
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe evolution of the M1 local structure during preparation of VMoNbTeO catalysts for ethane oxidative dehydrogenation to ethylene†
T. Yu Kardash *a,
E. V. Lazarevaa,
D. A. Svintsitskiya,
A. V. Ishchenkoa,
V. M. Bondarevaa and
R. B. Nederb
*a,
E. V. Lazarevaa,
D. A. Svintsitskiya,
A. V. Ishchenkoa,
V. M. Bondarevaa and
R. B. Nederb
aBoreskov Institute of Catalysis SB RAS, Pr. Ak. Lavrentieva, 5, 630090, Novosibirsk, Russia. E-mail: kardash@catalysis.ru
bFriedrich-Alexander University of Erlangen-Nuremberg, Staudtstr. 3., 91058 Erlangen, Germany
First published on 22nd October 2018
Abstract
The so-called M1 phase (the common formula (TeO)x(Mo, V, Nb)5O14) is a very promising catalyst for ethane oxidative dehydrogenation (ODE). It shows 90% selectivity to ethylene at 78% ethane conversion (400 °C, contact time – 5.5 s). The active crystal structure is formed under certain synthetic conditions in VMoNbTe mixed oxides. This paper is devoted to the analysis of how the local and average structure of the M1 phase is developed during the synthesis and what happens at particular synthetic steps. The analysis of the local structure was performed using the EXAFS and pair distribution function (PDF) methods. The EXAFS analysis of the initial VMoTe water solution and VMoNbTe slurry showed that Anderson-type heteropoly anions are formed in the solution and are preserved after fast spray-drying of the slurry. Nb cations do not enter the structure of the polyanions, but form an extended hydrated oxide matrix, where distorted NbO6 and NbO7 polyhedrons are connected to each other. The hydrated oxide matrix with captured polyanions provides the compositional homogeneity of the precursor. The distances in the second coordination shell are redistributed after thermal treatment at 310 °C. After being heated at T > 350°, the local structure of the M1 phase is organized and pentagonal domains are formed. These domains consist of a NbO7 pentagonal bipyramid and five MeO6 adjacent octahedra (Me = Mo, V). In the first stages, the building blocks are stacked along the [001] direction. The crystallization process results in the connection of the pentagonal domains to the extended polygonal grid. The formation of the regular grid with TeOx containing channels is accompanied by the increase in ethane conversion and ethylene selectivity of the catalysts.
1. Introduction
Industrial processing of associated petroleum gases and their components – methane, ethane and propane is in the focus of both fundamental and applied research. The growth of shale gas production and the need to decrease CO2 emissions are the main reasons for this.1,2 Oxidative dehydrogenation of ethane (ODE) is one of the possible processes for effective utilization of a natural gas feedstock.ODE has the potential to become an alternative to the existing pyrolysis processes for the production of ethylene, which is a commodity chemical widely used in industry. Pyrolysis is a high temperature (800 °C) process that thermally cracks a feedstock of ethane or liquid fuels. ODE transforms ethane to ethylene catalytically, under mild conditions (300–400 °C). VMoNbTe oxide catalysts show unsurpassed catalytic characteristics in the ethane oxidative dehydrogenation.3–6 According to ref. 1 and 7, the ODE process based on the VMoNbTe oxide catalysts show energy and cost savings in comparison to the pyrolysis process.
The catalytic activity and selectivity of such catalysts are attributed to the so-called M1 phase.8–12 The structure of the M1 phase is studied virtually at the atomic level.13–15 The M1 phase is a layered compound with a common formula (TeO)0.23(Mo, V, Nb)5O14 (see Fig. 1). Distorted octahedrons of MoO6- and VO6-types and NbO7 pentagonal bipyramids are connected to form extended grids. The grids are stacked along the [001] direction forming hexa- and heptagonal channels both of these are occupied by TeOx species depending on the Te content in a sample.
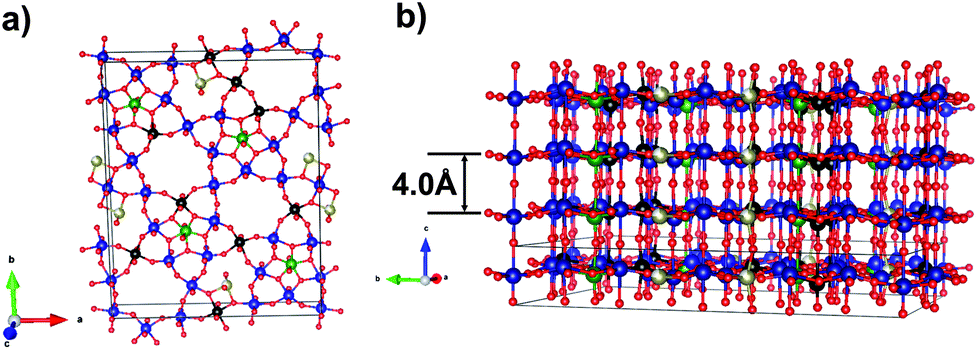 | ||
| Fig. 1 Schematic representation of the M1 phase in the (001) plane (a) and the stacking of the grids along the [001] direction (b). Mo atoms are shown in blue, Nb – green, V – black, Te – brown, O – red. Atomic coordinates taken from the ref. 14. | ||
The implementation of the VMoNbTeO catalysts in an industry has several challenges. One of them is a complex synthetic procedure, deviations from which result in significant changes of phase composition and structural disorder.16,17 To obtain the M1 phase, all the components should interact already at the step where a wet precursor is synthesized, which is reached in a narrow range of pH values.18–21 Chemical composition, drying and the conditions of heat treatment also have a strong influence on the phase composition of the samples. M1 oxide phase is prepared either by hydrothermal synthesis22–25 or by the “slurry” method, which implies mixing of aqueous solutions of initial reagents followed by solvent removal and thermal treatment in combined mode.26,27 The essential synthetic steps and required conditions are mostly known from empirical findings, whereas the structural transformations that occur at each step are still ill-understood. Controlled synthesis of M1 phase with predictable surface properties requires a deeper investigation of the reactions proceeding in the initial solutions and during crystallization.
The detailed analysis of chemical transformations that occur in the mixed solutions of the starting components was performed by Raman and NMR spectroscopy methods.28–30 It was shown that in the initial water mixed solution, that contains Mo, V and Te, different heteropoly anions (HPA) are formed and their composition, structure, a degree of condensation and protonation changes with component concentrations, temperature and pH.28 According to ref. 21 and 31 the optimal pH to obtain the best catalysts should be ≈3. At this pH values Anderson-type heteropoly anion [TeMo6O24]6− and polyoxoanion [TeV5Mo3O27]5− as the major species were detected.28 This pH value is reached when Nb oxalate solution is added to the VMoTe solution. It results in the formation of a VMoNbTe slurry. Although in ref. 29 and 30 it was detected that the structure of anionic species does not change after spray-drying of the slurry, the particular role of Nb at this synthesis stage is yet unclear.
The development of the M1 crystal structure during the thermal treatment of VMoNbTe dried precursor was studied by several groups.19,29,32 The successive synthetic procedure includes short activation in air flow at 310 °C with a subsequent heating in He flow up to 550–600 °C. The oxides precursors are semi-crystalline up to 550 °C when M1 crystallization stars. Such crystallization is a non-equilibrium process. Minor deviations from the optimal synthetic procedure might result in the formation of additional phases such as M2, Mo5O14, and MoO3. The M2 phase usually co-exists with the M1 phase in the active catalysts. Its structure is closely related with a hexagonal bronze.13 Mo5O14-type of the structure is usually formed in the VMoNb mixed oxides and has a similar structural arrangement to M1.33 The only difference between Mo5O14 and M1 in the arrangement of the polygonal grids is the absence of big heptagonal channels in the Mo5O14 structure.22
The goal of the present work is to study the development of the local structure of the M1 phase in the VMoNbTe oxide samples with cationic composition V0.3Mo1Nb0.12Te0.23, typical for highly active catalysts.34 We started with the V0.3Mo1Te0.23 solution and analyzed the local structure changes during all the synthetic steps using EXAFS and pair distribution function (PDF) methods.
Both methods provide information about the local structure for highly disordered materials, amorphous structures, and liquids. PDF is a powerful tool to study and describe the structure of both nanoparticles and highly disordered materials. PDF curve is a measure of the probability of finding pairs of atoms in the material separated by a distance r. As PDF data is obtained from a diffraction experiment, it provides all atom-pair correlations in the material.35 EXAFS data provides chemically resolved information on the local structure.
We used EXAFS spectroscopy for Mo–K and Nb–K edges to study the structural evolution from the mixed V0.3Mo1Te0.23 solution to V0.3Mo1Nb0.12Te0.23 slurry, amorphous spray-dried sample and highly disordered oxide precursor formed after calcination. The joint PDF analysis and EXAFS spectroscopy were applied to model the atomic structure of a highly disordered precursor of the M1 phase. X-ray photoelectron spectroscopy (XPS) was applied to address the changes in the oxidation state of the components (Mo, V, Nb and Te) during the thermal treatment of the dried precursor.
