
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: PVT1 depletion protects cartilage ATDC5 cells against LPS-induced inflammatory injury by regulating the miR-24/ADAMTS5 axis†
Wenjun Li *,
Gejun Liu and
Xing Wu
*,
Gejun Liu and
Xing Wu
Department of Orthopedics, Shanghai Tenth People's Hospital of Tongji University, Shanghai City, 200072, China. E-mail: etyyhwb@126.com; Tel: +86-186-2129-9525
First published on 7th November 2018
Abstract
Background: Osteoarthritis is a common chronic arthritis among adults and cartilage dysfunction is largely responsible for osteoarthritis development. Long noncoding RNAs (lncRNAs) have been reported to be related to osteoarthritis progression. However, the mechanism that underlies the effect of lncRNA plasmacytoma variant translocation 1 (PVT1) on inflammatory injury in cartilage ATDC5 cells remains elusive. Methods: The quantity of PVT1 and microRNA-24 (miR-24) was detected in human cartilage ATDC5 cells after transfection of si-PVT1, si-con, PVT1 or pcDNA and lipopolysaccharide (LPS) treatment for 12 h. Inflammatory injury was investigated using cell viability, apoptosis and secretion of inflammatory cytokines. The interaction of miR-24 with PVT1 or a disintegrin-like metalloproteinase with thrombospondin motifs 5 (ADAMTS5) was probed by bioinformatics, luciferase activity, RNA pull down and Ago1 RNA immunoprecipitation (RIP) assays. The effect of PVT1 and miR-24 on ADAMTS5 expression was evaluated in ATDC5 cells by western blotting (WB). Results: Treatment of LPS induced elevated PVT1 and reduced miR-24 expression in ATDC5 cells. Moreover, LPS inhibited cell viability, increased apoptosis and inflammatory cytokine production. However, PVT1 depletion attenuated LPS-induced inflammatory injury in ATDC5 cells. In addition, miR-24 directly bound to PVT1 and its deficiency reversed the effect of PVT1 deletion in LPS-treated ATDC5 cells. Furthermore, ADAMTS5 was a target of miR-24 and aberrant expression of ADAMTS5 was regulated by PVT1 and miR-24. Conclusion: PVT1 abrogation protected against LPS-induced inflammatory injury in ATDC5 cells by coordinating with the miR-24/ADAMTS5 axis, opening up a novel avenue for osteoarthritis therapeutics.
Introduction
Osteoarthritis is a chronic inflammatory disease of the joints, which afflicts many elderly people.1 Although multiple factors have been implicated in osteoarthritis, no approved treatment is completely effective.2 Chondrocytes are the only cell type in mature cartilage, the integrity of which plays an important role in osteoarthritis progression.3 Moreover, chondrocyte apoptosis has been reported to participate in the development of osteoarthritis.4 Hence, the mechanism that underlies chondrocyte inflammatory injury is promising for the treatment of osteoarthritis.Emerging finding has recently highlighted that long noncoding RNAs (lncRNAs) play essential roles in diverse diseases by functioning as biomarkers and therapeutic targets.5 The expressions of extracellular genomic including lncRNAs are aberrant and serve as biomarkers for detection, diagnosis and monitoring of osteoarthritis.6 LncRNAs have an impact on osteoarthritis progression through regulating chondrocytes function, factors, apoptosis as well as angiogenesis.7 For instance, lncRNA maternally expressed gene 3 (MEG3) depletion protects against lipopolysaccharides (LPS)-induced inflammatory injury via regulating microRNA-203 (miR-203) in chondrocytic ATDC5 cells.8 LncRNA differentiation antagonizing non-protein coding RNA (DANCR) induces cell proliferation and reduces apoptosis by mediating miR-577/sphingosine kinase 2 (SphK2) axis in osteoarthritis chondrocytes.9 Likewise, lncRNA zinc finger antisense 1 (ZFAS1) has been suggested to be associated with chondrocytes proliferation, apoptosis and migration, providing promising therapeutics for osteoarthritis.10 Apart from lncRNA MEG3, DANCR and ZFAS1, lncRNA homeobox transcript antisense RNA (HOTAIR) facilitates LPS-induced inflammatory injury in ATDC5 cells through regulating miR-124 expression.11 LncRNA plasmacytoma variant translocation 1 (PVT1) has been regarded as an oncogene in multiple cancers, providing a novel approach for cancer diagnosis and therapy.12 Moreover, PVT1 plays a pivotal role in LPS-induced septic acute kidney injury, revealed by promoting inflammatory response by regulating tumor necrosis factor-α (TNF-α), c-Jun N-terminal kinase (JNK) and nuclear factor-κB (NF-κB) signaling pathway.13 Besides, PVT1 has been suggested to participate in chondrocyte apoptosis in osteoarthritis development via functioning as a sponge for miR-488-3p.14 However, a broader view of PVT1 regulating osteoarthritis progression is required.
In addition, functional lncRNAs are known to act as miRNA sponges, resulting in reversing their regulatory effect on mRNAs.15 Dysregulated miRNAs have been involved in inflammation, metabolism, apoptosis, autophagy and chondrogenesis in osteoarthritis.16 Former work suggests miR-24 is associated with the differentiation of interleukin-1β (IL-1β)-treated chondrocyte by regulating matrix remodeling in osteoarthritis.17 Moreover, miR-24 has essential roles in chondrocytes apoptosis, proliferation and inflammatory cytokines production in rats with osteoarthritis.18 Nevertheless, many details about miR-24 regulating inflammatory injury in osteoarthritis remain poorly understood.
A disintegrin-like and metalloproteinase with thrombospondin motifs (ADAMTS) proteins play crucial physiological role in human disorders through participating in varying biological pathways.19 Moreover, ADAMTS5 is a key enzyme and has an important role in cartilage destruction, indicating a promising therapeutic intervention in osteoarthritis.20 The available evidence indicates that ADAMTS5 is responsible for the deterioration of osteoarthritic cartilage.21 Moreover, ADAMTTS5 has been regarded as promising biomarker for chondrogenic differentiation and pathology of osteoarthritis.22 In the current study, we developed ATDC5 cells as osteoarthritis model in vitro. Here we first demonstrated that PVT1 might participate in LPS-induced inflammatory injury in cartilage ATDC5 cells and act as a sponge for miR-24 to regulating ADAMTS5 expression.
