μ-AsS3)] subunit is formed. The subunit propagates via centrosymmetric operation at Mn1, forming a trinuclear [{Mn(N2H4)}2{Mn(μ-N2H4)2(μ-AsS3)2}] SBU (Fig. 6). By the same connectivities with four N2H4 (containing N9–N12) molecules, As(2)S3, Mn(3)2+ and Mn(4)2+ form another trinuclear [{Mn(N2H4)}2{Mn(μ-N2H4)2(μ-AsS3)2}] SBU. The two SBUs are alternately connected by two μ-N2H4 (containing N3–N6) bridges into a neutral infinite [{Mn(N2H4)}2{Mn(μ-N2H4)2(μ-AsS3)2}(μ-N2H4)2]n chain (Fig. 6). In the neutral chain, hydrazine molecules adopt two types of coordination modes of monodentate terminal mono-N2H4 and bidentate bridging μ-N2H4 to Mn(II) centers. All Mn2+ ions are in octahedral geometries with MnS4N2 (Mn(1)2+, Mn(4)2+) and MnS2N4 (Mn(2)2+, Mn(3)2+) donor sets. In 4, the [{Mn(N2H4)}2{Mn(μ-N2H4)2(μ-AsS3)2}(μ-N2H4)2]n chains run parallel, and interact with each other via intermolecular N–H⋯N and N–H⋯S hydrogen bonds (Table S8†), forming layers parallelling with the (111) plane (Fig. S12†). The layers are further connected by the N–H⋯S hydrogen bonds into a 3D frame work (Fig. S13†).
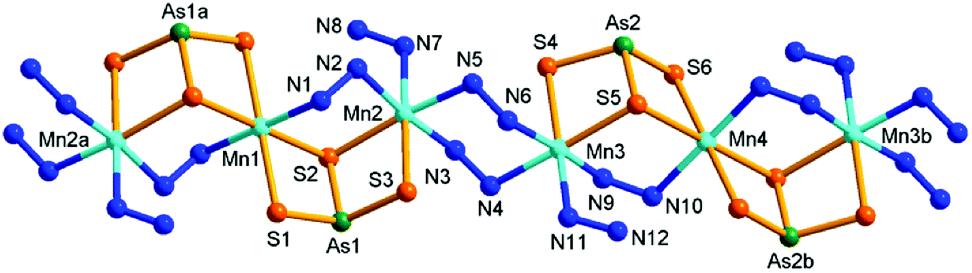 | ||
| Fig. 6 Structure of 4 with the labeling scheme. Hydrogen atoms are omitted for clarity. Symmetry codes: (a) −x, −y + 1, −z + 2; (b) −x + 1, −y + 3, −z + 1. | ||
Compound 5 crystallizes in the monoclinic space group P2/n with two formulae in the unit cell. There are one and a half crystallographically independent Mn, two halves As, and four S atoms (Fig. 7a). As(1)3+ and As(2)3+ ions are both coordinated with four S2− anions to form tetrahedral AsS4 units. Mn(1)2+ is coordinated by four N atoms from four μ-N2H4 bridging ligands and two S atoms of two AsS4 units, while Mn(2)2+ ion is coordinated by four N atoms from four μ-N2H4 bridging ligands and is chelated by two S atoms of the As(1)S4 unit. Both Mn(1) and Mn(2) form distorted octahedra MnN4S2. The As–S, Mn–S, and Mn–N bond lengths are in the range of the corresponding bond lengths in the Mn(II)-thioarsenate(V) of 2 (Tables S3 and S6†). In 5, Mn(1) and Mn(2) are connected by six μ-N2H4 bridging ligands into a infinite [Mn3(N2H4)6]n chain. The As(1)S4 unit bridges Mn(1) and Mn(2) centers with a μ3-1κS1:2κ2S1,S2:3κS2 bridging mode, forming a [Mn3(μ-N2H4)6(μ3-AsS4)3+]n cationic chain (Fig. 7b). The cationic chains are linked by the [As(2)S4]3− unit at Mn(1) to form the neutral [Mn3(μ-N2H4)6(μ3-AsS4)(μ2-AsS4)]n layer perpendicular to the b axis (Fig. 7c). The As(2)S4 unit adopts a bidentate μ-1κS1:2κS2 bridging mode. Large circle [{Mn4(μ-N2H4)3}2(μ2-AsS4)2] is formed in the layer. It is built up from eight Mn2+ ions joined by twelve μ-N2H4 and two μ-AsS4 bridges (Fig. 7d). The [Mn3(μ-N2H4)6(μ3-AsS4)(μ2-AsS4)]n layers run parallel to the (101) plane, and interact with each other via intermolecular N–H⋯S hydrogen bonds (Table S8†), forming a 3D framework (Fig. S14 and S15†).