2. Experimental
2.1. Sample preparation
For preparation of solutions, the following starting materials were used: ammonium paramolybdate (PMA) (NH4)6Mo7O24·4H2O, ammonium metavanadate (MVA) NH4VO3 and telluric acid (TA) H6TeO6. All the compounds were the best available reagent grade and were used without further purification.For preparation of the solution with a Mo/V/Te molar ratio (S1 sample) corresponding to the optimum catalyst composition V0.3Mo1Nb0.12Te0.23, PMA (1.77 g), MVA (0.351 g), and TA (0.528 g) were dissolved in distilled water (10 mL) at 80 °C with stirring, followed by cooling to ca. 40 °C. The pH of the resulting solution was ≈6.
For obtaining a V0.3Mo1Nb0.12Te0.23Nb0.12 gel (G1 sample), a Nb oxalate solution (2.5 mL, 46.5 mg mL−1 Nb; C2O42−/Nb = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was added to a V0.3Mo1Nb0.12Te0.23 solution.
:1) was added to a V0.3Mo1Nb0.12Te0.23 solution.
The gel with a bulk composition V0.3Mo1Te0.23Nb0.12 was dried using a spray-drying procedure (sample D1) or under ambient conditions at room temperature (sample D2). A lab spray-dryer (Buchi-290) was used; the inlet temperature was 220 °C and the outlet temperature was 180 °C.
The solid resulting after spray-drying was divided into several parts. One sample was calcined in air at 310 °C for 1 hour (H310 sample). Other samples were heated in two steps: first in air at 310 °C for 1 hour and then in He flow at 350 °C (H350), 400 (H400), 450 (H450) and 550 °C (H550). Heating in He flow was conducted for 2 h for every sample.
2.2 Characterization techniques
X-ray diffraction was performed using Bruker D8 Advance diffractometer and CuKα-radiation. The diffraction intensities were measured using a LynxEye position sensitive detector. XRD patterns were collected in the 2θ range of 5–70°, with a 0.02° step size and a 3 s collection time. Phase analysis was performed using the ICDD PDF-2 database. The Rietveld refinement was performed using a Topas v.4.3 (Bruker) software.XPS measurements were performed on a ES-300 photoelectron spectrometer (KRATOS Analytical) using an MgKα (hν = 1253.6 eV) X-ray source (the power of 180 W). Spectrometer calibration was performed using values of BE(Au4f7/2) = 84.0 eV and BE(Cu2p3/2) = 932.7 eV for Ar-sputtered gold and copper foils, respectively. Before measurements all samples were pressed into a pellet followed by the fixation using a conducting carbon tape. No charging effect was observed during spectrum acquisition. Processing of spectral data (curve fitting, area calculation, difference spectra, etc.) was performed using the homemade XPS-Calc program, which has been tested on a number of systems.36–38 The procedure of curve fitting was performed using an approximation based on a combination of the Gaussian and Lorentzian functions with the subtraction of Shirley-type background.
Mo–K and Nb K X-ray absorption spectra were recorded on an EXAFS station at the Siberian Synchrotron Radiation Center (Novosibirsk) using the standard transmission procedure. The spectra were recorded at electron energy of 2 GeV in the VEPP-3 ring and current of 80 mA. The spectrometer had a Si(111) double monochromator and proportional chambers as detectors. Data processing and analysis were performed using Athena and Artemis software.39 For all samples, the oscillating part of the EXAFS spectrum (χ(k)) was analyzed in the form of k2χ(k) in the wave number range k = 2.5–12.5 Å−1.
X-ray PDF data were collected at beam line 11-ID-B, APS, Argonne at an energy of 58.2636 keV, i.e. λ = 0.2128 Å. Data collection was at room temperature as well. The samples were stored in 0.18 cm polyimide capillaries (Cole-Parmer, Illinois). Data treatment was performed with PDFgetX3.40 Building and refinement of PDF for nanoparticles was done using DISCUS program.41 To visualize structural models, VESTA 3 software was used.42
TEM data was obtained using JEM-2010 (accelerating voltage of 200 kV, resolution of 1.4 Å) and JEM-2200FS (field electron emission cathode, accelerating voltage of 200 kV, resolution of 0.8 Å) transmission electron microscopes. Particles were deposited by dispersing the sample suspension in ethanol on a copper substrate using an ultrasonic dispergator. The local analysis of the elemental composition of the samples was performed with the use of a Phoenix EDAX spectrometer with a Si(Li) detector and an energy resolution of about 130 eV.
2.3 Catalytic testing
The ethane oxidative dehydrogenation (ODE) was carried out in a fixed-bed glass tubular reactor with on-line chromatographic analysis of the reaction mixture components. Catalyst fraction 0.25–0.50 mm was used. Experiments were performed at atmospheric pressure and temperature of 400 °C using feed consisting of a mixture C2H6:O2:N2 = 10:10:80 (% vol). Ethane conversion and selectivity to the reaction products were calculated as in ref. 31 Additional details on catalytic experiment are in ESI.†
3. Results and discussion
3.1 XRD analysis of structural transformations
Table 1 shows the bulk chemical composition of VMoNbTeO samples after spray-drying and heating in He at different temperatures.| Sample | Tcal, °C | SBET, m2 g−1 | Chemical composition | ||
|---|---|---|---|---|---|
| V/Mo | Nb/Mo | Te/Mo | |||
| D1 | 220 | — | 0.30 | 0.11 | 0.23 |
| H350 | 350 | 4.5 | 0.30 | 0.11 | 0.23 |
| H400 | 400 | 5.0 | 0.30 | 0.11 | 0.20 |
| H450 | 450 | 6.7 | 0.30 | 0.11 | 0.21 |
| H500 | 500 | 7.3 | 0.30 | 0.11 | 0.18 |
| H550 | 550 | 5.4 | 0.30 | 0.11 | 0.17 |
We used XRD data for a preliminary characterization of the solid intermediates obtained by spray-drying, heating in He flow and the final crystalline sample.
Fig. 2 displays the XRD patterns of the VMoNbTeO samples after spray-drying (D1 sample), short-term heating in air at 310 °C (H310) and heated in He flow (H350, H450 and H550). The diffraction patterns for the H400 and H500 samples were similar to the XRD pattern of the H450 sample. According to the XRD data, D1 sample is an amorphous solid. The following heating in He below 550 °C results in a formation of disordered solids (H310, H350, H450 and H500). The diffraction pattern of these samples is characterized by the presence of two peaks at 2θ = 22.20° and 45.2° and a broad diffuse scattering at 2θ ≈ 27°. The crystallization of the bulk M1 phase occurs after heating in He at 550 °C (H550). Curve 4 shows the Rietveld fit of the experimental pattern. The structural parameters of the fitting model were taken from the ref. 14 The full-profile Rietveld refinement of the diffraction data confirmed that the sample is a single phase. Table S1 (ESI†) shows the fitting results and structural parameters of the crystalline phase.
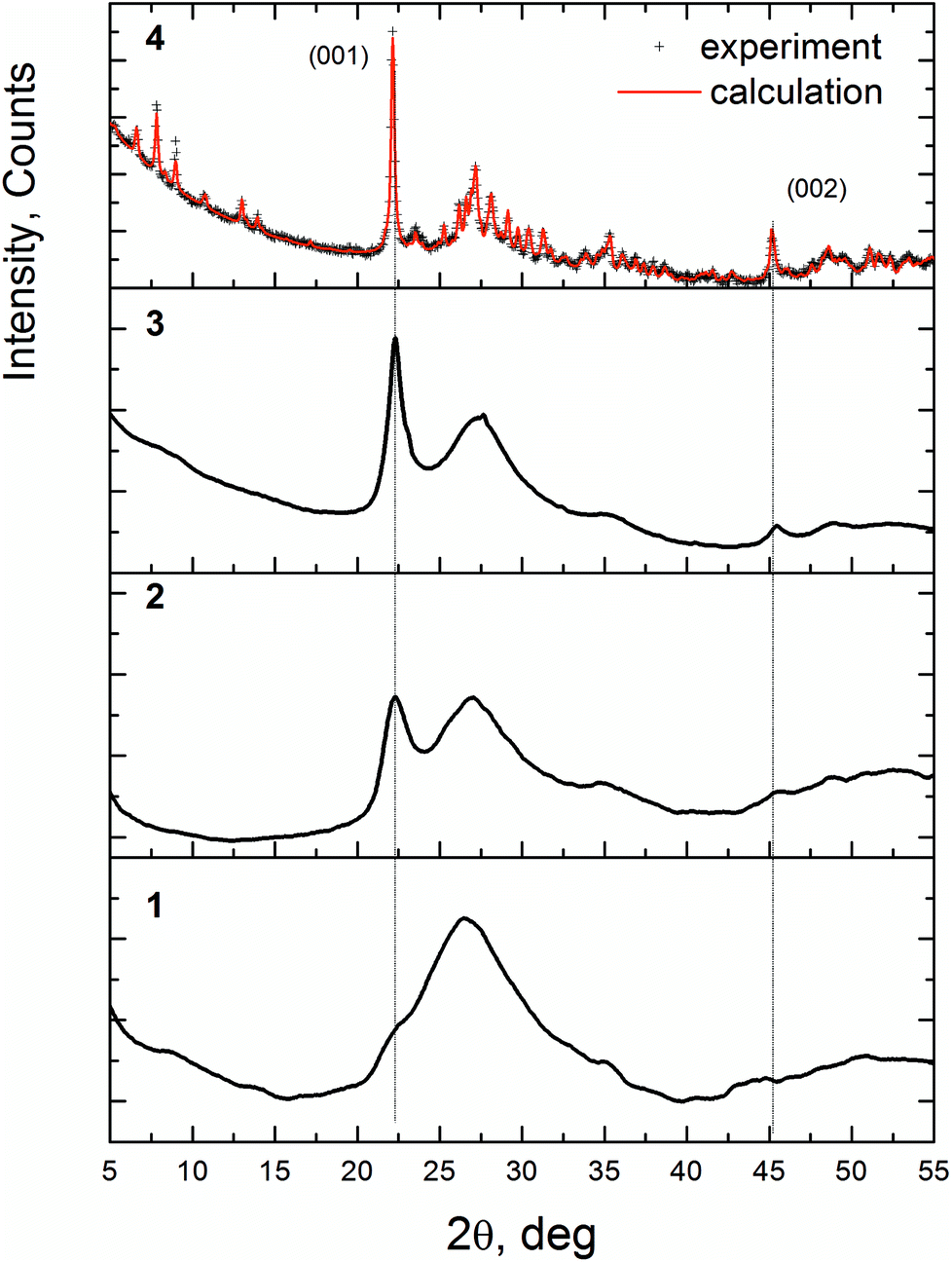 | ||
| Fig. 2 XRD patterns for the V0.3Mo1Nb0.12Te0.23 samples after spray-dying and thermal treatment. 1 – D1 sample (spray-dried precursor), 2 – H350, 3 – H450, 4 – H550. The red curve shows the Rietveld fit for the crystalline M1 phase. | ||
The peaks at 2θ = 22.20° and 45.2°, observed for the H350 and H450 samples, after crystallization become (001) and (002) reflections of the M1 structure. It indicates that at the first stage of crystallization the long-range order appears in the [001] direction, which corresponds to the interlayer distance of 4.0 Å (see Fig. 1b). So, we will identify these samples as “semi-crystalline materials” to indicate the presence of the ordering in the [001] direction and disorder in other directions. The crystallization process is connected with ordering in the (001) plane when hk0 reflections appear on the XRD pattern.
