
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancement of nitrogen self-doped nanocarbons electrocatalyst via tune-up solution plasma synthesis†
SeungHyo Lee *a and
Nagahiro Saitoab
*a and
Nagahiro Saitoab
aGraduate School of Materials Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Japan. E-mail: seunghyo0121@gmail.com
bGreen Mobility Collaborative Research Center, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Japan
First published on 16th October 2018
Abstract
The development of a metal-free carbon based electrocatalyst for the oxygen reduction reaction (ORR) is an essential issue for energy conversion systems. Herein, we suggest a tune-up solution plasma (SP) synthesis based on a simple one-step and cost-effective method to fabricate nitrogen self-doped graphitic carbon nanosheets (NGS) as an electrocatalyst. This novel strategy using a low-pass filter circuit provides plasma stability and energy control during discharge in pyridine, determining the graphitic structure of nanocarbons doped with nitrogen. Notably, NGS have a relatively high surface area (621 m2 g−1), and high contents of nitrogen bonded as pyridinic-N and pyrrolic-N of 55.5 and 21.3%, respectively. As an efficient metal-free electrocatalyst, NGS exhibit a high onset potential (−0.18 V vs. Ag/AgCl) and a 3.8 transferred electron pathway for ORR in alkaline solution, as well as better long-term durability (4% current decrease after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s of operation) than commercial Pt/C (22% current drop). From this point of view, the nitrogen self-doped graphitic carbon nanosheet material synthesized using the tune-up SP system is a promising catalyst for the ORR, as an alternative to a Pt catalyst for application in energy conversion devices.
000 s of operation) than commercial Pt/C (22% current drop). From this point of view, the nitrogen self-doped graphitic carbon nanosheet material synthesized using the tune-up SP system is a promising catalyst for the ORR, as an alternative to a Pt catalyst for application in energy conversion devices.
1. Introduction
Efficient electrocatalysts for the oxygen reduction reaction (ORR) have attracted widespread attention for achieving optimal performance for application in energy storage and conversion devices, such as fuel cells (FC) and metal-air batteries (MABs).1–4 However, catalysts on the cathode suffer from several critical obstacles, and the possible commercialization of FC and MABs is difficult due to their inherently sluggish kinetics and high overpotential in the ORR. Currently, platinum (Pt) and platinum supported on carbon materials (Pt/C) are regarded as the best solutions to these problems because of a four-electron reduction pathway in the ORR process, but their prohibitive cost, low reserves, and poor methanol tolerance have interrupted the development of their large-scale commercialization.1,5–7 In this regard, ongoing research efforts have been devoted to searching to replace Pt with a Pt-free catalyst with highly efficient performance, selectivity, and durability, such as a non-precious metal supported on nanocarbons,3,8,9 or metal-free heterogeneous nanocarbons.3,7,10,11 Among these, metal-free heterogeneous nanocarbons have been regarded as one of the most prominent alternatives for an ORR electrocatalyst when heteroatoms (e.g. N, B, P, S, and I) are incorporated into the carbonaceous skeleton.12–20 In particular, N-functionalized nanocarbons represent the most investigated catalyst for efficient ORR activity, because of their comparable catalytic efficiency, low cost, and stability in an alkaline medium.7,11 In detail, N-functionalized nanocarbons can play a crucial role in the ORR by imparting higher positive charge density to neighboring carbon atoms and facilitating the dissociation of the O–O bond on the neighboring C atoms.7,12,15,21–27A two-dimensional (2D) carbon nanosheet, which is a sheet with few to multi-layers, exhibits intrinsic characteristics such as a large surface area, excellent electrical conductivity, and high mechanical/chemical stability.28,29 Moreover, a number of catalytic sites with an efficient pathway for the ORR can be created by the planar structure of a 2D carbon nanosheet. In particular, carbon nanosheets containing heteroatoms, especially nitrogen, have been demonstrated using practical experiments and theoretical calculations to effectively improve the ORR activity in an alkaline electrolyte.12,15,22,25,30–34 However, the synthesis procedure and conditions are relatively complicated and strict, resulting in a high fabrication cost.
Solution Plasma (SP), a non-equilibrium discharge in liquids at atmospheric pressure and room temperature, has emerged as a useful synthetic method for various nanomaterials such as nanoparticles,35–37 nanocarbons,38 heterogeneous nanocarbons,39–41 and transition metal–carbon composites.40 Among these nanomaterials, the most interesting achievement is the formation of heterogeneous nanocarbons. The SP synthesis approach has not only provided new insight into the fabrication and design of nanocarbons but has also been further extended to a series with heteroatom doping simply by selecting organic precursors containing the desired heteroatoms.41 However, the conventional SP synthesis is still at an early stage in the field of carbon fabrication and, in particular, it is difficult to control the morphology, structure, and bonding configuration between carbon and the doped heteroatoms. To overcome these limitations, we have developed a tune-up SP system which is composed of a low-pass filter circuit to improve the discharge stability by controlling the current oscillations (Fig. S1†).42
Herein, we propose a new synthetic approach for nitrogen self-doped nanocarbons by the tune-up SP system. Notably, our synthesis strategy has the advantage of producing milligrams of material within ten minutes without any pre or post treatment, and it is a one-step process, unlike other processes.1,7,16,25 Nitrogen doped graphitic carbon nanosheets (NGS) were synthesized by this new tune-up SP approach and nitrogen doped amorphous carbon nanoparticles (NAP) were synthesized by conventional SP. As-prepared NGS exhibit a graphitic structure with a high surface area and mesoporous structure, as well as an ORR activity that is comparable with that of commercial Pt/C in alkaline media, and show excellent stability.