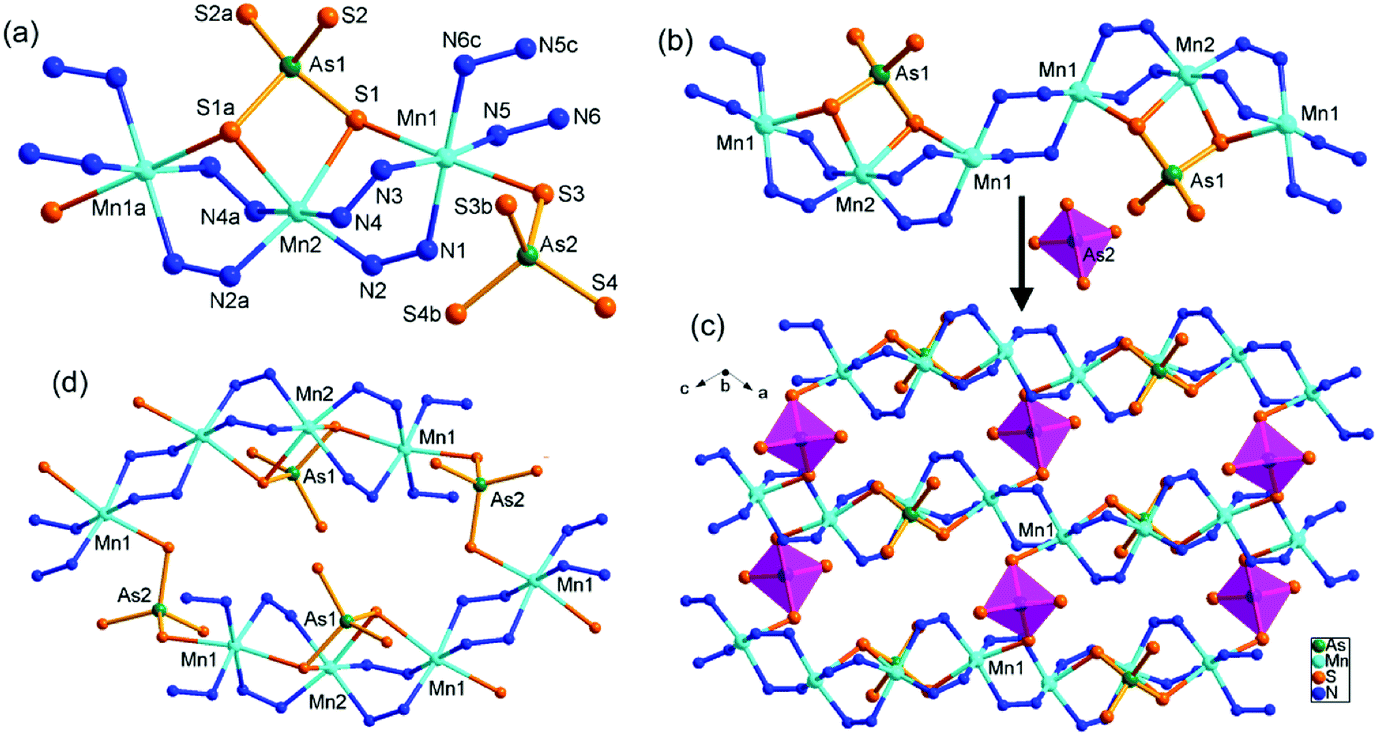 | ||
| Fig. 7 (a) The asymmetric structural unit of 5 with the labeling scheme, showing coordination environments of each Mn2+ ion. Hydrogen atoms are omitted for clarity. Symmetry codes: (a) −x + 1/2, y, −z + 3/2; (b) −x + 1/2, y, −z + 5/2; (c) −x, −y + 2, −z + 2. (b) The [Mn3(μ-N2H4)6(μ3-AsS4)3+]n chain. (c) The layer constructed by the [Mn3(μ-N2H4)6(μ3-AsS4)3+]n chains and μ2-AsS4 tetrahedral linkers. (d) The [{Mn4(μ-N2H4)3}2(μ2-AsS4)2] circle in 5. Hydrogen atoms are omitted for clarity. | ||
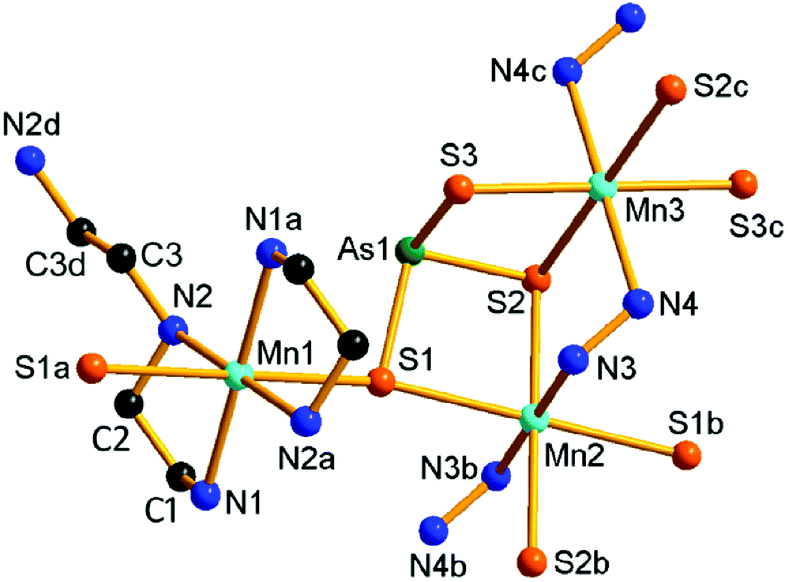
Compound 6 crystallizes in the triclinic space group P. The As5+ ion is coordinated with four S2− anions, forming an AsS4 tetrahedron. The Mn(1)2+ ion binds a NH3 molecule forming a [Mn(NH3)]2+ unit. The [Mn(NH3)]2+ units are joined by the AsS4 tetrahedra into a [{Mn(NH3)(μ3-AsS4)}22−]n anionic chain (Fig. 8). The AsS4 tetrahedron acts as tridentate ligand in μ3-1κ2S1,S2:2κ2S1,S3:3κS1 bridging mode. The Mn(1)2+ ion lies in an octahedral geometry involved in five S atoms from three AsS4 tetrahedra and a NH3 molecule. The As–S and Mn–S bond lengths are consistent with the corresponding bond lengths found in 5 (Tables S6 and S7†). The Mn(2)2+ ion is coordinated by six NH3 molecules to form a [Mn(NH3)6]2+ complex cation. The [{Mn(NH3)(μ3-AsS4)}22−]n chains run parallel along the a axis. The [Mn(NH3)6]2+ complex cations are located between the chains, and interact with the anionic chains with N–H⋯S hydrogen bonds (Fig. S16†).