3.2 XPS study
Fig. 3 presents curve-fitted Nb3d spectra for different VMoNbTeO samples: from the spray-dried precursor (D1 sample) up to oxide heated in He at 550 °C (H550). Table 2 summarizes the XPS data for all studied samples. A surface chemical composition of studied samples during all synthetic steps is shown in ESI, Table S2.† For the D1 sample, the element atomic ratios were different from the bulk. In the course of heating, the surface V/Mo and Nb/Mo rations were approaching the bulk values. Te/Mo surface ration was increasing during heating in He, which shows the surface enrichment by Te in the crystalline H550 sample.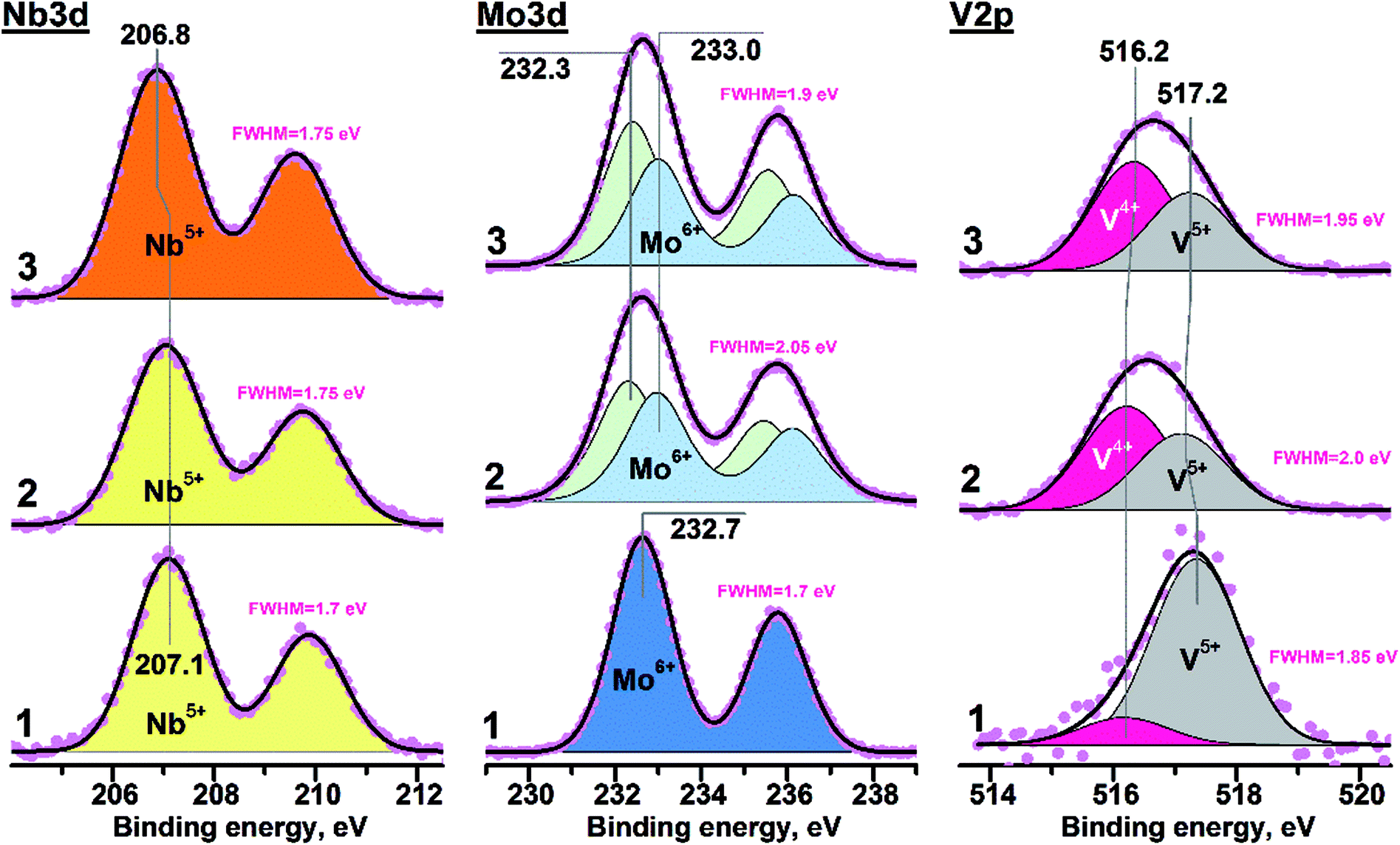 | ||
| Fig. 3 Photoelectron Nb3d, Mo3d and V2p spectra: 1 – D1 sample; 2 – H400 sample; 3 – H550 sample. The FWHM values (full width at half maximum) are presented for experimental spectral lines. | ||
| Sample | Nb3d | Mo3d | V2p | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nb5+ | FWHM, eV | Mo5+ | Mo6+ | FWHM, eV | V4+ | V5+ | FWHM, eV | |||||
| BE, eV | BE, eV | % | BE, eV | % | BE, eV | % | BE, eV | % | ||||
| a The partial reduction of vanadium was observed during spectra acquisition. | ||||||||||||
| D1 | 207.1 | 1.7 | — | — | 232.6 | 100 | 1.7 | 516.2 | 13a | 517.3 | 87 | 1.7 |
| H310 | 207.05 | 1.9 | 232.1 | 33 | 232.8 | 67 | 1.8 | 516.3 | 44a | 517.3 | 56 | 1.85 |
| H350 | 207.1 | 1.75 | 232.3 | 53 | 233.0 | 47 | 1.7 | 516.2 | 56 | 517.1 | 44 | 1.75 |
| H400 | 207.1 | 1.75 | 232.3 | 52 | 233.0 | 48 | 1.75 | 516.2 | 58 | 517.1 | 42 | 1.7 |
| H450 | 206.95 | 1.75 | 232.3 | 53 | 233.0 | 47 | 1.7 | 516.2 | 55 | 517.1 | 45 | 1.7 |
| H500 | 206.9 | 1.75 | 232.3 | 55 | 233.0 | 45 | 1.6 | 516.3 | 57 | 517.1 | 43 | 1.7 |
| H550 | 206.8 | 1.75 | 232.4 | 57 | 233.0 | 43 | 1.7 | 516.2 | 58 | 517.1 | 42 | 1.6 |
The value of FWHM (full width at half maximum) for all experimental Nb3d lines was close ∼1.7 eV, indicating the presence of only single niobium state. The D1 sample was characterized by the Nb3d5/2 peak position near 207.1 eV. Heating in helium at T ≤ 400 °C didn't result in any evident spectral changes for Nb3d line (for example, broadening or shift). The shift of Nb3d5/2 peak ∼0.3 eV towards smaller binding energies (BE) was observed for the H450, H500 and H550 samples (see Table 1). The Nb3d spectra obtained for the H450, 500 and 550 samples were similar to each other. The BE(Nb3d5/2) value close to ∼207 eV corresponds to oxidized niobium in form of Nb5+.43,44 So, the niobium state on the surface of all studied samples can be considered as Nb5+.
The BE(Nb3d5/2) value in case of Nb2O5 is varied in the range 207.2–207.5 eV,45,46 while corresponding value for Nb5+ in the Mo5O14- or M1-like structures decreases down to 206.8–206.9 eV.33,47,48 Observed Nb3d5/2 shift during high-temperature heating was probably related to the change of local coordination environment accompanied by the insertion of niobium in M1-like structure of VMoNbTe mixed oxide. Note that such a process occurred only at T > 400 °C.
