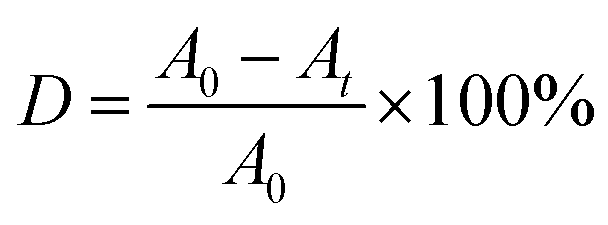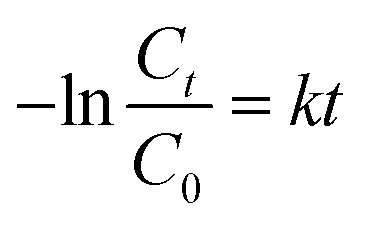DOI:
10.1039/C8RA06778C
(Paper)
RSC Adv., 2018,
8, 36745-36753
Preparation and photocatalytic application of a S, Nd double doped nano-TiO2 photocatalyst†
Received
13th August 2018
, Accepted 22nd October 2018
First published on 31st October 2018
Abstract
Nowadays, water pollution is getting more and more severe in society, and recently the rational use of photocatalytic technology to treat sewage has become a hot spot for research. Because of the low-cost and environmental friendliness of nano-TiO2, it has attracted widespread attention. In this paper, we dope sulfur and neodymium into nano-titanium dioxide via a sol–gel method. The synthesized S, Nd-codoped-TiO2 was characterized via transmission electron microscopy, scanning electron microscopy, X-ray diffraction, X-ray photoelectron spectroscopy, EDS and N2 adsorption–desorption isotherms. Then the real-life dye reactive red (X-3B) was used as the target degradant to compare the levels of practicality of single-doped and double-doped photocatalysts. The results showed that the photocatalytic activity of S, Nd-codoped-TiO2 was significantly higher than that of Nd–TiO2. The double-doped photocatalyst was transformed from anatase to rutile, and the bandgap was reduced considerably. The responding ability to visible light also increased, so S, Nd-codoped-TiO2 has obvious advantages and has better degradation efficiency for the target degradation products. Under xenon lamp irradiation and pH = 4 conditions, the degradation of reactive red using the new catalyst reached 93.2%. The new catalyst has high practicality and also indicates a new direction for wastewater treatment.
1. Introduction
With the rapid development of the dying and weaving industries, dye wastewater has become one of the most challenging problems for human beings. Traditional sewage treatment mainly includes physical degradation, chemical degradation, and biodegradation. However, these methods are time-consuming and consume much energy. The discovery of photocatalytic technology has completely altered the dilemma of disposing of dye wastewater and has made a difference in the protection of human water resources.1–8
Since the innovation of photocatalytic technology in 1972, public interest in photocatalysis has continued unabated.9 With the deepening of research, photocatalysis technology can not only produce hydrogen, but also effectively degrade water pollution. This brings benefits to societies with serious water pollution. In photocatalyst experiments applied to water pollution degradation, TiO2 is the most widely used compound. This is because TiO2 has the advantages of low cost, low toxicity, and excellent chemical stability.10–14 When light is incident on the semiconductor TiO2, if the energy of the photon generated by the light is higher than the band gap level of the semiconductor TiO2, an electron is excited from the valence band to the conduction band. However, the photocatalytic performance of TiO2 is poor due to the extremely short recombination time of photogenerated electrons and electron holes. Therefore, prolonging the photogenerated electron and hole recombination time has become a hot spot in research into TiO2 photocatalysts. Due to the wide bandgap of TiO2 (Eg = 3.2 eV), it can only be excited by ultraviolet radiation at a wavelength of 387 nm; it can also agglomerate easily with poor dispersion and has a low utilization rate for sunlight. Therefore, researchers have adopted modified or retouched TiO2 to improve defects and improve photocatalytic efficiency.15–20 In 2017, Balter Trujillo-Navarrete et al. explained the reasons for lattice distortion caused by Nd–TiO2, which affects the recombination rate of photoelectrons and holes and improves photocatalytic performance.21 In 2018, Thirupathi Boningari used a one-step method to synthesize S–TiO2 and confirmed that S doping could effectively reduce the optical band gap of TiO2. The photo-responsive wavelength of TiO2 can not only be extended beyond the visible region, but also the absorption of light in the ultraviolet region can be maintained.22 However, many experiments have confirmed that the single doping of TiO2 does not completely increase the utilization of titanium dioxide. Therefore, double-doped photocatalysts and multi-doped photocatalysts have become hot spots for research.23–25 A large number of studies have found that the rare earth element neodymium is more active in terms of chemical properties, cheaper and more readily available, and can be well combined with titanium dioxide, so it has attracted the attention of scholars. The contribution of non-metallic sulfur to the light absorption capacity of titanium dioxide is well known, so combining the advantages of both would be a very meaningful exercise.
Based on these analyses, we first prepared a single-doped photocatalyst Nd–TiO2 with a low electron–hole recombination rate using a sol–gel method. Then, adopting the same process, Nd and S are co-doped into the TiO2 structure to obtain a bifunctional catalyst with a low electron recombination rate and wide photoresponse range. The S element is doped while controlling the Nd content. The purpose of doing this is to obtain photocatalysts with two advantages. Degradation using Nd–TiO2 and S, Nd-codoped-TiO2 photocatalysts with diverse contents were investigated using active red as the target degradation product. The results are positive. The double-doped photocatalyst has significantly higher photodegradation capability than the single-doped photocatalyst, and the double-doped photocatalyst has a broader light response range. This research has provided valuable theoretical exploration of and technical support for the large-scale treatment of dye wastewater.
2. Experimental
2.1 Reagents and apparatus
Thiourea, tetrabutyl titanate and neodymium nitrate were purchased from Tianjin Fuchen Reagent Factory of China. Reactive red was a product of Tianjin Kaitong Reagent Factory of China. All reagents were commercially available and were not subjected to secondary purification. Infrared spectroscopy was performed using a Perkin Elmer Nicolet-50X FTIR spectrometer (JEOL Ltd., Japan). X-ray diffraction analysis was performed using a Dmax-IIIC X-ray diffractometer (JEOL Ltd., Japan). Structural analysis was performed using a S-4300 field emission scanning electron microscope and a H7650 transmission electron microscope (HITACHI Ltd., Japan). UV-visible spectra of the samples were recorded on a UV-visible DRS instrument (DRS; UV-2450, Shimadzu Co.) using BaSO4 as the standard reference. Adsorption/desorption analysis was performed using an Autosorb 1 physical adsorption–desorption instrument (QUANTACHROME Ltd., America). XRD analysis was performed using a D8 Focus X-ray powder diffractometer (German Bruker-Axs). The sample was fluorescently characterized using an F-4500 fluorescence spectrophotometer, manufactured by Hitachi, Japan. All pH values were measured using a pH indicator manufactured by Beijing Sartorius.
