
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface design and preparation of multi-functional magnetic nanoparticles for cancer cell targeting, therapy, and imaging
Jing Qu†
a,
Zhijie Tian†b,
Qiuyue Wanga,
Si Penga,
Jian-bin Luo a,
Qing-han Zhou*a and
Juan Lin*b
a,
Qing-han Zhou*a and
Juan Lin*b
aCollege of Chemical and Environment Protection, Southwest Minzu University, First Ring Road, 4th Section No. 16, Chengdu, Sichuan 610041, China. E-mail: zhqinghan@163.com; Fax: +86-28-85524382; Tel: +86-28-85522269
bSchool of Biomedical Sciences and Technology, Chengdu Medical College, No. 783 Xindu Road, Chengdu, Sichuan 610500, China. E-mail: linjuan.scu@gmail.com
First published on 16th October 2018
Abstract
Recently, theranostic candidates based on superparamagnetic iron oxide nanoparticles (SPIONs) providing the combination of therapy and diagnosis have become one of the most promising system in cancer research. However, poor stability, premature drug release, lack of specific tumor cell targeting, and complicated multi-step synthesis processes still hinder them for potential clinical applications. In this research, the multi-functional magnetic nanoparticles (MNPs-DOX) were prepared via a simple assembly process for targeted delivery of doxorubicin (DOX) and enhanced magnetic resonance (MR) imaging detection. Firstly, the multi-functional copolymer coating, polyamidoamine (PAMAM), was designed and synthesized by Michael addition reaction, where N,N-bis(acryloyl)cystamine served as backbone linker, and DOX, dopamine (DA), and polyethylene glycol (PEG) acted as comonomers. The PAMAM was then directly assembled to the surface of SPIONs by the ligand exchange reaction with SPIONs forming the MNPs-DOX. The hydrophilic PEG moieties provide the nanoparticles with colloidal stability and good-dispersity in aqueous solution. Comparing with the quick release of free DOX, the drug release behavior of MNPs-DOX exhibited a sustained drug release. Because the chemical cleavage of disulfide in the polymer backbone, a high cumulative drug release up to 60% in GSH within 48 h was observed, rather than only 26% in PBS (pH 7.4) without GSH. The MR imaging detection experiment showed that the MNPs-DOX had an enhanced T2 relaxivity of 126 mM−1 S−1 for MR imaging. The results of the cytotoxicity assays showed a remarkable killing effect of cancer cells by MNPs-DOX due to the FA tumor-targeting ligand, comparing with non-targeted drug molecules. All the results showed that the as prepared multi-functional magnetic nanoparticles may serve as a promising theranostic candidate for targeted anticancer drug delivery and efficient detection through MR imaging in medical application.
1. Introduction
In recent years, superparamagnetic iron oxide nanoparticles (SPIONs) have been extensively developed as a promising candidate for both magnetic resonance (MR) imaging and tumor-targeting drug delivery in theranostics.1–4 However, the practical uses of SPIONs have been disappointing because of poor water-solubility, acid erosion, oxidation, and severe aggregation under in vivo conditions.5–8 Therefore, recently, to avoid these disadvantages functional polymers were designed and used as the coating materials to develop the applications of SPIONs-based magnetic nanoparticles in aqueous solution. Because hydrophilic polymer coatings can remarkably enhance the structural stability of SPIONs to endure the complex biological environment for efficient delivery of drug, incorporation of SPIONs into hydrophilic polymer coatings have been improved to be a simple and effective method to stabilize magnetic nanoparticles for in vivo medical uses.9–13 Recently, the magnetic nanoparticles with hydrophilic polymer coating with excellent good biostability, excellent biodegradability, prolonged blood circulation, and MR imaging contrast effect were widely studied for medical application.14,15 However, non-degradable structure of the nanoparticle is still a big shortcoming of this kind of nanoparticle, because the excessively stabilized nanoparticle will prevent the drug release at aimed sites and accumulate in the cells or tissues causing a long-term toxicity.16–18 Therefore, SPIONs with biodegradable coating are suggested to be promising candidates for clinic use in theranostics.19,20Polymer with reduction-sensitivity were proved to be an effective vehicle for intracellular and triggered drug release owing to 1000 times higher concentrations of glutathione (GSH, about 2–10 mM) in the various subcellular organelles in cytoplasm than in the extracellular fluids (about 2–20 μM).21,22 Therefore, the nanoparticles with reduction-sensitive polymer coating can be biodegraded in presence of the reducing agent such as GSH due to the chemical cleavage of disulfide bond via a thiol/disulfide exchange process, which can achieve controllable drug delivery for efficient cancer therapy.23,24
On the other hand, short-term stability that caused by massive dilution or disruptions under in vivo chemical condition, such as pH, temperature, and ion concentration was often observed for SPIONs based magnetic nanoparticles because of the weak bond between SPIONs and polymer coatings. Bio-stable magnetic nanoparticles are urgently required to achieve long circulation time in vivo.25–27 In recent years, catechol-containing molecules are proved to have the property to adhere to almost any material of either organic or inorganic origin. The catechol functional group in 3,4-dihydroxy-L-phenylalanine (L-DOPA) and its analog dopamine (DA) could form strong bonds on inorganic/organic materials surfaces such as coordination of metal ions, formation of π-electrons, and hydrogen bond interactions.28–30 Thus, catechol-containing molecules have been widely used to immobilize SPIONs into polymer coating to avoid unexpected disassembly. For example, Chen et al. developed a magnetic nanoparticle with enhanced MR imaging and pH-sensitive drug release kinetics by a simple and controllable method in cancer theranostics.31 Huang et al. developed a novel SPION and silica based theranostic nanoplatform for MR imaging and magnetically guided/cancer targeted drug delivery, which exhibited a sustained drug release and high superparamagnetism.32 McDonagh et al. reported a magnetic theranostic system functionalized with L-DOPA that can gradually degrade in biological media, which offers a time-dependent switch in the MR contrast and allows for complementary imaging and enhanced diagnosis.33 Sasikala et al. use poly(2-hydroxyethyl methacrylate-co-dopamine methacrylamide) p(HEMA-co-DMA) to functionalize the SPIONs as well as to conjugate borate containing anticancer drug bortezomib for the synergistic anticancer treatment by pH responsive anticancer drug delivery and hyperthermia.34 Recently, our group reported a reduction-sensitive cross-linked micellar magnetic nanoparticle demonstrating a controllable drug release and effective MR imaging detection.35 However, most magnetic nanoparticles can only passively accumulate at the tumor site. To enhance the uptake of drug-loaded nanoparticles into the targeting cells, recently, active targeting drug delivery has gained great interest in cancer therapy. To our best knowledge, there was still no report on multi-functional magnetic nanoparticle with in vivo bio-stability, controllable drug delivery, specific tumor cell targeting, and enhanced MR imaging detection in theranostic system so far.
Herein, our object here is to design and synthesize a multi-functional polymer coating, and then fabricate a multi-functional magnetic nanoparticle via a single assembly step as a theranostic candidate for cell targeting drug delivery and MR imaging (Scheme 1). To accomplish this, the polymer coating was designed as a linear multi-functional copolymer, polyamidoamine (PAMAM). The PAMAM was synthesized with N,N-bis(acrylate)cystamine (BACy), dopamine (DA), DOX, folic acid-polyethylene glycol (PEG-FA), and PEG by Michael addition reaction, where BACy served as the backbone linker, DA moiety as anchor to immobilize SPIONs, DOX as anti-cancer drug unit, PEG provided colloidal stability, and folic acid (FA) as cell targeting ligand. Finally, the multi-functional magnetic nanoparticles (MNPs-DOX) were prepared via a ligand exchange reaction between the PAMAM coating and SPIONs. Based on the experimental results, this theranostic magnetic nanoparticle exhibited bio-stability, controllable drug delivery, specific tumor cell targeting, and remarkable cancer killing effect. On the other hand, the magnetic nanoparticles exhibited effective r2 relaxivity for contrast-enhanced MR imaging. It was suggested that the as prepared MNPs-DOX can be used as a promising nano-platform that can incorporate various therapeutic drugs and diagnostic nanoparticles to prepared various of functional nanoparticles for clinically theranostic application.