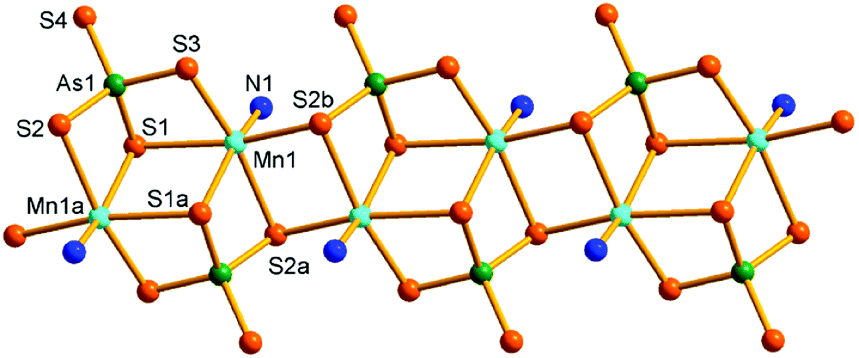 | ||
| Fig. 8 Structure of the [{Mn(NH3)(μ3-AsS4)}22−]n anionic chain in 6 with the labeling scheme. Hydrogen atoms are omitted for clarity. Symmetry codes: (a) −x, −y + 1, −z + 1; (b) −x + 1, −y + 1, −z + 1. | ||
In compounds 1–6, the AsS3 and AsS4 units coordinate to the Mn(II) centers in different bridging coordination modes, which are summarily illustrated in Fig. 9. As shown in Fig. 9, all S atoms of the AsS3 trigonal pyramid take part in coordination to the Mn(II) center (Fig. 9a–c). The AsS3 unit chelates two Mn2+ ions in a μ2-1κ2S1, S2:2κ2S1, S3 mode in compound 4 (Fig. 9c). It acts as a μ3-AsS3 bridging ligand with a μ3-1κ2S1,S2:2κ2S1,S3:2κ2S2,S3 mode in compound 1 (Fig. 9a), and a μ3-1κ1S1:2κ2S1,S2:2κ2S2,S3 mode in compound 3 (Fig. 9b), to join three different Mn2+ ions. The tetrahedral AsS4 unit can bind Mn(II) center with one, two or three S atoms (Fig. 9d–g). The AsS4 unit adopts a terminal mode with one S atom to join two Mn(II) centers in compound 2 (Fig. 9d). It links two Mn(II) centers with a μ-1κ1S1:2κ1S2 mode (Fig. 9e), and three Mn(II) centers with a μ3-1κS1:2κ2S1,S2:3κS2 mode in compound 5 (Fig. 9g). The AsS4 unit in 6 joins three Mn(II) centers with a μ3-1κ1S1:2κ2S1,S2:2κ2S2,S3 mode (Fig. 9f), which is in the same coordination mode with the AsS3 unit in compound 3 (Fig. 9b). Different modes μ2-1κ2S1,S2:2κ2S1,S3, μ3-1κ2S1,S2:2κ2S2,S3:2κ2S3,S4 and μ3-1κ1S1:2κ2S1,S2:2κ1S3 for the AsS4 unit are observed in compounds [NH4]8[Mn2(AsS4)4],14a [Mn3L3(AsS4)2]·H2O (L = 2,2′-bipy, phen),9b,24 and [Mn(1,2-dap)2]{[Mn(1,2-dap)]2(AsS4)2},21c respectively.