2.2 The preparation of S, Nd–TiO2
Tetrabutyl titanate (25 mL) and glacial acetic acid (4 mL) were dissolved in 34 mL of anhydrous ethanol, and this mixture was stirred continuously for 30 minutes. The pH of the solution was then adjusted using dilute hydrochloric acid solution. The pH of the mixture was adjusted to 3. The resulting mixture was placed in an ultrasonic bath and irradiated for 10 minutes under a sound wave of 40 kHz to make the mixing more uniform. Four portions of weighed neodymium nitrate (0.166 g each) were added to distilled water (4 mL) and anhydrous ethanol (17 mL), then thiourea (0 g, 0.170 g, 0.340 g or 0.51 g) was added. Then the pH was adjusted to 3 with an aqueous solution of hydrochloric acid. The mixture was sonicated at the same intensity. Under magnetic stirring, the neodymium nitrate mixture was added dropwise to the tetrabutyl titanate solution at 1–2 drops per second. After magnetic stirring for 10 h, it was let stand for 10 h. The mixture was then placed in a drying oven at 70 °C and dried for 2 h, then it was moved into a muffle furnace and calcined for 2 h (450 °C). Based on the above method, we synthesized photocatalysts with different S content values.
2.3 Photocatalytic activity tests
The photocatalytic activities of the S, Nd–TiO2 nanocomposites were evaluated by measuring the absorbance of reactive red solution added to the photocatalysts under sunlight irradiation. In the experiment, the light source was condensed under a cylindrical glass cannula, and each of the outer tubes contained 50 mL of reactive red solution with an initial concentration of 10 mg L−1. 15 mg of photocatalyst was added to each test tube. The solid–liquid mixture was magnetically stirred in the dark for 30 min to bring the photocatalyst to adsorption–desorption equilibrium. Then the mixture was placed vertically under a 300 W xenon lamp equipped with an optical transmission filter (λ = 365 nm). 5 mL of the mixture was centrifuged at intervals of 20 min to separate the solid photocatalyst from the solution. The absorbance was measured using a spectrophotometer (TU-1901) at the maximum absorption wavelength, 540 nm, of the reactive red solution, and the decolorization rate was calculated. The solution decolorization rate (D) was calculated via the following formula:| |
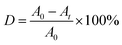 | (1) |
where A0 represents the absorbance of the reactive red solution before illumination, and At represents the absorbance of the reactive red solution after irradiation.
3. Results & discussion
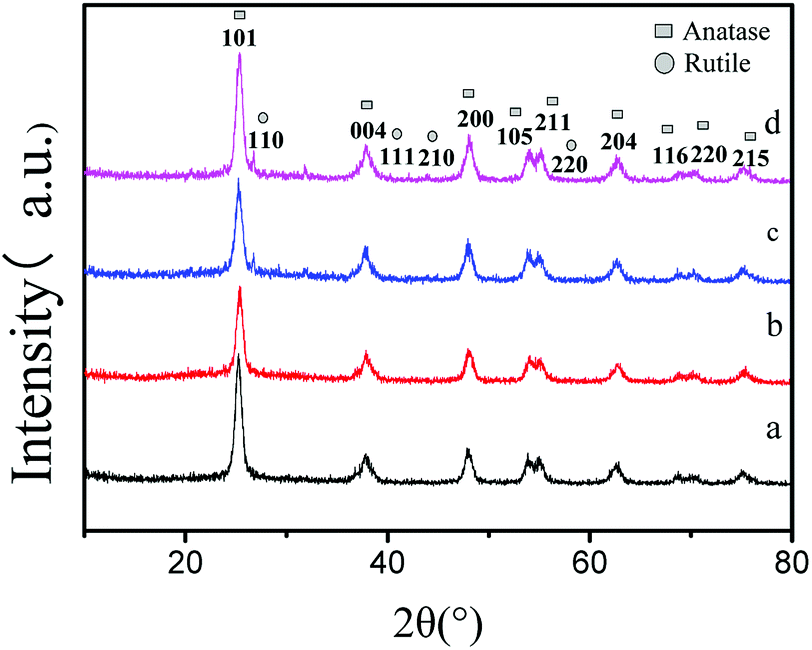
X-ray diffraction patterns of the double-hybrid photocatalyst S, Nd–TiO2 with different sulfur content are shown in Fig. 1. The S, Nd–TiO2 samples show diffraction peaks at 2θ = 25.2°, 38.0°, 48.0°, 54.3°, 55.3°, 63.1°, 68.7°, 70.1°, and 74.9°, corresponding to the (101), (004), (200), (105), (211), (204), (116), (220), and (215) planes of anatase titania. These were confirmed to be from anatase titanium dioxide by comparison with JCPDS card #84-1286. In addition, the peaks at 2θ = 27.0°, 41.0°, 43.8°, and 56.6° can be ascribed to the (110), (111), (210), and (220) planes of a rutile titania structure. These were confirmed to be from rutile titanium dioxide by comparison with JCPDS card #75-1753. Fig. 1a shows the Nd–TiO2 photocatalyst. This only shows anatase TiO2 characteristics. Fig. 1b–d are double doped photocatalysts with different S doping amounts. The photocatalyst having a sulfur doping amount of 2% exhibited only anatase characteristics. The photocatalysts having sulfur doping amounts of 4% and 6% exhibited mixed crystal forms. In the experiment, we observed that with an increase in the amount of non-metallic sulfur doping, anatase type titanium dioxide gradually changed to rutile type. This may be due to the combined action of sulfur and neodymium to convert the photocatalyst to high stability rutile TiO2. Although the doping of Nd can inhibit the crystalline phase transition of nano-TiO2 and increase its thermal stability, an increase in the sulfur content will weaken the effects of neodymium. Because the band gap of the rutile phase is smaller than the forbidden bandwidth of the anatase phase, the absorption spectrum is red-shifted, and the response to visible light is enhanced. Therefore, the double-doped photocatalyst will have significantly higher degradability than the single-doped photocatalyst. The average grain size was calculated using the Scherrer formula: D = kλ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ (D is the average grain size; k is a constant of 0.89; λ is the X-ray wavelength, equal to 0.154056 nm; β is the full width at half maximum; and θ is the diffraction angle). We can estimate the diameter of Nd–TiO2 and S, Nd–TiO2 particles. The average particle diameter of Nd–TiO2 was 13.18 nm. 2% S, Nd–TiO2 has a particle diameter of 17.66 nm, 4% S, Nd–TiO2 has a particle diameter of 18.11 nm, and 6% S, Nd–TiO2 has a particle diameter of 18.50 nm. It can be seen that Nd-doped TiO2 shows reduced particle size, and the particle size of double-doped TiO2 is less affected.
cosθ (D is the average grain size; k is a constant of 0.89; λ is the X-ray wavelength, equal to 0.154056 nm; β is the full width at half maximum; and θ is the diffraction angle). We can estimate the diameter of Nd–TiO2 and S, Nd–TiO2 particles. The average particle diameter of Nd–TiO2 was 13.18 nm. 2% S, Nd–TiO2 has a particle diameter of 17.66 nm, 4% S, Nd–TiO2 has a particle diameter of 18.11 nm, and 6% S, Nd–TiO2 has a particle diameter of 18.50 nm. It can be seen that Nd-doped TiO2 shows reduced particle size, and the particle size of double-doped TiO2 is less affected.