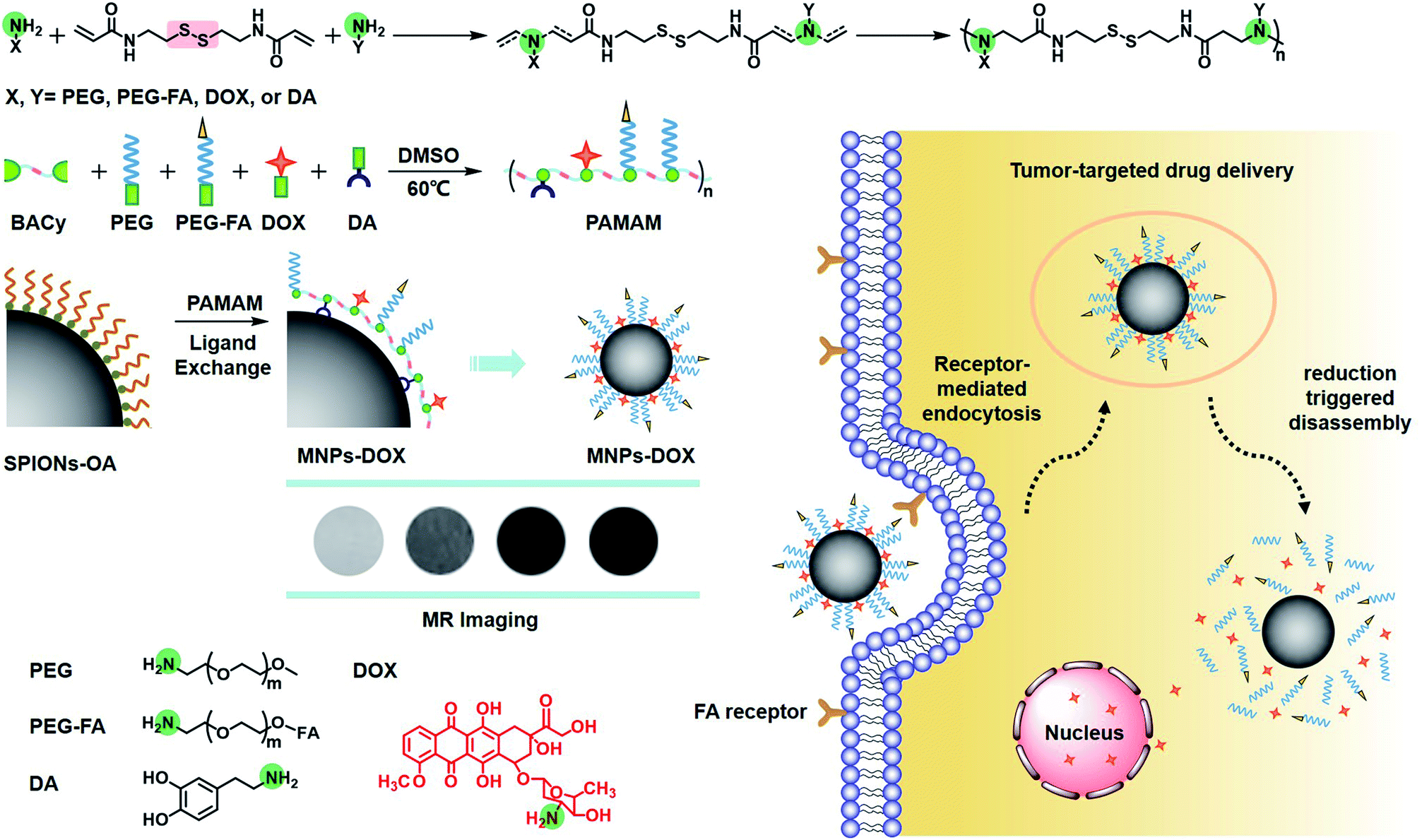 | ||
| Scheme 1 Schematic illustration of the synthesis of PAMAM, nanoparticle surface modification, and intracellular trafficking pathway of redox-sensitive MNPs-DOX. The intracellular trafiicking pathway includes steps of receptor-meditated cellular internalization, endo/lysosomal escape, reduction triggered disassembly, and drug release. Amine groups are highlighted in green. | ||
2. Experimental
2.1 Materials
Methoxy poly(ethylene glycol)amine (mPEG-NH2, Mn = 2000, Ponsure), folic acid poly(ethylene glycol)amine (FA-PEG-NH2, Mn = 2000, Ponsure), acryloyl chloride (98%, J&K Chemical), cystamine dihydrochloride (96%, J&K Chemical), dopamine hydrochloride (98%, Aladdin), GSH (98%, Aladdin), DOX (98%, Beijing HuaFeng United Technology Co. Ltd.) were used as received. Dichloromethane (DCM), dimethyl sulfoxide (DMSO), tetrahydrofuran (THF), ethylene acetate (EA), n-hexane, ethanol, and ether were used after purified.2.2 Synthesis of PAMAM
BACy was synthesized following a previously published procedure.36 A typical procedure for preparing the PAMAM is as following: molar ratio of comonomers was set at 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :38:12:16:3 (BACy:DA:DOX:PEG-HN2:FA-PEG-NH2), 130 mg BACy, 72 mg DA, 70 mg DOX, 328 mg PEG-HN2, and 52 mg FA-PEG-NH2 were added to a 25 mL flask. 50 μL TEA and 10 mL DMSO were added into the flask. The reaction proceeded at 50 °C for 44 h under the protection of nitrogen gas. The raw product was gathered by precipitation in ice-cooled diethyl ether, and then the product was purified by a dialysis method using a dialysis tube with molecular weight cut-off (MWCO) of 3500 for 3 days to remove the excess molecules. Finally, the solution was frozen and lyophilized into a dark red solid.
:38:12:16:3 (BACy:DA:DOX:PEG-HN2:FA-PEG-NH2), 130 mg BACy, 72 mg DA, 70 mg DOX, 328 mg PEG-HN2, and 52 mg FA-PEG-NH2 were added to a 25 mL flask. 50 μL TEA and 10 mL DMSO were added into the flask. The reaction proceeded at 50 °C for 44 h under the protection of nitrogen gas. The raw product was gathered by precipitation in ice-cooled diethyl ether, and then the product was purified by a dialysis method using a dialysis tube with molecular weight cut-off (MWCO) of 3500 for 3 days to remove the excess molecules. Finally, the solution was frozen and lyophilized into a dark red solid.
2.3 Preparation method of MNPs-DOX and characterization
SPIONs was synthesized following a previously published method.37 The PAMAM copolymer was designed to be coated onto SPIONs surface via a simple ligand exchange reaction. The as prepared PAMAM (10 mg) and SPIONs (20 mg) and were dissolved in DMF (25 mL). 10 mL of distilled water was added to the above mixture solution under vigorous sonication, and the resulting colloid was stirred vigorously for 24 h under N2 ambient to obtain the raw product of MNPs-DOX. After that, the MNPs-DOX was separated by centrifugation method and then washed with distilled water for three times. The raw product was further purified by dialysis method using a dialysis tube with MWCO of 3500 to remove the unbound copolymer. Then the solution was frozen and lyophilized into a red-brown powder. The magnetic nanoparticles without DOX, MNPs, were also prepared by the same procedure as the MNPs-DOX.The chemical structure of PAMAM and related molecules were obtained by 1H NMR spectra using a Bruker 400 MHz spectrometer with CDCl3 or DMSO as solvent with tetramethylsilane (TMS) as the internal standard. FTIR data were gathered in solid state and taken from 400 to 4000 cm−1 utilizing a resolution at 4 cm−1 resolution on a PE Spectrum One FTIR spectrophotometer under ambient. In DLS experiments the nanoparticle size (Dh) and distribution (PDI) of MNPs-DOX were determined using a Malvern Zetasizer Nano-ZS90 apparatus equipped with a 4.0 mW laser operating at λ = 633 nm. All DLS tests of samples were measured under ambient and conducted in a 1.0 mL quartz cuvette. The nano-morphologies of micelles were observed by TEM (Hitachi H-600, Japan). The sample solution was directly dropped onto carbon-coated copper grids and dried at room temperature without staining before measurement. Magnetic behaviors of the MNPs-DOX and SPIONs were evaluated by a vibrating sample magnetometer (VSM) (MPMS-XL-7, Quantum Design Company, USA).
2.4 MR imaging detection
The effect of an MR imaging contrast agent can be evaluated by its longitudinal and transverse relaxivities, r1 and r2, which reflect the ability of the contrast agent to alter T1 (spin-lattice relaxation) and T2 (spin–spin relaxation), respectively. All samples were positioned in a Varian (Palo Alto, California, USA) 4.0 cm inner diameter quadrature coil and relaxation data were acquired at 7 T using a Varian Inova imaging and spectroscopy system. A single slice, multi-echo spin echo sequence was used to measure T2 relaxation times (TR = 2000 ms, TE = 11–110 ms (10 echoes, 11 ms increments), SL = 2 mm, FOV = 100 × 100 mm, MA = 128 × 128). Relaxation times were obtained by fitting the multi-echo data to a monoexponential decay curve using linearized least-squares optimization.2.5 Drug loading and release profile
In vitro release study was performed at 37 °C. Typically, MNPs-DOX and free DOX at a concentration of 1 mg mL−1 (2 mL) in a dialysis membrane tube (MWCO 3500) were incubated in two different media: PBS (10 mM, pH 7.4) with 10 mM GSH, and PBS (10 mM, pH 7.4) without GSH under gentle stirring. At specified time intervals, the DOX content in the samples was analyzed with a UV absorbance at 480 nm. All DOX drug release data were averaged over three measurements. The concentration of released DOX was calculated according to a standard curve obtained from DOX/DMF solutions at a series of DOX concentrations.2.6 In vitro cell assay
The cancer cell viability was evaluated by a Cell Counting Kit-8 (CCK-8) assay. The HeLa cells (human cervical carcinoma cells, cancer cells) and HUVEC cells (human umbilical vein endothelial cells, normal cells) were kindly provided by Chengdu Lilai Biotechnology Co. Ltd. The HeLa cells and HUVEC cells were cultured in DMEM (dulbecco's modified eagle medium) at 37 °C in a humidified atmosphere with 5% CO2. The cells were supplemented with 10% heat-inactivated fetal bovine serum. Then, the logarithmic growth phase cells were washed with PBS, digested with trypsin, and centrifugalized to remove liquid supernatant. Cells were seeded into 96-well plate at a density of 5 × 105 cells per well and incubated for 24 h to allow cell attachment. After that, the cell culture medium was removed, and 100 μL of different concentrations of free DOX, MNPs, or MNPs-DOX were added into each well. The cells were incubated with different samples for several hours. At different time intervals, 10 mL of CCK-8 solution were added into each well, and the cells were incubated for another 3 h. After incubation, the cell viability was investigated by measuring the absorbance at 450 nm by using a micro-plate reader (Thermo Fisher Multiskan FC, USA).2.7 Confocal laser scanning microscopy
The cellular uptake of different samples was monitored by Confocal Laser Scanning Microscope (CLSM) technique. The cells were incubated in an UV disinfected 6-well plate at a density of 5 × 105 cells per well. After 24 h incubation period, the culture medium was replaced with samples of the free DOX or MNPs-DOX. For 2 h and 48 h incubation, the suspension was removed, and fresh DMEM and PBS were used to wash the samples before fluorescence investigation. The samples were then mixed with fluorescent mounting medium to be observed by CLSM (Motic AE31, Japan) by using an imaging software.3. Results and discussion
3.1 Preparation and characterization of the MNPs-DOX
In this work, the synthesis route of PAMAM and formation of MNPs-DOX were shown in Scheme 1. The PAMAM was synthesized by Michael addition reaction, where DMSO was used as reaction solvent. The PAMAM contains five functional units: BACy served as the backbone linker for reduction-sensitivity, DA as metal binding unit to anchor the SPIONs, DOX as the anti-cancer drug unit, PEG as hydrophilic corona to ensure colloidal stability, and FA as cell targeting ligand. Finally, SPIONs were immobilized into PAMAM coating to prepare the theranostic nanoparticles, MNPs-DOX, via a simple ligand exchange assembly process between catechol group in DA unit with the oleic acid layer on SPIONs. As anticipated, the MNPs-DOX system can achieve specific tumor cell targeting, triggered drug release, and enhanced contrast for MR imaging detection.The chemical structures of the synthesized BACy and PAMAM copolymer were characterized by 1H NMR. The representative 1H NMR spectrum of BACy in CDCl3 was depicted in Fig. 1a. The resonance signals at δ = 2.88 (e), 3.65 (d), 5.61–6.36 (a and b), and 6.66 ppm (c) are ascribed, respectively, to –CH2– (e and d) neighboring to the disulfide bond in the BACy molecule, vinyl group (a and b), and –NH– (c), respectively. The 1H NMR spectrum of PAMAM copolymer (in DMSO) was also depicted in Fig. 1b. The resonance signals at 2.71–2.89 (a), 2.91–3.09 (f), 3.25 (d), 3.33–3.63 (b and c), and 8.10–8.43 ppm (e) were belonged to the methylene group (a) in polymer backbone, α-methylene group (f) in DA units, the methyl end group (d) in PEG, the glycol units in PEG (b and c), and phenolic hydroxyl (e) in DMA units, respectively. Additionally, the presence of the FA molecule in the PAMAM copolymer was confirmed by the appearance of weak signals at 6.64–8.66 ppm, which corresponded with the aromatic protons of FA. Moreover, the conjugation of DOX onto the PAMAM copolymer was confirmed by the presence of characteristic DOX peaks at 1.24, 3.94, 4.58, and 5.61 ppm. It was indicated that the multi-functional copolymer, PAMAM, was successfully synthesized based on the 1H NMR spectrum. As shown in Fig. 2, FT-IR spectra were also used to confirm the chemical structure of SPIONs, PAMAM copolymer, and MNPs-DOX. As shown in Fig. 2a, the absorption bands around 580 cm−1 belonged to the Fe–O stretching vibration of Fe3O4. In Fig. 2b, the vibrational band C–S in BACy in polymer backbone occurred at 1277 cm−1 indicating that the successful synthesis of PAMAM copolymer. In Fig. 2c, Fe–O of Fe3O4, C–S in BACy in polymer backbone were distinctively observed in MNPs-DOX at 583 and 1272 cm−1, indicating that the SPIONs were successfully immobilized into the PAMAM copolymer to form MNPs-DOX. The functional polymer coating (PAMAM) was successfully synthesized by Michael addition reaction, and the magnetic nanoparticles, MNPs-DOX, were further prepared via a simple ligand exchange assembly process.