The Mo3d spectrum of the D1 sample was characterized by the BE(Mo3d5/2) value equal to 232.7 eV and integral halfwidth of 1.7 eV (Fig. 3b). It corresponds to Mo6+ state in the MoO3 structure.49,50 Heating in helium at 400 and 550 °C (H400 and H450 samples) resulted in the significant spectral broadening of Mo3d line (FWHM = 1.9–2.1 eV), indicating the contribution from two components. Corresponding Mo3d spectra were curve-fitted into two doublets with BE(Mo3d5/2) values of 232.3 and 233.0 eV. It was reliably shown that the VMoNbTe oxide with M1 structure contains the combination of Mo5+ and Mo6+ species.13,47 Thus, components with BE(Mo3d5/2) values of 232.3 and 233.0 eV were attributed to Mo5+ and Mo6+ states, respectively. The surface Mo5+ ratio for samples heated at 400 and 550 °C was close to ∼52 and ∼57%, respectively. The shift of Mo3d5/2 peak from 232.7 to 233.0 eV in case of Mo6+ state can be related to the change of molybdenum coordination environment during the formation of M1 structure.
An increased FWHM value was the reason for curve-fitting of V2p into two components with BE(V2p3/2) near ∼516.2 and ∼517.2 eV (see Fig. 3c). Such components were attributed to V4+ and V5+ states.51,52 In case of the D1 sample the small V4+ contribution appeared immediately during spectra acquisition (see Fig. S1, ESI†), while the predominant presence of V5+ state was observed on its initial surface. The H400 and H550 samples were characterized by an unchangeable surface state during XPS measurements. The V4+ ratio at the surface of such samples was close to ∼58%. The combination of V4+ and V5+ states is a characteristic feature of VMoNbTe oxides with M1 structure.13,53 So, it can be suggested that the heating up to 400 °C is enough for insertion of vanadium into final mixed oxide.
For all solid samples the maximum of Te3d5/2 peak was located near 576.5 eV (see Fig. S2, ESI†) corresponded to Te4+ species.50 So, the main tellurium state on the surface of VMoNbTe oxides is an oxidized form of Te4+.
3.3 EXAFS study
EXAFS analysis of Mo and Nb local structures was performed because of their higher scattering power in comparison to V and Te. Furthermore, Mo is an element that determines the structure, whereas Nb role is still debatable.In order to interpret the spectra of the initial solution, an EXAFS spectrum for a water solution of the (NH4)6[TeMo7O24] salt was measured (see Fig. 4, curve 1). Curves 2–4 in the Fig. 4 show the pseudo-radial distribution functions (RDF) obtained via Fourier transformation of the Mo–K EXAFS spectra for the S1 sample (V0.3Mo1Te0.23 solution), and dried powders D1 and D2 correspondently. RDF curve for the G1 sample was similar to the RDF of the D1 sample and is not shown. Curves 5–7 in the Fig. 4 show RDFs for the H350, H450 and H550 samples. The RDFs are not phase-shift corrected and the peaks are shifted by approximately ≈0.4 Å with respect to the crystallographic distances.
 | ||
| Fig. 4 Pseudo-radial distribution functions (RDF) obtained via Fourier transformation of Mo–K edge EXAFS spectra. 1 – The water solution of (NH4)6TeMo6O24; 2 – S1; 3 – D1; 4 – D2; 5 – H350, 6 – H450 and 7 – H550. | ||
For an interpretation of EXAFS spectra in the mixed Mo-based compounds, the severe distortion of MoO6 octahedrons, where the central atom is shifted from the center towards one of the edges, should be considered. Due to the distortion, the Mo–O lengths in the first coordination sphere vary from 1.8 to 2.4 Å (see Fig. 5a) which causes a wide variation of metal–metal distances in the second coordination shell.54 A similar distortion is typical for Nb5+ and V5+ oxides. Therefore, the inclusion of all the paths in the modeling of EXAFS spectra is impossible, because it would produce an excessive number of correlated parameters. Because of this, we performed only qualitative interpretation of EXAFS data on the determination of mean interatomic distances within the atomic groups and comparison of different samples.
 | ||
| Fig. 5 The structural arrangement and typical interatomic distances for (a) the MoO6 octahedron, (b) Anderson-type HPA and (c) the pentagonal domain in the M1 structure. | ||
Fig. 5b shows the structure of the [TeMo7O24]6− anion. The RDFs for the (NH4)6[TeMo7O24] salt and the S1 sample (see Fig. 4, curve 2) show three characteristic peaks of the Anderson anion: the first and the second peak could be attributed to Mo–O distances in the distorted polyhedron, the third peak corresponds to the mean Mo–Me (Me = V, Mo) distance of 3.3 Å. This distance corresponds to the distance between adjacent octahedrons as shown in the Fig. 5b. According to ref. 21 vanadium could substitute Mo in the Anderson heteropoly anion, therefore, vanadium could also contribute to the second shell distances. No additional peaks are present on the RDF curves, indicating that anions are not included in the ordered structures.
The local structure of the Anderson anion is preserved during spray-drying in the D1 sample (see Fig. 4, curve 3). However, for the D2 sample, we observe a decrease in the intensity of the Mo–Me peak (see Fig. 4, curve 4). Thus, the prolonged aging of the wet precursor (G1 sample) in air under ambient conditions caused the destruction of the Anderson anion.
The subsequent heating in He flow at higher temperature results in the appearance of new Mo–Me peaks corresponding to the interatomic distances (see Fig. 4, curves 6 and 7) that are characteristic of the local structure of the M1 phase.23 The distance that corresponds to the interlayer distance of 4.0 Å appears for samples, heated at temperature ≥450 °C.
The RDF curve for Mo–K edge of the H550 sample (see Fig. 4, curve 7) can be described by six groups of interatomic distances: three groups of metal–oxygen distances and three groups of metal–metal distances. The typical distances of the M1 phase within the {(Nb)Me5} domain are shown in Fig. 5c. The Mo–O distances correspond to the bond lengths in the distorted MoO6 octahedra: short 1.7 and 2.3 Å in the [001] direction and average 2.0 Å in the (001) plane.13 The short distance 1.7 Å is clearly resolved only for the H550 sample, indicating the structural ordering. For the H350 and H450 samples, distances at 1.7 and 2.0 Å are not resolved. Mo–Me (Me = Mo, V, Nb) distances indicate different types of polyhedral connection in the M1 structure. The short distance 3.3 Å corresponds to the mean distance between the pentagonal bipyramid and the edge-sharing octahedrons, the longer 3.8 Å – between corner-sharing octahedrons in the (001) plane. The 4.0 Å peak corresponds to the distance between polygonal grids along the [001] direction (see Fig. 1b).13
Thus, for the H350 and H450 samples, we observe a reconstruction of the metal–metal coordination shell. There appears a peak, which corresponds to the distance between the pentagonal bipyramid and the adjacent octahedrons. The crystallization of the M1 phase results in an increase of the intensities of other Me–Me peaks indicating the appearance of the long-range ordering in the polygonal grid.
The RDFs for the Nb–K edge of the sample V0.3Mo1Te0.23Nb0.12 at various synthetic steps are shown in Fig. 6. The Nb source for synthesis is Nb-oxalate solution, which is shown by the RDF curve is in Fig. 6 (curve 1). The RDF of an Nb-oxalate solution is characterized by the single Nb–O peak, whereas peaks in the second coordination sphere are not pronounced. The general formula of a niobium oxalate solution could be described as NH4[NbO(C2O4)2(H2O)2]·nH2O. The characteristic structural unite of Nb-oxalate has a pentagonal bipyramidal structure with a short Nb![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond55 and bidentate oxalate-ligands.
O double bond55 and bidentate oxalate-ligands.