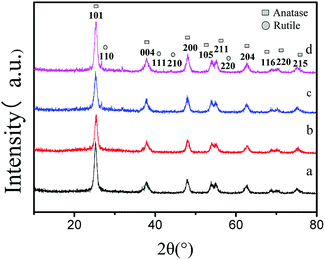 |
| | Fig. 1 XRD patterns of (a) Nd–TiO2, (b) 2% S, Nd–TiO2, (c) 4% S, Nd–TiO2, and (d) 6% S, Nd–TiO2. | |
The particle size of pure TiO2 is around 50 nm, but the doping of Nd leads to a decrease in TiO2 grain size. An SEM image in Fig. 2 shows as-prepared Nd–TiO2 (Fig. 2a) with a size of about 15 nm. An SEM image in Fig. 2 also shows as-prepared S, Nd–TiO2 (Fig. 2b) with a size of about 20 nm. The doped TiO2 is spherical, which effectively reduces the issue of agglomeration and improves the dispersibility. The results show that both single and double doping can better improve the defects of TiO2. Double-doped photocatalyst particles are significantly smaller in diameter than single-doped examples, which provides a basis for the better adsorption of dye molecules.
 |
| | Fig. 2 SEM images of (a) Nd–TiO2, and (b) S, Nd–TiO2. | |
The TEM images in Fig. 3 show aggregated particles of Nd–TiO2 (Fig. 3a) and S, Nd–TiO2 (Fig. 3b–d), with particle sizes of 20 nm. In the Nd–TiO2 and S, Nd-codoped-TiO2 photocatalysts, the black Nd is more homogeneously dispersed in the TiO2, which obviously improves the defect of TiO2 agglomerating easily. Fig. 3b–d show double doped photocatalysts with different S concentrations. In Fig. 3c, the 4% S doped double doped photocatalyst disperses more evenly. Compared with the other photocatalysts, the doping of Nd and S has less effect on TiO2, and all crystal grains are mainly spherical. The gap between crystal grains is homogeneous and the particle diameter is about 20 nm.
 |
| | Fig. 3 TEM images of (a) Nd–TiO2, (b) 2% S, Nd–TiO2, (c) 4% S, Nd–TiO2, and (d) 6% S, Nd–TiO2. | |
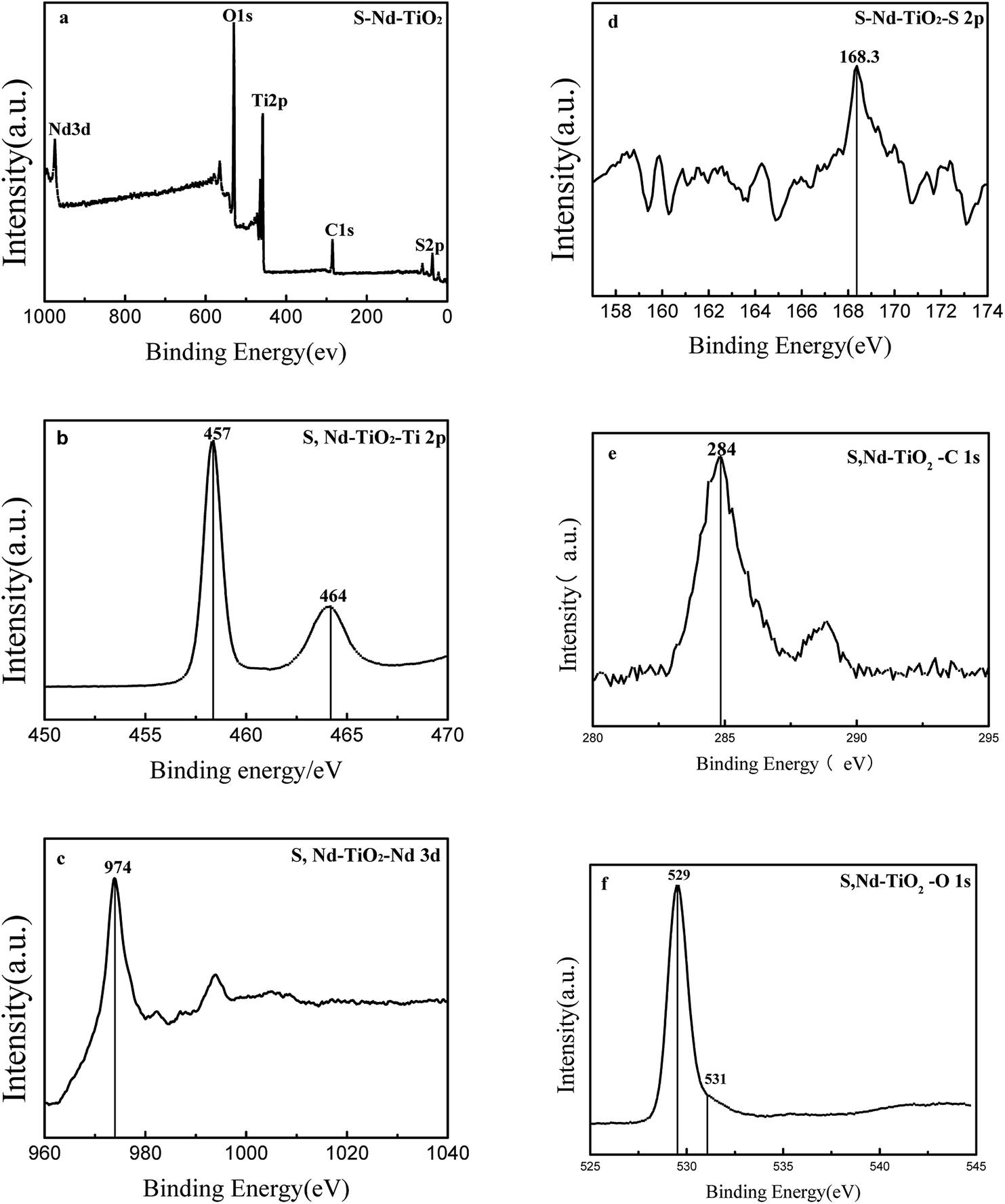
Fig. 4 shows XPS analysis of the S, Nd–TiO2 photocatalyst. Since the photocatalytic activity of TiO2 is affected by the type and content of doping elements, the chemical composition of S, Nd–TiO2 is analyzed via XPS spectroscopy to detect whether S and Nd are co-doped into the TiO2 lattice. The XPS spectrum shows that the photocatalyst S, Nd–TiO2 is mainly composed of five elements: S, Nd, Ti, O and C. The Ti and O elements are derived from nano-titanium dioxide, the S and Nd elements are derived from the doping solution, and C is derived from organic precursor calcination residue. The binding energies of the Ti 2p1/2 peak and the Ti 2p3/2 peak are 464 eV and 457 eV, respectively; they differ by 7 eV, indicating that Ti is mainly present in the form of Ti4 +. The Ti 2p3/2 binding energy peak of pure TiO2 is 459 eV. This indicates that the doping of TiO2 by the two elements S and Nd causes the chemical environment of Ti to change, resulting in a chemical shift. 531 eV is a characteristic peak of a surface hydroxyl oxygen. This exists in the form of an Nd–O bond or Nd2O3 at 529 eV. The metal Nd has a large radius and is difficult to dope into TiO2. Regarding the doping of sulfur, S 2p has a peak at 168 eV, indicating the doping of S in the TiO2 lattice, possibly in the form of O–Ti–S. This is why S-doped TiO2 has a visible light response. For S, Nd–TiO2 particles, the binding energy of Nd 3d is located at 974 eV. In the S, Nd–TiO2 photocatalyst, the Ti content is 19.53%, the O content is 53.01%, the Nd content is 1.08%, the S content is 0.12%, and the C content is 26.25%. Of these, carbon is mainly derived from the calcination of the photocatalyst, and the carbon content is too high due to the XPS analysis background. Therefore, we have reason to believe that Nd and S are successfully doped into the TiO2 lattice.