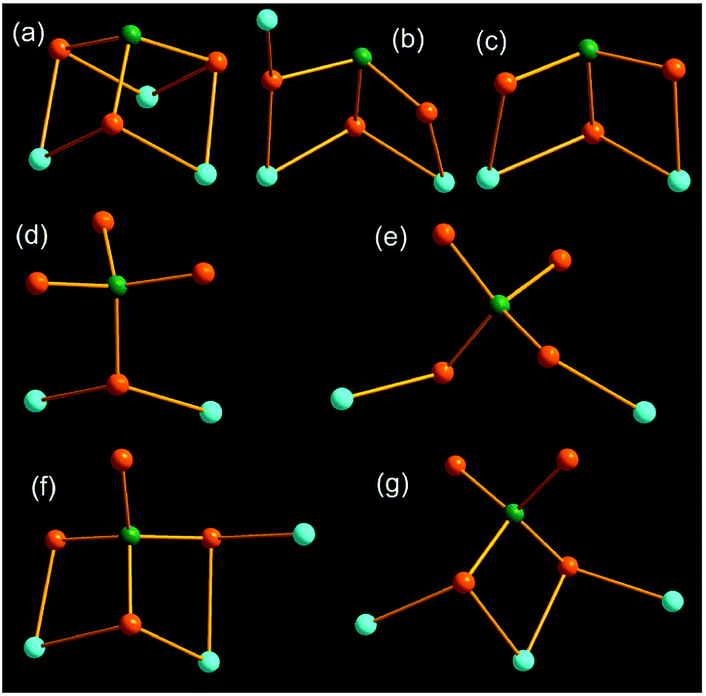 | ||
| Fig. 9 Coordination modes of the AsS3 and AsS4 units to Mn(II) centers in compounds 1 (a), 2 (d), 3 (b), 4 (c), 5 (e, g), 6 (f). The As, S and Mn atoms are drawn as green, orange and cyan spheres. | ||
Optical properties
The solid state near-IR/UV-Vis reflectance spectra of compounds 1–6 above were measured at room temperature on powder samples. The reflectance spectra were converted to optical absorption data by the Kubelka–Munk function,24 which are shown in Fig. 10. The band gaps can be estimated from the steep absorption edge at 2.32 (1), 2.44 (2), 2.19 (3), 2.39 (4), 2.35 (5), and 2.47 (6) eV, which are in accordance with their colors, respectively. The band gaps of 2, 4 and 6 are comparable to that of the Mn(II)-thioarsenates [Mn(NH3)6][(N2H4)2Mn2As2S8] (Eg = 2.46 eV).14a Compounds 1–6 exhibit a blue shift compared with those of polymeric Mn(II)-thioarsenates [Mn2(phen)(As2S5)]n (Eg = 2.01 eV) and [Mn3(phen)3(AsS4)2]n·nH2O (Eg = 1.97 eV) decorated by rigid amine phen (phen = 1,10-phenanthroline).25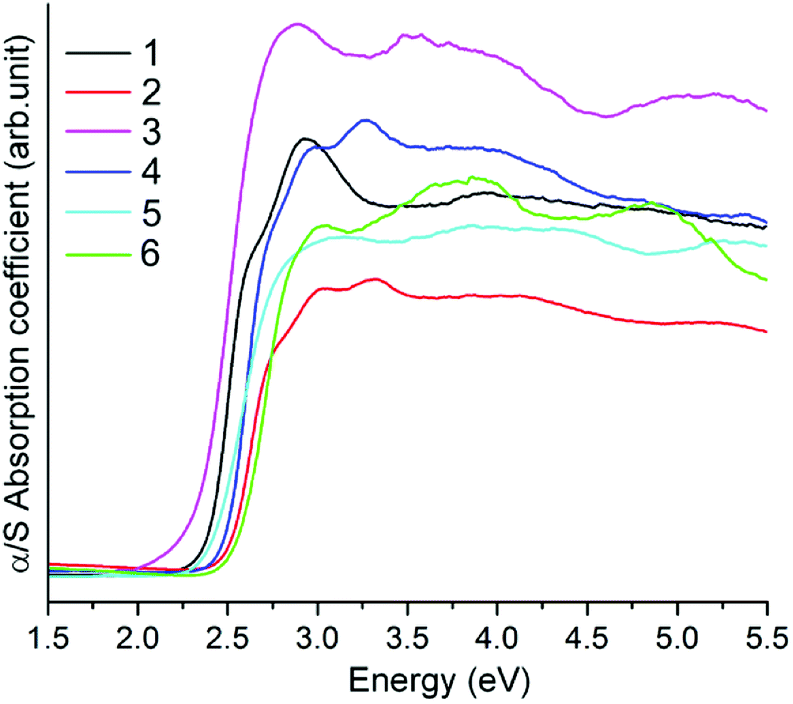 | ||
| Fig. 10 Solid state optical absorption spectra of compounds 1–6. | ||
Magnetic properties