Materials and methods
Cell culture and treatment
Human cartilage ATDC5 cells and HEK 293T cells were obtained from American Tissue Culture Collection (ATCC, Manassas, VA, USA). All cells were cultured at 37 °C in 5% CO2 using Roswell Park Memorial Institute (RMPI)-1640 culture medium (Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (Gibco), 1% penicillin and streptomycin (Invitrogen, Carlsbad, CA, USA) during the study. For lipopolysaccharide (LPS, Sigma, St. Louis, MO, USA) stimulation, cells were insulted by varying concentrations of LPS (0, 1, 5 10 μg ml−1) for 12 h.3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay
Cell viability was investigated by MTT assay. ATDC5 cells were seeded into 96-well plates with 1 × 104 cells per well. After different treatments, MTT (Sigma) was introduced for 4 h at 37 °C. Subsequently, dimethyl sulfoxide (DMSO, Sigma) was administrated with the cells until formazan were completely dissolved. The absorbance was measured at 570 nm with a microplate reader (Bio-Rad, Hercules, CA, USA).Flow cytometry
Cell apoptosis was evaluated by flow cytometry through Annexin V-FITC/propidium iodide (PI) staining (Sigma) according to the manufacturer's protocols. Treated ATDC5 cells were collected and resuspended in binding buffer after washed with PBS. Then Annexin V-FITC and PI were introduced into cell suspension for 20 min in the dark. The positive cells were examined using a flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) and analyzed by Image Lab software (Bio-Rad, Hercules, CA, USA).Enzyme linked immunosorbent assay (ELISA)
After treatments, the cell culture medium was collected from 24-well plates and the levels of interleukin-1β (IL-1β), IL-6, IL-8 and tumor necrosis factor-α (TNF-α) were quantified with a microplate reader using ELISA Kit (Invitrogen) following the manufacturer's instructions. Intensity of color was assayed at 450 nm with reference wave length at 620 nm.Cell transfection
The PVT1-overexpression vector (PVT1) were cloned into pcDNA and identified by Genepharma (Shanghai, China). The siRNA for PVT1 (si-PVT1), scrambled control (si-con), miR-24 mimic, miR-24 inhibitor and their negative controls (miR-NC or anti-miR-NC) were obtained from Genepharma. The sequences were showed as follows: si-PVT1 (sense, 5′-GCUUGGAGGCUGAGGAGUUTT-3′; antisense, 5′-AACUCCUCA GCCUCCAAGCTT-3′); si-con (sense, 5′-UUCUCCGAACGUGUCACGUTT-3′; antisense, 5′-ACGUGACACGUUCGUAGAATT-3′); miR-24 mimic (sense, 5′-UG GCUCAGUUCAGCAGGAACAG-3′; antisense, 5′-GUUCCUGCUGAACUGA GCCAUU-3′); miR-24 inhibitor (5′-CUGUUCCUGCUGAACUGAGCCA-3′); miR-NC: (sense, 5′-UUCUCCGAACGUGUCACGTT-3′; antisense, 5′-ACGUGACACG UUCGGAGAATT-3′); anti-miR-NC (5′-GAGGCTAGACCATACGTAGTA-3′). Transient transfection with the 50 nM of oligonucleotides or 2 μg of plasmids in ATDC5 cells was conducted using Lipofectamine 2000 (Invitrogen) according to manufacturer's instructions. After the incubation for 48 h at 37 °C in 5% CO2, transfected cells were collected for further analyses.Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was isolated using Trizol reagent (Invitrogen) following the manufacturer's instructions. Subsequently, quality of RNA was determined using a NanoDrop Spectrophotometer (NanoDrop, Wilmington, DE, USA) and 500 ng of total RNA was used for complementary DNA (cDNA) synthesis by TaqMan Reverse Transcription Kit or TaqMan microRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Then cDNA was diluted and used for qRT-PCR using SYBR green detection kit (Toyobo, Tokyo, Japan) following the amplification instructions. And the results were evaluated with 2−ΔΔCt method using β-actin or U6 small RNA as housekeeping gene. All primers were obtained from Invitrogen: PVT1 (forward, 5′-TTGGCACATACAGCCATCAT-3′; reverse, 5′-GCAGTAAAAG GGGAACACCA-3′), ADAMTS5 (forward, 5′-GAGCCAAGGGCACTGGCTAC TA-3′; reverse, 5′-CGTCACAGCCAGTTCTCACACA-3′), β-actin (forward, 5′-AGCAGCATCGCCCCAAAGTT-3′; reverse, 5′-GGGCACGAAGGCTCATCATT-3′), miR-24 (forward, 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCA CTGGATACGACCTGTTCCT-3′; reverse, 5′-TGCGCTGGCTCAGTTCAGCAG-3′), U6 (forward, 5′-GCTTCGGCAGCACATATACTAAAAT-3′; reverse, 5′-CGC TTCACGAATTTGCGTGTCAT-3′).Luciferase assays
Online software miRcode and starBase predicted putative binding sites of miR-24 with PVT1 or ADAMTS5. The wild-type or mutant miR-24-binding sites of 3′ untranslated regions (3′-UTR) sequences of PVT1 were amplified and cloned into the XhoI and XbaI sites in pmirGlO luciferase reporter vector or control vector (Promega, Madison, WI, USA) to generate the wide-type plasmids (PVT1-WT) or mutant-type plasmids (PVT1-MUT), respectively. WT or MUT reporter plasmids were co-transfected with miR-24 mimic or miR-NC in HEK 293T cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocols. Then lysed cells were exploited in the luciferase activities analysis using Dual-Luciferase Assay Kit (GeneCopoeia, Rockville, MD, USA) after 48 h and renilla luciferase activity was used as internal control. Likewise, WT or MUT miR-24-binding sites of 3′-UTR of ADAMTS5 were cloned into the XhoI and XbaI sites in pmirGlO luciferase reporter vector to obtain ADAMTS5-WT or ADAMTS5-MUT plasmids. MiR-24 mimic or PVT1 as well as their negative control were co-transfected with ADAMTS5-WT or ADAMTS5-MUT plasmids into HEK 293T cells for luciferase activity analysis as mentioned above.RNA pull down assay
RNA pull down analysis was conducted to probe the interaction between PVT1 and miR-24 using RNA-Protein Pull Down Kit (Thermo Fisher, Wilmington, DE, USA). MiR-24 or PVT1 was labelled with biotin and transfected into ATDC5 cells. Biotinylated miR-24 without complementary sites of PVT1 and biotinylated PVT1 without complementary sites of miR-24 acted as negative control (Bio-NC). The cell lysates were incubated with streptavidin magnetic beads (Invitrogen) for 1 h. Then the complex was eluted with biotin elution buffer and PVT1 or miR-24 level was detected by qRT–qPCR.RNA immunoprecipitation (RIP)
Argonaute 1 (Ago1) RIP was used to probe the link between miR-24 and ADAMTS5 using Magna RIP Kit (Millipore, Billerica, MA, USA) following the protocol. In brief, ATDC5 cells with miR-24 or miR-NC transfection were lysed and added to magnetic beads (Thermo Fisher) bounded with anti Ago1 or IgG antibody. Following the washes with PBS, the RNA in beads complexes was isolated and detected by qRT-PCR.Western blots (WB)
Cell proteins were prepared using cell lysis buffer (Thermo Fisher) supplemented with protease inhibitor and then quantified by bicinchoninic acid protein (BCA) assay kit (Sigma) according to the instructions. Denatured samples at 98 °C for 5 min were separated by SDS-PAGE gel and then transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). Following the block in 5% non-fat milk for 2 h at room temperature, membranes were incubated with primary antibodies against ADAMTS5 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2000) or β-actin (1:5000) (Abcam, Cambridge, UK) overnight at 4 °C. β-actin was used as housekeeping protein during this study. Subsequently, membranes were incubated with horseradish peroxidase (HRP) conjugated secondary antibodies (Abcam) for 2 h at room temperature. The visualization of protein bands was realized using enhanced chemiluminescence (ECL) reagent (GE Healthcare, Amersham, UK) and densitometry analysis was performed with Image Lab software.
:2000) or β-actin (1:5000) (Abcam, Cambridge, UK) overnight at 4 °C. β-actin was used as housekeeping protein during this study. Subsequently, membranes were incubated with horseradish peroxidase (HRP) conjugated secondary antibodies (Abcam) for 2 h at room temperature. The visualization of protein bands was realized using enhanced chemiluminescence (ECL) reagent (GE Healthcare, Amersham, UK) and densitometry analysis was performed with Image Lab software.