2. Experimental section
2.1 Materials
A tungsten electrode (Φ 0.8 mm, 99.9% purity) was obtained from Nilaco Corporation. Pyridine (C5H6N, ≥99.5% purity), ethanol (C2H5OH, ≥99.5% purity) and 0.1 M potassium hydroxide aqueous solution (0.1 M KOH, ≥99.5% purity) were purchased from Kanto Chemical Co., Inc. Nafion® DE 521 solution (5 wt% in a mixture of lower aliphatic alcohols and water), and 20 wt% Pt on Vulcan XC-72 (20% Pt/C) were purchased from Sigma-Aldrich. Ultrapure water (18.2 MΩ cm−1 at 25 °C) was provided by an Advantec RFD 250NB system. All chemicals were of analytical grade and used without further purification.2.2 Synthesis of NGS and NAP
To generate the plasma in pyridine for the synthesis of NGS, a tune-up SP system was set up, as shown in Fig. 1. The nanocarbons synthesized using the conventional SP system were denoted as NAP. Two tungsten electrodes shielded with an insulating ceramic tube were fixed at the center of a Teflon® reactor with a gap distance of 1.0 mm. The plasma was generated between these electrodes using a bipolar pulsed power supply (MPS-R06K01C-WP1-6CH, Kurita) used in two cases with or without a low-pass filter circuit made in our laboratory. The low-pass filter circuit consists of a parallel capacitor of 2 nF, a variable resistor series from 50 to 150 Ω, and a variable inductor set at 100 μH to adjust the plasma stability. Using the tune-up SP system, the stability of the plasma was considerably improved by eliminating the energy fluctuations which determine the structure of the synthesized nanocarbons. Fig. S1† shows a characterization of the plasma by its electrical energy according to a current–voltage (I–V) curve. The optimum pulse duration and pulse repetition frequency were 0.5 μs and 30 kHz, respectively. The discharge was generated in a volume of 200 mL of pyridine under stirring. After discharging for 30 min, the obtained black powdered material was separated from the unreactive pyridine using a membrane filter paper (0.1 μm, PTFE) and a suction pump, and it was washed several times with ethanol until the remaining solvent after washing was colorless. The acquired nanocarbon material was dried at 60 °C in an oven for 12 h.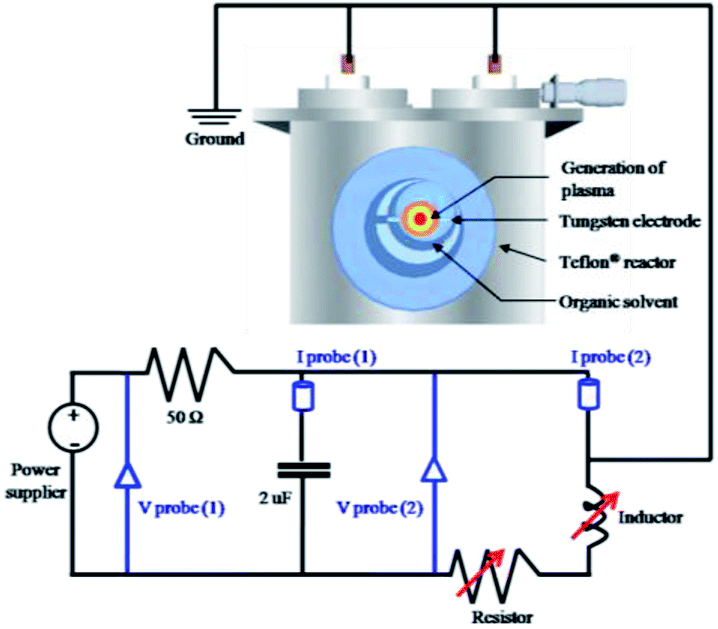 | ||
| Fig. 1 Schematic setup of the tune-up SP system with a low-pass filter circuit for NGS synthesis. The tune-up SP system consists of a low-pass filter circuit with a parallel capacitor of 2 nF, a variable resistor series from 50 to 150 Ω, and a variable inductor set at 100 μH to adjust the plasma stability. Details of measurements are included in the ESI.† | ||
2.3 Characterization
Transmission Electron Microscopy, Selected Area Electron Diffraction, and Electron Dispersive Spectroscopy (TEM, SAED, and EDS, JEOL JEM-2500SE at 200 kV) observations were conducted for the study the microstructure, shape and size, and the elemental composition of the synthesized nanocarbon material. An image obtained from Nanoscope IIIa Multimode Atomic Force Microscopy (AFM, Veeco) was used to measure the thickness of an NGS sample prepared by dropping the dispersed nanocarbon material in ethanol on an Si/SiO2 substrate. The X-ray diffraction (XRD) patterns of all samples were performed using Cu Kα (0.154 nm) X-rays source in the range from 15° to 60° with 0.02° step and 2° min−1 scan speed. Raman spectra were recorded with a Raman spectrometer (JASCO NRS-100) equipped with a laser with a wavelength of 532.5 nm. Nitrogen adsorption–desorption isotherms were measured on a Belsorp-mini II sorption analyzer (MicrotracBEL Corp.) to investigate the specific surface area, pore volume, and pore size distribution of all samples. The specific surface area was measured by the Brunauer–Emmett–Teller (BET) method at a relative pressure between 0.05 and 0.30 and liquid nitrogen temperature. The pore volume and pore size distribution were determined by the Barrett–Joyner–Halenda (BJH) method. The chemical bond structures were examined by X-ray photoelectron spectroscopy (XPS, Ulvac-PHI 5000 versaprobe II) using Al Kα radiation (1486.6 eV) as an X-ray source. The emission current and the anode voltage of the source were set at 0.1 mA and 15 kV, respectively. The binding energy (BE) was calibrated using the containment carbon (C 1s = 284.5 eV). Each binding energy was referenced from the NIST XPS Database.43 The elemental analysis (EA) of C, H, and N was performed using an elemental analyzer (PerkinElmer 2400 Series). The electrical conductivity was measured with a device based on a four probe method with a resistivity meter (2001 multimeter, TFT Corp., Keithley Instruments) and DC constant power supply (model 692A, Metronix Corp.). The samples were inserted into a Teflon cylinder with 0.5 cm diameter. The prepared samples were compressed at 0.6 MPa between two brass electrodes.2.4 Electrochemical characterization
The electrochemical measurements were carried out on an HZ-5000 electrochemical analyzer (Hokudo Denko Inc.) equipped with a rotating disk electrode (RDE). A Pt wire and an Ag/AgCl electrode filled with saturated KCl solution were used as the counter and reference electrodes, respectively. All the samples for the electrochemical measurements were coated onto a glassy carbon (GC) electrode with 3 mm diameter and prepared as follows: 50 μL of Nafion solution was added to 0.5 mL of ethanol, and then 5 mg of finely ground catalyst was dispersed in the as-prepared solution by at least 30 min sonication to form a homogeneous suspension. Then a volume of 5 μL from the suspension was loaded onto the GC electrode and dried at room temperature. Before measurements, N2 or O2 gasses were purged into 0.1 M KOH electrolyte for at least 1 hour. Cyclic voltammetry (CV) was conducted at a scan rate of 50 mV s−1 from −1.0 to 0.3 V (vs. Ag/AgCl). Linear sweep voltammetry (LSV) was performed with a scan rate of 10 mV s−1 from −1.0 to 0.3 V (vs. Ag/AgCl). The rotation speed was varied from 100 to 3600 rpm. Commercial 20% Pt/C was selected as the performance benchmark. The electrochemical stability was measured by the chronoamperometric response in 0.1 M KOH saturated with oxygen for 10000 s at −0.4 V. All the polarization curves were corrected for background currents, which were determined in the N2-saturated electrolyte.
3. Results
The morphological structures of NGS and NAP were investigated by TEM. As shown in Fig. 2(a), NGS show typical sheet-like morphology. The corresponding SAED pattern shown in the inset in Fig. 2(a) reveals well-defined diffraction spots which mean that the NGS are crystallized. The HR-TEM image further indicates that the NGS are composed of multi-layer graphitic nanosheets, as shown in Fig. 2(b). Well-contoured edges originating from the graphitic layer clearly appear with a d space of the 002 crystal plane of approximately 0.350 nm, which is close to that of single-crystal graphite with d = 0.335 nm.44,45 | ||
| Fig. 2 (a) Wide-field TEM images of NGS; the corresponding SAED pattern (inset) shows well-defined diffraction spots which mean that the NGS are crystallized. (b) HR-TEM image of NGS indicates well-contoured edges originating from the graphitic layer with a d space of 0.350 nm. (c) Dark-field TEM image of NGS, and the corresponding carbon, nitrogen, and oxygen element mapping images. | ||
The phase identification of the samples was performed by XRD, as shown in Fig. 3. The NGS present two prominent peaks, near 2θ ≈ 25.4° and 42.9° which indicate 002 and 100/101 diffraction planes of graphite, respectively. Using Bragg's law, the interlayer distance of NGS was calculated to be approximately 0.351 nm from the 002 diffraction peak, which is slightly larger than that of single-crystal graphite 0.335 nm. This result also agrees with the TEM results. The AFM image (Fig. S2†) demonstrates that the thickness of most NGS samples is 2.97 ± 0.94 nm, corresponding to 10 layers according to the theoretical thickness of single-layer graphene (0.34 nm). The EDS mapping images of NGS correspond to the elemental composition (Fig. 1(c)). Carbon and nitrogen atoms are distributed throughout the carbon frame, indicating the homogenous incorporation of nitrogen into the graphitic carbon nanosheets. Water impurities in pyridine and slight oxidation while drying the samples might explain the presence of oxygen in the composition of the synthesized materials.
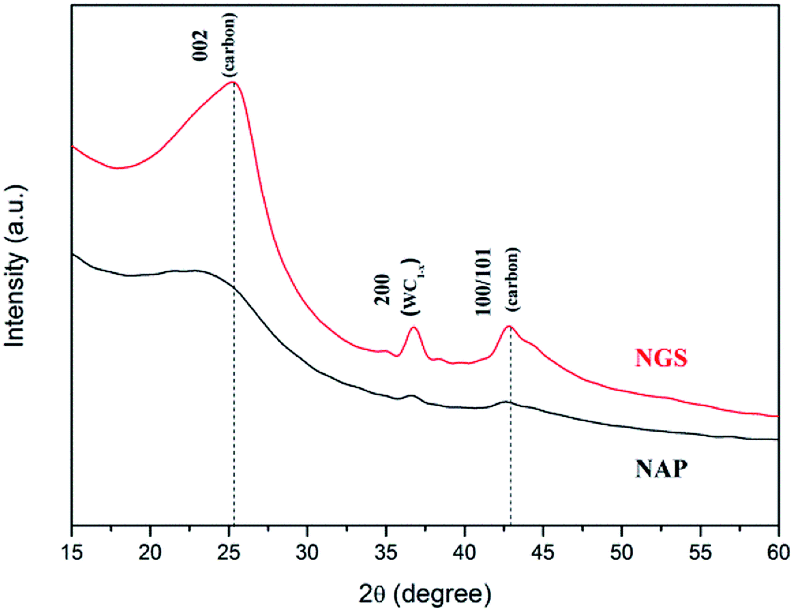 | ||
| Fig. 3 X-ray diffraction (XRD) patterns of NGS and NAP. Using Bragg's law, the interlayer distance of NGS was calculated to be approximately 0.351 nm from the 002 diffraction peak, which is slightly larger than the 0.335 nm of single-crystal graphite. | ||
In contrast, NAP samples reveal the formation of aggregates of small carbon particles with a diameter of approximately 10–30 nm, as shown in Fig. S3(a).† The corresponding SAED pattern (the inset of Fig. S3(a)†) presents a bright center and diffuse ring pattern without any diffraction spots, confirming its amorphous nature.46,47 The two major peaks in the XRD pattern are also broad, which means amorphous and disordered carbon. From these results, we can conclude that NAP synthesized by conventional SP is an amorphous form of carbon nanoparticles in accordance with many other works.38–40
Raman spectroscopy has been used to conduct further structural investigation. Raman spectra of both NGS and NAP show two bands, at approximately ∼1595 cm−1 for the G band and ∼1349 cm−1 for the D band, as shown in Fig. 4. The G band is attributed to one of the two E2g modes of the stretching vibrations in the sp2 domains of perfect graphite. The D band refers to the disorder and imperfection of sp2 hybridized carbon48,49 The second-order D band (2D) at ∼2712 cm−1 is a typical feature of graphitic carbon.50 The integrated intensities ratio between the G band and D band (IG/ID) depends linearly on the crystallite size (La) of the nanocarbons, which was calculated from the equation: La (nm) = (2.4 × 10−10)λl4(ID/IG)−1, where λl is the laser wavelength in nanometer units.51 The IG/ID of NGS is 1.05 which is much higher than the value for NAP (0.55), so that the crystallite size of NGS (95.9 nm) is bigger than that of NAP (50.6 nm), implying that the carbon nanosheets have long-range order and a high degree of graphitization. This result correlates with the SAED above and HR-TEM data that show that NGS have a graphitic structure while NAP contain an amorphous carbon structure. In the Raman spectrum of NGS, the intensity of the D band is still high compared with a few layers of graphene, which means that NGS have more defect sites which can influence the electrocatalytic activity for ORR because of the enhanced charge density on carbon atoms.
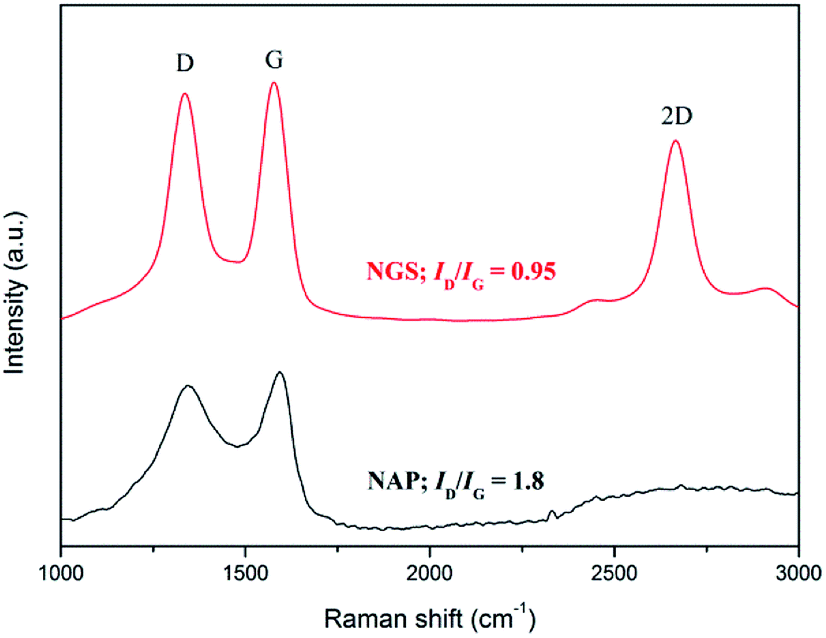 | ||
| Fig. 4 Raman spectra with the corresponding ID/IG ratios of NGS and NAP. The ID/IG of NGS is about 0.95 which is much lower than that of the value for NAP (1.8). This result correlates with the SAED above and HR-TEM data that show that NGS has a high degree of graphitization while NAP contains an amorphous carbon structure. | ||
Moreover, the ratio of WD and ID/(ID + IG) was correlated to the better-ordered parameter for the disordered carbon materials. The ratios of ID/(ID + IG) for NGS and NAP were about 49.9 and 48.3, respectively. These values indicate that the graphitization of NGS and NAP were almost the same, and were less than for activated carbons and more than for conventional graphite. The values of WD for NGS and NGA were 64 cm−1 and 127 cm−1. The values of WD for NGS and NGA were closer to those of conventional graphite and activated carbons. Thus, the structure of NGS has become microcrystalline compared to that of conventional graphite and was similar to that of microcrystalline graphite. The structure of NGA was more disordered and had lower graphitization than activated carbons.52
Specific surface area (SSA) and porosity, which are necessary for understanding ORR activity, were investigated by N2 sorption analysis. The BET surface area of NAP was measured to be 48 m2 g−1. The BET surface area of NGS synthesized using the tune-up SP system was greatly increased and reached as high as 621 m2 g−1, as shown in Fig. 5. All samples exhibit a type IV adsorption/desorption isotherm with a hysteresis loop at relative pressures ranging from 0.4 to 0.9, suggesting the characteristic of a mesoporous structure.53,54 The corresponding pore size distributions of all samples derived by the BJH method indicate a broad distribution on the meso–macro length scale (the inset of Fig. 5).