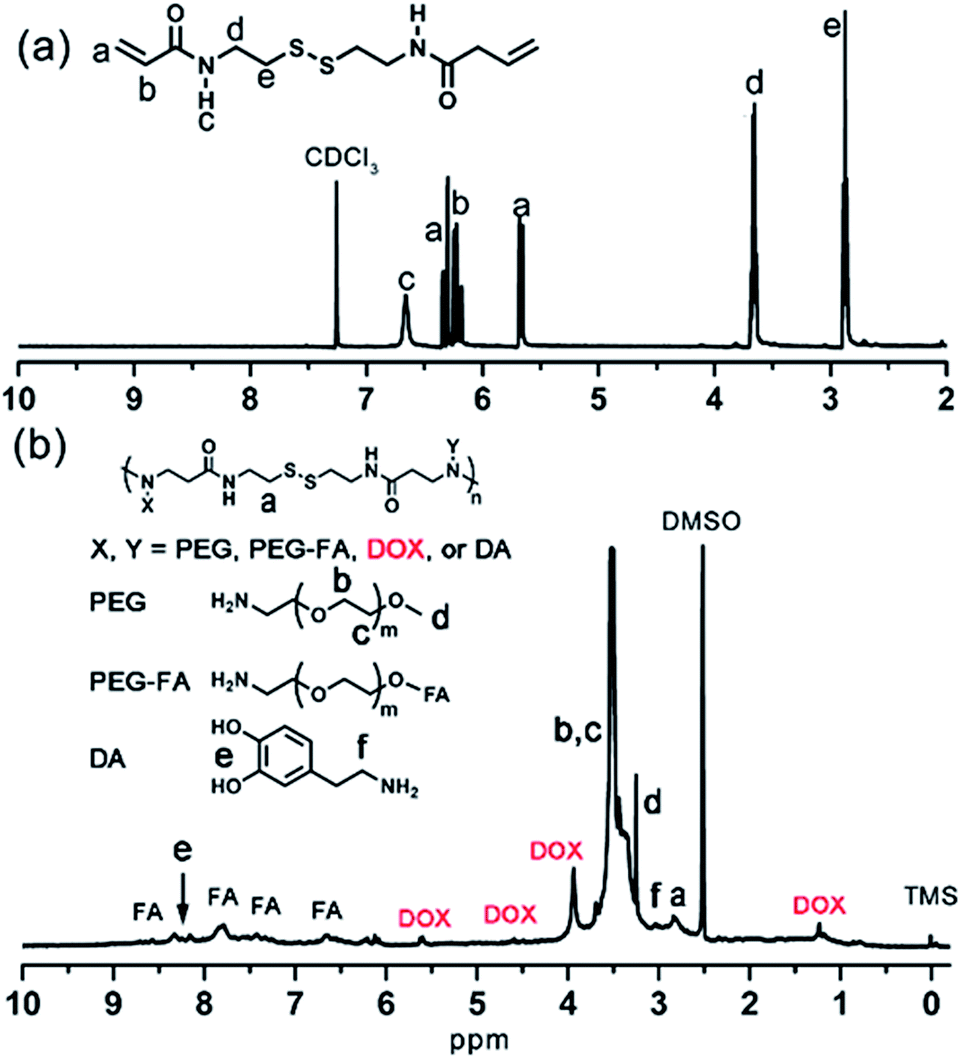 | ||
| Fig. 1 1H NMR spectrum of (a) BACy in CDCl3 and (b) PAMAM in DMSO. | ||
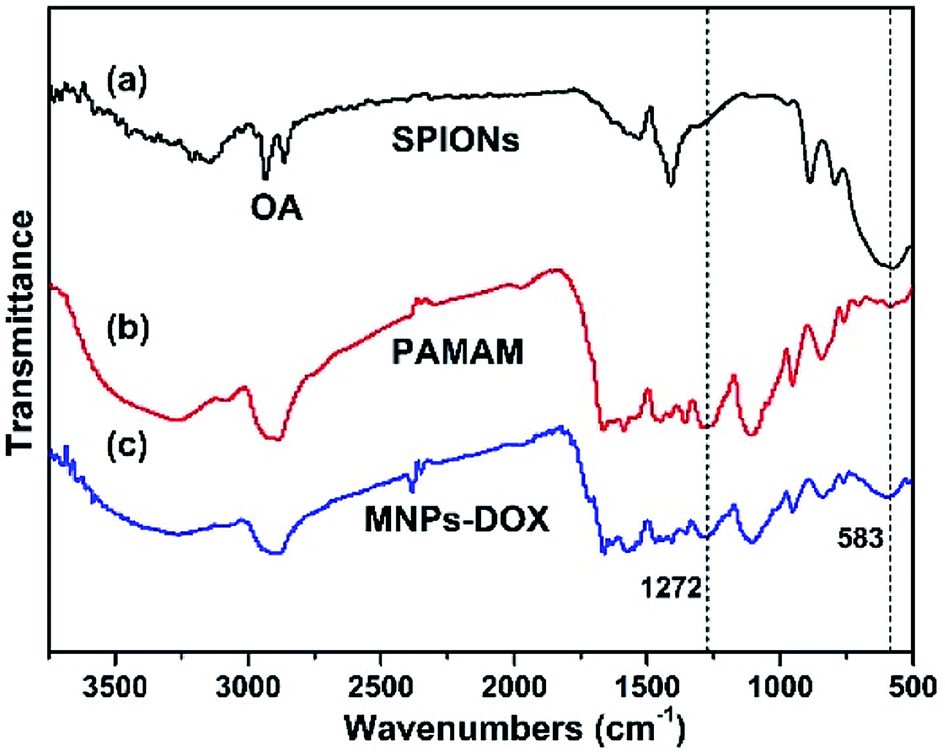 | ||
| Fig. 2 FT-IR spectra of (a) SPIONs, (b) PAMAM copolymer, and (c) MNPs-DOX. | ||
The MNPs-DOX were prepared by introducing different functional molecules into the PAMAM copolymer backbone via Michael addition reaction. Herein, the polymer component weight ratios were referred to the feeding and loading ratios before and after polymerization, respectively. As shown in Fig. 3a, the small difference between the feeding and loading of monomers indicated an efficient conversion during the polymerization reaction indicating that the drug/polymer composition in the end products can be precisely controlled by altering the feeding ratio of monomers. MNPs-DOX was also characterized by UV-vis and fluorescence spectroscopy to further confirm and quantify the DOX loading. Because free DOX emits red fluorescence at 590 nm, the spectrum depicted in Fig. 3b confirms that MNPs-DOX conserved the fluorescent property of DOX with the absorbance and emission maxima at 480 and 590 nm, respectively. By using the absorbance measured from MNPs-DOX, the drug loading was quantified that the weight of DOX accounts for 12.0% of the total drug entrapped polymer weight, and 67.4% for encapsulation efficiency. In brief, this system offers a nano-platform based on a controllable synthesis, which can be tailored to incorporate therapeutic drugs with precisely controlled dose for cancer therapy.