The magnetic susceptibilities of 1 and 2 were investigated for polycrystalline samples in the temperature range of 2–300 K under an applied field of 1000 Oe. Temperature dependence of χm and χmT (χm is the magnetic susceptibility per Mn(II) ion) curves for compounds 1 and 2 are shown in Fig. 11. For compound 1, the χmT value is equal to 3.82 cm3 K mol−1 at 300 K, which is smaller than the spin-only value (4.375 cm3 K mol−1) for a single high-spin Mn(II) ion, indicating strong antiferromagnetic (AF) exchange interactions in 1. The diamagnetic bridges μ2-S and μ2-NH2NH2 between the Mn(II) ions give rise to the superexchange magnetic interactions, as observed in the chalcogenides Ba2MnS3,26 [Mn2(2,2′-bipy)As2S5],9b and [Mn2(NH2NH2)2SnS4].17a When the temperature is lowered, the χmT value decreases gradually and reaches a value of 0.074 cm3 mol−1 K at 2 K. Meanwhile, the magnetic susceptibility (χm) increases with lowering temperature until χm reaches a maximum value of 0.0380 mol cm−3 and then decreases gradually, which exhibits antiferromagnetic ordered state with Néel temperature of 26 K. The 1/χm vs. T curve in the temperature range 100–300 K obeys the Curie–Weiss law with C = 5.42 cm3 K mol−1 and θ = −126.45 K (Fig. S17†). The large negative θ value further confirms the strong AF interactions among the Mn(II) ions in compound 1. The triangular arrangement of the Mn(II) ions can induce spin frustration in 1. The parameter, which is defined as f = |θ|/TN, is widely used to measure the relative degree of the spin frustration. The f value of 4.86 indicates weak spin-frustration in 1.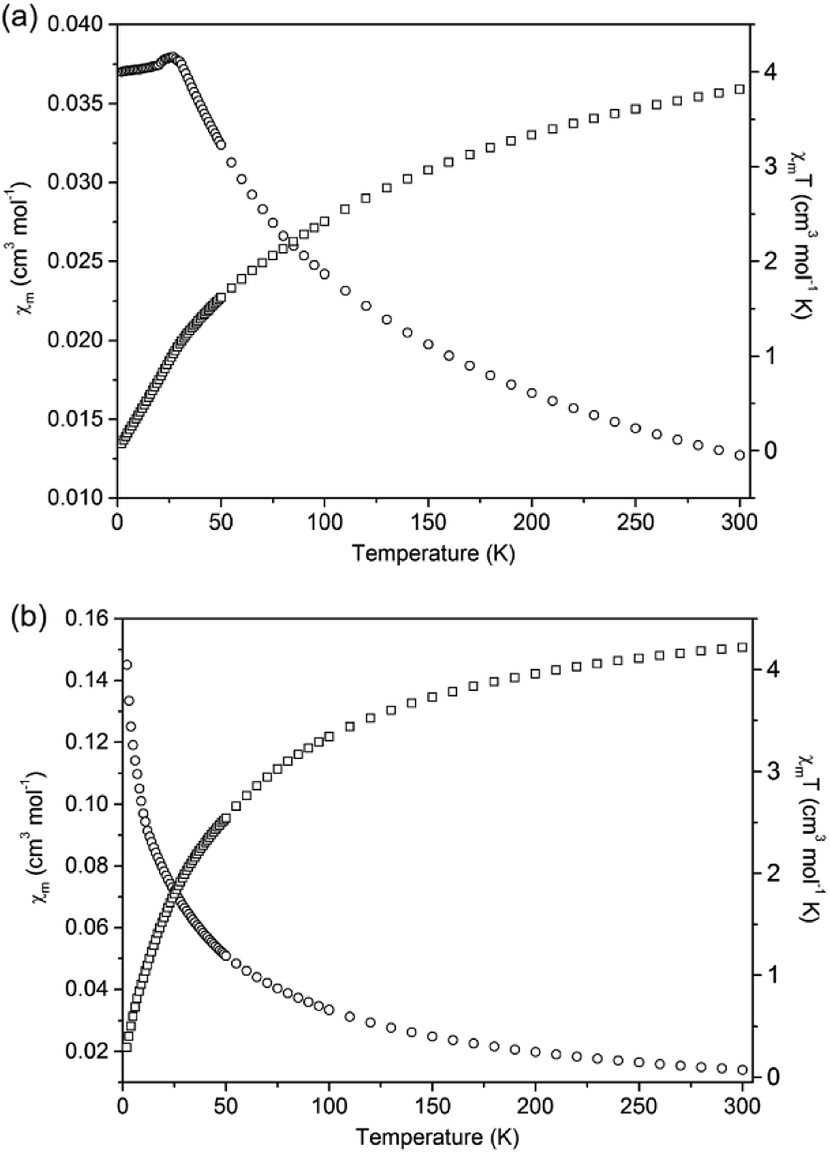 | ||