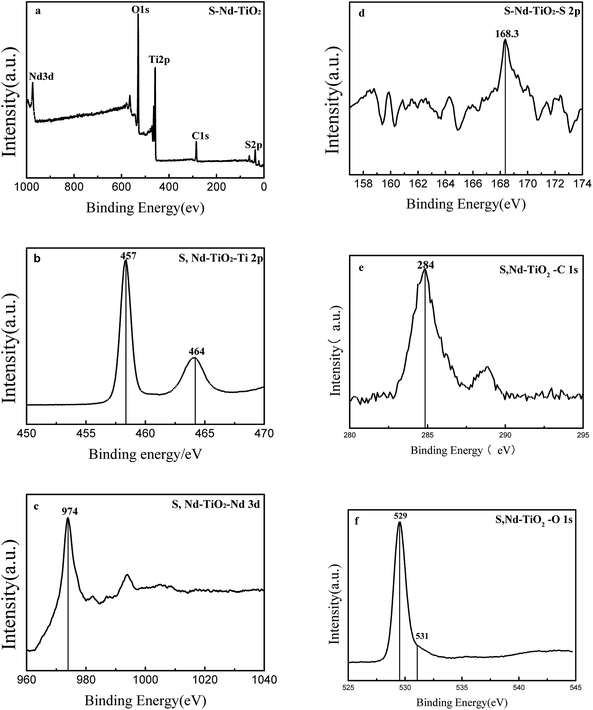 |
| | Fig. 4 XPS spectra of (a) S, Nd–TiO2: (b) the Ti 2p band, (c) the Nd 3d band, (d) the S 2p band, (e) the C 1s band, and (f) the O 1s band. | |
Fig. 5 shows EDS spectra of the S, Nd-codoped-TiO2 photocatalyst. Analyzing these, the four elements Ti, O, Nd and S are derived from the preparation of the photocatalyst, and the C element is derived from the calcination of the precursor. The results show that the element content is in the order of Ti > C > O > Nd > S, from high to low. It can be found from the test that in the double-doped photocatalyst, the content of the sulfur element is significantly lower than that of the doping amount, and the content of the Nd element is level with the doping amount. This may be because Nd3+ is difficult to insert into the TiO2 crystal lattice, and S6+ can replace Ti4+ in the TiO2 crystal lattice, so the S element content detected by EDS and XPS is low.
 |
| | Fig. 5 EDS images of S, Nd–TiO2. | |
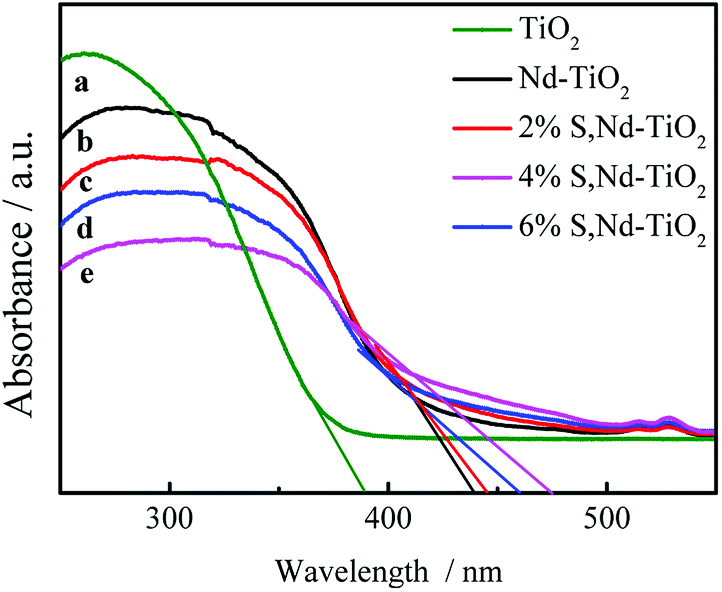
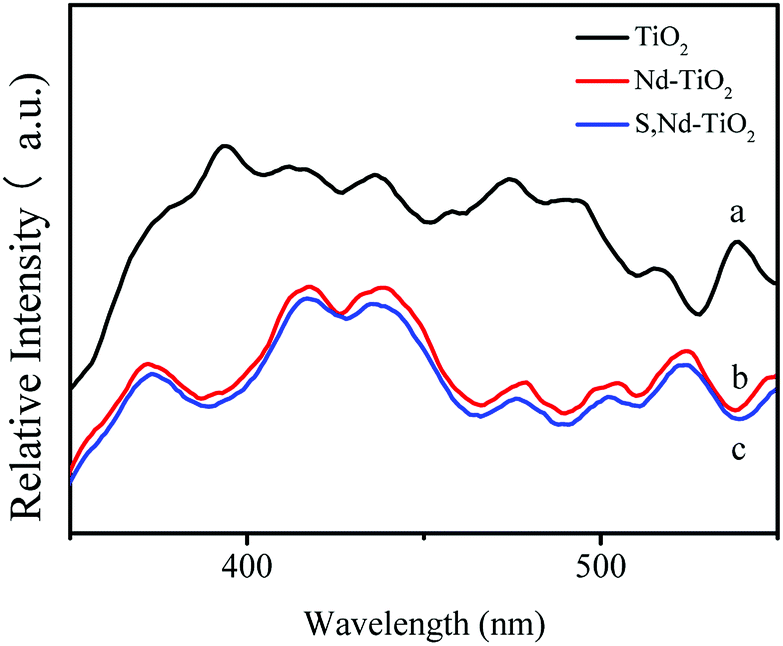
DRS absorption spectra provided information about the wavelength region in which the catalyst absorbs light (Fig. 6). In the ultraviolet region, TiO2 has the largest light absorption capabilities, which are significantly higher than those of the double-doped photocatalyst. The synthesized S, Nd–TiO2 composites exhibited significantly stronger absorption compared to TiO2 in the visible region. The synthesized 4% S, Nd–TiO2 composite exhibited a clear absorption edge at approximately 476 nm in the visible light region, and a band gap energy of 2.61 eV was estimated via the equation: λg = 1240/Eg. Similarly, the forbidden band width of pure TiO2 is 3.16 eV. In the infrared region, the four photocatalysts exhibited good light absorption capacity, and S, Nd double-doped photocatalysts showed significantly higher values than the single-doped photocatalyst. Pure TiO2 can only absorb ultraviolet light, so the peak in the infrared region is caused by Nd doping. With an increase in S content, the peak increases correspondingly, indicating that S doping can promote the absorption of visible light by the photocatalyst. The experimental results show that the TiO2 and Nd–TiO2 calcined samples are white, while for the S, Nd–TiO2 samples, as the S content increases, the light yellow color gradually deepens, which is consistent with corresponding literature reports.26,27
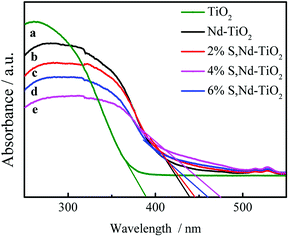 |
| | Fig. 6 DRS spectra of single and double-doped photocatalysts: (a) TiO2; (b) Nd–TiO2; (c) 2% S, Nd–TiO2; (d) 6% S, Nd–TiO2; and (e) 4% S, Nd–TiO2. | |
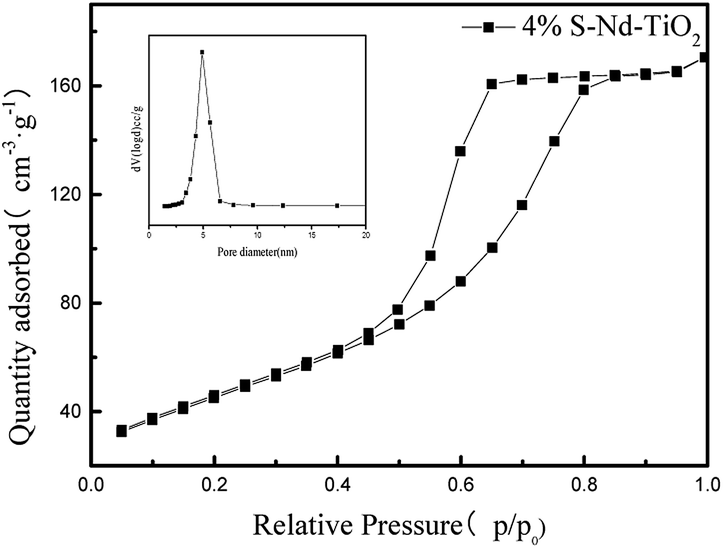
The nitrogen adsorption and desorption isotherms in Fig. 7 were used to characterize the structure of S, Nd–TiO2. It shows a type V isotherm. S, Nd–TiO2 shows a hysteresis loop at P/P0 = 0.45–0.8, indicating that the new catalyst has mesopores, and there is no obvious inflexion point at P/P0 = 0.05, which suggests that there are no micropores or very few micropores in the new material. From pore size distribution curve analysis, it is shown that the pore sizes of S, Nd–TiO2 are mainly distributed between 30–50 nm, which is consistent with the TEM analysis results. The pore size of TiO2 is between 50 and 90 nm, and the specific surface area is small. When S and Nd are doped into TiO2, the pore size decreases and the pore volume decreases accordingly. The specific surface area was significantly higher than that of TiO2, and the specific surface area was about 3 times that of pure TiO2. The specific surface area of the double-doped photocatalyst is 166.8 m2 g−1, and the specific surface area of pure TiO2 is 56.5 m2 g−1. This shows that doping with Nd and S can suppress the lattice growth of TiO2, increase the specific surface area, and increase the photocatalytic activity (Table 1).
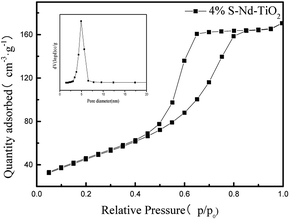 |
| | Fig. 7 The nitrogen adsorption–desorption isotherm and pore size distributions curve of 4% S, Nd–TiO2. | |
Table 1 The specific surface area and pore size distribution parameters of TiO2 and S, Nd–TiO2
| Sample |
Surface area (m2 g−1) |
Pore volume (cm3 g−1) |
Crystallite size (nm) |
P/P0 |
| TiO2 |
56.50 |
0.9918 |
38.29 |
0.99915 |
| S, Nd–TiO2 |
166.82 |
0.2644 |
20.56 |
0.99578 |
Fig. 8 shows fluorescence spectra analysis of the semiconductor TiO2 and the new catalysts. This was used to effectively investigate the recombination of electrons and holes on the surface of TiO2 before and after modification. The results show that the fluorescence intensity of pure TiO2 is large, and electrons and holes are easily recombined. With the doping of neodymium and sulfur, the fluorescence intensity of the composite decreases and the electron–hole recombination rate decreases. The doping of sulfur has no effect. The reason for this phenomenon is that the rare earth metal Nd forms a Ti–O–Nd bond around TiO2. This is uniformly dispersed in the form of an oxide on the surface of TiO2 or in the octahedral gaps of the TiO2 lattice. In order to compensate for this charge imbalance, the surface of TiO2 will adsorb more hydroxyl groups and promote the photocatalytic oxidation of TiO2. This not only effectively separates photogenerated electrons and holes, but also generates more active hydroxyl groups, improving the photocatalytic performance of nano-TiO2.
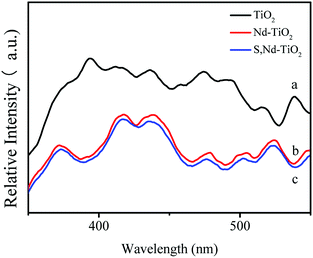 |
| | Fig. 8 PL fluorescence spectra analysis of each sample: (a) TiO2; (b) Nd–TiO2; and (c) S, Nd–TiO2. | |
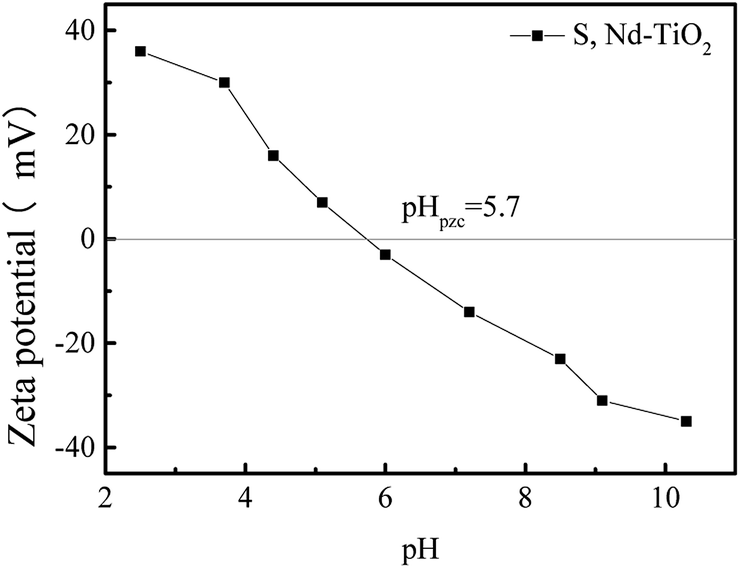
Fig. 9 shows the zeta potential values of S, Nd–TiO2 photocatalysts under different pH conditions. The point at which the potential is zero is called the isoelectric point (pHpzc). The adsorbent surface charge is neutral when pH = pHpzc. The electrostatic force between the surface of the adsorbent and the ions is negligible. The double-doped photocatalyst S, Nd–TiO2 has an isoelectric point of about 5.7. When the pH is less than 5.7, the zeta potential is greater than zero and the catalyst surface is positively charged. When the pH is greater than 5.7, the zeta potential is less than zero and the catalyst surface is negatively charged. Reactive red is an anionic dye, and the double-doped photocatalyst shows better adsorption effects due to electrostatic attraction.
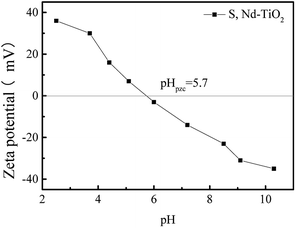 |
| | Fig. 9 The point of zero charge (pHpzc) for the S, Nd–TiO2 catalyst. | |
4. Photocatalytic activity
4.1 Photocatalyst degradation tests
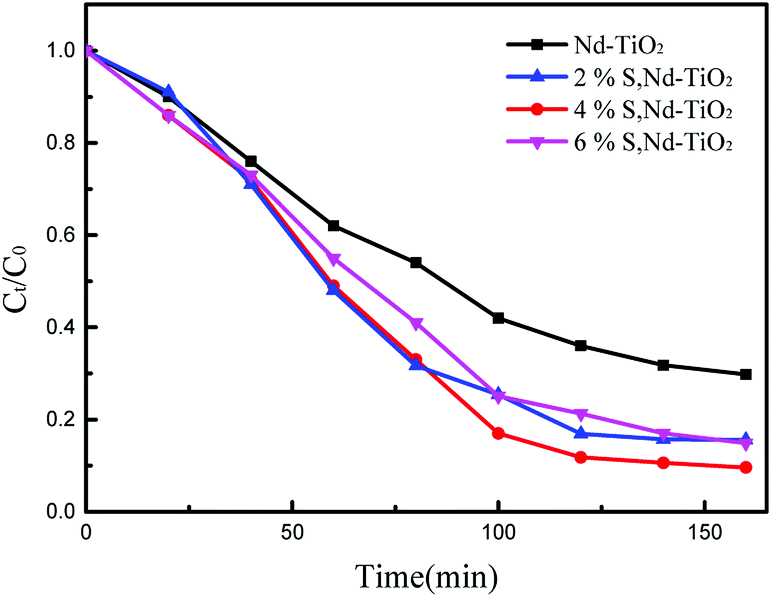
The photocatalytic activities of the S, Nd–TiO2 nanocomposites were investigated by measuring the relationship between the degradation rate of reactive red solution irradiated by a xenon lamp and the irradiation time. In the degradation formula, C is the initial concentration of reactive red and Ct is the concentration after a fixed reaction time. The comparative experiment is degradation experiments conducted with the photocatalyst Nd–TiO2. Stirring under dark conditions is indispensable prior to photocatalytic degradation. A quantitative amount of photocatalyst was added to the target degradant solution and magnetically stirred for 30 min to achieve adsorption and analytical equilibrium. For all samples, adsorption appeared to be of little use and the reactive red solution concentration did not change. But as the irradiation time increased, all samples degraded the reactive red solution. After 120 min using the single-doped photocatalyst, the degradation rate of reactive red reached about 90%. Comparing the single-doped and double-doped photocatalysts, the double-doped photocatalysts have better abilities to degrade reactive red than the single-doped photocatalyst. So, the S doping described can significantly improve the catalyst activity. In addition, the photocatalytic activities of the single-doped photocatalyst Nd–TiO2 and the double-doped photocatalyst S, Nd–TiO2 were analyzed via the Langmuir–Hinshelwood kinetic model. The kinetic formula is as follows:| |
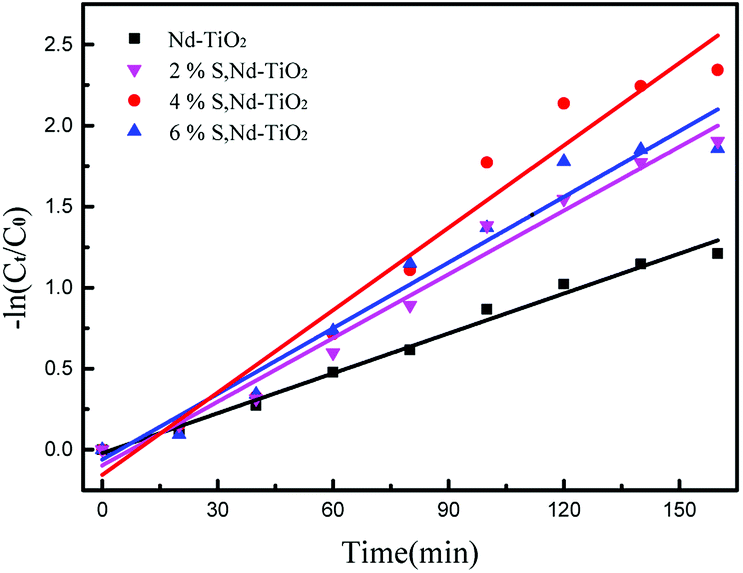
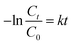 | (2) |
In the formula, C0 is the initial concentration before the reactive red solution is degraded, and Ct is the concentration of the reactive red solution at different time points. The linear relationships between (Ct/C0) and −ln(Ct/C0) and the irradiation time are shown in Fig. 10 and 11. The rate constants of S, Nd–TiO2 were higher than that of Nd–TiO2. The above results indicated that 2% S, Nd–TiO2, 4% S, Nd–TiO2, and 6% S, Nd–TiO2 exhibited higher photocatalytic activity than Nd–TiO2. This is because Nd element doping prolongs the time of recombination of electrons and holes compared to pure TiO2. Compared with Nd–TiO2, the doping of S, Nd–TiO2 broadens the light absorption range of nano-TiO2. And 4% S, Nd–TiO2 presented a slightly higher rate constant than 2% S, Nd–TiO2 and 6% S, Nd–TiO2. The degradation percentage of the reactive red solution by 4% S, Nd–TiO2 photocatalyst after 160 minutes was 90.4%. The reactive red solution is almost completely decolorized, and the double-doped photocatalyst exhibits very high photocatalytic ability. The degradation rate of reactive red using Nd–TiO2 was 0.0082 min−1. The degradation rates of reactive red using 2% S, Nd–TiO2 and 6% S, Nd–TiO2 were 0.0131 min−1 and 0.0135 min−1, respectively. Of the different samples, the 4% S, Nd–TiO2 photocatalyst had the highest degradation rate towards reactive red solution, 0.0169 min−1. The rate constants of the S, Nd–TiO2 nanocomposites were higher than that of Nd–TiO2 (0.0082 min−1). The above results indicate that 2% S, Nd–TiO2, 4% S, Nd–TiO2 and 6% S, Nd–TiO2 exhibited higher photocatalytic activity than Nd–TiO2, which should be attributed to their bigger light absorption ranges and excellent adsorption performances. And 4% S, Nd–TiO2 presented a slightly higher rate constant than 2% S, Nd–TiO2 and 6% S, Nd–TiO2. In experiments, we chose the optimal doping amount of Nd as a basic quantity. The doping of different S content values (2%, 4% and 6%) widens the light absorption range of TiO2. Too little S cannot modify TiO2 very well, and too much S cannot enter the lattice of TiO2, so 4% S, Nd–TiO2 has the best photocatalytic efficiency.
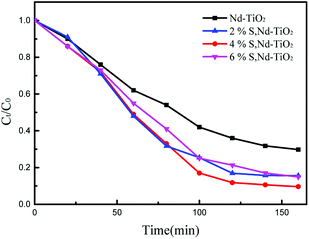 |
| | Fig. 10 Photocatalytic degradation of reactive red over S, Nd–TiO2. | |
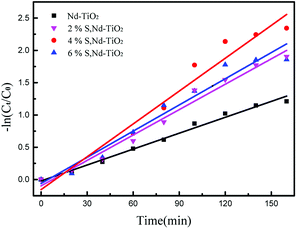 |
| | Fig. 11 Plots of −ln(Ct/C0) versus irradiation time for the degradation of reactive red solution over S, Nd–TiO2. | |
4.2 Effect of pH on the degradation rates of the catalysts
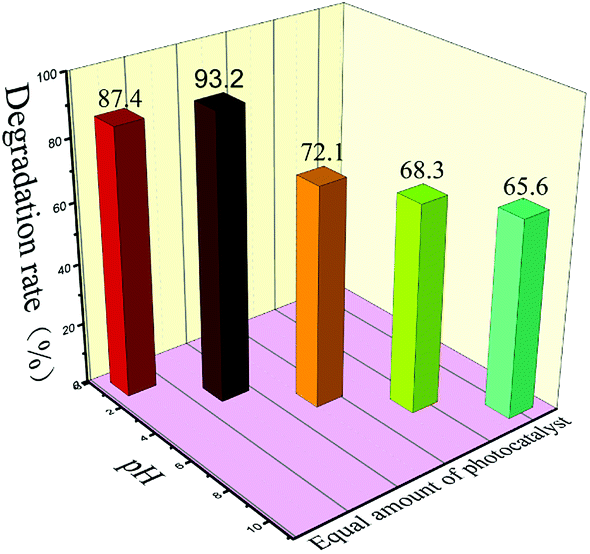
Whether CH+ in solution affects photocatalytic experiments is a problem we must consider. So, we conducted photocatalytic tests at different pH values. We chose 4% S, Nd–TiO2 as a photocatalyst to perform pH tests. After the catalyst was added to solution, the pH of the solution was adjusted to 2, 4, 6, 8, or 10. After a certain amount of reactive red solution was sampled every 20 minutes, the absorbance was measured, and the degradation rate was calculated. We conclude that the double-doped photocatalyst has a wide pH response range, and the catalyst has the best degradation efficiency at pH = 4. Under acidic conditions, the catalyst can better absorb dye molecules on the surface. In the presence of hydroxyl radicals and the presence of suitable amounts of H+, the reactive red (X-3B) molecule is eventually decomposed into small groups (NO3−, N2, H2O, CO2, etc.). When it is exposed to light, the catalyst shows good ability to degrade reactive red. Under alkaline conditions, the reactive red dye molecules cannot be stably present, so the degradation rates of the catalysts are low. However, when the solution is alkaline, the double-doped photocatalysts still show more than 65% degradation. Therefore, the double-doped photocatalyst will have a wider pH response range and higher application value.
4.3 Photocatalytic repetition experiments
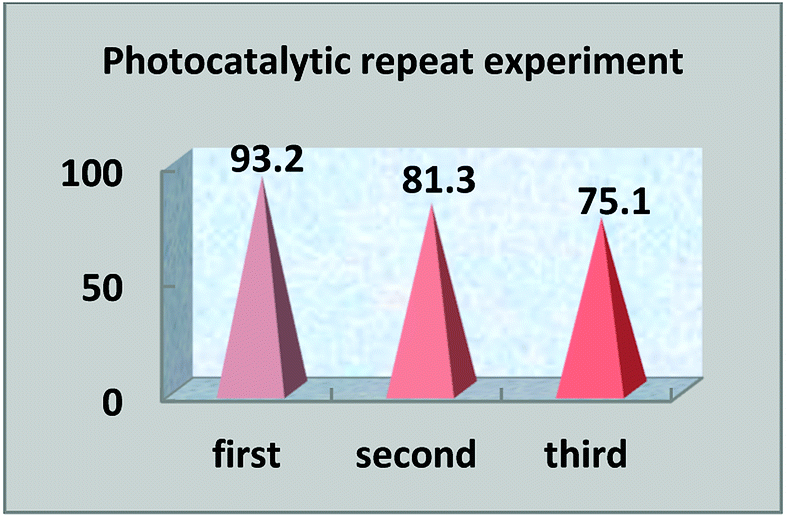
The multiple use of photocatalysts is important for practical value. Photocatalysts that retain their photocatalytic abilities after repeated degradation experiments are extremely rare. In this experiment, after the end of the first photocatalysis experiment, the upper liquid was poured out and the lower solid catalyst was collected and dried. The photocatalyst was replenished before the next photocatalytic experiment, so that the photocatalytic experiment was carried out with the same amount of photocatalysts. Under the same light degradation conditions, the same concentration of reactive red solution was degraded again, and the degradation rate was calculated again. The photocatalytic degradation experiment was repeated three times to judge the reusability. In the experiment, we found that the photocatalyst S, Nd–TiO2 has good reusability. As the photocatalyst is reused, the photocatalytic efficiency is lowered. The first cycle photocatalytic degradation amount reached 93%, the second cycle photocatalytic degradation amount reached 81%, and the third cycle photocatalytic degradation amount reached 75%. From the above results, it is shown that the double-doped photocatalyst S, Nd–TiO2 has a high practical reusability value in active red solution (Fig. 12 and 13).
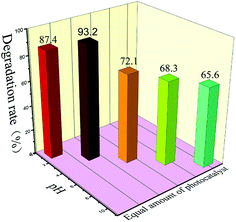 |
| | Fig. 12 The photocatalytic degradation of reactive red solution by S, Nd-codoped-TiO2 under different pH conditions. | |
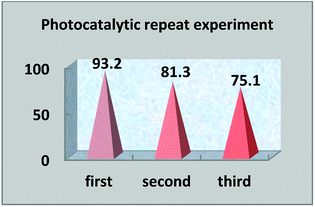 |
| | Fig. 13 Photocatalyst reusability efficiency. | |
4.4 Photocatalytic mechanism
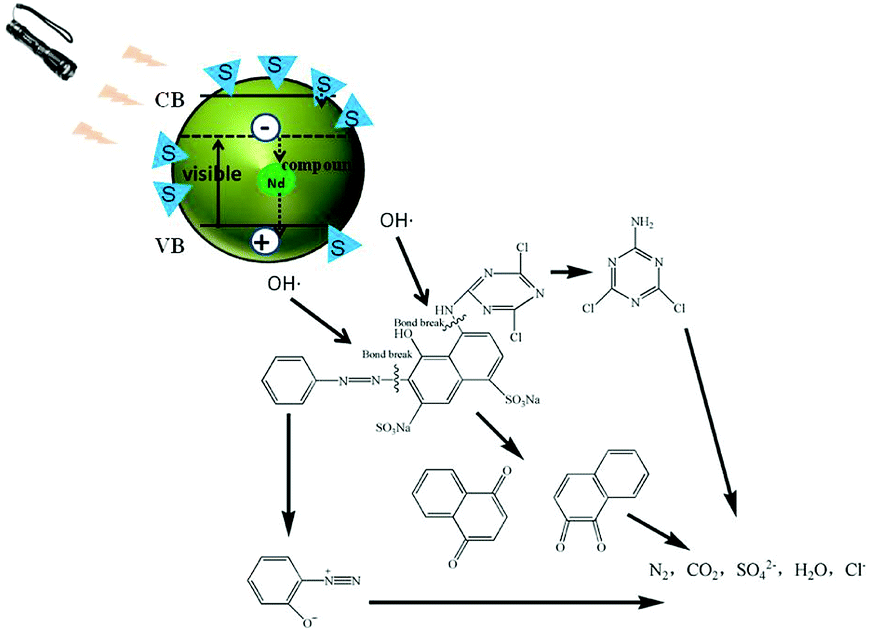
It is well known that the electron and hole recombination times determine the photocatalytic activities of catalysts, and the doping of Nd can effectively prolong the recombination time. The doping of an S element can effectively broaden the TiO2 light absorption range and move this to the visible light region. Therefore, the S, Nd–TiO2 double-doped photocatalyst combines the above advantages and exhibits excellent photocatalytic activity through testing. In the experiments, photocatalysis produces a large number of hydroxyl radicals, which can decompose organic matter into small molecules. The decomposed small molecule products of reactive red solution after illumination are shown in Fig. 14. This coincides with the principle of hydroxyl radical degradation of organic matter reported in a large amount of literature. Single-doped photocatalysts still have limitations regarding the degradation of dyes and need improvement. Double-doped photocatalysts combine the two advantages and ultimately decompose the dye molecules into CO2 and H2O. This composite material has successfully improved the defects of TiO2, and the low-consumption method has allowed the degradation of a practical dye solution, which provides a certain forward-looking route for real industry applications.
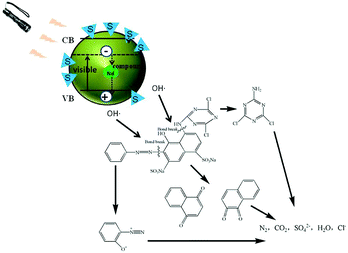 |
| | Fig. 14 Photocatalytic mechanism diagram. | |
5. Conclusions
S, Nd-codoped-TiO2 was prepared via a sol–gel method. The results of catalyst characterization show that the double-doped photocatalyst can effectively broaden the photoresponse range of TiO2. From the EDS and XPS results, Nd and S were successfully doped into TiO2. The S, Nd-codoped-TiO2 photocatalyst has a large specific surface area: SBET = 166.82 m2 g−1. Compared with many studies, our synthesized double-doped photocatalysts have higher degradation efficiencies.28–30 The new photocatalysts improve the dispersibility of TiO2 and enhance its characteristics regarding ease of agglomeration. The degradation results show that the photodegradation of reactive red solution reached 93.2% within 160 min using the S, Nd co-doped photocatalyst. The degradation of reactive red solution using Nd–TiO2 after 160 minutes was 70%. The double-doped photocatalysts have significantly higher degradation abilities than the single-doped photocatalyst. The reason is that the doping of S and Nd not only broadens the light absorption range of TiO2, but also reduces the recombination rate of electrons and holes. And the new photocatalyst has high stability and reusability. The double-doped photocatalyst is a promising photocatalytic material, which provides a great approach for solving water pollution issues with low consumption and high efficiency.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China [grant number 21776144].
Notes and references
- S. Zong, W. Wei, H. Cui, Z. Jiang, X. Lü, M. Zhang and J. Xie, Mater. Res. Innovations, 2015, 19, 361–367 CrossRef CAS.
- T. Ochiai, T. Fukuda, K. Nakata, T. Murakami, D. A. Tryk, Y. Koide and A. Fujishima, Appl. Electrochem., 2010, 40, 1737–1742 CrossRef CAS.
- M. S. Dieckmann and K. A. Gray, Water Res., 2016, 30, 1169–1183 CrossRef.
- D. González, E. Cueto, F. Chinesta, P. Díez and A. Huerta, J. Phys. Chem. C, 2016, 117, 1955–1962 Search PubMed.
- Z. Zhu, H. Pan, M. Murugananthan, J. Gong and Y. Zhang, Appl. Catal., B, 2018, 232, 19–25 CrossRef CAS.
- H. K. Seo, C. M. Elliott and S. G. Ansari, J. Nanosci. Nanotechnol., 2012, 12, 6996–7001 CrossRef CAS PubMed.
- H. Barndõk, D. Hermosilla, L. Cortijo, C. Negro and Á. Blanco, J. Adv. Oxid. Technol., 2016, 15, 125–132 Search PubMed.
- J. Keleher, J. Bashant, N. Heldt, L. Johnson and Y. Li, World J. Microbiol. Biotechnol., 2002, 18, 133–139 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- K. M. Reza, A. Kurny and F. Gulshan, Appl. Water Sci., 2017, 7, 1569–1578 CrossRef CAS.
- S. Garcia-Segura and E. Brillas, J. Photochem. Photobiol., C, 2017, 31, 1–35 CrossRef CAS.
- V. Binas, D. Venieri, D. Kotzias and G. Kiriakidis, Journal of Materiomics, 2017, 3, 3–16 CrossRef.
- D. Zhao and C. F. Yang, Renewable Sustainable Energy Rev., 2016, 54, 1048–1059 CrossRef CAS.
- L. Hu, G. Zeng, G. Chen, H. Dong, Y. Liu, J. Wan, A. Chen, Z. Guo, M. Yan, H. Wu and Z. Yu, J. Hazard. Mater., 2016, 301, 106–118 CrossRef CAS PubMed.
- M. Reli, P. Huo, M. Šihor, N. Ambrožová, I. Troppová, L. Matějová, J. Lang, L. Svoboda, P. Kuśtrowski, M. Ritz, P. Praus and K. Kočí, J. Sol-Gel Sci. Technol., 2017, 43, 8564–8573 Search PubMed.
- S. Demirc, T. Dikici, M. Yurddaskal, S. Gultekin, M. Toparli and E. Celik, Appl. Surf. Sci., 2016, 390, 591–601 CrossRef.
- W. Yan, S. M. Mahurin, Z. Pan, S. H. Overbury and S. Dai, J. Am. Chem. Soc., 2005, 127, 10480–10481 CrossRef CAS PubMed.
- Z. Xu and J. Yu, Nanoscale, 2011, 3, 3138–3144 RSC.
- C. Wang, L. Yin, L. Zhang, N. Liu, N. Lun and Y. Qi, ACS Appl. Mater. Interfaces, 2010, 2, 3373–3377 CrossRef CAS PubMed.
- B. Trujillo-Navarrete, M. del Pilar Haro-Vázquez, R. María Félix-Navarro, F. Paraguay-Delgado, H. Alvarez-Huerta, S. Pérez-Sicairos and E. Alonso Reynoso-Soto, Journal of Rare Earths, 2017, 35, 259–270 CrossRef CAS.
- T. Boningari, S. N. R. Inturi, M. Suidan and P. G. Smirniotis, Chem. Eng. J., 2018, 339, 249–258 CrossRef CAS.
- A. Charanpahari, S. S. Umare, S. P. Gokhale, V. Sudarsan, B. Sreedhar and R. Sasikala, Appl. Catal., A, 2012, 443–444, 96–102 CrossRef CAS.
- A. Charanpahari, S. S. Umare and R. Sasikala, Appl. Surf. Sci., 2013, 282, 408–414 CrossRef CAS.
- F. Dong, H. Wang, G. Sen, Z. Wu and S. C. Lee, J. Hazard. Mater., 2011, 187, 509–516 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- Y. H. Xu, C. Chen, X. L. Yang, X. Li and B. F. Wang, Appl. Surf. Sci., 2009, 255, 8624–8628 CrossRef CAS.
- C. H. Cao, Adv. Mater. Res., 2012, 488–489, 221–225 CAS.
- W. M. Hou and Y. Ku, J. Alloys Compd., 2011, 509, 5913–5918 CrossRef CAS.
- W. J. Zhang, M. L. Hu and H. B. He, Adv. Mater. Res., 2012, 457–458, 563–566 CAS.
Footnote |
| † CCDC #84-1286 and #75-1753. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra06778c |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Liming Bai and
Xinling Ao
*,
Liming Bai and
Xinling Ao