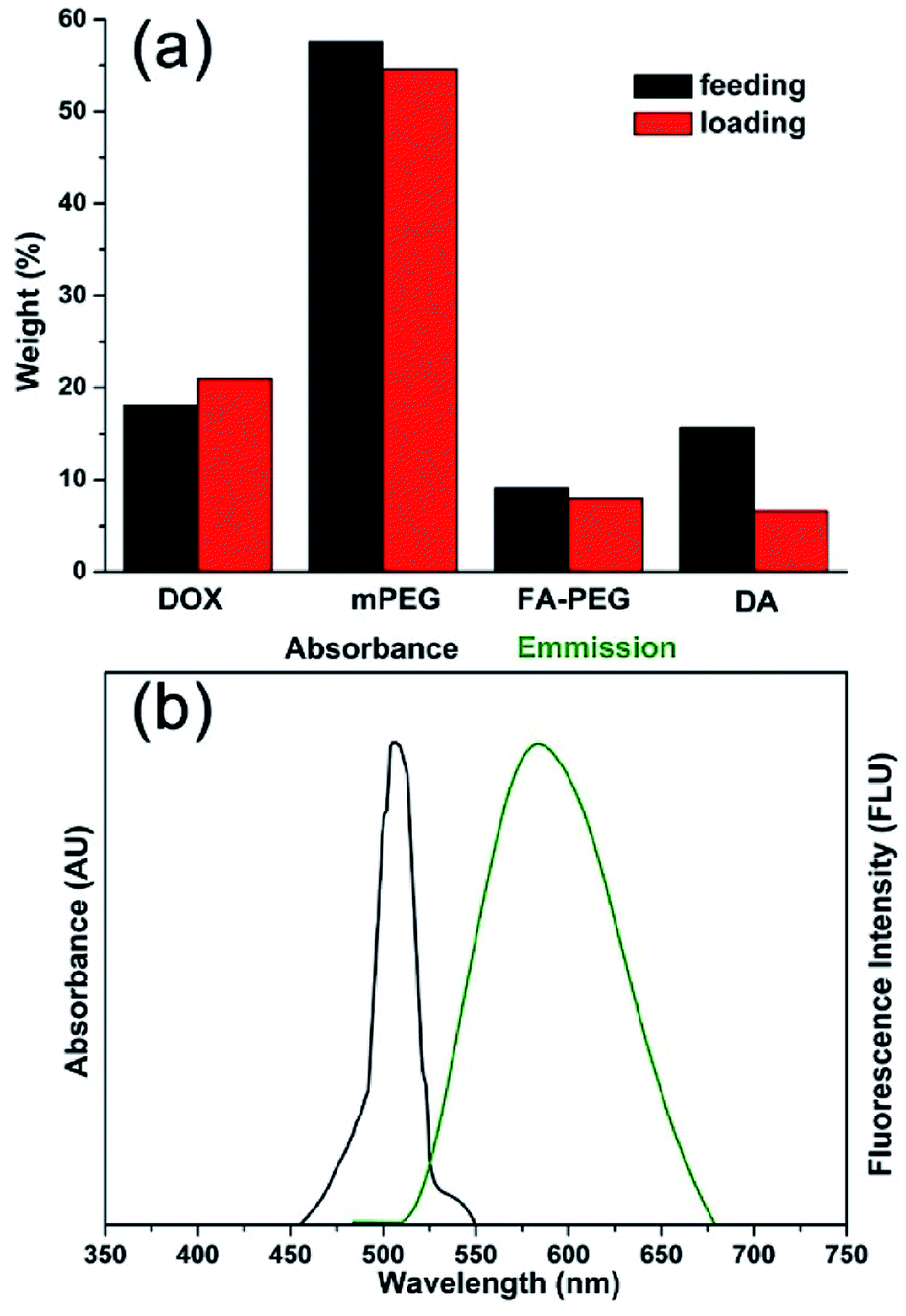 | ||
| Fig. 3 Properties of MNPs-DOX. (a) Comparison chart of DOX, PEG, PEG-FA, and DA feeding and loading ratios in PAMAM copolymer. (b) Absorbance and fluorescence spectra of DOX in MNPs-DOX. | ||
The as prepared oleic acid coated SPIONs were composed of Fe3O4 magnetite nanoparticles with a size of about 15 nm (Fig. 4a). Due to their hydrophobic property of the oleic acid layer they were dispersed well in chloroform. After addition of the as prepared PAMAM copolymer, the copolymer was self-assembled onto the surface of SPIONs via a ligand exchange reaction between DA moieties and the oleic acid layer of SPIONs to form MNPs-DOX. The MNPs-DOX were dispersed well in water because the hydrophilic PEG chains in the PAMAM backbone could provide colloidal stability. As shown in Fig. 4b and c, TEM photos showed that the PAMAM copolymer micelles with a hollow-sphere structure were well dispersed in water solution with an average hydrodynamic diameter of 70 nm, while the MNPs-DOX have an average hydrodynamic diameter of 120 nm after ligand exchange reaction. The as prepared SPIONs, PAMAM copolymer micelles, and water-soluble MNPs-DOX were also characterized by DLS measurement in aqueous solution (shown in Fig. 4d), and the hydrodynamic size of SPIONs, PAMAM copolymer micelles, and MNPs-DOX were about 40, 83, and 160 nm, respectively. No significant changes in size were observed for weeks in aqueous solution, indicating that the MNPs-DOX retained good stability. Additionally, Fig. 4e showed that hydrophobic SPIONs were dispersed in chloroform while MNPs-DOX were soluble in water. As shown in Fig. 5, the as prepared MNPs-DOX were successfully prepared and exhibited nano-sized spherical morphology, good water-dispersity, and excellent stability for potential use in drug delivery.
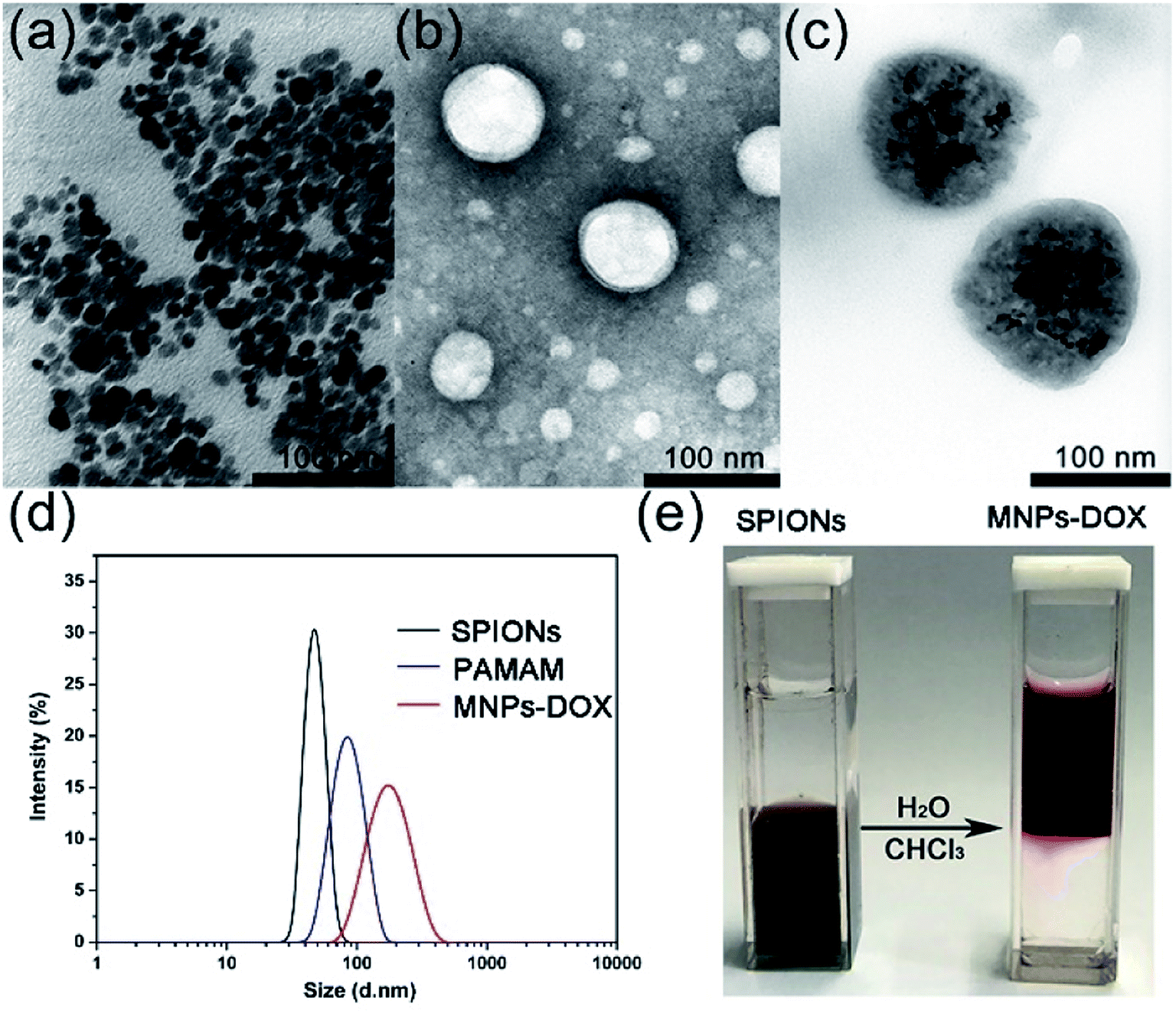 | ||
| Fig. 4 TEM photos of (a) SPIONs, (b) PAMAM copolymer micelles, and (c) MNPs-DOX after the ligand exchange reaction; (d) DLS data showing the change of hydrodynamic size (in diameter) of SPIONs before, and after ligand exchange reaction with PAMAM copolymer; (e) photograph of hydrophobic SPIONs dispersed in chloroform (left) and water-soluble MNPs-DOX (right). | ||
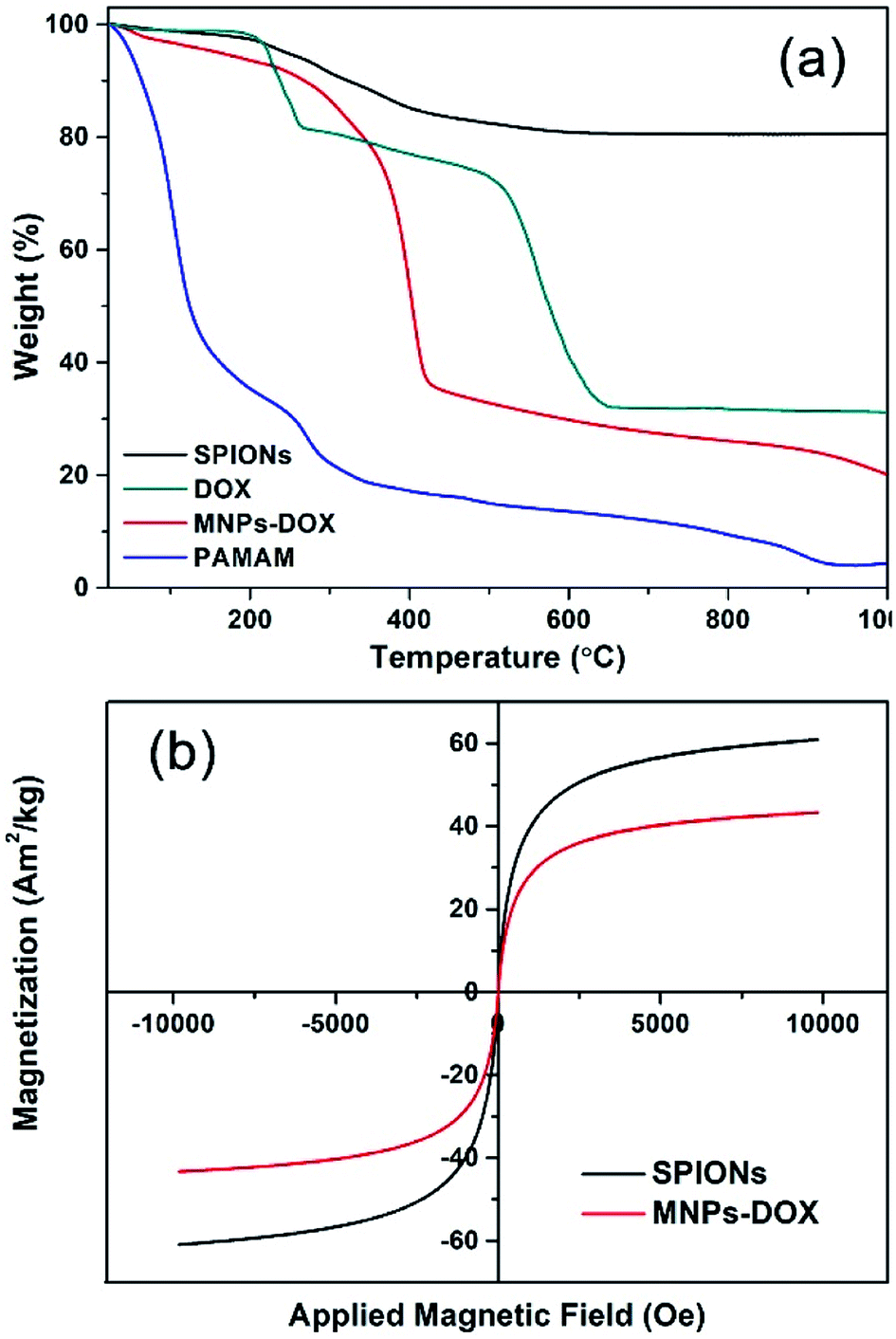 | ||
| Fig. 5 (a) TGA curves of SPIONs, DOX, MNPs-DOX, and PAMAM copolymer. (b) Magnetization studies of SPIONs (black) and MNPs-DOX (red). | ||
The TGA curves of SPIONs, MNPs-DOX, and PAMAM copolymer were depicted in Fig. 5a. The TGA curves of SPIONs showed that the weight loss over temperature ranging from room temperature to 1000 °C is about 19.4%, which is attributed to the escape of physically adsorbed water and the oleic acid layer on the surface. For MNPs-DOX, the weight loss over temperature ranging from room temperature to 1000 °C is associated with the escape of the physically adsorbed water and organic polymers, which is about 80.5%. From the TGA curve of MNPs-DOX, organic polymers on MNPs-DOX began to degrade rapidly at the temperature higher than 250 °C. For the PAMAM copolymer the organic polymers were almost completely decomposed at the temperature of 1000 °C, which has a weight loss of 95.5%. Therefore, the weight loss of MNPs-DOX can be used to estimate the weight proportions of the MNPs-DOX, that is, the weight proportion of SPIONs in MNPs-DOX is about 19.5 wt%. This result further improved that PAMAM was successfully modified on SPIONs. On the other hand, the magnetic behavior of SPIONs and MNPs-DOX were evaluated with VSM. The magnetization of SPIONs and MNPs-DOX was shown as a function of the variation of magnetic field (shown in Fig. 5b). The saturation magnetization was around 60.8 and 43.3 A m2 kg−1 for SPIONs and MNPs-DOX, respectively. The saturation magnetization of SPIONs and MNPs-DOX was totally attributed to the actual content of magnetic particles in the material, while the organic shells have no contribution to the magnetic performance of the nanoparticles. The slightly decrease of saturation magnetization for MNPs-DOX can be ascribed to the polymer coating outside the SPIONs. However, it was indicated that the self-assembly process did not affect the superparamagnetism of SPIONs based on the zero coercivity and remanence of the magnetization curve, that was, MNPs-DOX still remained the superparamagnetic nature.