| Fig. 11 Curves of χmT and χm versus T for compounds 1 (a) and 2 (b). | ||
At 300 K, the χmT value of compound 2 is equal to 4.22 cm3 K mol−1 in agreement with the spin-only value (4.375 cm3 K mol−1) for a single high-spin Mn(II) ion. When the temperature is lowered, the χmT value decreases gradually and reaches a value of 0.31 cm3 mol−1 K at 2 K. Meanwhile, χm continuously increases from 0.0141 cm3 mol−1 to a value of 0.155 cm3 mol−1 at 2 K. The 1/χm vs. T curve in the temperature range 50–300 K obeys the Curie–Weiss law with C = 4.51 cm3 K mol−1 and θ = −19.85 K (Fig. S18†). The negative θ value confirms the weak antiferromagnetic interactions among the Mn(II) ions. The calculated effective magnetic moment (μeff) for one Mn(II) ion in compound 2 is 5.81 μB (Fig. S18†), which is close to the expected spin-only value of a free Mn(II) ion (5.92 μB). It is worthy to note that the negative Weiss constant of 1 is larger than that of 2. Besides the more Mn–S–Mn superexchange magnetic interactions in 1, the shorter Mn(II)⋯Mn(II) distance in 1 aids the AF exchange interactions.9d The distances between adjacent Mn(II) ions in 2 are 3.774 Å and 6.458 Å based on crystal structure. Whilst, compound 1 contains shorter Mn(II)⋯Mn(II) distances in the range of 3.568–3.792 Å, causing a stronger AF coupling interaction in 1.