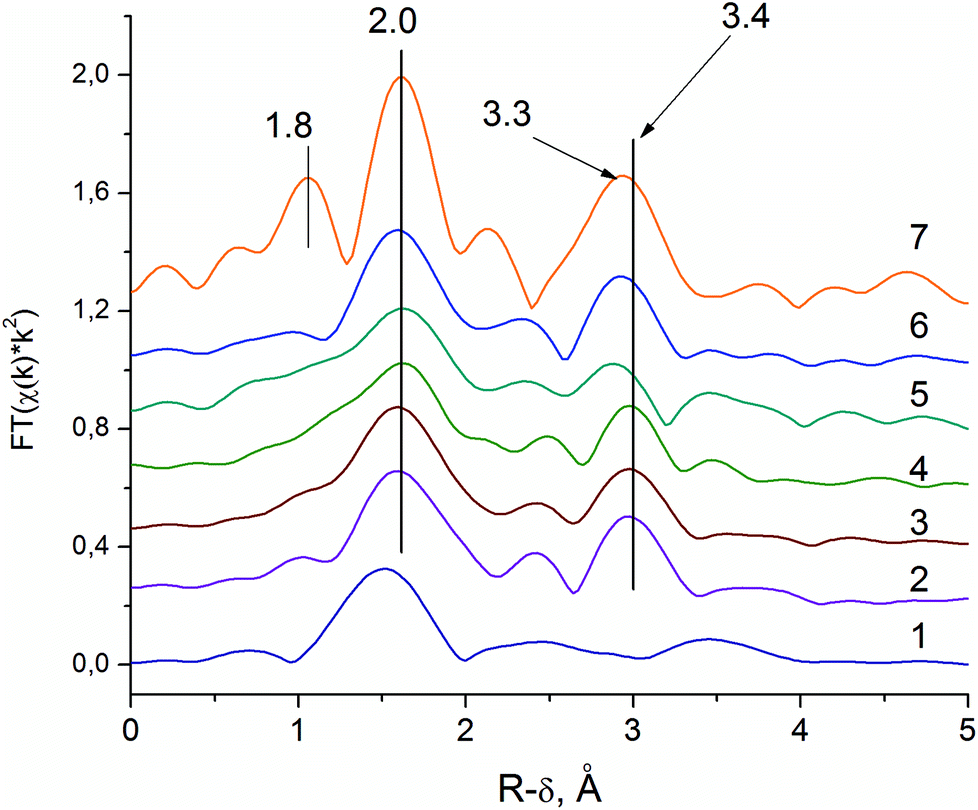 | ||
| Fig. 6 Pseudo-radial distribution functions (RDF) obtained via Fourier transform of Nb–K edge EXAFS spectra. 1 – Nb-oxalate solution; 2 – G1 gel; 3 – D1 sample; 4 – D2 sample; 5 – H350; 6 – H450; 7 – H550. | ||
The addition of Nb-oxalate solution to the S1 solution causes the formation of the orange-colored gel and significant changes in the Nb local structure (G1 sample, Fig. 6, curve 2). For the G1 sample, the Nb–O peak is shifted to the longer distances indicating the Nb–O bond length typical for Nb oxides, which are longer than those of the oxalate complex.55,56 It indicates the formation of an amorphous hydrous niobium oxide Nb2O5·nH2O, which contains distorted NbO6, NbO7 and NbO8 polyhedra.55
The characteristic feature of the G1 sample is the appearance of the pronounced peak that corresponds to the Nb–Nb distance. The fitting of the spectra showed that its peak corresponds to the distance of 3.4 Å, typical for Nb–Nb octahedrons,56 connected by common edges.
The integration of Nb cations into the structure of the Anderson anion by the substitution of Mo cations in the HPA structure (see Fig. 5b) would result in an appearance of the similar Nb–Me distances as on the Mo-RDF of the G1 and D1 samples, namely 3.30 Å. However, Nb–Nb distances are longer than Mo–Mo distances in the D1 and G1 samples. So, it could be concluded that Nb cations do not enter the structure of the HPA. The fitting results and used parameters are shown in the ESI (see Fig. S3 and Table S3†).
The difference between Nb local structure and Mo local structure within the VMoNbTeO gel is also revealed as a different behavior upon its drying. The Nb local environment does not change either during spray-drying or long aging in the ambient air (D1 and D2 samples, Fig. 6, curves 3 and 4). However, the long aging causes the significant decrease of intensity of the Mo–Mo (or Mo–V) peak on the Mo-RDF, which is connected with a destruction of the HPA (D1 and D2 samples, Fig. 5, curves 3 and 4). If Nb cations were included in the HPA structure, we would observe the decrease in Me–Me intensities for the both Nb–K and Mo–K RDFs.
The Nb-RDF for the H350 sample shows broadening of all the peaks (see Fig. 6, curve 5). It is an indication of the strong structural disorder of the Nb local structure that takes place after the sample is heated in He flow.
The Nb-RDF for the H450 sample (see Fig. 6, curve 6), is similar to the RDF curve as H550 sample. It shows that the local Nb structure in the semi-crystalline sample is similar to those of M1 crystal structure. The absence of the 1.8 Å peak and lower intensities of the RDF peaks corresponds to a disordering of the structure. However, the main characteristic features of the M1 phase is the 3.3 Å distance, presented on the RDF curve of the semi-crystalline sample. It is known from the previous HAADF-STEM studies, that Nb occupies pentagonal columns of the M1 structure.57 The similar Nb–K EXAFS spectra were obtained for a phase-pure M1 structure by Sanfiz et al.23 Our data support this finding.
The data obtained by EXAFS show that the {(Nb)Me5} (Me = Mo, V) domain is already formed in the semi-crystalline samples despite the long-range ordering along the [001] direction only.
3.4 X-ray PDF study
As the detailed modeling of the EXAFS data is complicated due to the presence of a number of different distances even in a crystalline sample, we performed a PDF analysis and structure simulation for a semi-crystalline sample (H450). The PDF advantage to EXAFS is the possibility of direct measuring of the interatomic distances.X-ray PDFs for the H450 and H550 samples are shown in Fig. 7a and b. The PDF data shows that the local structures of the samples with crystalline M1 phase and semi-crystalline precursor are similar. The first three peaks at 1.8, 2.0 and 2.3 Å correspond to different Me–O (Me = V, Nb, Mo) interatomic distances in the highly distorted MeO6 octahedrons for both samples. The PDF maxima near 3.3, 3.7 and 4.0 Å match three different types of Me–Me distances in the M1 structure. The appearance of such distances is in good agreement with the Mo–K and Nb–K edge EXAFS data (see Fig. 4, curve 7 and Fig. 7, curve 6).
 | ||
| Fig. 7 (a) X-ray PDF data for the sample with composition V0.3Mo1Nb0.12Te0.23 after heating in he at 450 °C and 550 °C. (b) Experimental PDFs in a wide range of distances and calculated PDF for a pentagonal domain from Fig. 5c. | ||
The difference in the long-range order between the samples heated at 450 and 550 °C is displayed in the PDF region above 7–8 Å. For the semi-crystalline sample, we observe the fall of peak intensities above 8 Å distance range. The crystalline sample shows evident G(r) maxima beyond the distance range of 20 Å shown in Fig. 7. The semi-crystalline sample has no intensive peaks at the distances >20 Å.
The observed features of the local structure are similar to those previously reported by our group in ref. 58, where the development of the Mo5O14-like local structure was studied in the three-component VMoNb system. To simulate the structure of a semi-crystalline VMoNb oxide, a model based on the pentagonal domain was developed.
A similar procedure was applied in current work to simulate the structure of a VMoNbTe oxide of the M1 phase. The unit cell of the M1 structure was expanded along a, b, and c directions to build a sufficiently large crystal. During the second step, an ellipsoid particle was cut from the big crystal. The procedure is illustrated in Fig. 8. The pentagonal bipyramid was placed in the center of the ellipsoid. The long axis of the ellipsoid corresponds to the [001] direction in the M1 crystal.
 | ||
| Fig. 8 Nanodomain structure building from the M1 structure. | ||
Fig. 7b shows the calculated PDF from the ellipsoidal particle with the length of 7 nm along the [001] direction and width of 3 nm in a perpendicular direction. The simulated PDF shows good qualitative agreement with the experimental PDF.
According to the PDF data, although the semi-crystalline samples do not show a long-range order in the (001) plane before calcination at 550 °C, the M1 local structure is already formed. The main fragment of the local structure is a pentagonal bipyramid in the {(Nb)Me5} domain (Me = Mo, V). These domains are firstly stacked on the top of each other, building the ordering along the [001] direction. As a result, the (001) reflection appears on the XRD pattern.
The similar pentagonal domains were found as a main structural feature of semi-crystalline oxide phases in the VMoTe and VMoNb oxide systems.58,59 However, despite the structural similarity of the semi-crystalline phases in these systems, the phase composition after crystallization is different. In the V0.3Mo1Te0.23 oxide system, M2 phase and (Mo, V)5O14 phases are formed,59 while the calcination of the semi-crystalline V0.3Mo1Nb0.37 precursor results in the appearance of the (Mo, V, Nb)5O14 mixed oxide.33
3.5 TEM study
In order to confirm PDF and EXAFS structural data, the TEM analysis was performed. We started from the H310 sample, as D1 sample was very unstable under electron beam exposure. Fig. 9 shows the TEM image of the H310 sample. The images show big porous spongy particles in the sample. The EDX analysis shows the composition Mo1V0.29Te0.23Nb0.17, which corresponds well to the bulk chemical composition. The particles are amorphous, which corresponds well to the EXAFS and XRD data. | ||
| Fig. 9 TEM image of the H310 sample. | ||
Fig. 10 shows TEM images for the H450 sample. The sample is characterized by the presence of the disoriented crystallites within the amorphous matrix. The diffraction pattern taken from the area is marked in the Fig. 10b shows only 4.0 Å reflections. In the Fourier filtered image, it can be seen that there is no pronounced boundary between the amorphous structure of the matrix and the crystallites, i.e. crystallization occurs from the amorphous phase. Some structural features of the M1 phase can be recognized in the Fourier filtered image. However, these structural fragments are disordered within the matrix. This finding confirms the PDF observation on the similarity of the local structure in the crystalline M1 phase and its semi-crystalline precursor.
 | ||
| Fig. 10 TEM of the H450 sample. (a) The microstructure of the particles (b) the red square shows the sample area, where diffraction was measured (c) the Fourier filtered image of the sample area on the (b) image. | ||
Fig. 11 shows TEM images of the H550 sample. According to the XRD data, the crystallization of the M1 phase was observed for this sample. The TEM images show the large particles with an ordered crystalline lattice. The diffraction data shows the set of reflections that correspond to the interplanar distances in the M1 phase.
 | ||
| Fig. 11 TEM image of the H550 sample. The red square area was used to obtain a Fourier filtered image and diffraction data. | ||
3.6 Catalytic measurements
Catalytic properties of the VMoNbTeO samples after heating at different temperatures in the reaction of ethane oxidative dehydrogenation are presented in Table 3.Both ethane conversion and ethylene selectivity increase with the rise in temperature of the sample heating in He. The crystalline M1 phase occurred after the catalyst's heating at 550 °C. The crystalline M1 phase shows the highest ethane conversion and ethylene selectivity. H350 sample slowly changes its catalytic characteristic with time in a stream, therefore the initial ethane conversion and reaction products selectivity are shown in Table 3.