To monitor the reduction-sensitive behavior of the magnetic nanoparticles, the degradation experiments of the MNPs-DOX with redox agent (GSH) were performed. The size change of MNPs-DOX was monitored by DLS measurements in response to 10 mM GSH in PBS buffer (pH 7.4) at 37 °C. In Fig. 6a, a slow degradation was observed for MNPs-DOX in the first 1 h, in which the average hydrodynamic size increased from about 160 nm to about 190 nm. After 2 h, the hydrodynamic size increased to about 300 nm, reaching over 400 nm after 24 h. It was suggested that the chemical cleavage of the disulfide bonds had resulted in the degradation of the PAMAM coating and formation of a swelling nano-structure in aqueous solution. Additionally, smaller particles less than 50 nm were also observed indicating the surface-shedding of the MNPs-DOX into linear polymer chains. However, almost no size change was observed after 24 h without GSH under the same condition. Additionally, the nanoparticle stability of MNPs-DOX was also investigated by DLS in culture media (Fig. 6b). It was observed that the hydrodynamic size of MNPs-DOX in DMEM was about 160 nm right after the beginning of the DLS measurements, and no significant changes in size or aggregations were discerned for up to a week, indicating that the MNPs-DOX system could remain stable in culture media. The excellent stability of MNPs-DOX can be attributed to the hydrophilic PEG shell with coiled conformations that provides good colloidal stability, and the strong interaction between SPIONs and polymer coating. According to experimental results, it was indicated that the MNPs-DOX could be chemically degraded through disulfide-thiol exchange reaction, but remained stable in physiological condition, suggesting a potential application in the tumor microenvironment.
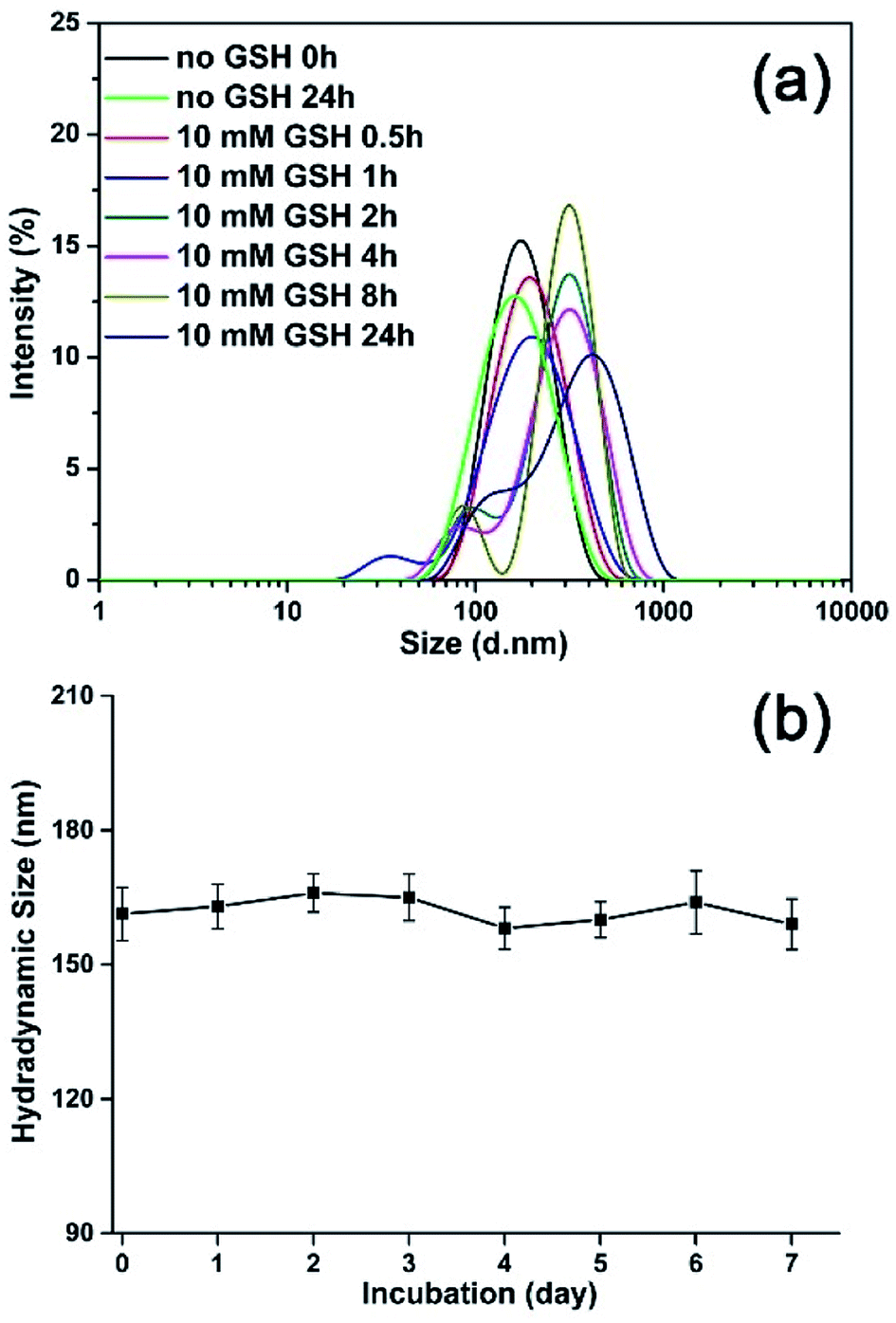 | ||
| Fig. 6 Investigations on the stability of MNPs-DOX: (a) the size change of MNPs-DOX in response to GSH by DLS measurement, and (b) the hydrodynamic size of MNPs-DOX in cell culture media (DMEM with 10% FBS) as a function of time. | ||
3.2 Relaxivity measurement
Generally, the effect of MRI contrast agents can be evaluated based on their r1 and r2 (the longitudinal and transverse relaxivities) that can reflect the ability of the MRI contrast agents to alter T1 and T2. SPIONPs are widely used as a T2 contrast agent, so the transverse relaxivities r2 are calculated through the linear least-squares fitting of 1/relaxation time (s−1) versus the iron concentration (mM Fe) to evaluate the effect of SPIONs based contrast agents. Fig. 7 showed the measurement of the T2 relaxivities for SPIONs and MNPs-DOX. As shown in Fig. 7a, the MNPs-DOX had a larger r2 (126 mM−1 s−1) than SPIONs (103 mM−1 s−1), as indicated by the slopes of the two linear lines at 7 T on an MRI scanner at room temperature. The higher r2 value of MNPs-DOX can be explained by the short distance between the clustered SPIONs inside the MNPs-DOX, which may permit for magnetic coupling between the SPIONs leading to a synergistic increase in r2. Therefore, the as prepared MNPs-DOX can serve as highly efficient T2 contrast agents.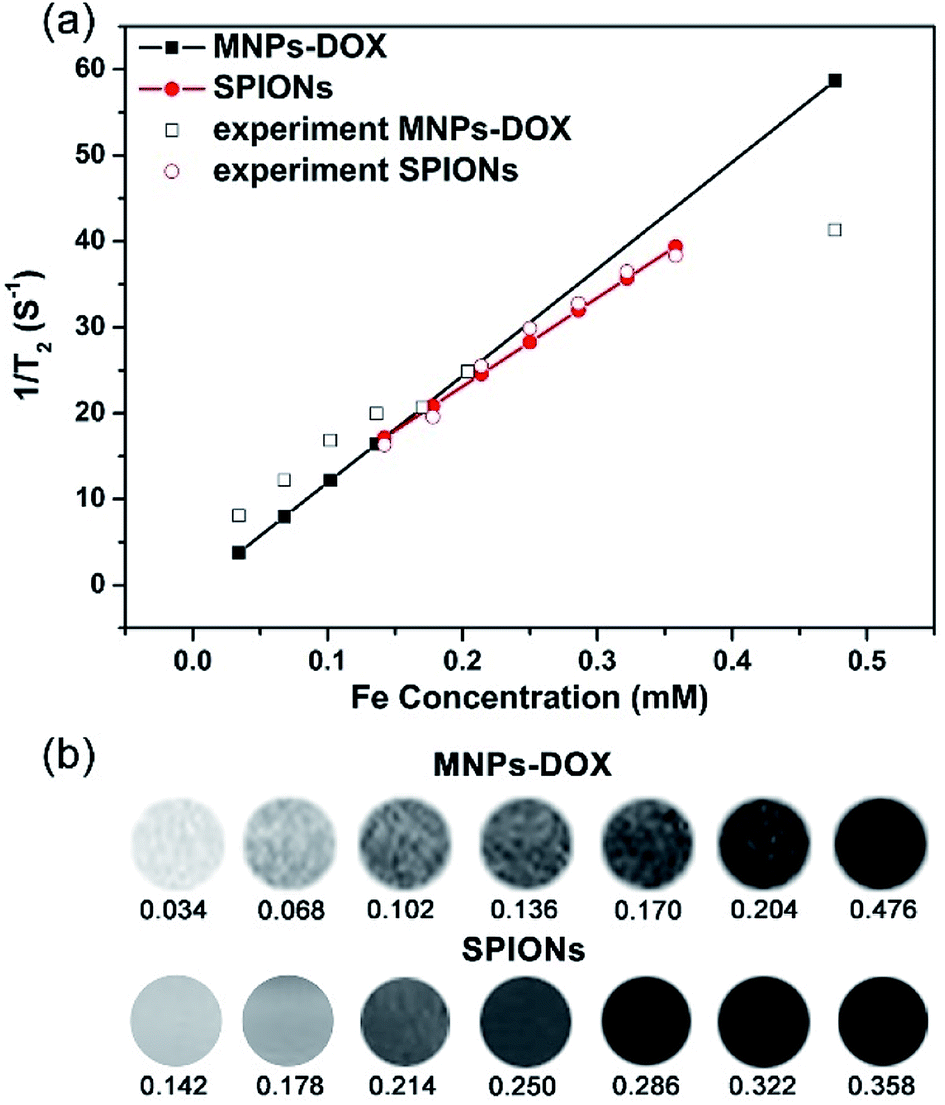 | ||