Experimental
Materials and methods
All starting chemicals except N2H4·H2O are of analytical grade and were used as received. N2H4·H2O is 98% aqueous solution. Elemental analyses were conducted using an EA1110-CHNS-O elemental analyzer. Powder X-ray diffraction (PXRD) patterns were collected on a D/MAX-3C diffractometer using graphite monochromatized CuKα radiation (λ = 1.5406 Å). Fourier infrared (FT-IR) spectra were recorded on a Nicolet Magna-IR 550 spectrometer using dry KBr discs over the 4000–400 cm−1 range. Room-temperature optical diffuse reflectance spectra of powder samples were obtained using a Shimadzu UV-3150 spectrometer. Absorption (α/S) data were calculated from reflectance using the Kubelka–Munk function α/S = (1 −R)2/2R, where R is the reflectance at a given energy, α is the absorption, and S is the scattering coefficient.24 Magnetic susceptibility measurements were performed with a Quantum Design MPMS-XL SQUID susceptometer in the temperature range 2–300 K under an applied field of 1000 Oe. The magnetic data were corrected for the sample holder and the diamagnetic contributions. Thermogravimetric analysis was conducted on a SDT 2960 microanalyzer and the samples were heated at a rate of 5 °C min−1 under a nitrogen stream of 100 mL min−1.Syntheses of 1–6
:1 (V/V)). Most of the black powder was suspended in the solution, which was then decanted leaving behind crystals. This procedure was repeated until the water remained clear. Then, the resulting yellow block crystals of 1 were filtered off, washed with ethanol, and stored under vacuum. Yield 72 mg (41% based on As). Elemental analysis calcd for C6H40N14S13Mn7As4 (1): C, 5.11; H, 2.86; N, 13.91. Found: C, 4.95; H, 2.82; N, 13.78%. IR data (KBr, cm−1): 3449 (m), 3252 (w), 3139 (w), 2946 (w), 2864 (w), 2085 (w), 1636 (s), 1583 (s), 1480 (s), 1379 (w), 1304 (m), 1190 (w), 1123 (w), 1077 (w), 986 (w), 949 (w), 880 (w), 816 (w), 669 (w), 619 (w), 591 (w), 458 (w), 431 (w).X-ray crystal structure determinations
Data were collected on a Rigaku Saturn CCD diffractometer at 293(2) K using graphite-monochromated Mo-Kα radiation with a ω-scan method to a maximum 2θ value of 50.70°. The intensity data sets were collected with a ω-scan method and reduced with the CrystalClear program.27 An empirical absorption correction was applied for compounds 1–6 using the multiscan technique. The structures were solved with direct methods using the program of SHELXS-97,28a and the refinement was performed on F2 using SHELXL-97.28b All the non-H atoms were refined anisotropically. The hydrogen atoms were added geometrically and refined using the riding model. Technical details of data acquisition and selected refinement results are summarized in Table S1.†Conclusions
It has been demonstrated that hydrazine is a successful reaction media for the thermal synthesis of thioarsenate. A series of 1-D, 2-D and 3-D polymeric Mn(II)-thioarsenates have been prepared by hydrazine-solvothermal methods. The templating effect of hydrazine is influenced by the second ligand or solvent, which causes variable coordination modes between the thioarsenate anions and TM cations. The syntheses and properties of the title compounds indicate the possibilities of syntheses of new TM chalcogenidometallates with tunable structures by selecting the different coordinating solvents in hydrazine. Further studies on constructing new hybrid chalcogenidometallates with TM complex cation by hydrazine-solvothermal methods are in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC, no. 21171123), and the project funded by the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions.Notes and references