For all synthesized catalysts the main reaction product in ODE is ethylene, and carbon oxides (CO, CO2) are by-products. The dependence of products selectivity on ethane conversion for the H500 and H550 samples is shown in Fig. 12. Selectivity to ethylene decreases, while CO and CO2 selectivities increase with ethane conversion growth. This is in agreement with literature data3,8–10,31 and reflects the triangular consecutive-parallel scheme of ODE reaction: ethylene and carbon oxides (both CO and CO2) are formed from ethane via parallel way and COx are formed from ethylene via the consecutive route. However, the increase in ethylene selectivity and ethane conversion, as shown in Table 3, reflect the catalytic properties of the samples with the different structure.
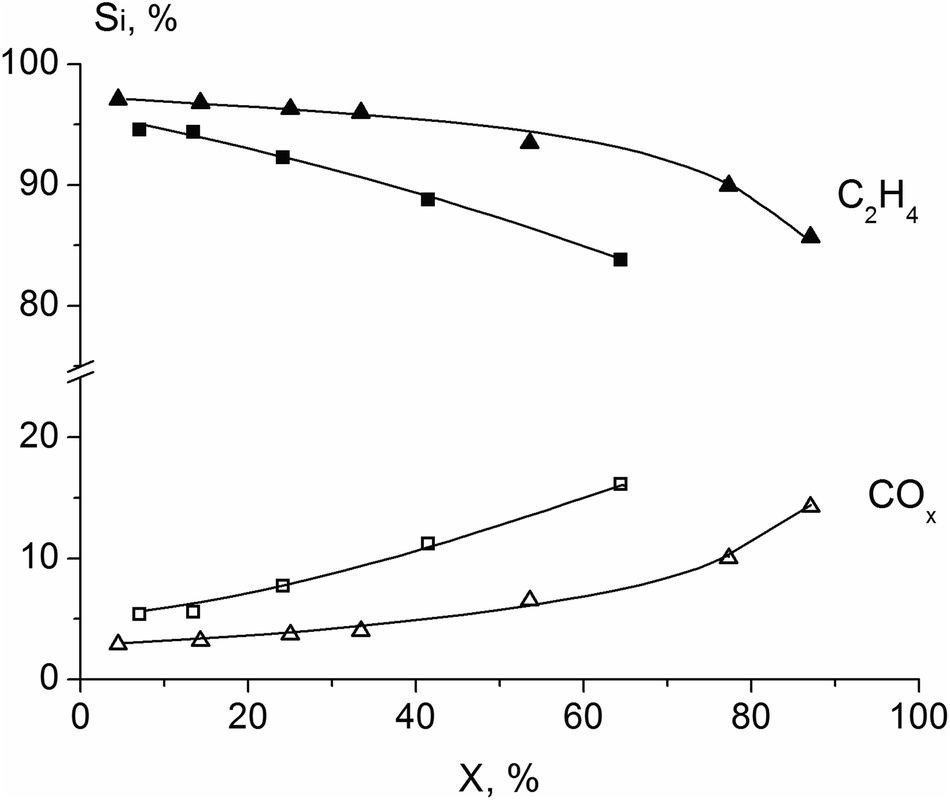 | ||
| Fig. 12 Dependence of products selectivity on ethane conversion for the sample calcined at 500 (■, □) and 550 °C (▲, Δ). Reaction temperature 400 °C, C2H6:O2:N2 = 10:10:80 (% vol). | ||
Our findings are in a good agreement with Konya et al.,10 who showed that crystalline M1 phase has superior catalytic properties in ODE reaction than “amorphous” Mo3VOx oxide.
The similar results of the fourfold increase in activity in VMoNbTeO catalysts were reported in ref. 20 for the reaction of propane ammoxidation to acrylonitrile. It was shown that reaction constant and acrylonitrile selectivity are the highest when the semi-crystalline sample is transformed into the crystalline M1 phase.
3.7 Development of the M1 structure
Although progress in describing of structural features of the active M1 phase in VMoNbTe oxide catalysts has already been achieved, the detailed understanding of its formation during the synthesis is still required. In this study, we analyze the transformations that occur in the course of a ‘slurry’ synthetic route with particular attention to Nb and its role in the structural development of the active phase.The ‘slurry’ method of synthesis of V0.3Mo1Nb0.12Te0.23 oxide catalysts includes the following sequential steps for the preparation of the final catalyst:18,19
(1). Preparation of V0.3Mo1Te0.23 aqueous solution from (NH4)6Mo7O24·4H2O, NH4VO3 and H6TeO6.
(2). Addition of niobium oxalate solution to the V0.3Mo1Te0.23 solution, which resulted in the formation of the V0.3Mo1Nb0.12Te0.23O gel (wet precursor).
(3). Spray-drying procedure giving dried precursor.
(4). Calcination at 310 °C in air, followed by heating in He flow at 550 °C.
The schematic representation of all synthetic steps and structural transformations that were detected in this work are summarized in Fig. 13.
 | ||
| Fig. 13 The schematic diagram to represent the structural transformations that occur during the synthesis of the M1 phase in the VMoTeNbO mixed oxide. | ||
The obtained Mo–K edge EXAFS data for the synthetic steps 1–3 is in a good agreement with the literature ones and our previous findings.28–30 The local structure that is typical for an Anderson-type heteropoly anion was detected for the initial V0.3Mo1Te0.23 solution. It remains unchanged in wet and dried precursors. It was shown that Nb cations do not enter the structure of the VMoTe heteropoly anions during the second preparation step and form a spongy hydrated NbOx matrix. The first coordination shell of Nb in this matrix is typical for NbO6 octahedrons, which are connected to each other by common edges. The local structure of the Nb in the gel is rather stable and does not change after prolonged drying in air, while Mo local structure changes significantly (D2 sample, see Fig. 4). Our data are in good agreement with in situ Raman data,30 which show no changes in the HPA structure upon addition of Nb-oxalate solution to the MoVTe solution.
The chemical interaction of all components in the amorphous precursor occurs when the initial polyanions are destroyed after the thermal treatment at 310 °C.
The heating in He flow at 400–450 °C results in the formation of the pentagonal domains {(Nb)Me5} according to the PDF and EXAFS data. Nb cations enter the centers of pentagonal bipyramids, whereas Mo cations enter the adjacent octahedrons.
According to XPS data, the heating of the spray-dried precursor (D1 sample) is accompanied by a reduction of molybdenum and vanadium cations caused by the decomposition of ammonia salts of the HPA-anion and oxalate anions. Major changes in comparison with the D1 sample were observed for the spray-dried precursor and sample heated in He at T > 400 °C. It correlates with the EXAFS and PDF data that show that the local structure of the M1 phase was already formed after heating at 400 °C.
The oxidation state of niobium cations during all the synthetic steps is attributed to the Nb5+ state. However, the position of Nb3d5/2 peak is highly sensitive to the local environment of niobium. The shift of Nb3d line indicating the rearrangement of the local structure was observed only after heating at T > 400 °C. So, the ordering of the local Nb structure occurred only after the pentagonal domains {(Nb)Me5} has been formed. The crystallization of M1 phase caused the further shift of the Nb3d line and resolving of the Nb–O different distances of the RDF curve of the EXAFS spectrum.
The {(Nb)Me5} domains are well known in the molybdenum polyoxometalate chemistry. They are the building blocks of Kepplerate anions, that are formed in reduced molybdates solutions under hydrothermal conditions.30 Furthermore, Kepplerate anions have been reported as building blocks in VMo,60,61 MoVSb,62 and VMoW63 oxide catalysts with M1 or Mo5O14-like structure. Similar pentagonal domains are formed after the hydrothermal treatment of the Nb-oxalate solution.64 Aberration-corrected STEM images of the distorted Nb oxide confirmed the presence of the same pentagonal building blocks as in the semi-crystalline precursor of the M1 phase.65
During the thermal treatment in helium, the M1 crystallization is firstly accompanied by the stacking of the structural fragments along the [001] direction. The next step is ordering in the (001) plane via connection of pentagonal fragments by single octahedra.
According to our previous findings, only Mo5O14-type of the structure with pentagonal domains is formed in the V0.3Mo1Nb0.37 oxide system.58 The addition of Te in the mixed oxide causes the formation of the M1 phase. However, the local structure of the semi-crystalline VMoNb and VMoNbTe oxides is similar.58 The major difference between these structures is a presence of big hexagonal and heptagonal channels.66 Note that the Te release during the thermal treatment of the M1 phase results in the formation of the (Mo, VNb)5O14 structure.67
The role of Nb in the crystallization process could be attributed to the stabilization of pentagonal bipyramids. Although the M1 phase could be obtained in VMo and VMoTe oxides by hydrothermal synthesis,10,68 the slurry method for such compositions gives only nanosized M1 crystallites.59 The heating of the nanosized crystallites at 550 °C results in destruction of the M1 phase and crystallization of M2 and (Mo, V)5O14 oxides.