| Fig. 7 (a) T2 relaxation rates (1/T2, s−1) as a function of iron concentration (mM) for the MNPs-DOX and SPIONs. (b) T2-weighted spin echo MR images of MNPs-DOX and SPIONs at different concentrations. | ||
3.3 Drug release behavior
To investigate the stimuli-triggered drug release of MNPs-DOX, the release experiments were performed under different conditions. The release behavior of the free DOX in PBS (pH 7.4) and the incorporated DOX from MNPs-DOX was monitored under PBS (pH 7.4, 37 °C) with and without 10 mM GSH, under gentle stirring. As shown in Fig. 8, comparing to the burst release of free DOX in the first 12 h, a sustained drug release from MNPs-DOX was observed. For MNPs-DOX, the DOX cumulative release was less than 26% at pH 7.4 for 48 h, indicating that the nanoparticles were remarkably stable with low premature drug release under physiological conditions. On the other hand, the release rate of the DOX from MNPs-DOX was much faster in stimuli conditions, that was, the cumulative release of drug increased to about 60% at pH 7.4 in the presence of 10 mM GSH. The effective drug release could be explained by the fact that following the chemical cleavage of disulfide bonds MNPs-DOX were undergoing a degradation process, and the degradation process could be facilitated in the intracellular reducing environment. When the drug diffusion channels were formed, the drug quickly released from the swelling nanoparticles causing a much higher drug release rate. Overall, the release behavior of the MNPs-DOX suggested that the as prepared theranostic nanoparticle with reduction-sensitivity could be used as a potential drug carrier for cancer therapy in the tumor microenvironment.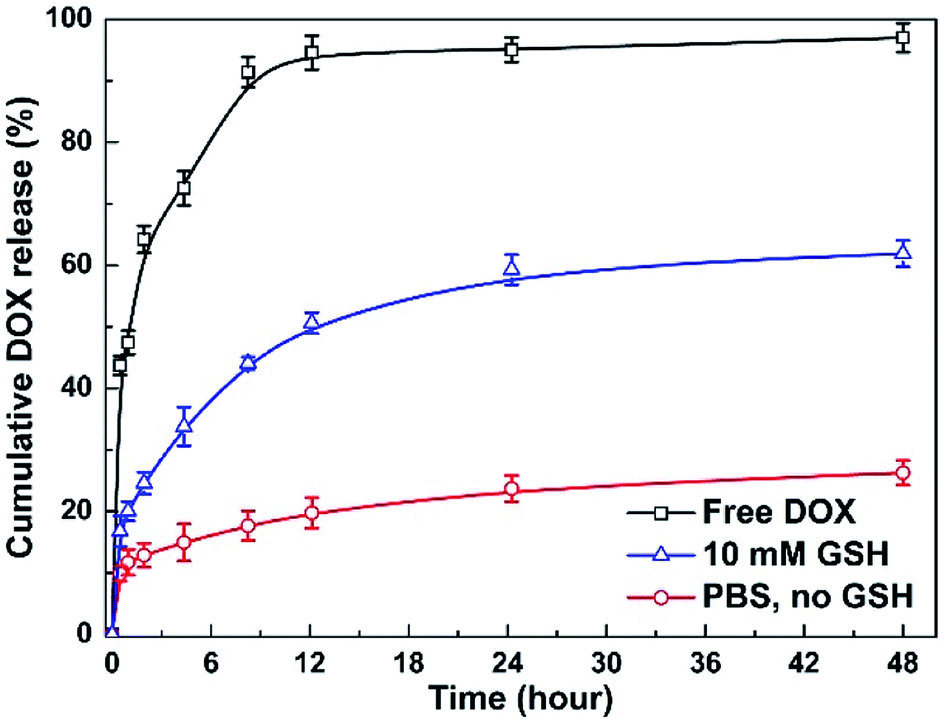 | ||
| Fig. 8 In vitro DOX release profiles of MNPs-DOX and free DOX under different conditions. | ||
3.4 In vitro cell assays
Based on the degradable structure of the nanoparticles, the MNPs-DOX can be degraded in intracellular media because of the presence of GSH in tumor cells. Therefore, the released DOX from the MNPs-DOX system will efficiently kill cancer cells. To confirm this expectation and investigate the killing effect of cancer cell, the in vitro cell assays were performed to evaluate the cellular uptake and intra-cellular drug release of MNPs-DOX by using CLSM technique. Additionally, the cytotoxicity of free DOX, MNPs, and MNPs-DOX against HUVEC (normal cells) and HeLa cells (cancer cells) was investigated by CCK8 assay.To monitor the cellular uptake behavior of the MNPs-DOX for HeLa cells, Fig. 9 showed the CLSM images of the cells incubated for 2 and 48 h with free DOX and MNPs-DOX. Comparing to the free DOX incubated with HeLa cells for 2 h in Fig. 9a, a little weaker DOX fluorescence was observed in the HeLa cells after 2 h incubation with MNPs-DOX in Fig. 9c. It was suggested that only small amount of released DOX from MNPs-DOX entered the HeLa cells because of the slow drug release kinetics of MNPs-DOX. For free DOX, more DOX molecules quickly entered the cells via endocytosis due to the quicker diffuse rate, which was consistent with the results of the in vitro drug release experiments. When HeLa cells were cultured with MNPs-DOX for 48 h, a brighter fluorescence was observed within the cell nuclei in Fig. 9d. It is indicated that more MNPs-DOX entered cells via endocytosis and more DOX was released than that observed at 2 h. Briefly, according to the results of the CLSM images it was indicated that it would take a period of time for MNPs-DOX to be endocytosed to cell and release drug molecules into the cells nuclei, while a quicker endocytosis process was observed for free DOX due to the fast diffuse of drug molecules during passing through the cell membrane. In general, the CLSM experiment confirmed the successfully drug delivery of the MNPs-DOX into cell nuclei, demonstrating the expected result.