-
(a) C. L. Cahill, Y. Ko and J. B. Parise, Chem. Mater., 1998, 10, 19–21 CrossRef CAS
; (b) H. Li, M. Eddaoudi, A. Laine, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 1990, 121, 6096–6097 CrossRef
-
(a) N. F. Zheng, X. H. Bu, H. Vu and P. Y. Feng, Angew. Chem., Int. Ed., 2005, 44, 5299–5303 CrossRef CAS PubMed
-
(a) M. Wu, T. J. Emge, X. Y. Huang, J. Li and Y. Zhang, J. Solid State Chem., 2008, 181, 415–422 CrossRef CAS
-
(a) S. Bag, P. N. Trikalitis, P. J. Chupas, G. S. Armatas and M. G. Kanatzidis, Science, 2007, 317, 490–493 CrossRef CAS PubMed
-
(a) T. K. Bera, J. I. Jang, J. H. Song, C. D. Malliakas, A. J. Freeman, J. B. Ketterson and M. G. Kanatzidis, J. Am. Chem. Soc., 2010, 132, 3484–3495 CrossRef CAS PubMed
- Q. C. Zhang, Y. Liu, X. H. Bu, T. Wu and P. Y. Feng, Angew. Chem., Int. Ed., 2008, 47, 113–116 CrossRef CAS PubMed
-
(a) N. F. Zheng, X. H. Bu and P. Y. Feng, Nature, 2003, 426, 428–432 CrossRef CAS PubMed
-
(a) M. J. Manos, K. Chrissafis and M. G. Kanatzidis, J. Am. Chem. Soc., 2006, 128, 8875–8883 CrossRef CAS PubMed
-
(a) Q. C. Zhang, X. H. Bu, Z. E. Lin, M. Biasini, W. P. Beyemann and P. Y. Feng, Inorg. Chem., 2007, 46, 7262–7264 CrossRef CAS PubMed
- R. L. Bedard, S. T. Milson, L. D. Vail, J. M. Bennett and E. M. Flanigen, Stud. Surf. Sci. Catal., 1989, 49, 375–387 CrossRef
-
(a) W. S. Sheldrick and M. Wachhold, Coord. Chem. Rev., 1998, 176, 211–322 CrossRef CAS
-
(a) J. Li, Z. Chen, R. J. Wang and D. M. Proserpio, Coord. Chem. Rev., 1999, 190, 707–735 CrossRef
-
(a) Y. Lin and S. Dehnen, Inorg. Chem., 2011, 50, 7913–7915 CrossRef CAS PubMed
-
(a) W. W. Xiong, E. U. Athresh, Y. T. Ng, J. F. Ding, T. Wu and Q. C. Zhang, J. Am. Chem. Soc., 2013, 135, 1256–1259 CrossRef CAS PubMed
- J. G. Speight, Lange's Handbook of Chemistry, CD&W Inc, Laramie, Wyoming, 16th edn, 2004, P. 1.173, P. 1.387 Search PubMed
-
(a) M. Yuan and D. B. Mitzi, Dalton Trans., 2009, 31, 6078–6088 RSC
-
(a) M. J. Manos and M. G. Kanatzidis, Inorg. Chem., 2009, 48, 4658–4660 CrossRef CAS PubMed
- P. P. Sun, S. Z. Liu, S. F. Li, L. M. Zhang, H. Sun and D. X. Jia, Inorg. Chem., 2017, 56, 6152–6162 CrossRef CAS PubMed
-
(a) X. Y. Huang, J. Li, Y. Zhang and A. Mascarenhas, J. Am. Chem. Soc., 2003, 125, 7049–7055 CrossRef CAS PubMed
-
(a) J. Zhao, J. J. Liang, J. F. Chen, Y. L. Pan, Y. Zhang and D. X. Jia, Inorg. Chem., 2011, 50, 2288–2293 CrossRef CAS PubMed
-
(a) M. L. Fu, G. C. Guo, L. Z. Cai, Z. J. Zhang and J. S. Huang, Inorg. Chem., 2005, 44, 184–186 CrossRef CAS PubMed
-
(a) J. L. Lu, F. Wang, Y. L. Shen, C. Y. Tang, Y. Zhang and D. X. Jia, J. Solid State Chem., 2014, 216, 65–72 CrossRef CAS
-
(a) J. J. Liang, J. Zhao, W. W. Tang, Y. Zhang and D. X. Jia, Inorg. Chem. Commun., 2011, 14, 1023–1026 CrossRef CAS
- W. W. Wendlandt and H. G. Hecht, Reflectance Spectroscopy, Interscience Publishers, New York, 1966 Search PubMed
- X. Wang, T. L. Sheng, S. M. Hua, R. B. Fu, J. S. Chen and X. T. Wu, J. Solid State Chem., 2009, 182, 913–919 CrossRef CAS
- I. E. Grey and H. Steinfink, Inorg. Chem., 1971, 10, 691–696 CrossRef
- CrystalClear, Version 1.35, Rigaku Corp., Tokyo, Japan, 2002 Search PubMed
-
(a) G. M. Sheldrick, SHELXS-97, Program for Crystal Structure Determination, University of Göttingen, Germany, 1997 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Selected bond lengths and angles, IR spectra, PXRD patterns, structural figures, magnetic measurement, and TG curves. CCDC 1856282–1856287 for 1–6. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra06335d |
| This journal is © The Royal Society of Chemistry 2018 |