Fig. 14 is a scheme showing how the chemical composition of the system containing V, Mo, Te, and Nb stimulates the formation of various crystalline products.
 | ||
| Fig. 14 The schematic representation of the development of the structurally-related M1, M2 and Mo5O14-type oxides in the VMo(Nb/Te) oxide systems via the transformations of pentagonal domains. | ||
According to the literature data, vanadium cations enter the centers of single octahedrons that connect pentagonal fragments into the polygonal network.14 A similar process of vanadium interaction with Kepplerate ion was described for VMoW oxides with Mo5O14-like structure.63,69
Thus, the VMoNb(Te) semi-crystalline oxide can be transformed either into the M1 structure, or into the M2 structure, or into the Mo5O14-type structure. The final structure of the polygonal grid is determined by the chemical composition and the thermal treatment conditions. The niobium cations are responsible for the stability of the pentagonal fragments,70,71 the tellurium – for the presence of hexa- or heptagonal channels.67
Structural flexibility explains the narrow pH range of the initial slurry, at which the most active VMoNbTeO catalysts with the largest amount of M1 phase are formed.21,28,31 This narrow pH interval corresponds to the isoelectric point of the Nb2O5·nH2O.28 The formation of the gel provides the uniform distribution of all components. Quick spray drying contributes to the preservation of this homogeneity in a precursor.20 The thermal treatment of the precursor in the range of 300–400 °C forms a nanostructured oxide with a local structure close to that in the M1 phase, crystallized at the high temperature.65
The obtained data show that the local structure, surface composition and oxidation state of the elements for the semi-crystalline VMoNbTe sample and corresponding crystallized M1 phase are similar. However, its catalytic properties in the ODE reaction differ significantly. The crystalline M1 structure is formed after the pentagonal domains are connected within a polygonal grid. It is accompanied by the appearance of ordered channels with TeOx units. The presence of such units is an essential feature of crystalline M1 phase in the VMoNbTe oxide.12 Some of the TeOx units are connected with V5+ species in the polygonal networks. Such Te–O–V5+ species are discussed as active sites for selective oxidation of C2–C3 alkanes8,11,72,73 on the basis of DFT calculations and in situ experiments. The importance of TeOx species in ODE reaction is also supported by our previous data. It was shown that Te-free (Mo, V, Nb)5O14 oxide is inactive in ODE reaction.33
The described mechanism of development of the crystalline M1 phase in the VMoNbTe oxide is supported by the ADF-STEM imaging data.74–76 It was demonstrated that the M1 phase contains many nanosized defects and nascent phases, including structurally disordered pentagonal domains co-joined in pairs, triplets, and quadruplets etc, which could be easily formed during the non-equilibrium crystallization process.
4. Conclusions
The development of the M1 phase of the cationic composition V0.3Mo1Nb0.12Te0.23 was studied at all steps of preparation by the slurry synthesis method. It was shown that heteropoly anions (HPA) with Anderson structure are formed after mixing the solutions of the initial reactants of Mo, V, and Te. The adding of niobium oxalate to solution results in the formation of a mixed VMoNbTe gel, where Nb cations do not enter the HPA structure and form hydrated NbOx matrix. The HPA structure is preserved in the gel and after spray-drying. Its destruction starts after heating at T ≥ 310 °C. It causes partial reduction of molybdenum and vanadium and the chemical interaction of all components. Such interaction results in the formation of the {(Nb)Me5} domains (Me = V, Mo), which are the building blocks of the M1 phase. Niobium occupies a position in the center of the pentagonal bipyramid during heating above 400 °C. Crystallization of the M1 phase occurs when the pentagonal clusters are connected with each other in the (001) planar. The crystallization process results in the appearance of the hexagonal channels containing TeOx units. Despite the similarity in the local structure of the semi-crystalline and crystalline samples, the crystallization results in the activity increase in oxidative dehydrogenation of ethane.The non-equilibrium conditions for the crystallization process and the structural similarity of the M1, M2 and Mo5O14-like precursors can be the reasons for the high sensitivity of the VMoNbTe-based catalyst structure to the preparation parameters and the chemical composition of the initial precursor.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Science Foundation, grant 17-73-20073.References
- A. M. Gaffney and O. M. Mason, Catal. Today, 2018, 285, 159–165 CrossRef.
- F. Cavani, N. Ballarini and A. Cericola, Catal. Today, 2007, 127, 113–131 CrossRef CAS.
- P. Botella, E. García-González, A. Dejoz, J. M. Lopez Nieto, M. Vazquez and J. Gonzalez-Calbet, J. Catal., 2004, 225, 428–438 CrossRef CAS.
- T. T. Nguyen, M. Aouine and J. M. M. Millet, Catal. Commun., 2012, 21, 22–26 CrossRef CAS.
- A. M. Gaffney, R. Ghosh, R. Song, C. Y. Yeh and T. Langner, US Pat. 8105972, 2012.
- J. M. L. Nieto, P. A. Botella, M. I. N. Vazquez and A. G. Dejoz, US Pat. 7319179, 2008.
- C. Baroi, A. M. Ga and R. Fushimi, Catal. Today, 2017, 298, 138–144 CrossRef CAS.
- B. Deniau, T. T. Nguyen, P. Delichere, O. Safonova and J. M. M. Millet, Top. Catal., 2013, 56, 1952–1962 CrossRef CAS.
- T. T. Nguyen, B. Deniau, P. Delichere and J.-M. M. Millet, Top. Catal., 2014, 57, 1152–1162 CrossRef CAS.
- T. Konya, T. Katou, T. Murayama, S. Ishikawa, M. Sadakane, D. Buttreyc and W. Ueda, Catal. Sci. Technol., 2013, 3, 380–387 RSC.
- M. Aouine, T. Epicier and J.-M. M. Millet, ACS Catal., 2016, 6, 4775–4781 CrossRef CAS.
- J. Valente, H. Armendáriz-Herrera, R. Quintana-Solórzano, P. del Ángel, N. Nava, A. Masso and J. M. L. Nieto, ACS Catal., 2014, 4, 1292–1301 CrossRef CAS.
- P. DeSanto, D. Buttrey, R. K. Grasselli, C. G. Lugmair, A. F. Volpe Jr, B. H. Toby and T. Vogt, Z. Kristallogr. Cryst. Mater, 2004, 219, 152–165 CAS.
- X. Li, D. J. Buttrey, D. A. Blom and T. Vogt, Top. Catal., 2011, 54, 614–626 CrossRef CAS.
- W. D. Pyrz, D. A. Blom, N. R. Shiju, V. V. Guliants, T. Vogt and D. J. Buttrey, J. Phys. Chem. C, 2008, 112, 10043–10049 CrossRef CAS.
- E. D. Finashina, A. V. Kucherov and L. M. Kustov, Russ. J. Phys. Chem. A, 2013, 87, 1983–1988 CrossRef CAS.
- F. Ivars, P. Botella, A. Dejoz, J. M. L. Nieto, P. Concepción and M. I. Vázquez, Top. Catal., 2006, 38, 59–67 CrossRef CAS.
- M. M. Lin, Appl. Catal., A, 2003, 250, 305–318 CrossRef CAS.
- G. Y. Popova, T. V. Andrushkevich, Y. A. Chesalov, L. M. Plyasova, L. S. Dovlitova, E. V. Ischenko, G. I. Aleshina and M. I. Khramov, Catal. Today, 2009, 144, 312–317 CrossRef CAS.
- G. Y. Popova, T. V. Andrushkevich, L. S. Dovlitova, G. a. Aleshina, Y. A. Chesalov, a. V. Ishenko, E. V. Ishenko, L. M. Plyasova, V. V. Malakhov and M. I. Khramov, Appl. Catal., A, 2009, 353, 249–257 CrossRef CAS.
- J. Oliver, J. M. López Nieto, P. Botella and A. Mifsud, Appl. Catal., A, 2004, 257, 67–76 CrossRef CAS.
- W. Ueda, D. Vitry and T. Katou, Catal. Today, 2005, 99, 43–49 CrossRef CAS.
- A. Celaya Sanfiz, T. W. Hansen, F. Girgsdies, O. Timpe, E. Rödel, T. Ressler, A. Trunschke and R. Schlögl, Top. Catal., 2008, 50, 19–32 CrossRef CAS.
- P. Botella, A. Dejoz, M. C. Abello, M. I. Vázquez, L. Arrúa and J. M. López Nieto, Catal. Today, 2009, 142, 272–277 CrossRef CAS.
- E. V. Ischenko, T. V. Andrushkevich, G. Y. Popova, V. M. Bondareva, Y. A. Chesalov, T. Y. Kardash, L. M. Plyasova, L. S. Dovlitova and A. V. Ischenko, in Studies in Surface Science and Catalysis, 2010, vol. 175, pp. 479–482 Search PubMed.
- Q. Xie, L. Chen, W. Weng and H. Wan, J. Mol. Catal. A: Chem., 2005, 240, 191–196 CAS.
- D. Zhonghua, W. Hongxin, C. Wenling and Y. Weishen, Chin. J. Catal., 2008, 29, 1032–1036 CrossRef.
- R. I. Maksimovskaya, V. M. Bondareva and G. I. Aleshina, Eur. J. Inorg. Chem., 2008, 2008, 4906–4914 CrossRef.