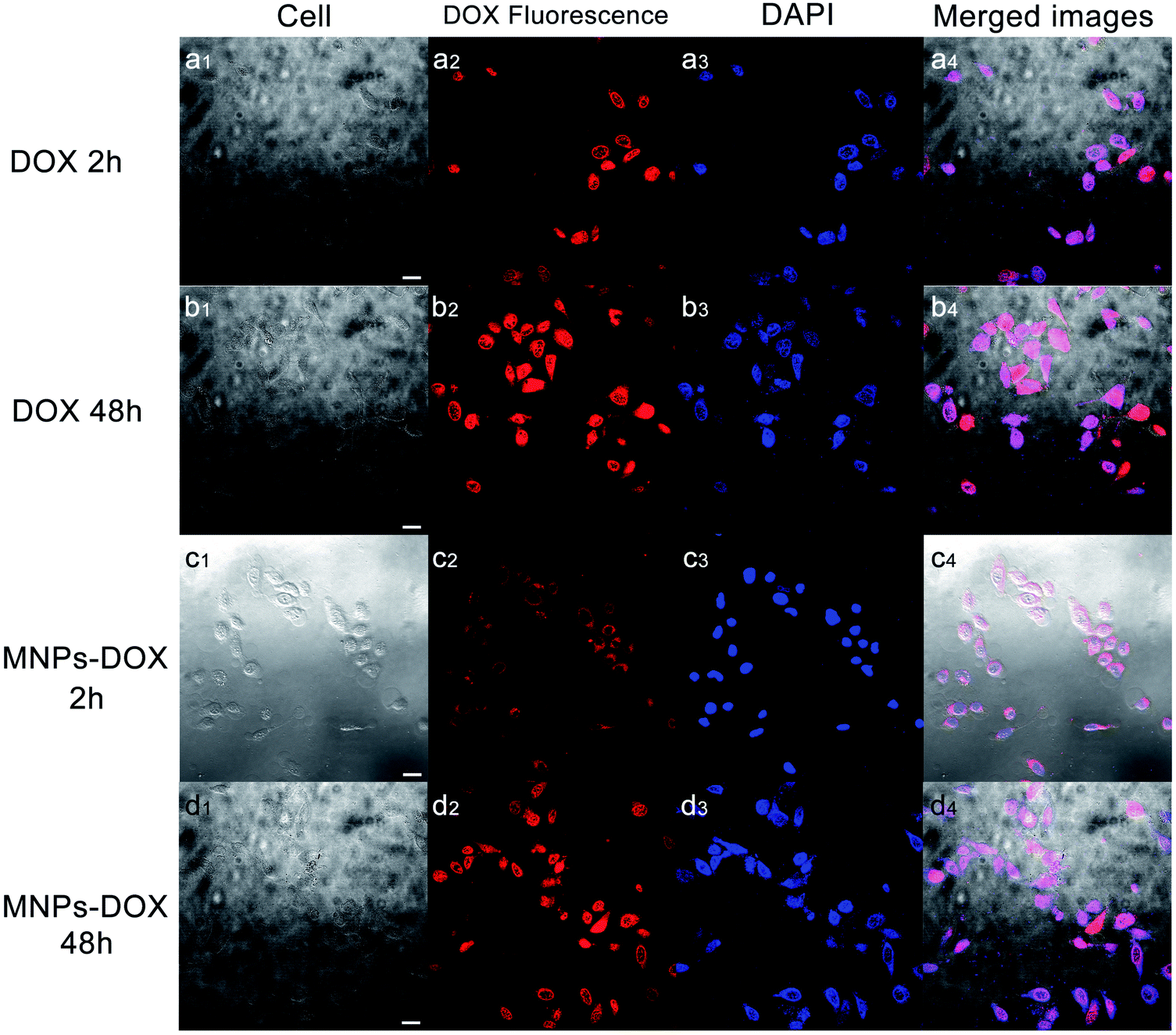 | ||
| Fig. 9 CLSM images of HeLa cells cultured with free DOX over incubation: (a1–a4) 2 h and (b1–b4) 48 h; Cells cultured with MNPs-DOX: (c1–c4) 2 h and (d1–d4) 48 h. (I) the bright field; (II) DOX fluorescence images; (III) stained by DAPI; (IV) merged images. The scale bar corresponds to 25 μm. | ||
The in vitro cytotoxicity tests were also performed for MNPs, MNPs-DOX, and free DOX against HUVEC and HeLa cells by using CCK8 assay. Firstly, the cytotoxicity of MNPs and MNPs-DOX was evaluated against HUVEC cells after 24 h incubation. As shown in Fig. 10a, it was indicated that MNPs and MNPs-DOX showed almost no cytotoxicity against HUVEC cells up to sample concentration of 80 μg mL−1 after 24 h incubation, which could be attributed to the good bio-compatibility of the PAMAM shell. In Fig. 10b and c, when against HeLa cells, the MNPs control showed almost non-cytotoxicity to HeLa cells because no drug was bonded to the nanoparticle. However, the cell viability of HeLa cells was effectively reduced when incubated with MNPs-DOX and free DOX. In additional, it was observed that the cell viability was dose-dependent that high drug concentration caused a remarkable killing effect of cancer cell. The amount of DOX from MNPs-DOX required to achieve IC50 for HeLa cells was 56.9 μg mL−1 at 24 h and 25.3 μg mL−1 at 48 h after incubation, which was higher than the IC50 of free DOX (17.2 μg mL−1 at 24 h, and 5.3 μg mL−1 at 48 h), indicating that MNPs-DOX had a lower killing effect against HeLa cells comparing with the free DOX. It can be explained by that the free DOX has a quicker diffuse rate when passing through cell membrane, while the MNPs-DOX has a slow release kinetics during the drug release process. In general, based on the above experimental results the MNPs-DOX demonstrate good biocompatibility, biodegradability, high drug loading, minimal premature drug release in physiological condition, and effective drug release in response to the tumor microenvironment, which makes the functional nanoparticle a promising candidate in cancer chemotherapeutics. The cellular killing efficiency of the free DOX and MNPs-DOX as a function of time has been performed and shown in Fig. 11. For free DOX, a higher cellular killing efficiency was observed within the first 9.5 h indicating a quicker diffuse rate of DOX molecules through the cell membrane, which was consistent with the results of the in vitro drug release and CLSM experiments. After 9.5 h incubation, the as prepared MNPs-DOX demonstrated a remarkable killing effect of cancer cell (shown in Fig. 11), while the free DOX showed a higher cell viability. It was indicated that more MNPs-DOX entered the cells via endocytosis due to the FA tumor-targeting ligand comparing with the non-targeted DOX molecules, which resulted in a better anti-cancer effect than DOX.
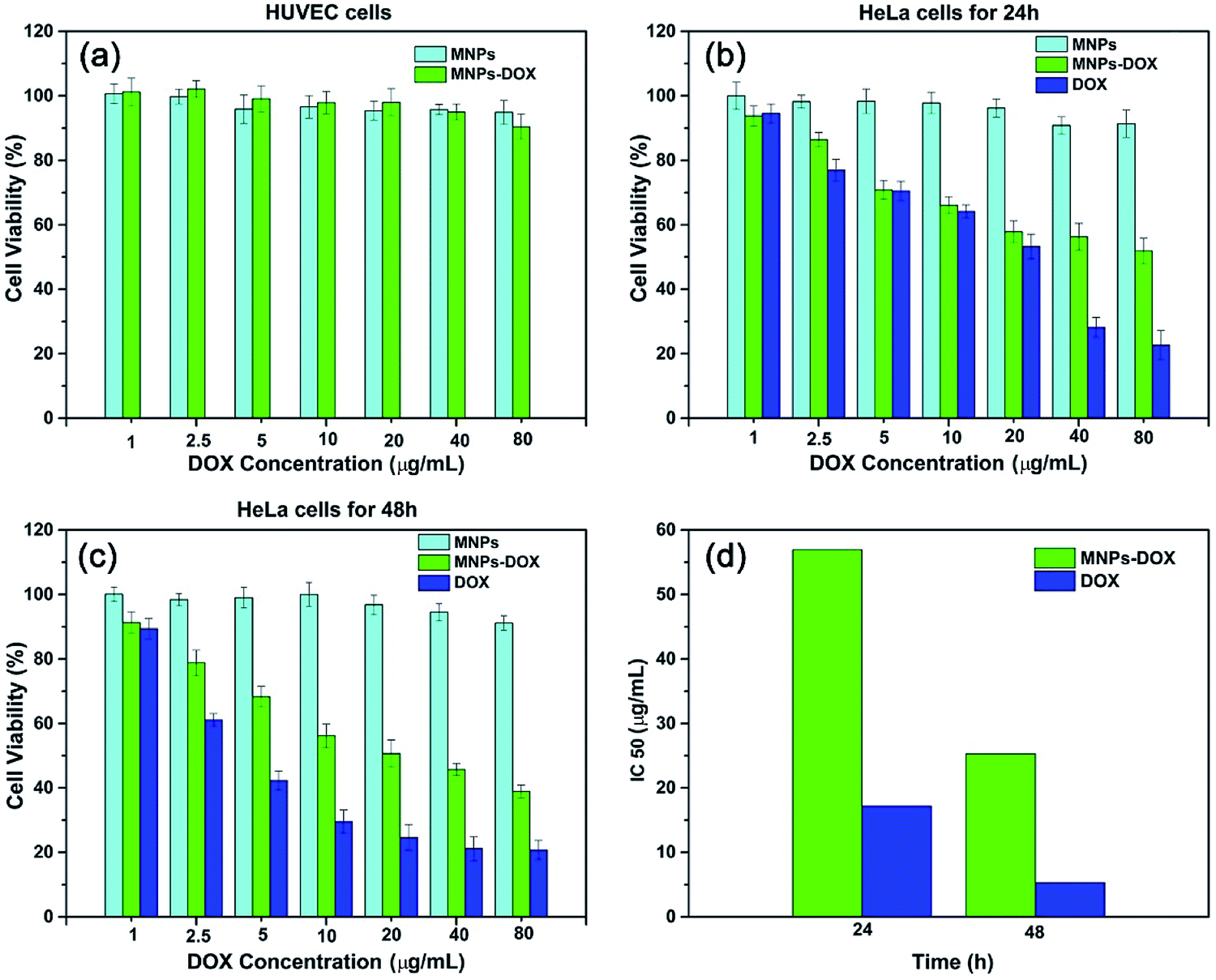 | ||
| Fig. 10 Cell survival assay. (a) Cytotoxicity of MNPs and MNPs-DOX to HUVEC cells following 24 h incubation. HeLa cells cultured with MNPs, MNPs-DOX, and free DOX for 24 h (b) and 48 h (c). (d) Graphic presentation for IC50 values of free MNPs-DOX and DOX at 24 and 28 h after treatment of HeLa cells. | ||
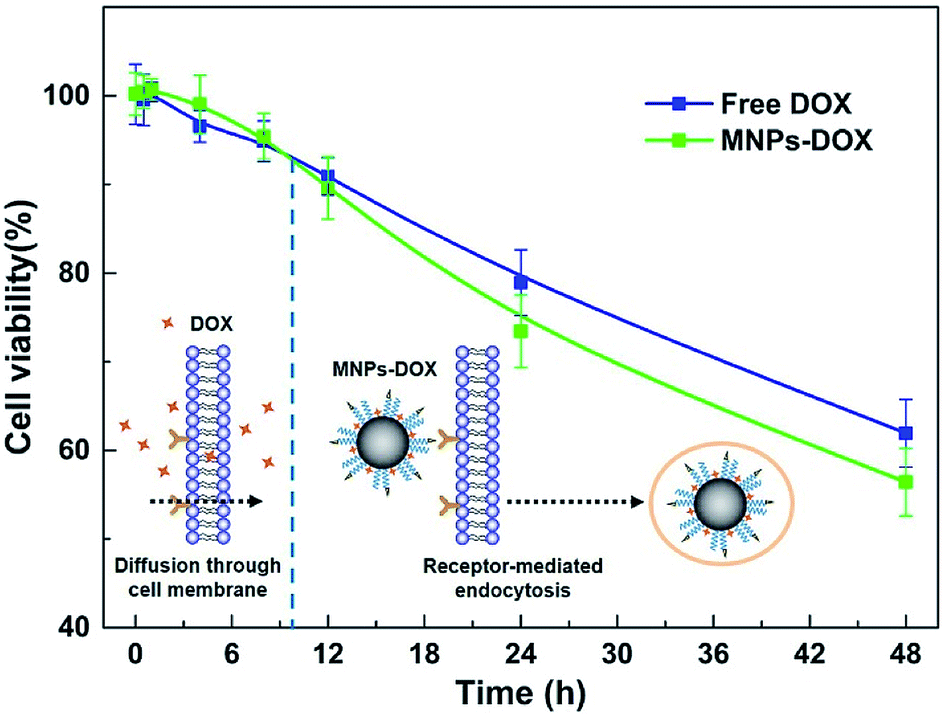 | ||
| Fig. 11 The cellular killing efficiency as a function of time for MNPs-DOX and free DOX. | ||
4. Conclusions
Briefly, a multi-functional magnetic nanoparticle was prepared via a simple assembly process for anti-cancer drug delivery and MR imaging detection. The MNPs-DOX exhibited minimized premature drug release in physiological condition but showed a remarkable killing effect against cancer cell. Comparing with non-targeted DOX molecules, a better anti-cancer effect was observed due to the FA tumor-targeting ligand. Additionally, the as prepared MNPs-DOX demonstrated enhanced T2 contrast comparing with the SPIONs. More importantly, the nanoparticle system with superparamagnetism, cell-targeting, excellent stability, good biocompatibility, and reduction-sensitivity will provide us a prosing platform via a simple preparation process that can incorporate various therapeutic drugs and diagnostic particles, which can be used as a promising theranostic candidate for targeted anticancer drug delivery and imaging diagnosis in clinic application.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Sichuan Science and Technology Program (18YYJC0265), and the Research Project of Sichuan Provincial Department of Education (16ZA0284), and the Functional Polymer Innovation Team Project, Southwest University for Nationalities (14CXTD04).References