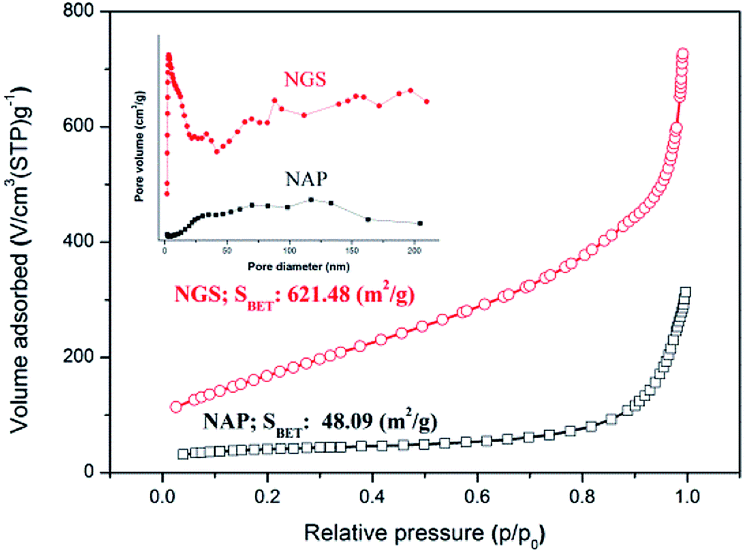 | ||
| Fig. 5 Nitrogen adsorption–desorption isotherm and pore size distribution (inset) of NGS and NAP. The BET surface area of NAP was measured to be 48 m2 g−1. The BET surface area of NGS synthesized using the tune-up SP system was greatly increased and reached as high as 621 m2 g−1. | ||
Although the active sites for ORR on nitrogen-doped carbon materials are still an open question, there is no doubt that the nitrogen containing functional groups facilitate the electron transfer to enhance ORR kinetics in a carbon matrix.12–15,22,27 XPS measurements were used to demonstrate that the ORR activity is dependent on the bonding configuration of nitrogen in the nanocarbon structure. The incorporation of nitrogen atoms in the carbon structure was confirmed by XPS and EA measurements. The survey XPS spectra of the NGS and NAP clearly show a peak at 400.0 eV for N 1s in addition to the dominant graphitic C 1s peak at 284.5 eV, which indicates the presence of nitrogen in the carbon structure (Fig. S4†). Importantly, no peak of the tungsten electrode was found in the spectra. The atomic contents of C, O, and N for NGS and NAP derived from the XPS analysis are summarized in Table S2.† The XPS data show that the nitrogen content of NGS (6.0 at%) was around twice as high as that of NAP (2.9 at%), which was almost the same as the results acquired from EA analysis (Table S3†).
High-resolution XPS measurements examined in more detail the chemical bonding configuration of the NGS and NAP on C 1s and N 1s peaks (Fig. 6(a)–(d)). The high-resolution asymmetric peak in the C 1s region presented in Fig. 6(a) and (c) indicates the presence of different types of carbon bonding: C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (284.5 eV), C–C bond (285.0 eV), C–N bond (285.9 eV), C–O bond (286.5 eV), and CO bond (287.0 eV),55–57 proving the graphitic carbon and the successful incorporation of nitrogen into the carbon structure. To clarify the effect of nitrogen incorporation, we analyzed the high-resolution N 1s peak, as shown in Fig. 6(b) and (d). The XPS N 1s peak can be deconvoluted into four components corresponding to pyridinic-N (398.9 eV), pyrrolic-N (400.4 eV), graphitic-N (400.9 eV), and pyridinic N-oxide (403.2 eV).58–60 The XPS results for NGS show higher relative contents of nitrogen bonded as pyridinic-N and pyrrolic-N at 55.5 and 21.3%, respectively, compared to NAP (47.2 and 8.5, respectively), as represented in Fig. 6(f). Also, the calculated atomic percentages of types of nitrogen bonding in the prepared samples are given in Table S4.† A significant increase in pyridinic-N and pyrrolic-N bonds is clearly confirmed, while there was no considerable change in graphitic-N and pyridinic oxide-N bonds in the material synthesized by the tune-up SP system. Pyridinic-N refers to nitrogen atoms possibly attached at the edges of the graphite planes, where each N atom is bonded to two carbon atoms and donates one of the lone pair electrons to the aromatic π conjugation. The pyrrolic-N atom is incorporated into the five-membered heterocyclic ring, which is bonded to two carbon atoms and contributes two p-electrons to the π system. The pyridinic-N and pyrrolic-N can be located at the graphite edge (Fig. S5†).61,62 According to many references,61–63 the formation of the edge-N, which is composed of pyridinic-N and pyrrolic-N, can reduce the adsorption energy of O2. NGS possess a higher content of edge-N than NAP, implying that the ORR performance might be enhanced.
C bond (284.5 eV), C–C bond (285.0 eV), C–N bond (285.9 eV), C–O bond (286.5 eV), and CO bond (287.0 eV),55–57 proving the graphitic carbon and the successful incorporation of nitrogen into the carbon structure. To clarify the effect of nitrogen incorporation, we analyzed the high-resolution N 1s peak, as shown in Fig. 6(b) and (d). The XPS N 1s peak can be deconvoluted into four components corresponding to pyridinic-N (398.9 eV), pyrrolic-N (400.4 eV), graphitic-N (400.9 eV), and pyridinic N-oxide (403.2 eV).58–60 The XPS results for NGS show higher relative contents of nitrogen bonded as pyridinic-N and pyrrolic-N at 55.5 and 21.3%, respectively, compared to NAP (47.2 and 8.5, respectively), as represented in Fig. 6(f). Also, the calculated atomic percentages of types of nitrogen bonding in the prepared samples are given in Table S4.† A significant increase in pyridinic-N and pyrrolic-N bonds is clearly confirmed, while there was no considerable change in graphitic-N and pyridinic oxide-N bonds in the material synthesized by the tune-up SP system. Pyridinic-N refers to nitrogen atoms possibly attached at the edges of the graphite planes, where each N atom is bonded to two carbon atoms and donates one of the lone pair electrons to the aromatic π conjugation. The pyrrolic-N atom is incorporated into the five-membered heterocyclic ring, which is bonded to two carbon atoms and contributes two p-electrons to the π system. The pyridinic-N and pyrrolic-N can be located at the graphite edge (Fig. S5†).61,62 According to many references,61–63 the formation of the edge-N, which is composed of pyridinic-N and pyrrolic-N, can reduce the adsorption energy of O2. NGS possess a higher content of edge-N than NAP, implying that the ORR performance might be enhanced.
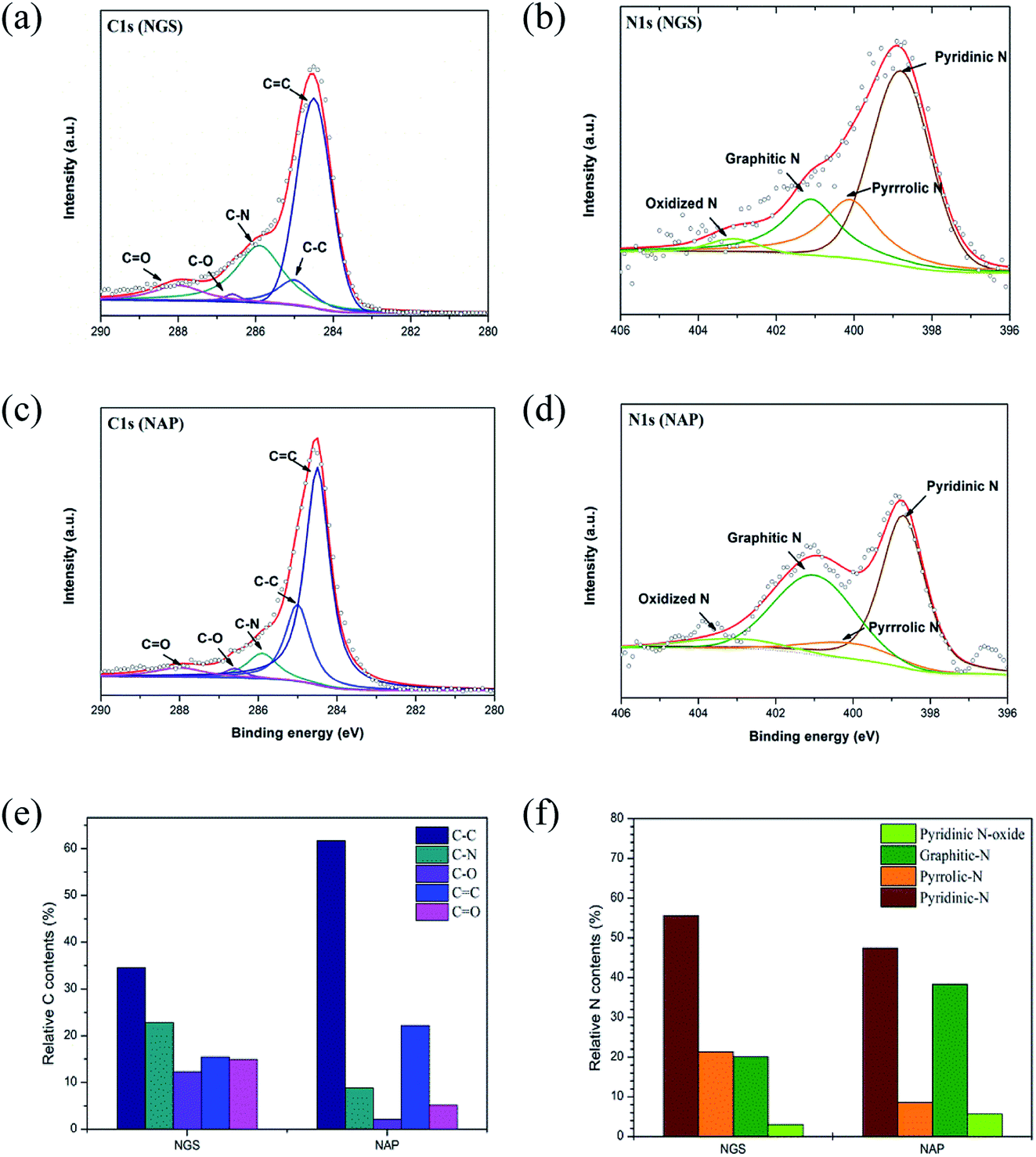 | ||
| Fig. 6 High-resolution XPS scans of NGS and NAP for C 1s and N 1s peaks are shown in (a), (b) and (c), (d), respectively. The distribution of N (e) and C species (f) in NGS and NAP, respectively. NGS shows higher relative nitrogen content bonded as pyridinic-N and pyrrolic N at 55.5 and 21.3%, respectively, compared to NAP (47.2 and 8.5%, respectively), which can play a crucial role in the ORR. | ||
To verify the catalytic activity, the ORR of all the synthesized nanocarbons was measured by CV, as shown in Fig. 7(a). All samples show a well-defined characteristic ORR peak in O2-saturated 0.1 M KOH solution. The peak potential of the ORR of the NGS sample at −0.25 V vs. Ag/AgCl reference electrode significantly shifts to a positive value in comparison with the NAP sample with its peak potential at −0.52 V vs. Ag/AgCl. Also, the NGS catalyst presents a higher cathodic current density than NAP, suggesting a considerably enhanced ORR activity due to the increased electrical conductivity. The electrical conductivity results are shown in Table S5.† Linear sweep voltammetry (LSV) measurements on the rotating disk electrode (RDE) were further performed to verify the electrocatalytic kinetics toward ORR. For comparison, a commercial 20 wt% Pt/C as reference catalyst was measured with the same mass loading on a GC electrode as the samples. Fig. 7(b) exhibits LSV curves obtained from a rotation rate of 1600 rpm, which obviously exhibit the differences in the ORR activity. A more positive onset potential (Eonset = −0.18 V), half-wave potential (E1/2 = −0.28 V) and higher limiting current density (LCD = 2.7 mA cm−2) are revealed for NGS than for NAP (Eonset = −0.21 V, E1/2 = 0.24 V and LCD = 1.8 mA cm−2). Although NGS are inefficient compared with commercial Pt/C for which Eonset = −0.03 V, E1/2 = −0.14 V, and LCD = 3.8 mA cm−2, they still show a possible use as a metal-free catalyst in ORR. To further understand the ORR mechanism of this catalyst, ORR polarization curves of NAP and NGS at various rotations from 100 to 3600 rpm were carried out (Fig. S6†). The Koutechy–Levich (K–L, see ESI†) formula was employed to calculate the average electron number (na) in the reduction reaction, deduced from the fact that the ORR current density is a function of the electrode rotation rate.64 According to the slope of the K–L plots (inset Fig. S6†), the na of NAP was found to be 2.9, which represents poor ORR activity, while the na of NGS was calculated to be 3.8, which is close to the 4 electron transfer number on a Pt/C catalyst (Fig. 7(c)). The value of 3.8 may suggest that NGS were mainly dominated by a four electron reaction pathway similar to that of Pt/C. On the other hand, the na = 2.9 of NAP may proceed predominantly with a pathway of two-electron and four-electron reactions simultaneously. The higher current density and the four electron process indicate that NGS is a kind of highly efficient ORR catalyst. Also, the ORR activity of NGS is closer to the highest values in previous reports, shown in Table S6,† from the number of electrons transferred, although these NGS can be synthesized compared to the other N-doped carbons. This high performance is due to the improved crystallinity of the carbon region, where electrons are transferred.
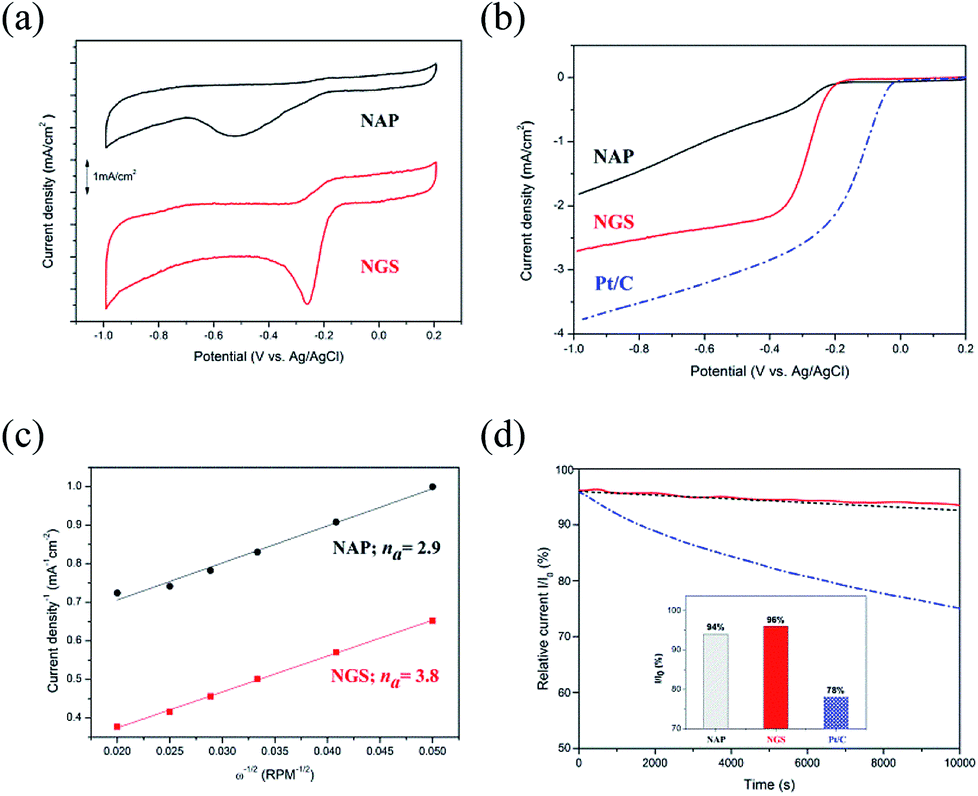 | ||
| Fig. 7 (a) CV curves of NAP and NGS in O2-saturated 0.1 M KOH electrolyte. (b) LSV curves on RDE of NAP and NGS, compared with 20 wt% Pt/C in O2-saturated 0.1 M KOH electrolyte with a sweep rate of 10 mV s−1 at a rotation rate of 1600 rpm after background correction by subtracting the measured current density in N2-saturated solution. (c) K–L plots of J−1 vs. ω−1/2 at 0.6 V for NGS and NAP. (d) Current–time (i–t) chronoamperometry response of NAP, NGS and Pt/C at −0.4 V in O2-saturated 0.1 M KOH solution during 10000 s. | ||
Consequently, based on the above characterization of the catalyst, the enhanced electrocatalytic performance of NGS can be attributed to the good electric conductivity, high surface area, porous structure, and a high content of edge-N species.65,66 The better electric conductivity of NGS results from the sheet-like morphology and graphitic structure compared with NAP, consequently determining a higher current density during ORR and assuring a good electron transfer to and from the catalytic site. The relatively high surface area (621.48 m2 g−1) and the mesoporous structure of the NGS catalyst mean a lot of active sites and have a direct influence on ORR activity and the mass transfer of O2. The analysis of N active sites was carried out by representing the onset potential together with the N species concentration, as in Fig. S7.† Importantly, the onset potential shifts more positively according to the increase in the edge-N contents, implying an enhanced ORR activity. DFT calculation32–34 and experimental results7,25,67,68 indicate that pyridinic-N and pyrrolic-N, which possess one lone pair of electrons in addition to one electron donated to the conjugated π bond, facilitate the high spin density and large atomic charge density on the adjacent carbon atoms on which O2 adsorption is enhanced compared to graphitic-N and pyridinic N-oxide. In conclusion, the high content of edge-N of NGS determines good ORR activity.
The stability of the NAP, NGS, and Pt/C catalysts was acquired by the i–t chronoamperometric response in O2-saturated 0.1 M KOH solution. As shown in Fig. 7(d), the current density of NGS decreases to 96% after 10000 s, which is higher than that of NAP (94%), due to the high content of pyridinic-N and pyrrolic N, as well as the graphitic carbon structure in the NGS (Fig. 6(f)).41 Although the electrocatalytic activity of NAP and NGS is not yet comparable to that of Pt/C, the ORR stability presents a better performance in alkaline media. The poor durability of Pt/C (78%) can be caused by migration, aggregation of Pt particles and dissolution, and detachment from the carbon support under the continuous potential.1,5–7 These problems can be solved by metal-free carbon based materials with their covalent bonding of a catalytically active heteroatom in the carbon framework.
4. Discussion
To elucidate the differences in material characteristics between the NGS and NAP according to plasma stability during the synthesis, we performed plasma diagnostics using the OES method, as shown in Fig. 8(a). The emission spectra present the degree of dissociation of pyridine in the two types of SP and the presence of the active species as radicals in the plasma.69 From the emission intensities, only the relative radical number density can be discussed. The Swan bands of the C2 molecule emission have much higher intensity in the case of plasma during the synthesis of NAP that of NGS, exhibiting a higher electron excitation energy of NAP (5000 K, 0.43 eV), as shown in Table S6,† which produces a higher degree of dissociation of pyridine, as shown in Fig. 8(a). The excitation temperature of C2 molecules in plasma was calculated to be 5000 K, which means that the organic compound was highly decomposed, so that the amorphous carbon structure was synthesized by carbonization. On the other hand, the decomposition of C2 molecules from the pyridine decreased when the SP system was tuned-up by the low-pass filter circuit. The C2 molecules were mainly produced at the interface between the plasma and the liquid since a large gradient of temperature from the plasma to the interface was relatively lower than that without the tune-up condition (conventional SP), as shown in Fig. 8(b) and S8.† These C2 molecules, which might exist as C2H2 and C2H4, lead the reaction, and subsequently, C2 molecules are mainly assembled at the interface, which can provide the graphitic structure.70,71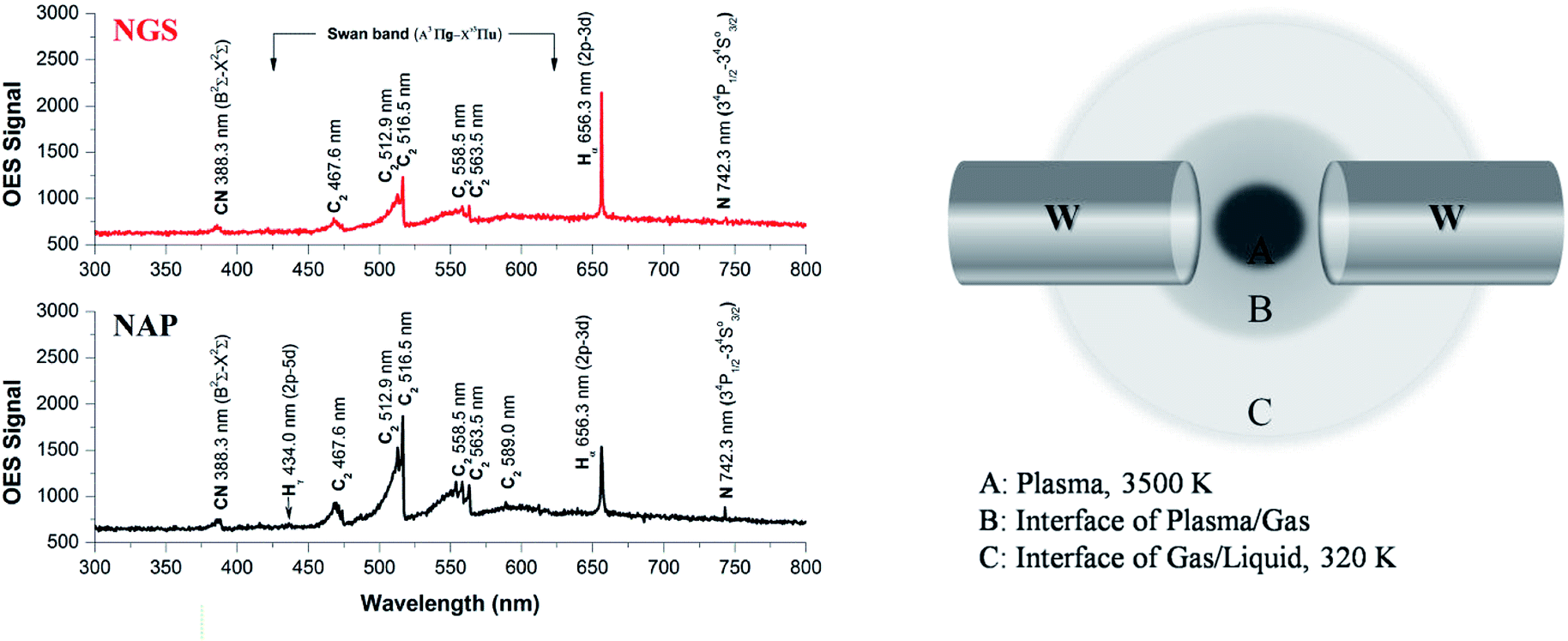 | ||
| Fig. 8 (a) The OES spectra of NAP (using the conventional SP system) and NGS (using a tune-up SP system). The Swan bands of the C2 molecule emission have much higher intensity in the case of plasma during the synthesis of NAP than that of NGS, exhibiting a higher degree of dissociation of pyridine. (b) Reaction areas according to temperature distributions during the synthesis of NGS. | ||
N species can also be explained in the same way as above. The high dissociation of pyridine leads to nitrogen species as CN and N radicals in the conventional SP system. However, the formed nitrogen leaves the plasma as a gas, resulting in a low content of nitrogen in the NAP samples (Tables S2 and S3†). In contrast, the low degree of dissociation of nitrogen in the tune-up SP system favors the preservation of the pyridine structure rather than breaking the aromatic ring. This reaction might create not only more nitrogen content in the graphitic carbon structure by aromatic ring assembly, but also create active sites with edge-N, such as pyridinic-N and pyrrolic-N, which are useful in the ORR (Fig. 6).
5. Conclusion
In summary, we suggest a novel route to synthesize nitrogen doped graphitic carbon nanosheets (NGS), using a tune-up solution plasma system. The strongest potential of SP for the N-doped carbon synthesis is to control the higher concentration of nitrogen, although the SP process has advantages over conventional thermal processes. The proposed method gives several advantages over the assembly of nanocarbons: (a) a one-step process, (b) an ambient reaction environment, (c) a straightforward and easy setup, (d) cost-effective production, (e) possible large-scale synthesis quantities, and (f) eco-friendly fabrication. In particular, the tune-up SP synthesis provides stability during the discharge in liquid, influencing the synthesized nanocarbon characteristics. The NGS material not only has a large surface area, high nitrogen content, and good electrical conductivity, but also has the appropriate active N species of a catalyst to enhance the ORR activity. In detail, the electrochemical measurements demonstrate that NGS show comparable catalytic activity and superior stability to Pt/C in alkaline medium, so that low-cost and large-scale nitrogen self-doped nanocarbon is a promising candidate for the next generation of electrocatalyst in metal-air batteries.Conflicts of interest
There are no conflicts to declare.References
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- M. A. Rahman, X. Wang and C. Wen, J. Electrochem. Soc., 2013, 160, A1759–A1771 CrossRef CAS.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- C. Sealy, Mater. Today, 2008, 11, 65–68 CrossRef CAS.
- R. R. Adzic, J. Z. K. Sasaki, M. B. Vukmirovic, M. Shao, J. X. Wang, A. U. Nilekar, M. Mavrikakis, J. A. Valerio and F. Uribe, Top. Catal., 2007, 46, 249–262 CrossRef CAS.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. (David) Lou and X. Wang, Nat. Energy, 2015, 444–452 Search PubMed.
- Z. Chen, D. Higgins, A. Yu, L. Zhang and J. Zhang, Energy Environ. Sci., 2011, 4, 3167–3192 RSC.
- Q. Li, R. Cao, J. Cho and G. Wu, Adv. Energy Mater., 2014, 4, 1301415 CrossRef.
- H. Liang, X. Zhuang, S. Brüller, X. Feng and K. Müllen, Nat. Commun., 2014, 5, 4973 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, M. Jaroniec, Y. Jin and S. Z. Qiao, Small, 2012, 8, 3550–3566 CrossRef CAS PubMed.
- J. Duan, S. Chen, M. Jaroniec and S. Zhang Qiao, ACS Catal., 2015, 5, 5207–5234 CrossRef CAS.
- M. Shao, Q. Chang, J. P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- L. Dai, Y. Xue, L. Qu, H. J. Choi and J. B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 125, 3192–3198 CrossRef.
- X. Zhou, J. Qiao, L. Yang and J. Zhang, Adv. Energy Mater., 2014, 4, 1301523 CrossRef.
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. Chen and S. Huang, ACS Nano, 2012, 6, 205–211 CrossRef CAS PubMed.
- R. Li, Z. Wei, X. Gou and W. Xu, RSC Adv., 2013, 3, 9978–9984 RSC.
- Y. Zhen, N. Huagui, Y. Zhi, Z. Xuemei, L. Zheng and H. Shaoming, Chem. Commun., 2012, 48, 1027–1029 RSC.
- K. N. Wood, R. O'Hayre and S. Pylypenko, Energy Environ. Sci., 2014, 7, 1212–1249 RSC.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 CrossRef PubMed.
- D. Yu, E. Nagelli, F. Du and L. Dai, J. Phys. Chem. Lett., 2010, 1, 2165–2173 CrossRef CAS.
- L. Qu, Y. Liu, J. B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- Y. Shao, S. Zhang, M. H. Engelhard, G. Li, G. Shao, Y. Wang, J. Liu, I. A. Aksay and Y. Lin, J. Mater. Chem., 2010, 20, 7491–7496 RSC.
- D. W. Wang and D. Su, Energy Environ. Sci., 2014, 7, 576–591 RSC.
- A. Fasolino, J. H. Los and M. I. Katsnelson, Nat. Mater., 2007, 6, 858–861 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 CrossRef CAS PubMed.
- S. F. Huang, K. Terakura, T. Ozaki, T. Ikeda, M. Boero and M. Oshima, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 235410 CrossRef.
- L. Zhang and Z. Xia, J. Phys. Chem. C, 2011, 115, 11170–11176 CrossRef CAS.
- E. J. Biddinger and U. S. Ozkan, J. Phys. Chem. C, 2010, 114, 15306–15314 CrossRef CAS.
- H. Kim, K. Lee, S. I. Woo and Y. Jung, Phys. Chem. Chem. Phys., 2011, 13, 17505–17510 RSC.
- M. A. Bratescu, O. Takai and N. Saito, J. Alloys Compd., 2013, 562, 74–83 CrossRef CAS.
- M. A. Bratescu, S. P. Cho, O. Takai and N. Saito, J. Phys. Chem. C, 2011, 115, 24569–24576 CrossRef CAS.
- Y. K. Heo, M. A. Bratescu, T. Ueno and N. Saito, J. Appl. Phys., 2014, 116, 024302 CrossRef PubMed.
- J. Kang, O. L. Li and N. Saito, Carbon, 2013, 60, 292–298 CrossRef CAS.
- D. Kim, O. L. Li and N. Saito, Phys. Chem. Chem. Phys., 2015, 17, 407–413 RSC.
- K. Hyun, T. Ueno, G. Panomsuwan, O. L. Li and N. Saito, Phys. Chem. Chem. Phys., 2016, 18, 10856–10863 RSC.
- S. H. Lee, Y. K. Heo, M. A. Bratescu, T. Ueno and N. Saito, Phys. Chem. Chem. Phys., 2017, 19, 15264–15272 RSC.
- Y. K. Heo, S. H. Lee, M. A. Bratescu, S. M. Kim, G. J. Lee and N. Saito, Plasma Processes Polym., 2017, 14, e1600163 CrossRef.
- NIST X-ray Photoelectron Spectroscopy Database, http://srdata.nist.gov/xps/, (accessed April 2017).
- B. E. Warren and P. Bodenstein, Acta Crystallogr., 1965, 18, 282–286 CrossRef CAS.
- Z. Q. Li, C. J. Lu, Z. P. Xia, Y. Zhou and Z. Luo, Carbon, 2007, 45, 1686–1695 CrossRef CAS.
- T. J. Konno and R. Sinclair, Acta Metall. Mater., 1995, 43, 471–484 CrossRef CAS.
- L. Kumari and S. V. Subramanyam, Mater. Res. Bull., 2006, 41, 2000–2006 CrossRef CAS.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B, 2000, 61, 14095–14107 CrossRef CAS.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- L. G. Cancado, K. Takai and T. Enoki, Appl. Phys. Lett., 2006, 88, 163106 CrossRef.
- A. Cuesta, P. Dhamelincourt, J. Laureynes, A. Martinez-Alonso and J. M. Tascon, Carbon, 1994, 32, 1523–1532 CrossRef CAS.
- X. Huang, K. Qian, J. Yang, J. Zhang, L. Li, C. Yu and D. Zhao, Adv. Mater., 2012, 24, 4419–4423 CrossRef CAS PubMed.
- J. C. Groen, L. A. A. Peffer and J. Pérez-Ramírez, Microporous Mesoporous Mater., 2003, 60, 1–17 CrossRef CAS.
- W. Wei, H. Liang, K. Parvez, X. Zhuang, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2014, 126, 1596–1600 CrossRef.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- P. H. Matter, L. Zhang and U. S. Ozkan, J. Catal., 2006, 239, 83–96 CrossRef CAS.
- J. R. Pels, F. Kapteijn, J. A. Moulijn, Q. Zhu and K. M. Thomas, Carbon, 1995, 33, 1641–1653 CrossRef CAS.
- R. J. J. Jansen and H. van Bekkum, Carbon, 1995, 33, 1021–1027 CrossRef CAS.
- F. Kapteijn, J. A. Moulijn, S. Matzner and H. P. Boehm, Carbon, 1999, 37, 1143–1150 CrossRef CAS.
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936–7942 RSC.
- E. J. Biddinger and U. S. Ozkan, J. Phys. Chem. C, 2010, 114, 15306–15314 CrossRef CAS.
- X. Fu, J. Jin, Y. Liu, Z. Wei, F. Pan and J. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 3930–3936 CrossRef CAS PubMed.
- R. E. Davis, G. L. Horvath and C. W. Tobias, Electrochim. Acta, 1967, 12, 287–297 CrossRef CAS.
- S. Kundu, T. C. Nagaiah, W. Xia, Y. Wang, S. V. Dommele, J. H. Bitter, M. Santa, G. Grundmeier, M. Bron, W. Schuhmann and M. Muhler, J. Phys. Chem. C, 2009, 113, 14302–14310 CrossRef CAS.
- N. P. Subramanian, X. Li, V. Nallathambi, S. P. Kumaraguru, H. C. Mercado, G. Wu, J. W. Lee and B. N. Popov, J. Power Sources, 2009, 188, 38–44 CrossRef CAS.
- C. V. Rao, C. R. Cabrera and Y. Ishikawa, J. Phys. Chem. Lett., 2010, 1, 2622–2627 CrossRef CAS.
- J. Wu, L. Ma, R. M. Yadav, Y. Yang, X. Zhang, R. Vajtai, J. Lou and P. M. Ajayan, ACS Appl. Mater. Interfaces, 2015, 7, 14763–14769 CrossRef CAS PubMed.
- R. W. B. Pearse and A. G. Gaydon, The Identification of Molecular Spectra, Chapman and Hall, New York, 1976 Search PubMed.
- T. Morishita, T. Ueno, G. Panomsuwan, J. Hieda, A. Yoshida, M. A. Bratescu and N. Saito, Sci. Rep., 2016, 6, 36880 CrossRef CAS PubMed.
- J. Senthilnathan, C. C. Weng, J. D. Liao and M. Yoshimura, Sci. Rep., 2013, 3, 2414 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06614k |
| This journal is © The Royal Society of Chemistry 2018 |