- P. Beato, A. Blume, F. Girgsdies, R. E. Jentoft, R. Schlögl, O. Timpe, A. Trunschke, G. Weinberg, Q. Basher, F. A. Hamid, S. B. A. Hamid, E. Omar and L. Mohd Salim, Appl. Catal., A, 2006, 307, 137–147 CrossRef CAS.
- M. Sanchez Sanchez, F. Girgsdies, M. Jastak, P. Kube, R. Schlögl and A. Trunschke, Angew. Chem., 2012, 124, 7306–7309 CrossRef.
- V. M. Bondareva, T. Y. Kardash, E. V Ishchenko and V. I. Sobolev, Catal. Chem. Petrochemical Ind., 2015, vol. 7 pp. 104–110 Search PubMed.
- F. Girgsdies, R. Schlögl and A. Trunschke, Catal. Commun., 2012, 18, 60–62 CrossRef CAS.
- T. Y. Kardash, L. M. Plyasova, V. M. Bondareva, T. V. Andrushkevich, L. S. Dovlitova, A. I. Ischenko, A. I. Nizovskii and A. V. Kalinkin, Appl. Catal., A, 2010, 375, 26–36 CrossRef CAS.
- H.-G. Lintz and S. P. Müller, Appl. Catal., A, 2009, 357, 178–183 CrossRef CAS.
- T. Egami and S. Billinge, Underneath the Bragg peaks: structural analysis of complex materials, Elsevier, Pergamon Materials Series, 2003 Search PubMed.
- D. A. Svintsitskiy, L. S. Kibis, A. I. Stadnichenko, V. I. Zaikovskii, S. V. Koshcheev and A. I. Boronin, Kinet. Catal., 2013, 54, 497–504 CrossRef CAS.
- D. A. Svintsitskiy, E. M. Slavinskaya, T. Y. Kardash, V. I. Avdeev, B. V. Senkovskiy, S. V. Koscheev and A. I. Boronin, Appl. Catal., A, 2016, 510, 64–73 CrossRef CAS.
- D. A. Svintsitskiy, L. S. Kibis, D. A. Smirnov, A. N. Suboch, O. A. Stonkus, O. Y. Podyacheva, A. I. Boronin and Z. R. Ismagilov, Appl. Surf. Sci., 2018, 435, 1273–1284 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- P. Juhás, T. Davis, C. L. Farrow and S. J. L. Billinge, J. Appl. Crystallogr., 2013, 46, 560–566 CrossRef.
- T. Proffen and R. B. Neder, J. Appl. Crystallogr., 1997, 30, 171–175 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653–658 CrossRef CAS.
- J. F. Moulder, W. F. Stickle and P. E. Sobol, K. D. Bomben, Handbook of X-ray photoelectron spectroscopy, Perkin-Elmer Corp, Eden Prairie, Minnesota, USA, 1992 Search PubMed.
- C. D. Wagner, A. V. Naumkin, A. Kraut-Vass, J. W. Allison and C. J. Powell, C.J. Rumble, NIST X-ray Photoelectron Spectroscopy Database, Version 3.5, Natl. Inst. Stand. Technol, Gaithersbg, 2003, http://srdata.nist.gov/xps Search PubMed.
- J. H. A. Darlinski, Surf. Interface Anal., 1987, 10, 223–237 CrossRef.
- M. Aufray, S. Menuel, Y. Fort, J. Eschbach, D. Rouxel and B. Vincent, J. Nanosci. Nanotechnol., 2009, 9, 4780–4785 CrossRef CAS PubMed.
- A. C. Sanfiz, T. W. Hansen, D. Teschner, P. Schnörch, F. Girgsdies, A. Trunschke, R. Schlögl, M. H. Looi and B. A. H. Sharifah, J. Phys. Chem. C, 2010, 114, 1912–1921 CrossRef CAS.
- M. Baca and J.-M. M. Millet, Appl. Catal., A, 2005, 279, 67–77 CrossRef CAS.
- Y. V Plyuto, I. V. Babich, I. V. Plyuto, A. D. Van Langeveld and J. A. Moulijn, Appl. Surf. Sci., 1997, 119, 11–18 CrossRef.
- P. Botella, J. M. López Nieto, B. Solsona, A. Mifsud and F. Marquez, J. Catal., 2002, 209, 445–455 CrossRef CAS.
- A. Trunschke, J. Noack, S. Trojanov, F. Girgsdies, T. Lunkenbein, V. Pfeifer, M. Hävecker, P. Kube, C. Sprung, F. Rosowski and R. Schlögl, ACS Catal., 2017, 7, 3061–3071 CrossRef CAS.
- M. Hävecker, S. Wrabetz, J. Kröhnert, L. I. Csepei, R. Naumann D'Alnoncourt, Y. V. Kolen’Ko, F. Girgsdies, R. Schlögl and A. Trunschke, J. Catal., 2012, 285, 48–60 CrossRef.
- R. K. Grasselli, D. J. Buttrey, P. DeSanto, J. D. Burrington, C. G. Lugmair, A. F. Volpe and T. Weingand, Catal. Today, 2004, 91–92, 251–258 CrossRef CAS.
- L. Kihlborg, Ark. Kemi, 1963, 21, 471–495 CAS.
- J. M. Jehng and I. E. Wachs, Chem. Mater., 1991, 3, 100–107 CrossRef CAS.
- J. E. L. Waldron, M. A. Green and D. A. Neumann, J. Phys. Chem. Solids, 2004, 65, 79–86 CrossRef CAS.
- J. Yu, J. Woo, A. Borisevich, Y. Xu and V. V. Guliants, Catal. Commun., 2012, 29, 68–72 CrossRef CAS.
- T. Kardash, L. M. Plyasova, D. I. Kochubey, V. M. Bondareva and R. B. Neder, Z. Kristallogr., 2012, 227, 288–298 CrossRef CAS.
- E. V. Ishchenko, T. V. Andrushkevich, G. Y. Popova, T. Y. Kardash, A. V. Ishchenko, L. S. Dovlitova and Y. A. Chesalov, Appl. Catal., A, 2014, 476, 91–102 CrossRef CAS.
- R. Canioni, C. Marchal-Roch, N. Leclerc-Laronze, M. Haouas, F. Taulèlle, J. Marrot, S. Paul, C. Lamonier, J.-F. Paul, S. Loridant, J.-M. M. Millet and E. Cadot, Chem. Commun., 2011, 47, 6413–6415 RSC.
- W. Ueda, M. Sadakane and H. Ogihara, Catal. Today, 2008, 132, 2–8 CrossRef CAS.
- M. Sadakane, K. Yamagata, K. Kodato, K. Endo, K. Toriumi, Y. Ozawa, T. Ozeki, T. Nagai, Y. Matsui, N. Sakaguchi, W. D. Pyrz, D. J. Buttrey, D. a Blom, T. Vogt and W. Ueda, Angew. Chem., Int. Ed. Engl., 2009, 48, 3782–3786 CrossRef CAS PubMed.
- S. Knobl, G. A. Zenkovets, G. N. Kryukova, R. I. Maksimovskaya, T. V. Larina, N. T. Vasenin, V. F. Anufrienko, D. Niemeyer and R. Schlogl, Phys. Chem. Chem. Phys., 2003, 5, 5343 RSC.
- T. Murayama, J. Chen, J. Hirata, K. Matsumoto and W. Ueda, Catal. Sci. Technol., 2014, 4, 4250–4257 RSC.
- K. Nakajima, J. Hirata, M. Kim, N. K. Gupta, T. Murayama, A. Yoshida, N. Hiyoshi, A. Fukuoka and W. Ueda, ACS Catal., 2018, 8, 283–290 CrossRef CAS.
- F. N. Naraschewski, C. Praveen Kumar, A. Jentys and J. A. Lercher, Appl. Catal., A, 2011, 391, 63–69 CrossRef CAS.
- Y. V. Kolen’ko, K. Amakawa, R. N. d'Alnoncourt, F. Girgsdies, G. Weinberg, R. Schlögl and A. Trunschke, ChemCatChem, 2012, 4, 495–503 CrossRef.
- W. Ueda, D. Vitry and T. Kato, Res. Chem. Intermed., 2006, 32, 217–233 CrossRef CAS.
- H. Hibst, F. Rosowski and G. Cox, Catal. Today, 2006, 117, 234–241 CrossRef CAS.
- M. Baca, A. Pigamo, J. L. Dubois and J. M. M. Millet, Catal. Commun., 2005, 6, 215–220 CrossRef CAS.
- P. Botella, B. Solsona, A. Martinez-Arias and J. Nieto, Catal. Lett., 2001, 74, 149–154 CrossRef CAS.
- J. Yu, Y. Xu and V. V. Guliants, Catal. Today, 2014, 238, 28–34 CrossRef CAS.
- M. J. Cheng and W. A. Goddard, J. Am. Chem. Soc., 2015, 137, 13224–13227 CrossRef CAS PubMed.
- D. A. Blom, T. Vogt, L. F. Allard and D. J. Buttrey, Top. Catal., 2014, 57, 1138–1144 CrossRef CAS.
- W. D. Pyrz, D. A. Blom, M. Sadakane, K. Kodato, W. Ueda, T. Vogt and D. J. Buttrey, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6152–6157 CrossRef CAS PubMed.
- T. Vogt, D. A. Blom, L. Jones and D. J. Buttrey, Top. Catal., 2016, 59, 1489–1495 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06424e |
| This journal is © The Royal Society of Chemistry 2018 |