Statistical analysis
All experiments were repeated more than three times and data were presented as the mean ± standard error of the mean (SEM). The statistical differences were evaluated by Student's t test or one-way analysis of variance (ANOVA) using SPSS 18.0 software (SPSS, Inc., Chicago, IL, USA). P values less than 0.05 were considered to be statistically significant.Results
LPS induces cell injury in ATDC5 cells
ATDC5 cells were stimulated with varying doses of LPS for ascertainable dosage, which was exploited in triggering inflammation response. Compared with untreated cells, the abundance of PVT1 was obviously enhanced in ATDC5 cells treated with 5 and 10 μg ml−1 LPS (Fig. 1A). Moreover, a progressive reduction of cell viability was observed in ATDC5 cells with treatment of LPS in a concentration dependent manner (Fig. 1B). Meanwhile, LPS induced severe cells apoptosis at 5 and 10 μg ml−1 compared with control group (Fig. 1C). Thus, 5 μg ml−1 LPS were used for subsequent experiments in this study. Following the stimulation of LPS, IL-1β production was greatly increased compared with untreated group (Fig. 1D). Likewise, similar enhancing tendency was described in term of IL-6, IL-8 as well as TNF-α secretion in ATDC5 cells after LPS insult (Fig. 1E–G). These findings uncovered that LPS successfully induced inflammatory injury in ATDC5 cells.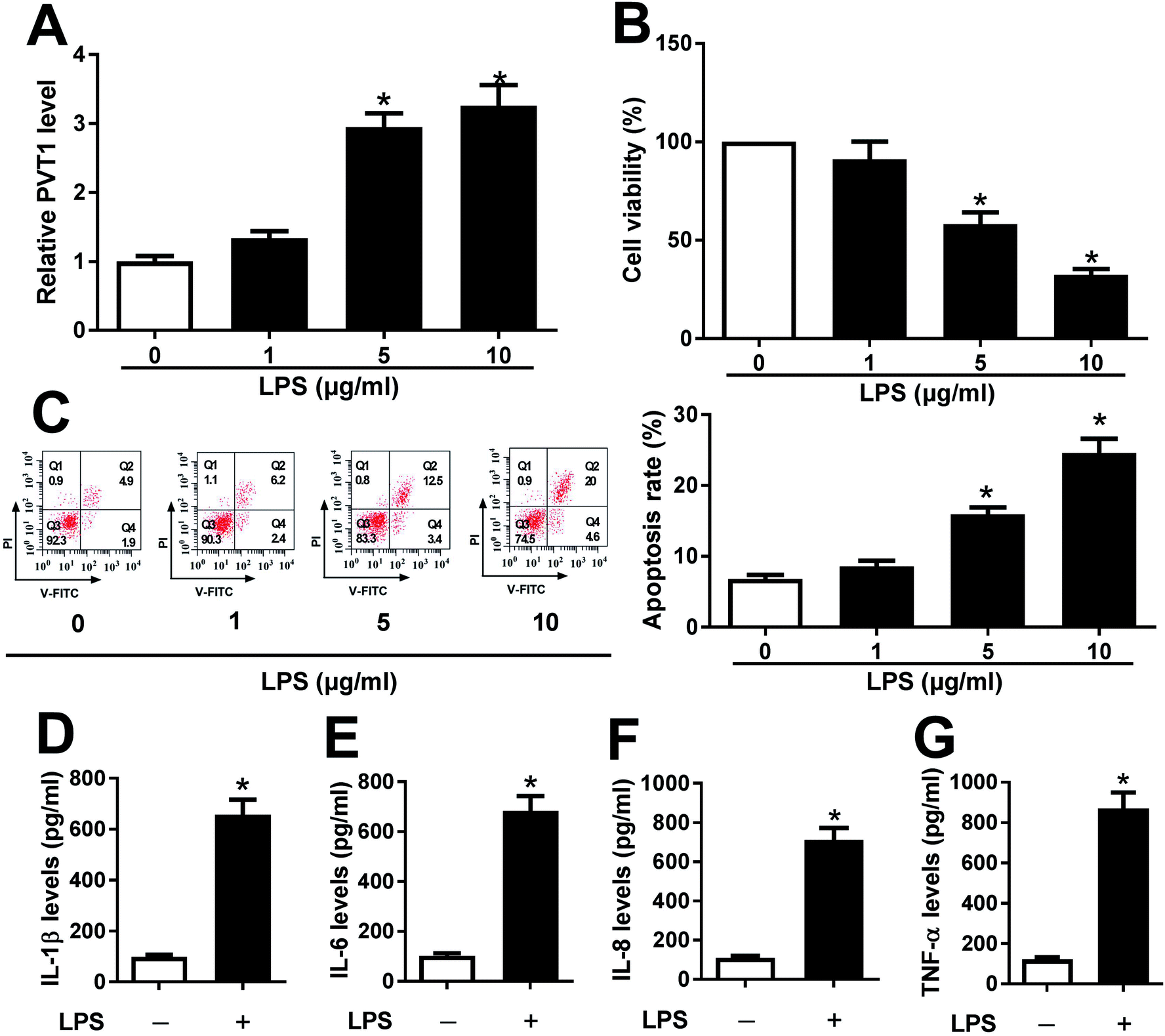 | ||
| Fig. 1 LPS induced cell injury in ATDC5 cells. ATDC5 cells were insulted with 0, 1, 5 or 10 μg ml−1 LPS for 12 h. (A) The expression of PVT1 was detected in LPS-treated ATDC5 cells compared with untreated group by qRT-PCR. (B) The effect of different concentrations LPS on cell viability was examined by MTT. (C) Cell apoptosis was investigated through V-FITC/PI staining by flow cytometry. (D–G) The levels of IL-1β, IL-6, IL-8 and TNF-α secretion were detected in ATDC5 cells with or without LPS treatment (5 μg ml−1) by ELISA. Data were expressed as mean ± SEM from three independent experiments. *p < 0.05. | ||
PVT1 depletion protects against LPS-induced cell injury
Since PVT1 expression was altered after LPS treatment, we next evaluated the effect of PVT1 knockdown on LPS-induced injury in ATDC5 cells. Si-PVT1 or si-con was transfected into ADTC5 cells to block PVT1 expression. Desired reduction of PVT1 abundance was observed in LPS-treated ADTC5 cells after si-PVT1 transfection compared with si-con treatment (Fig. 2A). Subsequently, cell viability, apoptosis and inflammatory cytokines production were evaluated in transfected cells after LPS insult. The absence of PVT1 contributed to cell viability compared with si-con after LPS induction (Fig. 2B). Conversely, cell apoptosis was significantly suppressed by PVT1 exhaustion in LPS-treated cells (Fig. 2C). Moreover, PVT1 knockdown led to a great loss of protein expression levels of IL-1β, IL-6, IL-8 as well as TNF-α in ATDC5 cells after LPS induction compared with si-con (Fig. 2D–G). All the results suggested that PVT1 depletion alleviated LPS-induced injury in ATDC5 cells.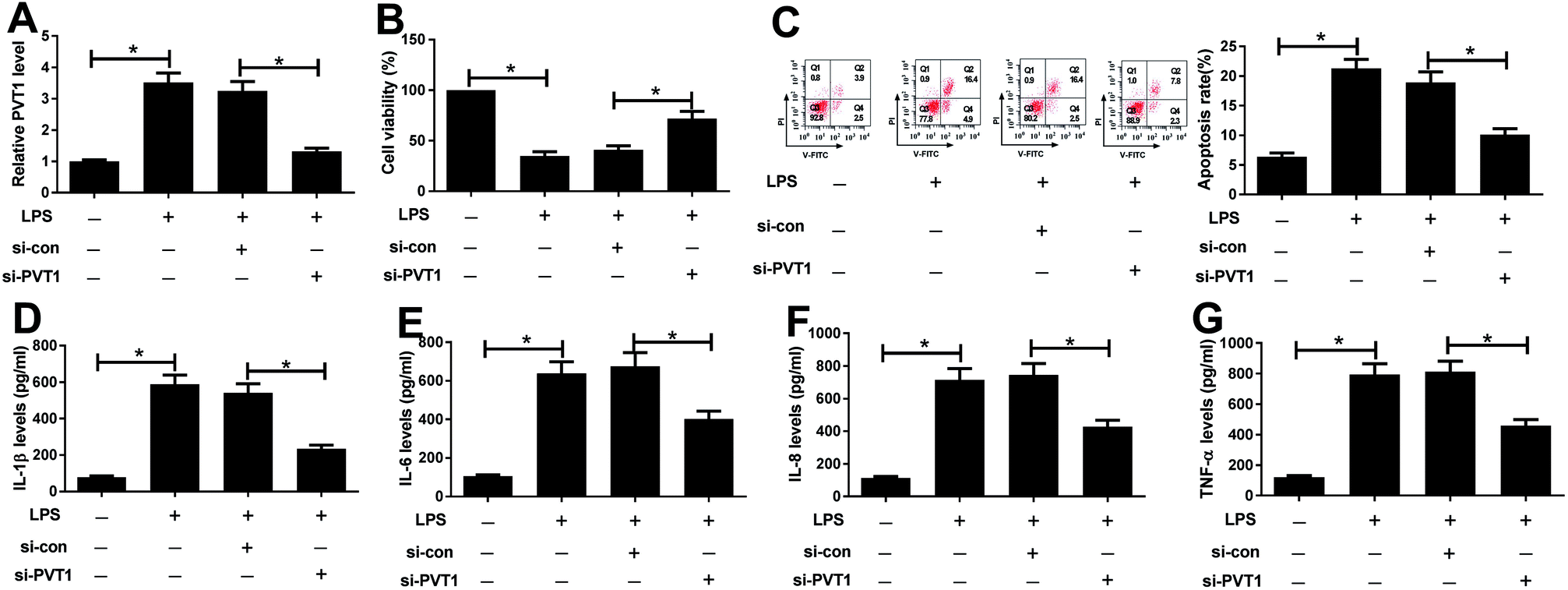 | ||
| Fig. 2 PVT1 exhaustion rescued LPS-induced cell injury in ATDC5 cells. ATDC5 cells were transfected with si-PVT1 or si-con and then stimulated with 5 μg ml−1 LPS. (A) The abundance of PVT1 was detected in ATDC5 cells with si-PVT1 transfection after LPS treatment by qRT-PCR. (B and C) The effect of PVT1 deletion on LPS-mediated cell viability and apoptosis was investigated in ATDC5 cells. (D–G) Inflammation factors levels were detected in cells with PVT1 knockdown and LPS stimulation by ELISA. Data were expressed as mean ± SEM from three independent experiments. *p < 0.05. | ||
PVT1 acts as a molecular sponge of miR-24
To probe a potential miRNA candidate for PVT1, bioinformatics analysis was conducted by miRcode and starBase. Potential binding sites of miR-24 and PVT1 were predicted at position 128902909–128902931, indicating miR-24 might be regulated by PVT1 (Fig. 3A). Hence, luciferase activity and RNA pull down analysis were performed to validate the prediction. Results showed that luciferase activity was great lower in miR-24 mimic and PVT1-WT-transfected HEK 293T cells, which were widely used for luciferase activity analysis due to the highly conservative miRNA-targeting mechanisms, whereas miR-24 had no effect on the luciferase activity in PVT1-MUT group (Fig. 3B). In turn, RNA pull down analysis was performed in ATDC5 cells using biotinylated miR-24 or PVT1, respectively. PVT1 was effectively pulled down by miR-24 with biotin label (Fig. 3C). Likewise, biotin-labeled PVT1 could bind abundant miR-24 compared with mimic alone (Fig. 3D). These results revealed that PVT1 was directly bound to miR-24 in ATDC5 cells.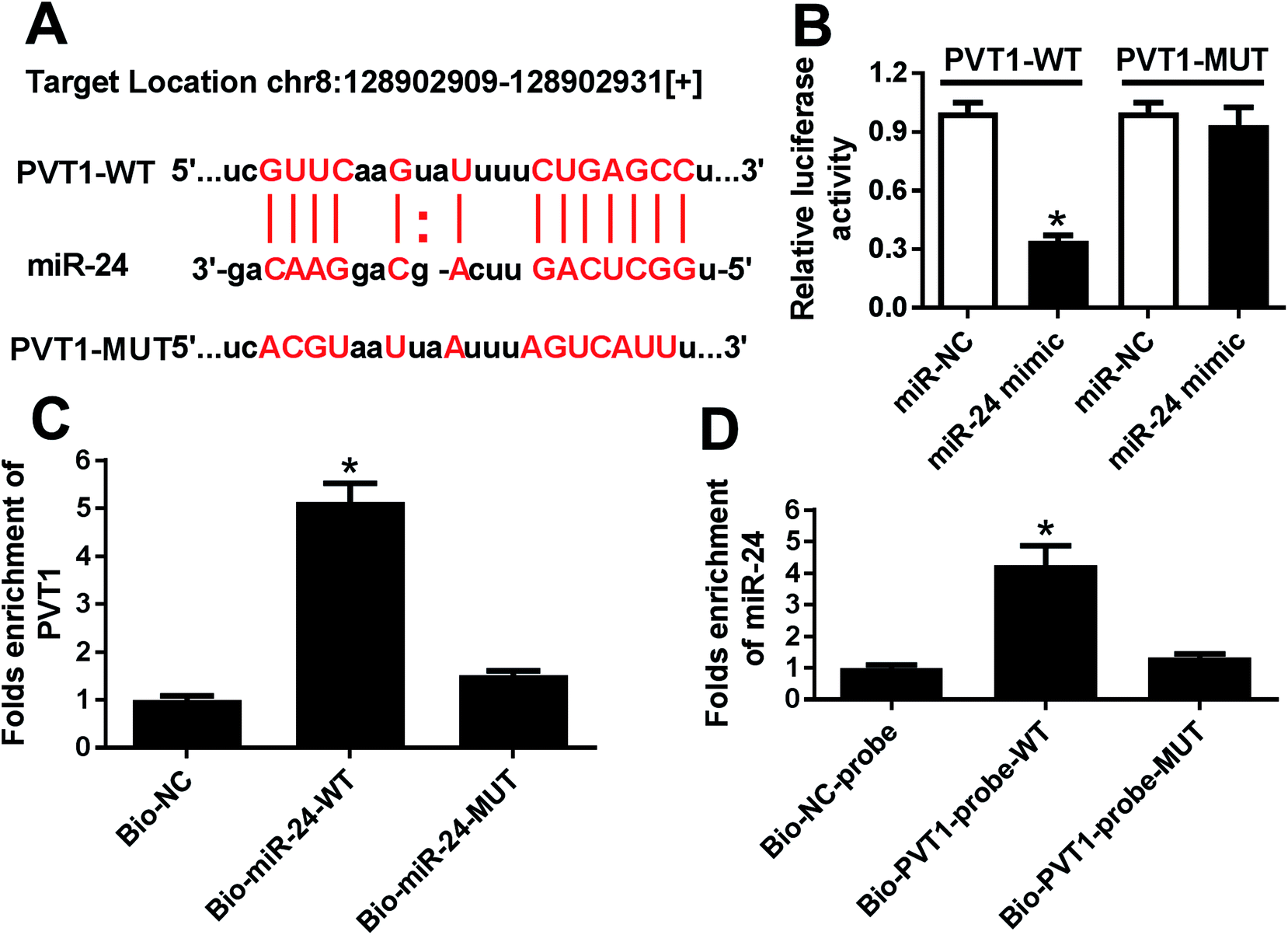 | ||
| Fig. 3 PVT1 was bound to miR-24. (A) Putative targeting sequences of PVT1 and miR-24 were predicted by miRcode and starBase. (B) Luciferase activity analysis was performed in HEK 293T cells co-transfected by miR-24 mimic and PVT1-WT or PVT1-MUT plasmids compared with negative control. (C and D) Biotin-based pull down analysis was conducted in ATDC5 cells after transfection and PVT1 or miR-24 level was analyzed by qRT-PCR. Data were expressed as mean ± SEM from three independent experiments. *p < 0.05. | ||
MiR-24 expression is impaired in ATDC5 cells after LPS induction
Seeing that miR-24 was mediated by PVT1, we further explored the alteration of miR-24 expression in ATDC5 cells after LPS treatment. A progressive decrease of miR-24 abundance was observed in cells with different concentrations of LPS stimulation and marked reduction was induced by 5 and 10 μg ml−1 LPS compared with untreated group (Fig. 4A). Then we probed the effect of PVT1 on miR-24 level in ATDC5 cells through transfection with PVT1 overexpression plasmids or si-PVT1. Addition of PVT1 resulted in a 70% loss of miR-24 expression compared with pcDNA alone, whereas PVT1 exhaustion induced 3.2-fold increase of miR-24 abundance in ATDC5 cells (Fig. 4B). These findings suggested that miR-24 expression was inhibited by LPS insult or PVT1 overexpression.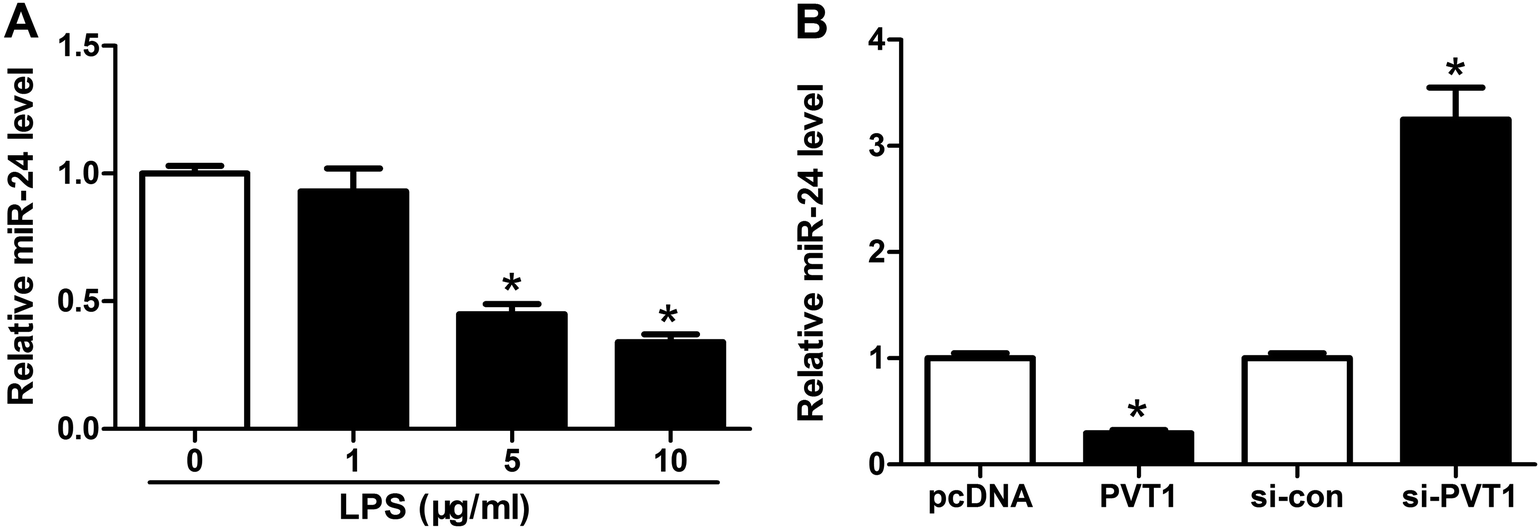 | ||
| Fig. 4 MiR-24 expression was impaired after LPS treatment and regulated by PVT1 in ATDC5 cells. (A) The expression of miR-24 was detected in ATDC5 cells after varying concentrations LPS stimulation by qRT-PCR. (B) The effect of PVT1 overexpression or knockdown on miR-24 level was examined by qRT-PCR. Data were expressed as mean ± SEM from three independent experiments. *p < 0.05. | ||
MiR-24 knockdown ablates PVT1 depletion-mediated protection in ATDC5 cells
To probe whether miR-24 was associated with PVT1 mediated LPS-induced cell injury, miR-24 inhibitor, anti-miR-NC or si-PVT1 was introduced into ATDC5 cells. After LPS treatment, cell viability, apoptosis and inflammatory cytokines expression were detected in ATDC5 cells, respectively. Abrogation of miR-24 inhibited cell viability in cells with presence of si-PVT1 and LPS compared with anti-miR-NC group (Fig. 5A). Moreover, inhibitory effect on apoptosis mediated by si-PVT1 was reversed following miR-24 knockdown in ATDC5 cells (Fig. 5B). Meanwhile, miR-24 inhibitor blocked the protection of si-PVT1 on LPS-treated ATDC5 cells, revealed by elevated secretion levels of IL-1β, IL-6, IL-8 and TNF-α compared an-miR-NC group (Fig. 5C–F). These data indicated the miR-24 knockdown abated the protective function of PVT1 exhaustion on LPS-treated ATDC5 cells.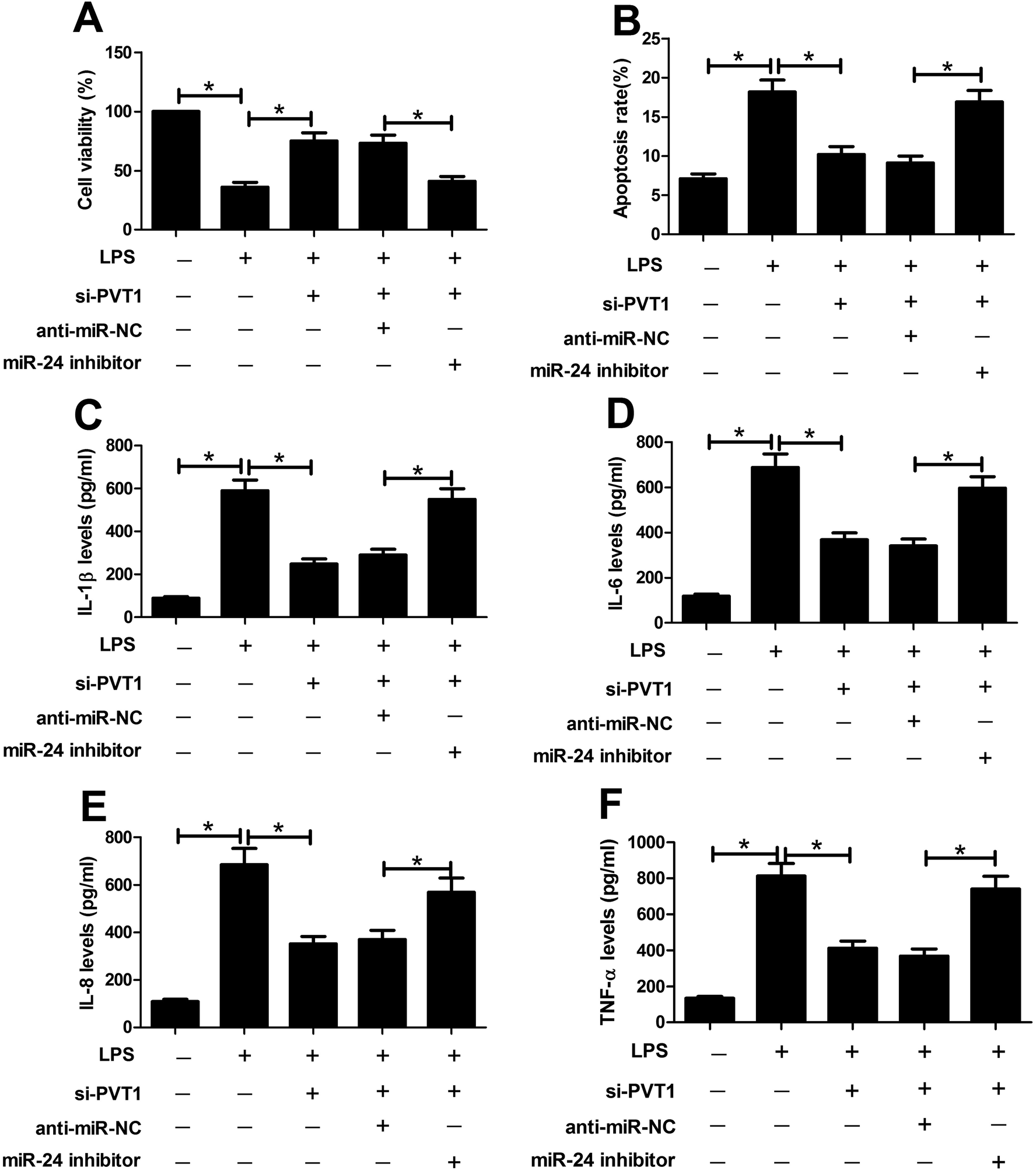 | ||
| Fig. 5 Abrogation of miR-24 attenuated si-PVT1-mediated protection in LPS-treated ATDC5 cells. (A and B) The effect of miR-24 inhibitor on cell viability or apoptosis was investigated in ATDC5 cells with si-PVT1 transfection after LPS treatment by MTT and flow cytometry, respectively. (C–F) MiR-24 deficiency affected IL-1β, IL-6, IL-8 and TNF-α secretion levels in ATDC5 cells with si-PVT1 and LPS treatment by ELISA. Data were expressed as mean ± SEM from three independent experiments. *p < 0.05. | ||
ADAMTS5 may be a target of miR-24
To further explore the potential mechanism that underlies the role of miR-24, one promising target gene of miR-24 is expected in ATDC5 cells. MiRcode and starBase online predicted the putative binding sites of miR-24 and ADAMTS5, revealing ADAMTS5 might be regulated by miR-24 (Fig. 6A). To evaluate the prediction, luciferase activity and Ago1 RIP were conducted. Luciferase activity assay described miR-24 induced an obvious loss of luciferase activity in HEK 293T cells with the presence of ADAMTS5-WT compared with miR-NC treatment, but both of them caused no alteration of luciferase activity in cells containing ADANTS5-MUT (Fig. 6B). Besides, the abundance of Ago1-enriched ADAMTS5 was significantly increased in ATDC5 cells with miR-24 transfection compared with miR-NC group, whereas IgG showed little enrichment (Fig. 6C). Moreover, the effect of miR-24 on ADAMTS5 protein expression was detected in ATDC5 cells with miR-24 mimic or inhibitor transfection. Addition of miR-24 inhibited ADAMTS5 expression, while inhibitor caused an opposite effect (Fig. 6D). These results suggested that ADAMTS5 was mediated by miR-24 in ATDC5 cells.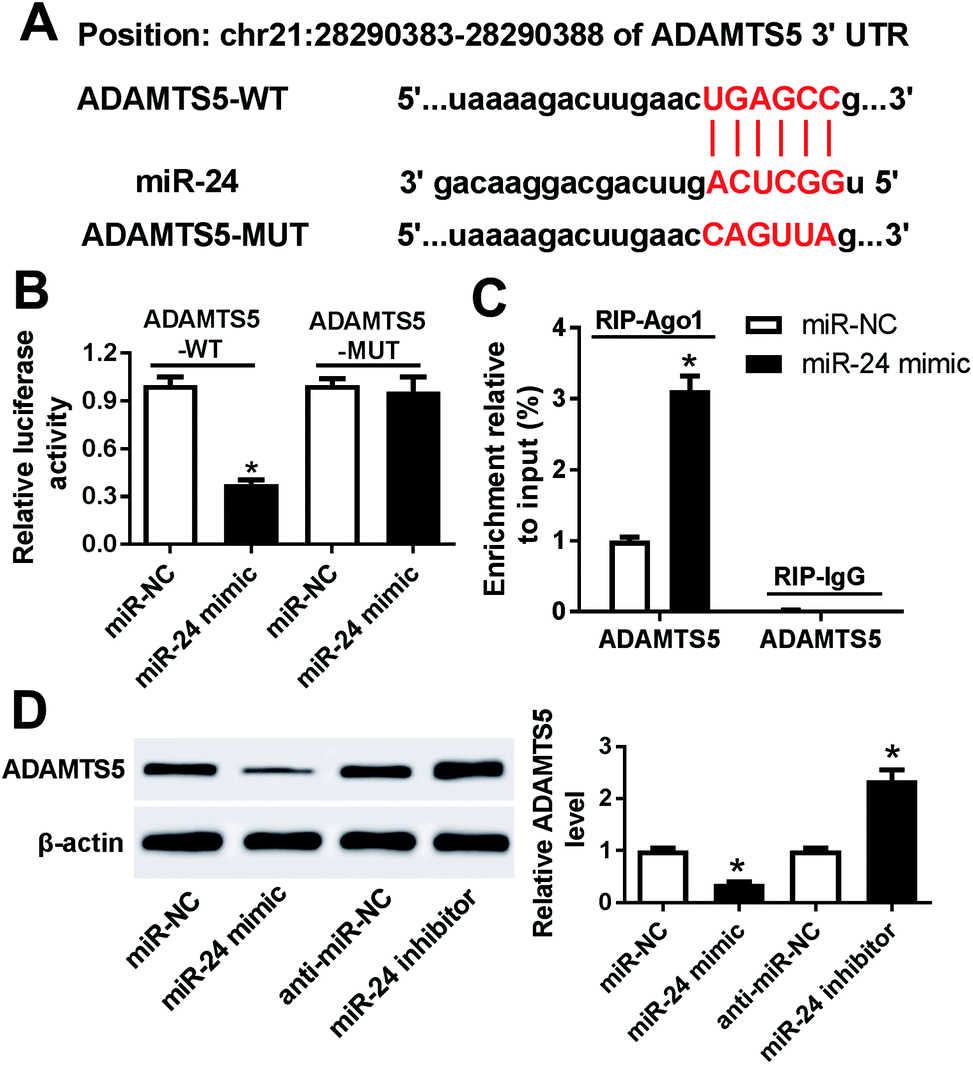 | ||
| Fig. 6 ADAMTS5 was a target of miR-24. (A) The potential binding sites of miR-24 and ADAMTS5 was predicted by miRcode and starBase. (B) Luciferase activity was investigated in HEK 293T cells with miR-24 mimic transfection compared with miR-NC. (C) Ago 1 RIP was conducted in ATDC5 cells. (D) The effect of miR-24 on ADAMTS5 expression was investigated in ATDC5 cells by WB. Data were expressed as mean ± SEM from three independent experiments. *p < 0.05. | ||
ADAMTS5 is regulated by PVT1 and miR-24
Given that lncRNA functioned as competing endogenous RNA (ceRNA) of miRNAs to derepress targeted mRNAs, we further investigated whether PVT1 was associated with ADAMTS5. The abundance of ADAMTS5 was enhanced in ATDC5 cells after treatment of varying concentrations of LPS compared with in untreated cells (Fig. 7A). Subsequently, luciferase activity and WB analyses were used to probe the interaction of PVT1 and miR-24 with ADAMTS5 via transfected with miR-24 mimic, PVT1 overexpression plasmids or pcDNA alone. Introduction of PVT1 induced higher luciferase activity than pcDNA alone in HEK 293T cells co-transfected with miR-24 mimic and ADAMTS5-WT (Fig. 7B). Moreover, PVT1 overexpression promoted ADAMTS5 protein expression, whereas miR-24 mimic addition counteracted the effect of PVT1 in ATDC5 cells (Fig. 7C). Besides, si-PVT1 and miR-24 inhibitor were introduced into ATDC5 cells. Results showed that PVT1 exhaustion induced reduced ADAMTS5 level at protein level, whereas absence of miR-24 cause an opposite effect on ADAMTS5 protein expression (Fig. 7D). These results uncovered ADAMTS5 might be regulated by PVT1 and miR-24.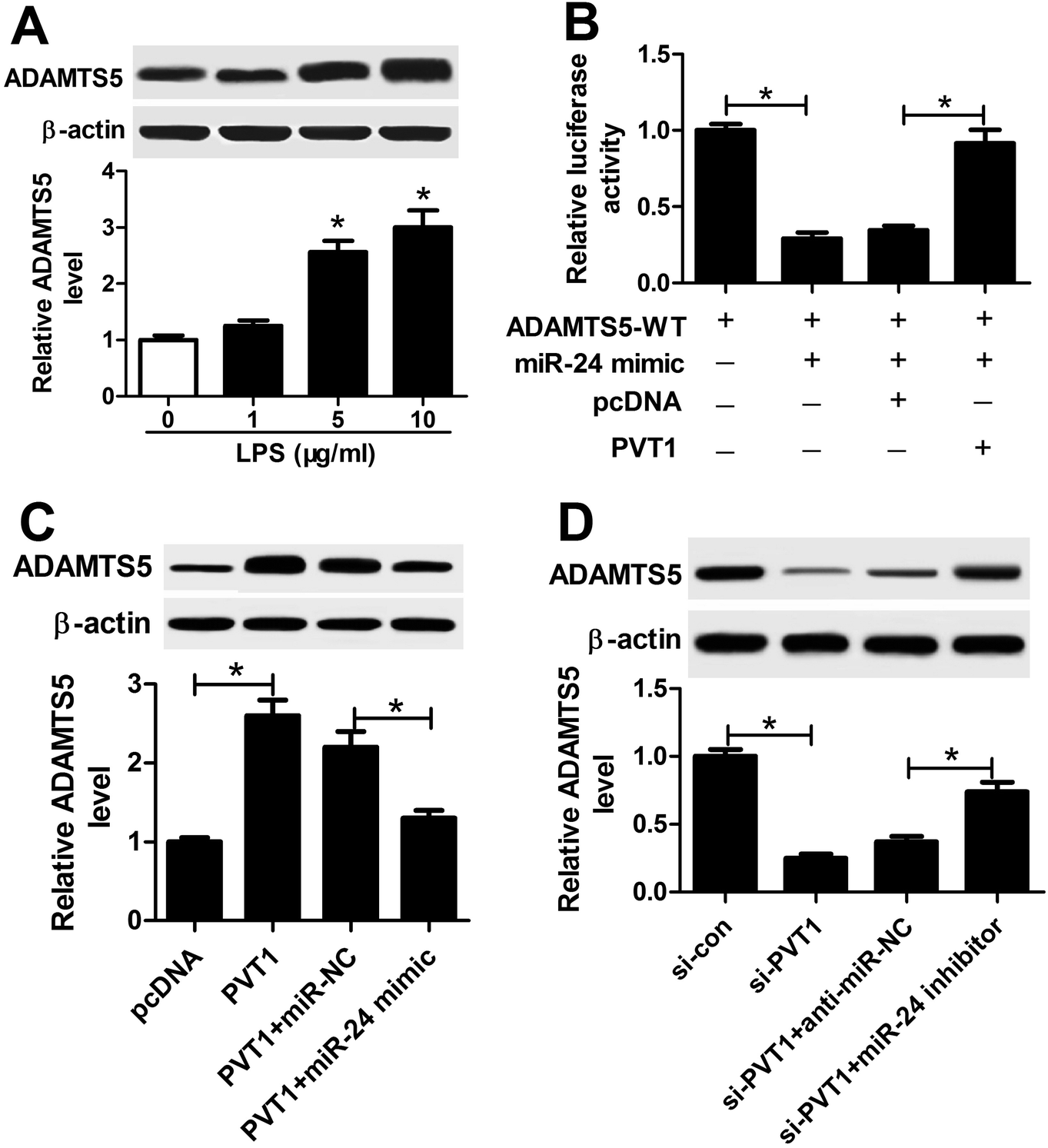 | ||
| Fig. 7 Both of PVT1 and miR-24 were associated with ADAMTS5 expression in ATDC5 cells. (A) ADAMTS5 expression was detected in ATDC5 cells after varying concentrations LPS treatment by WB. (B) Luciferase activity of ADAMTS5 was investigated in ATDC5 cells with miR-24 or combined with PVT1 transfection. (C) The effect of PVT1 and miR-24 on ADAMTS5 expression was examined in ATDC5 cells by WB. (D) The effect of si-PVT1 and miR-24 inhibitor on ADAMTS5 expression was investigated in ATDC5 cells by WB. Data were expressed as mean ± SEM from three independent experiments. *p < 0.05. | ||
Discussion
Osteoarthritis is reported as a chronic inflammatory disease of joints, and inflammatory cytokines production induces cartilage degradation in turn.23 Novel therapeutic targets are needed because of lack of curative treatments for osteoarthritis. LPS is a soluble factor which can be used to provoke inflammatory response in chondrocytes.24 Thus, LPS was used to construct osteoarthritis model by stimulating ATDC5 cells in vitro. We found LPS treatment induced a loss of cell viability, promoted apoptosis and inflammatory cytokines secretion in ATDC5 cells, indicating cell model of inflammatory injury was established in ATDC5 cells by LPS treatment.Recently, lncRNAs have been reported to play key roles in osteoarthritis. For instance, lncRNA HOTAIR expression was increased in osteoarthritis tissues and cells, addition of which promoted LPS-mediated cell viability, apoptosis of ATDC5 cells through activating NF-κB, janus-activated kinase-1 (JAK1) and signal transducer and activator of transcription 3 (STAT3) pathways by regulating miR-124 expression.11 Inhibition of lncRNA RP11-445H22.4 attenuated LPS-induced injury, uncovered by enhancing cell viability, decreasing apoptosis and inflammatory cytokines expression in ATDC5 cells.25 Moreover, lncRNA reprogramming (ROR) was impaired and its overexpression promoted cell viability of osteoarthritis chondrocytes.26 Besides, lncRNA ZFAS1 overexpression contributed to cell viability, proliferation as well as migration, and suppressed cell apoptosis in osteoarthritis chondrocytes.10 Likewise, enrichment of lncRNA growth arrest-specific transcript 5 (GAS5) abated LPS-induced injury in ATDC5 chondrocytes through blocking NF-κB and notch signaling pathways.27 These findings suggested the importance of lncRNA and LPS was responsible for establishment of osteoarthritis in vitro. Hence, we hypothesized PVT1 might be also associated with osteoarthritis processes. In this study, we also found that PVT1 was highly expressed after LPS insult, suggesting PVT1 might play essential role in osteoarthritis development. This is consistent with the finding that suggested PVT1 abundance was enhanced in osteoarthritis tissues or cells compared with normal control.14 Hence, we next explored the effect of PVT1 on LPS-induced inflammatory injury. We found PVT1 knockdown protected against LPS-induced inflammatory injury in ATDC5 cells, revealed by increase of cell viability and decrease of apoptosis and cytokines production. Considering these findings, we displayed PVT1 exhaustion might be regarded as a potential biomarker of limiting injury in osteoarthritis.
Notably, functional lncRNAs is exerted through mediating miRNAs in many conditions.15 For example, miR-301a was reported to be required for RP11-445H22.4 regulating LPS-induced injury in osteoarthritis.25 PVT1 contributed to cell proliferation through mediating miR-126 in lung cancer.28 Moreover, PVT1 might act as a sponge for miR-488-3p to aggravate chondrocyte apoptosis in osteoarthritis.14 MiRNAs are small noncoding RNA and regulate many targets by binding to the 3′-UTR of mRNA, participating in various inflammatory process, including osteoarthritis.29 Such as miR-125b, it inhibited LPS-induced inflammatory injury through regulating cell viability and apoptosis by targeting macrophage inflammatory protein-1α (MIP-1α).30 MiR-24 was reported to bind to lncRNA AC026166.2-001 and regulated cell proliferation and migration in laryngeal squamous cell carcinoma.31 Besides, miR-24 also bound to lncRNA cancer susceptibility candidate 2 (CASC2) and were involved in cell viability and apoptosis of hepatocellular carcinoma cells.32 Notably, available evidence indicated miR-24 was downregulated in chondrocyte treated by IL-1β or from rats with osteoarthritis and elevated miR-24 inhibited apoptosis, promoted cell proliferation.17,18 Hence, we probed a potential miRNA of PVT1 and found the possibility of binding of PVT1 and miR-24. Moreover, miR-24 was negatively regulated by PVT1 or LPS treatment and miR-24 deficiency counteracted si-PVT1-mediated protection in LPS-treated ATDC5 cells, which is also consistent with former efforts.17,18 Therefore, we suggested PVT1 inhibition rescued LPS-induced inflammatory injury by regulating miR-24.
Previous study have suggested that ADAMTS5 might be driven by proinflammatory cytokines and served as therapeutic targets in osteoarthritis.33 ADAMTS5 was reported to be targeted by miR-15a in chondrocytes, which contributed to development of osteoarthritis.34 In osteoarthritis, ADAMTS5 expression was enhanced compared with that in normal cartilage, which might be associated with the alteration of lncRNAs in osteoarthritis.35 Similarly, we also found ADAMTS5 was elevated at protein level in ATDC5 cells after LPS stimulation. In addition, ADAMTS5 was mediated by PVT1 and miR-24 in our study, uncovering ADAMTS5 might play essential role in PVT1-mediated effect on inflammatory injury. However, more details about ADAMTS5 treating osteoarthritis remain to be evaluated in further study. Moreover, clinical data and in vivo model are required for PVT1 investigation in development of osteoarthritis in future.
Conclusions
In conclusion, PVT1 was up-regulated and associated with LPS-induced inflammatory injury in ATDC5 cells. PVT1 knockdown inhibited LPS-induced injury through enhancing cell viability, reducing apoptosis and inflammatory cytokines production in LPS-treated ATDC5 cells. During the study, the interaction of miR-24 with PVT1 and ADAMTS5 was validated, respectively. Moreover, miR-24 deficiency abated PVT1 abrogation-mediated protective effect of ATDC5 cells on inflammatory injury induced by LPS. It was promising that PVT1 could be exploited in a novel therapeutic avenue involved in osteoarthritis treatment.Conflicts of interest
All authors declare that there are no financial or non-financial conflicts of interests.Acknowledgements
Not applicable.Notes and references
- A. Picorelli, A. Hatton, E. Gane and M. Smith, Gait Posture, 2018, 65, 89–99 CrossRef PubMed.
- M. Cutolo, F. Berenbaum, M. Hochberg, L. Punzi and J. Reginster, Semin. Arthritis Rheum., 2015, 44, 611–617 CrossRef PubMed.
- T. Aigner, B. Kurz, N. Fukui and L. Sandell, Curr. Opin. Rheumatol., 2002, 14, 578–584 CrossRef CAS PubMed.
- Z. Zamli and M. Sharif, Int. J. Rheum. Dis., 2011, 14, 159–166 CrossRef PubMed.
- N. Huynh, B. Anderson, F. Guilak and A. McAlinden, Connect. Tissue Res., 2017, 58, 116–141 CrossRef CAS PubMed.
- E. Budd, G. Nalesso and A. Mobasheri, Expert Rev. Mol. Diagn., 2018, 18, 55–74 CrossRef CAS PubMed.
- W. Chen, X. Yu, W. Yang, C. Wang, W. He, Y. Yan, J. Zhang and W. Wang, Cell Proliferation, 2017, 50, e12313 CrossRef PubMed.
- Z. Wang, X. Chi, L. Liu, Y. Wang, X. Mei, Y. Yang and T. Jia, Biomed. Pharmacother., 2018, 100, 240–249 CrossRef CAS PubMed.
- X. Fan, J. Yuan, J. Xie, Z. Pan, X. Yao, X. Sun, P. Zhang and L. Zhang, Biochem. Biophys. Res. Commun., 2018, 500, 658–664 CrossRef CAS PubMed.
- D. Ye, W. Jian, J. Feng and X. Liao, Biomed. Pharmacother., 2018, 104, 825–831 CrossRef CAS PubMed.
- G. Duan, S. Song and S. Niu, Life Sci., 2018, 30416–30418 Search PubMed.
- M. Cui, L. You, X. Ren, W. Zhao, Q. Liao and Y. Zhao, Biochem. Biophys. Res. Commun., 2016, 471, 10–14 CrossRef CAS PubMed.
- W. Huang, X. Lan, X. Li, D. Wang, Y. Sun, Q. Wang, H. Gao and K. Yu, Int. Immunopharmacol., 2017, 47, 134–140 CrossRef CAS PubMed.
- Y. Li, S. Li, Y. Luo, Y. Liu and N. Yu, DNA Cell Biol., 2017, 36, 571–580 CrossRef CAS PubMed.
- M. Cesana, D. Cacchiarelli, I. Legnini, T. Santini, O. Sthandier, M. Chinappi, A. Tramontano and I. Bozzoni, Cell, 2011, 147, 358–369 CrossRef CAS PubMed.
- G. Sondag and T. Haqqi, Curr. Rheumatol. Rep., 2016, 18, 56 CrossRef PubMed.
- D. Philipot, D. Guérit, D. Platano, P. Chuchana, E. Olivotto, F. Espinoza, A. Dorandeu, Y. Pers, J. Piette, R. Borzi, C. Jorgensen, D. Noel and J. Brondello, Arthritis Res. Ther., 2014, 16, R58 CrossRef PubMed.
- Y. Wu, W. Liu, L. Zhang, X. Liu, Y. Wang, B. Xue, B. Liu, R. Duan, B. Zhang and Y. Ji, J. Cell. Biochem., 2017, 119, 7944–7958 CrossRef PubMed.
- T. Mead and S. Apte, Matrix Biol., 2018, 71–72, 225–239 CrossRef CAS PubMed.
- P. Verma and K. Dalal, J. Cell. Biochem., 2011, 112, 3507–3514 CrossRef CAS PubMed.
- Y. Takahata, E. Nakamura, K. Hata, M. Wakabayashi, T. Murakami, K. Wakamori, H. Yoshikawa, A. Matsuda, N. Fukui and R. Nishimura, FASEB J., 2018 DOI:10.1096/fj.201800259R.
- X. Zhou, D. Luo, H. Sun, Y. Qi, W. Xu, X. Jin, C. Li, Z. Lin and G. Li, J. Cell. Biochem., 2018, 119, 2579–2587 CrossRef CAS PubMed.
- F. Berenbaum, Osteoarthr. Cartilage, 2013, 21, 16–21 CrossRef CAS PubMed.
- M. Liu, J. Sun, S. Tsai, S. Sheu and M. Chen, Nutr. Res., 2010, 30, 57–65 CrossRef CAS PubMed.
- T. Sun, J. Yu, L. Han, S. Tian, B. Xu, X. Gong, Q. Zhao and Y. Wang, Cell. Physiol. Biochem., 2018, 45, 832–843 CrossRef CAS PubMed.
- Z. Yang, Y. Tang, H. Lu, B. Shi, Y. Ye, G. Xu and Q. Zhao, J. Cell. Biochem., 2018, 119, 8432–8440 CrossRef CAS PubMed.
- F. Li, J. Sun, S. Huang, G. Su and G. Pi, Cell. Physiol. Biochem., 2018, 45, 1241–1251 CrossRef CAS PubMed.
- H. Li, S. Chen, J. Liu, X. Guo, X. Xiang, T. Dong, P. Ran, Q. Li, B. Zhu, X. Zhang, D. Wang, C. Xiao and S. Zheng, Biochem. Biophys. Res. Commun., 2018, 495, 2350–2355 CrossRef CAS PubMed.
- V. Trachana, E. Ntoumou, L. Anastasopoulou and A. Tsezou, Mech. Ageing Dev., 2018, 171, 15–23 CrossRef CAS PubMed.
- J. Jia, J. Wang, J. Zhang, M. Cui, X. Sun, Q. Li and B. Zhao, Cell. Physiol. Biochem., 2018, 45, 2305–2316 CrossRef CAS PubMed.
- Z. Shen, W. Hao, C. Zhou, H. Deng, D. Ye, Q. Li, L. Lin, B. Cao and J. Guo, Sci. Rep., 2018, 8, 3375 CrossRef PubMed.
- J. Fan, F. Zeng, Y. Le and L. Xin, J. Cell. Biochem., 2018, 119, 6391–6397 CrossRef CAS PubMed.
- J. Bondeson, S. Wainwright, C. Hughes and B. Caterson, Clin. Exp. Rheumatol., 2008, 26, 139–145 CAS.
- X. Lu, J. Lin, J. Jin, W. Qian and X. Weng, Int. J. Mol. Med., 2016, 37, 509–516 CrossRef CAS PubMed.
- D. Xing, J. Liang, Y. Li, J. Lu, H. Jia, L. Xu and X. Ma, Orthopaedic Surgery, 2014, 6, 288–293 CrossRef PubMed.
Footnote |
| † CCDC 128902909–128902931. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra06547k |
| This journal is © The Royal Society of Chemistry 2018 |