- P. B. Santhosh and N. P. Ulrih, Cancer Lett., 2013, 336, 8–17 CrossRef CAS PubMed.
- G. Huang, H. Chen, Y. Dong, X. Luo, H. Yu, Z. Moore, E. V. Bey, D. A. Boothman and J. Gao, Theranostics, 2013, 3, 116–126 CrossRef CAS PubMed.
- M. K. Yu, D. Kim, I. H. Lee, J. S. So, Y. Y. Jeong and S. Jon, Small, 2011, 7, 2241–2249 CrossRef CAS PubMed.
- A. Singh and S. K. Sahoo, Drug Discovery Today, 2014, 19, 474–481 CrossRef CAS PubMed.
- J. Chen, M. Shi, P. Liu, A. Ko, W. Zhong, W. Liao and M. M. Xing, Biomaterials, 2014, 35, 1240–1248 CrossRef CAS PubMed.
- C. Fang, F. M. Kievit, O. Veiseh, Z. R. Stephen, T. Wang, D. Lee, R. G. Ellenbogen and M. Zhang, J. Controlled Release, 2012, 162, 233–241 CrossRef CAS PubMed.
- Y. Huang, K. Mao, B. Zhang and Y. Zhang, Mater. Sci. Eng., C, 2017, 70, 763–771 CrossRef CAS PubMed.
- I. Karimzadeh, M. Aghazadeh, M. R. Ganjali, P. Norouzi, S. Shirvani-Arani, T. Doroudi, P. H. Kolivand, S. A. Marashi and D. Gharailou, Mater. Lett., 2016, 179, 5–8 CrossRef CAS.
- L. Li, L. Shang, K. Chen, Q. Wang, J. Luo, Q. Zhou and J. Lin, J. Nanosci. Nanotechnol., 2017, 17, 4532–4541 CrossRef CAS.
- Y. Liu, G. Feng, X. Guo, Z. Wu, Y. Chen, W. Xiang, J. S. Li and B. Zhong, J. Alloys Compd., 2018, 748, 100–110 CrossRef CAS.
- K. Zhu, Z. Deng, G. Liu, J. Hu and S. Liu, Macromolecules, 2017, 50, 1113–1125 CrossRef CAS.
- L. M. Huang, L. D. Li, L. Shang, Q. H. Zhou and J. Lin, React. Funct. Polym., 2016, 107, 28–34 CrossRef CAS.
- Y. P. Liu and J. S. Li, J. Bioresour. Bioprod., 2016, 1, 7–17 Search PubMed.
- Y. Shi, C. F. V. Nostrum and W. E. Hennink, ACS Biomater. Sci. Eng., 2015, 1, 393–404 CrossRef CAS.
- W. J. Stark, P. R. Stoessel, W. Wohlleben and A. Hafner, Chem. Soc. Rev., 2015, 44, 5793–5805 RSC.
- Q. Hu, C. J. Rijcken, G. E. Van, P. Brundel, H. Kostkova, T. Etrych, B. Weber, M. Barz, F. Kiessling, J. Prakash, G. Storm, W. E. Hennink and T. Lammers, J. Controlled Release, 2016, 244, 314–325 CrossRef CAS PubMed.
- C. Yao, J. Liu, X. Wu, Z. Tai, Y. Gao, Q. Zhu, J. Li, L. Zhang, C. Hu, F. Gu, J. Gao and S. Gao, J. Controlled Release, 2016, 232, 203–214 CrossRef CAS PubMed.
- Q. Hu, C. J. Rijcken, R. Bansal, W. E. Hennink, G. Storm and J. Prakash, Biomaterials, 2015, 53, 370–378 CrossRef CAS PubMed.
- Q. Liu, L. Song, S. Chen, J. Gao, P. Zhao and J. Du, Biomaterials, 2017, 114, 23–33 CrossRef CAS PubMed.
- K. Zhu, Z. Deng, G. Liu, J. Hu and S. Liu, Macromolecules, 2017, 50, 1113–1125 CrossRef CAS.
- Q. H. Zhou, J. Lin, L. D. Li and L. Shang, Colloid Polym. Sci., 2015, 293, 2291–2300 CrossRef CAS.
- K. Yang, L. Feng and Z. Liu, Adv. Drug Delivery Rev., 2016, 105, 228–241 CrossRef CAS PubMed.
- Y. Zhang, D. Yang, H. Chen, W. Q. Lim, F. S. Z. Phua, G. An, P. Yang and Y. Zhao, Biomaterials, 2018, 163, 14–24 CrossRef CAS PubMed.
- T. Kang, F. Li, S. Baik, W. Shao, D. Ling and T. Hyeon, Biomaterials, 2017, 136, 98–114 CrossRef CAS PubMed.
- J. Wang, B. Zhang, L. Wang, M. Wang and F. Gao, Mater. Sci. Eng., C, 2015, 48, 416–423 CrossRef CAS PubMed.
- X. Ma, A. Gong, B. Chen, J. Zheng, T. Chen, Z. Shen and A. Wu, Colloids Surf., B, 2015, 126, 44–49 CrossRef CAS PubMed.
- G. Kandasamy and D. Maity, Int. J. Pharm., 2015, 496, 191–218 CrossRef CAS PubMed.
- G. Marcelo, A. Munozbonilla, J. Rodriguezhernandez and M. Fernandezgarcia, Polym. Chem., 2013, 4, 558–567 RSC.
- V. A. Li, d. A. D. Jimenez, M. Henriksen-Lacey, G. Giammona, M. Licciardi and L. M. Liz-Marzan, J. Mater. Chem. B, 2016, 4, 1150–1155 RSC.
- A. Gulzar, S. Gai, P. Yang, C. Li, M. B. Ansari and J. Lin, J. Mater. Chem. B, 2015, 3, 8599–8622 RSC.
- C. Fang, F. M. Kievit, O. Veiseh, Z. R. Stephen, T. Wang, D. Lee, R. G. Ellenbogen and M. Zhang, J. Controlled Release, 2012, 162, 233–241 CrossRef CAS PubMed.
- L. Huang, L. Ao, W. Wang, D. Hu, Z. Sheng and W. Su, Chem. Commun., 2015, 51, 3923–3926 RSC.
- B. H. McDonagh, G. Singh, S. Hak, S. Bandyopadhyay, I. L. Augestad, D. Peddis, I. Sandvig, A. Sandvig and W. R. Glomm, Small, 2016, 12, 301–306 CrossRef CAS PubMed.
- A. R. Sasikala, A. Ghavaminejad, A. R. Unnithan, R. G. Thomas, M. Moon, Y. Y. Jeong, C. H. Park and C. S. Kim, Nanoscale, 2015, 7, 18119–18128 RSC.
- L. Shang, Q. Wang, K. Chen, J. Qu, Q. H. Zhou, J. B. Luo and J. Lin, RSC Adv., 2017, 7, 47715–47725 RSC.
- J. Qu, Q. Y. Wang, K. L. Chen, J. B. Luo, Q. H. Zhou and J. Lin, Colloids Surf., B, 2018, 170, 373–381 CrossRef CAS PubMed.
- L. Shang, Q. Y. Wang, K. L. Chen, J. Qu, J. Lin, J. B. Luo and Q. H. Zhou, J. Bioresour. Bioprod., 2017, 2, 1–3 Search PubMed.
Footnote |
| † The authors Jing Qu and Zhijie Tian contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |