DOI:
10.1039/C8RA06926C
(Paper)
RSC Adv., 2018,
8, 32899-32908
Self-curing triphenol A-based phthalonitrile resin precursor acts as a flexibilizer and curing agent for phthalonitrile resin†
Received
18th August 2018
, Accepted 11th September 2018
First published on 24th September 2018
Abstract
Major problems currently limiting the widespread application of phthalonitrile resins are the high precursor melting point and volatility of the curing agent. Herein, a novel self-curing triphenol A-based phthalonitrile resin precursor (TPPA-Ph) was successfully synthesized by reacting α,α,α′-tris(4-hydroxyphenyl)-1-ethyl-4-isopropylbenzene (TPPA) with 4-nitrophthalonitrile (NPh) via nucleophilic substitution. The presence of residual phenolic hydroxyl groups in the TPPA-Ph precursor promoted the curing reaction of phthalonitrile resin in the absence of an additional curing reagent. Self-cured TPPA-Ph resins exhibited relatively low melting points (less than 100 °C), high thermal stability, and a wide processing window (116 °C). Furthermore, the TPPA-Ph precursors contained phenolic hydroxyl and cyano groups that can be used as flexibilizers and curing agents to optimize other phthalonitrile resins. Resorcinol-based phthalonitrile resin (DPPH) cured with various amounts of TPPA-Ph possessed excellent thermal and thermo-oxidative stability with a 5% weight loss temperature exceeding 530 °C, Tgs above 380 °C, and a wide processing window and time. Therefore, as a novel precursor and curing agent for phthalonitrile resins, the triphenol A-based phthalonitrile resin is an ideal resin matrix for high-performance composites with broad application prospects in aerospace, shipping, machinery, and other high-tech fields.
Introduction
Materials play important roles in daily life. With increasing demand for materials, traditional materials no longer meet basic needs, with polymer materials receiving increasing use and recognition. Among many heat-resistant high-performance matrix resins, phthalonitrile resin stands out owing to its unique and comprehensive properties. Phthalonitrile resin is a class of high performance thermosetting resins with a phthalonitrile structure at the end of the polymer chain. In addition to its excellent heat resistance and heat-resistant oxygen-aging properties, phthalonitrile resin has excellent mechanical properties, high flame retardancy, low water absorption, chemical resistance, and unique optical and electrical properties that can be used in composite matrices, coating, adhesives,1 and structural applications.2–4 Phthalonitrile resin also has broad application prospects in aerospace, shipping, electronics,5–7 machinery, automobiles, and other high-tech fields.8,9
The Keller research group has been researching phthalonitrile resin since 1980, resulting in great progress. A series of simple bisphenol-based phthalonitriles10 have been designed and synthesized. However, the curing rate of the phthalonitrile monomer is very slow, taking nearly one hundred hours to produce a significant gel at 280 °C. Subsequently, Keller and coworkers found that mixing phthalonitrile monomers and aromatic amines effectively promoted the curing of phthalonitrile monomers, with the curing rate related to the alkalinity of the aromatic amines.11 Curing additives that can catalyze the production of high-performance phthalonitrile resin include strong organic acids,12 organic amines,13 organic acid/amine salts,14 metals,15 and metal salts.16 Currently, aromatic diamines, such as 1,3-bis(3-aminophenoxy)benzene (m-APB), diamino diphenyl sulfone (DDS), and 1,3-bis(3,4-dicyanophenoxy)benzene (BDB), are commonly used as curing agents for phthalonitrile resins. Although the amount of aromatic diamine curing agent added can adjust the rate of phthalonitrile curing, the aromatic amines17 are easily decomposed. Adding too much curing agent produces voids in the material during high-temperature treatment, which affects the mechanical properties of the material. To resolve this problem, a stable curing agent is needed to prevent volatilization during high-temperature curing and, therefore, improve the mechanical performance of phthalonitrile resin.
Meanwhile, another problem of phthalonitrile resin is the high melting point of the phthalonitrile monomer, which is caused by its larger molecular rigidity (usually greater than 200 °C).18–21 For example, bisphenol A-based phthalonitrile has a melting point of 195 °C, while the melting point of biphenyldiol-based phthalonitrile is 235 °C.17 However, phthalonitrile resin curing can be conducted after the phthalonitrile monomer has been melted. Therefore, the processing temperature window from the melting point to the gel point is too narrow, making it difficult to process and mold the phthalonitrile resin. Therefore, the synthesis of a phthalonitrile monomer with a low melting point is key to the development of phthalonitrile resin.
In brief, the major problems currently limiting the widespread application and development of phthalonitrile resin are the high precursor melting point, curing agent volatility, high curing temperatures, and long curing times.22,23 Therefore, research into new resin precursors and novel curing agents can effectively solve these issues.24–26 In our laboratory, we have demonstrated introducing phthalazinone groups into phthalonitrile resins enhances their thermal stability.27 Meanwhile, the new curing agent has been screened to improve the phthalonitrile resin processing properties.28,29
Herein, a new type of multifunctional phthalonitrile resin (TPPA-Ph) containing a triphenol A structure was prepared using α,α,α′-tris(4-hydroxyphenyl)-1-ethyl-4-isopropylbenzene (TPPA) as a feedstock in a one-pot reaction. Using TPPA as a monomer was advantageous for the construction of phthalonitrile resins. Firstly, TPPA-based phthalonitrile precursor TPPA-Ph has a relatively low melting point due to the TPPA monomer containing an alkyl flexible center and isopropyl benzene flexible group. Secondly, by regulating the reaction point, the phthalonitrile resin system retains a certain amount of phenolic hydroxyl groups after the reaction, which can activate phthalonitrile resin self-curing without an additional curing agent. Therefore, the formation of voids due to the addition of external curing agent is avoided, resulting in an excellent phthalonitrile resin. Thirdly, phenolic hydroxyl groups in the precursor can also be used as a flexibilizer to optimize other phthalonitrile resins, which improves the processing performance of phthalonitrile resin. The cure kinetics, and thermal and dynamic mechanical properties, were also investigated.
Experimental
Materials
All materials were obtained from commercial suppliers and used without further purification. α,α,α′-Tris(4-hydroxyphenyl)-1-ethyl-4-isopropylbenzene (TPPA) was supplied by Tokyo Chemical Industry Development Co., Ltd. 4-Nitrophthalonitrile (NPh) was purchased from Wuhan Yinguan Chemical Co., Ltd. Anhydrous potassium carbonate (K2CO3) was obtained from Tianjin Damao Chemical Reagent Factory, ground into powder in a mortar, and dried in vacuum prior to use. Resorcinol was purchased from Sinopharm Chemical Reagent Co., Ltd. Dimethyl sulfoxide (DMSO) was supplied by Tianjin Fuyu Fine Chemical Co., Ltd.
Synthesis of self-curing TPPA-based phthalonitrile resin precursor (TPPA-Ph)
TPPA-Ph was synthesized by regulating the reaction conditions to control the phenolic hydroxyl group content in the system. Typically, TPPA (10 g), NPh (4.28 g), pulverized anhydrous K2CO3 (4.88 g), and DMSO (100 mL) were added into a 250 mL round-bottom flask with magnetic stirring. The resulting solution was heated at 40 °C for 24 h under a nitrogen atmosphere. When the reaction ended, the product was poured into deionized water (1000 mL) and adjusted to neutral pH with dilute HCl solution. The resultant off-white solid was washed with deionized water until the filtrate was neutral and dried at 70 °C for 24 h in a vacuum oven. Similar to this process, different TPPA-Ph precursor systems were synthesized by changing the reaction conditions, such as reactant proportion and reaction temperature.
Synthesis of resorcinol-based phthalonitrile resin precursor (DPPH)
Resorcinol (11 g), NPh (35.46 g), pulverized anhydrous K2CO3 (20.71 g), and DMSO (110 mL) were added into a 250 mL three-necked flask. The resulting solution was stirred at 30 °C for 48 h under a nitrogen atmosphere. The product mixture was then poured into hot deionized water and adjusted to neutral pH with dilute HCl solution. The wet filter cake was washed with deionized water and ethanol until the filtrate was neutral, and dried at 50 °C for 24 h in a vacuum oven.
Thermal curing of phthalonitrile resins
TPPA-Ph was stirred at 100 °C in a reaction vessel until melting and pouring into a mold for thermal curing. The curing process was performed at 200 °C for 2 h, 250 °C for 2 h, 350 °C for 2 h, and 400 °C for 2 h in a muffle furnace.
DPPH and TPPA-Ph (mass ratios of 10%, 20%, 25%, and 30%) were ground evenly and stirred in a reaction vessel at 180 °C until melting. The mixtures were then cured at 200 °C for 2 h, 250 °C for 2 h, 300 °C for 2 h, 350 °C for 2 h, 375 °C for 2 h, and 400 °C for 2 h in a muffle furnace. As a reference, 5% (mass ratio) DDS was mixed with DPPH and cured under identical conditions.
Characterizations
1H NMR spectra were obtained using a Bruker Avance II 400M nuclear magnetic resonance (NMR) spectrometer, using deuterated dimethylsulfoxide (DMSO-d6) as solvent and operating at 25 °C. Fourier transform infrared (FTIR) spectra were collected on a Thermo Nicolet Nexus 470 FTIR spectrometer using an attenuated total reflectance (ATR) accessory in the range 4000–650 cm−1 at room temperature. High-performance liquid chromatography (HPLC) was performed with a HP-1100 liquid chromatography system. Differential scanning calorimetry (DSC) was performed with a Mettler Toledo DSC1 calorimeter under nitrogen flow (50 mL min−1) at different heating rates (5, 10, 15, and 20 °C min−1) over the temperature range 25–350 °C. DSC scans showed the curing temperature for maximum peaks (Tp) and the curing kinetics of TPPA-Ph with different compositions. Thermogravimetric analysis (TGA) curves were obtained using a Mettler TGA1 Instrument at a heating rate of 10 °C min−1 under a nitrogen or air purge (50 mL min−1) from 30 to 800 °C. Rheological behavior was investigated using a TA Instrument AR-2000 rheometer. Measurements were made at a frequency of 1 Hz under a strain of 0.02 N and employed at a heating rate of 3 °C min−1 from room temperature to 400 °C. Samples for rheometric testing were compressed into a cylinder (diameter, 25 mm; thickness, 1 mm). Dynamic mechanical analysis (DMA) was performed using a TA Instruments Q800 dynamic mechanical analyzer at a frequency of 1 Hz from room temperature to 380 °C with a heating rate of 3 °C min−1.
Curing kinetic analysis
The kinetic analysis of thermal transformations is based on the assumption that the transformation rate (dα/dt) during a reaction is the product of two functions, one depending completely on the temperature (T), and the other depending completely on the fraction transformed (α):30| |
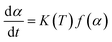 | (1) |
where K(T) is a temperature-dependent reaction rate constant, and f(α) is a differential conversion function dependent on the curing mechanism.31
The temperature-dependent function can be easily determined using the Arrhenius equation:30
| |
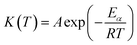 | (2) |
where
A is the pre-exponential factor,
Eα is the apparent activation energy,
R is the universal gas constant, and
T is the absolute temperature.
The frequently applied methods of determining Eα are the Kissinger, Ozawa, and Friedman methods for non-isothermal reactions. Using the Starink method is recommended due to higher accuracy of Eα according to the ICTAC kinetic project.32
| |
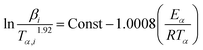 | (3) |
where
Tα,i is the temperature at which the extent of conversion
α is reached under the
ith heating rate, and
β is the heating rate.
Results and discussion
Preparation and structure of TPPA-Ph precursors
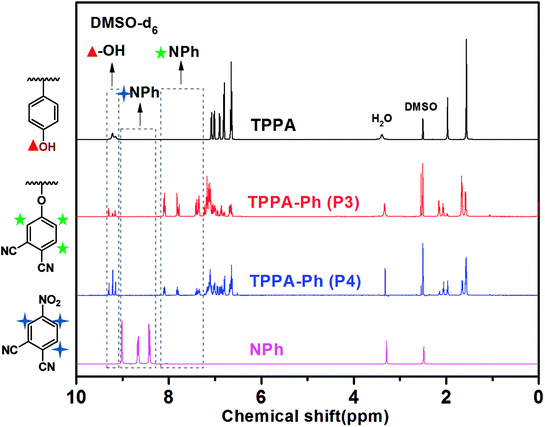
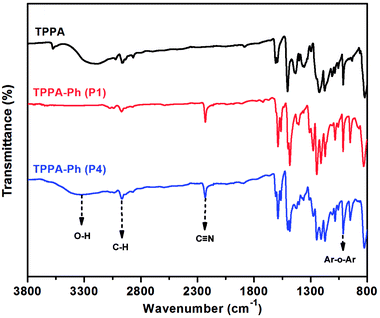
As shown in Scheme 1, the TPPA monomer consists of an alkyl flexible center, isopropyl benzene flexible group, and three phenolic hydroxyl groups with different reactivities. In a polar aprotic solvent, the three phenolic hydroxyl groups of TPPA were reacted with NPh under using an inorganic base as catalyst to afford the multifunctional TPPA-based phthalonitrile resin precursors. By controlling the reaction conditions, such as reactant proportion and reaction temperature, we obtained TPPA-Ph precursors with different phenolic hydroxyl contents, as shown in Table 1. Comparing the 1H NMR spectra of the raw material and the TPPA-Ph precursors indicated that the presence of certain residual phenolic hydroxyl groups (δ = 9.12–9.34 ppm) was key to the self-curing nature of TPPA-based phthalonitrile resin precursor systems (Fig. 1).33,34 However, characterizing each proton peak was difficult due to the resultant products being mixtures, as shown by 1H NMR analysis. In the FT-IR (ATR) spectra, absorption bands at around 3400 cm−1, 2970 cm−1, 2229 cm−1, and 1012 cm−1 were attributed to the stretching vibrations of phenolic hydroxyl groups, carbon–hydrogen bonds, cyano groups, and aromatic ether groups, as shown in Fig. 2. Notably, the peak at 3400 cm−1 was very weak in the case of TPPA-Ph (P1) due to the low content of phenolic hydroxyl groups. Both the 1H NMR spectra and FT-IR (ATR) spectra demonstrated the successful synthesis of self-curing TPPA-based phthalonitrile resin precursors.
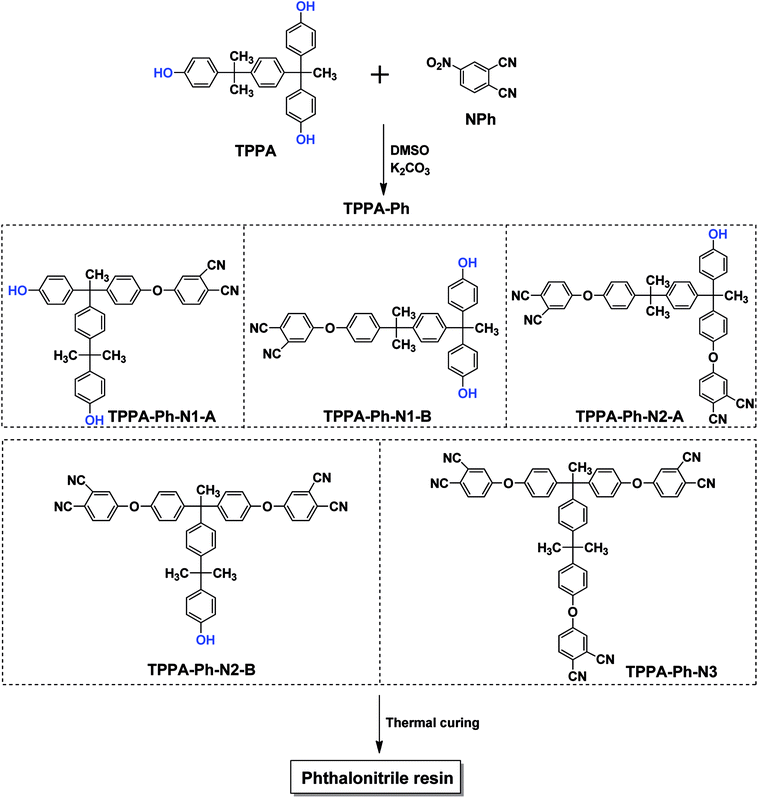 |
| | Scheme 1 Synthesis of multifunctional TPPA-based phthalonitrile resin precursor (TPPA-Ph). | |
Table 1 Component contents in TPPA-Ph products
| TPPA-Ph |
TPPA-Ph-N1 (%) |
TPPA-Ph-N2 (%) |
TPPA-Ph-N3 (%) |
| P1 |
0.0 |
9.8 |
90.2 |
| P2 |
11.6 |
42.3 |
46.1 |
| P3 |
28.0 |
29.5 |
42.5 |
| P4 |
44.0 |
43.1 |
12.9 |
 |
| | Fig. 1 1H NMR spectra of TPPA, NPh, and TPPA-Ph precursors with different compositions. | |
 |
| | Fig. 2 FT-IR (ATR) spectra of TPPA and TPPA-Ph precursors with different compositions. | |
The presence of residual phenolic hydroxyl groups in TPPA-Ph promoted curing of the phthalonitrile resin. The phenolic hydroxyl content, structure, and distribution of the TPPA-Ph precursors were determined using HPLC and HPLC-MS (Fig. 1S†). These results indicated that five compounds were present in total, whose structures were classified as TPPA-Ph-N1–3, and a total of three types according to the different phenolic hydroxyl contents. There were two mono-substituted compounds (TPPA-Ph-N1-A and TPPA-Ph-N1-B), two bis-substituted compounds (TPPA-Ph-N2-A and TPPA-Ph-N2-B) and one tri-substituted compound (TPPA-Ph-N3) in the TPPA-Ph precursor systems. Different distributions of the TPPA-Ph precursor systems were synthesized by changing the reaction parameters, such as reactant proportion and reaction temperature (Table 1). Increasing the molar ratio of NPh to TPPA and the reaction temperature led to a decrease in the proportion of mono-substituted compounds (TPPA-Ph-1; from 44.0% to 0.0%), while the proportion of the tri-substituted compound (TPPA-Ph-3) was increased from 12.9% to 90.2%. Using a TPPA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :K2CO3:NPh molar ratio of 1:1.5:1.05 and a temperature of 25 °C, the TPPA-Ph precursor system contained 44.0% of the mono-substituted compounds (TPPA-Ph-N1) and 43.1% of the bis-substituted compounds (TPPA-Ph-N2), which afforded the majority of phenolic hydroxyl groups that act as curing centers to facilitate TPPA-based phthalonitrile resin curing.
:K2CO3:NPh molar ratio of 1:1.5:1.05 and a temperature of 25 °C, the TPPA-Ph precursor system contained 44.0% of the mono-substituted compounds (TPPA-Ph-N1) and 43.1% of the bis-substituted compounds (TPPA-Ph-N2), which afforded the majority of phenolic hydroxyl groups that act as curing centers to facilitate TPPA-based phthalonitrile resin curing.
Performance of self-curing TPPA-based phthalonitrile resin
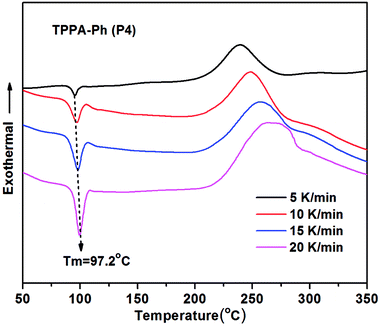
Non-isothermal DSC analysis was used to investigate the curing kinetics of TPPA-Ph precursors at heating rates of 5, 10, 15, and 20 K min−1 under a nitrogen atmosphere. As shown in Fig. 3, TPPA-Ph (P4) melted at 97.2 °C and the exothermic peak at 248.9 °C confirmed that TPPA-Ph curing had proceeded successfully without an additional curing reagent. According to basic kinetic theory,35,36 the shape of the curing kinetic curves can be used to determine the reaction path. Graphs that plot conversion a as a function of curing temperature (Fig. 2S†) using data obtained from dynamic DSC (Fig. 3) show typical sigmoid shapes that are characteristic of curing reactions that follow an autocatalytic mechanism.37 The Starink method is normally recommended for conducting isoconversional analysis of dynamic DSC data. Therefore, ln(βi/Tα,i1.92) vs. 1/Tα values for the same fractional extent of conversion for a series of dynamic DSC experiments conducted at various heating rates can be plotted to afford a straight line with a gradient of −1.0008Eα/R. Plots were obtained for TPPA-Ph (P4) for a conversion range of 0.1–0.9, with a good linear relationship observed in both cases, as shown in Fig. 3S.† Repeating the procedure, Eα values corresponding to various a values can then be obtained from the DSC curing curves, with the change in apparent activation energy with respect to the degree of conversion shown in Fig. 4S.† The average value of Eα was 127.7 kJ mol−1 for the TPPA-Ph (P4) precursor, which was similar to those reported previously for this type of resin.38 Notably, TPPA-Ph (P4) showed a gradual decrease in activation energy at conversion levels greater than 0.5 in Fig. 4S,† which can be attributed to changes in the rate-determining step of a diffusion-controlled process.39
 |
| | Fig. 3 DSC curing kinetics curves for self-curing TPPA-Ph (P4) under a nitrogen atmosphere at various heating rates. | |
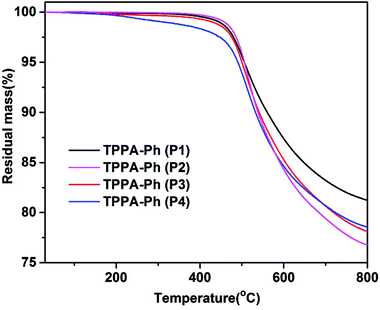
The self-curing reactions of TPPA-Ph precursors were conducted under temperature-programmed conditions (200 °C for 2 h, 250 °C for 2 h, 350 °C for 2 h, and 400 °C for 2 h). The thermal stabilities of the resulting TPPA-Ph resins were investigated using TGA analysis over the temperature range 30–800 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere. The data, including the temperature at 5% mass loss (Td5%) and the char yields at 800 °C, are summarized in Table 2. As shown in Fig. 4, the self-cured TPPA-Ph (P1) resin had the highest 5% weight loss temperature (Td5%) of 506.1 °C, exhibiting a char retention of 81.2% after heating to 800 °C under a nitrogen atmosphere. Cured TPPA-Ph resins with different distributions had distinct thermal stabilities, with a decrease in phenolic hydroxyl content in TPPA-Ph precursors resulting in an increase in Td5% (see Table 2). Although the presence of residual phenolic hydroxyl groups in TPPA-Ph precursors can promote resin curing, a higher molar ratio of residual phenolic hydroxyl groups to cyano groups will reduce the crosslinking density and thermal stability of the phthalonitrile resin. This suggested that the curing rate and thermal properties of self-cured TPPA-Ph resins can be easily controlled by adjusting the phenolic hydroxyl content in the TPPA-Ph precursors.
Table 2 Thermal properties of self-cured TPPA-Ph resins after curing at 400 °C
| TPPA-Ph |
Molar ratio of residual OH/CN |
Td5% (°C) |
Char yield at 800 °C (%) |
| P1 |
0.03 |
506.1 |
81.2 |
| P2 |
0.22 |
505.7 |
76.8 |
| P3 |
0.35 |
500.3 |
78.1 |
| P4 |
0.55 |
490.6 |
78.5 |
 |
| | Fig. 4 TGA traces of self-cured resins with different TPPA-Ph contents under a nitrogen atmosphere. | |
The rheological properties of TPPA-Ph (P2) without an additional curing reagent were also investigated. As shown in Fig. 5, the TPPA-Ph (P2) precursor showed a rapid decrease in viscosity at around 100 °C. When the temperature was raised to 140 °C, the viscosity dropped to 50 Pa S, which is considered in the machinable range. The viscosity was stable in the range 1–0.1 Pa S with increasing temperature, while the minimum viscosity was 1.615 Pa S, which confirmed that the melt of TPPA-Ph resin has satisfactory fluidity. The viscosity levels increased gradually above 256 °C, meaning that the processing temperature window was 116 °C. Therefore, TPPA-Ph resins have a wide processing window and excellent processing fluidity.
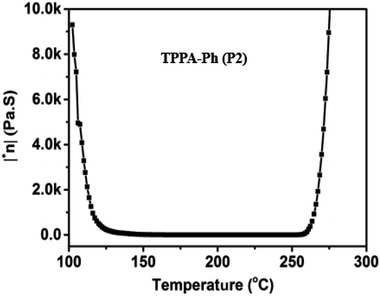 |
| | Fig. 5 Complex viscosity (η*) as a function of temperature for TPPA-Ph (P2). | |
Performance of TPPA-Ph system-cured DPPH resins
TPPA-Ph precursor systems contain phenolic hydroxyl groups and cyano groups that can also be used as curing agents and flexibilizers to optimize the processing performance of phthalonitrile resins. Herein, resorcinol-based phthalonitrile resin precursor DPPH (synthetic route and 1H NMR spectrum shown in Scheme 1S and Fig. 5S,† respectively) was mixed with TPPA-Ph (P4) in different mass ratios of 10%, 20%, 25%, and 30% and cured at 200 °C for 2 h, 250 °C for 2 h, 300 °C for 2 h, 350 °C for 2 h, 375 °C for 2 h, and 400 °C for 2 h to afford a black green solid. As shown in Fig. 6, after thermal curing, the absorption peak at around 2229 cm−1, attributed to vibration of the cyano group, was diminished. Furthermore, new peaks appeared at around 1010 cm−1, 1355 cm−1, 1519 cm−1, and 1713 cm−1, which evidenced the formation of phthalocyanine, s-triazine and isoindoline structures, respectively. This suggested that the presence of residual phenolic hydroxyl groups in the TPPA-Ph precursor successfully promoted the curing of resorcinol-based phthalonitrile resin.
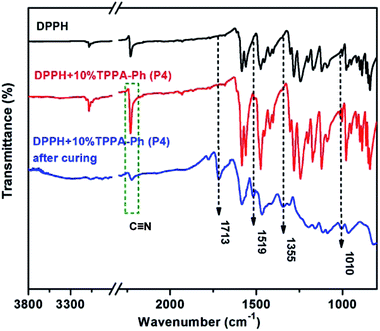 |
| | Fig. 6 FT-IR(ATR) spectra of DPPH and DPPH with 10% TPPA-Ph content before and after curing. | |
The thermal and thermo-oxidative properties of the DPPH resins cured with TPPA-Ph (P4) were evaluated using TGA over the temperature range 30–800 °C at heating rates of 10 °C min−1, as shown in Fig. 7, and the key data are shown in Table 3. In general, the DPPH resins cured with various contents of TPPA-Ph (P4) possessed excellent thermotolerance, with the temperatures corresponding to 5% weight loss all exceeding 528 °C under either nitrogen or air atmospheres, while char yields were all above 76% when heated to 800 °C under a nitrogen atmosphere. Although the TPPA-Ph (P4) content was 10%, the 5% and 10% weight loss temperatures were the highest in any atmosphere, and were also higher than those of DPPH cured with 5% DDS as reference.
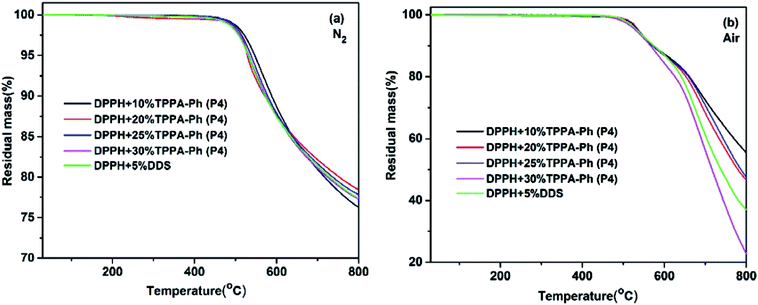 |
| | Fig. 7 TGA traces of DPPH resins cured with DDS and different TPPA-Ph contents under (a) nitrogen and (b) air atmospheres. | |
Table 3 Thermal properties of DPPH resins cured with DDS and different TPPA-Ph (P4) contents
| Curing agent |
In N2 |
In air |
| Td5% (°C) |
Char yield at 800 °C (%) |
Td5%(°C) |
Char yield at 800 °C (%) |
| 5%DDS |
530.9 |
77.4 |
534.4 |
45.3 |
| 10%TPPA-Ph |
547.0 |
76.3 |
537.3 |
55.5 |
| 20%TPPA-Ph |
528.8 |
78.4 |
537.2 |
46.6 |
| 25%TPPA-Ph |
538.0 |
77.8 |
534.6 |
47.5 |
| 30%TPPA-Ph |
532.3 |
77.3 |
531.2 |
22.8 |
The influence of long-term oxidative aging at high temperature under an air atmosphere is also an important consideration for phthalonitrile resin use. The conditions were simulated by subjecting DPPH resins cured with TPPA-Ph (P4) to stepwise heating at a series of temperatures between 200 and 375 °C at 8 h intervals under air. Fig. 8 shows the percentage residual mass after weight loss at various temperatures. A cumulative weight loss of 1.68–3.84% was observed for a TPPA-Ph (P4) content range of 10–30%. This performance was similar to that of the resin with DDS as curing agent. Based on the above results, the DPPH resins cured with TPPA-Ph (P4) showed outstanding thermal stability and thermo-oxidative stability.
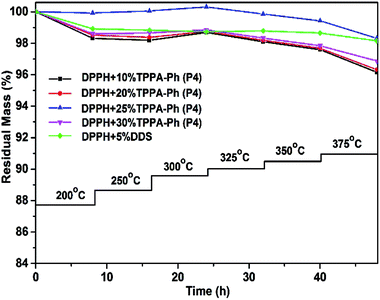 |
| | Fig. 8 Long-term oxidative aging plots of DPPH phthalonitrile resins cured with DDS and different TPPA-Ph (P4) contents. | |
Rheological behavior plays an important role in confirming the processability of resins. The processing window and time required for curing were further investigated by examining the complex viscosity (η*) of DPPH resins cured with TPPA-Ph (P4) as a function of temperature from 160 °C to 370 °C. As shown in Fig. 9a, the viscosity of the resins was influenced by the TPPA-Ph (P4) content, with TPPA-Ph (P4) contents of 10%, 20%, 25%, and 30% showing rapid complex viscosity increases at 330 °C, 290 °C, 270 °C, and 260 °C, respectively. At temperatures above 175 °C, a minimum melt complex viscosity of around 0.2 Pa S was observed for all DPPH resins. The low melt viscosity of the resins at higher temperatures was evidence of their superb melt fluidity and stability. However, for the DDS curing system, the lowest viscosity was around 0.08 Pa S at 191 °C, and the curing window was narrower than that of the TPPA-Ph system. In Fig. 9b, complex viscosity measurements as a function of time for DPPH resins with different TPPA-Ph (P4) contents were performed at 250 °C, respectively. The processing window and time of the DPPH resins could be adjusted by the TPPA-Ph (P4) content. Based on the above results, the DPPH resins with TPPA-Ph (P4) showed good processability with low complex viscosity and a wide processing window at moderate temperatures.
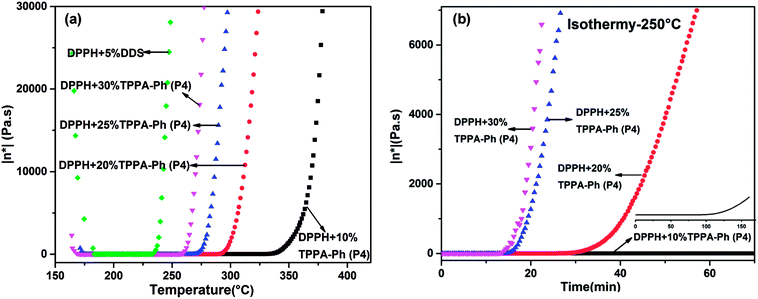 |
| | Fig. 9 Complex viscosity (η*) as a function of (a) temperature and (b) time for cured DPPH phthalonitrile resins with DDS and different TPPA-Ph (P4) contents. | |
The dynamic mechanical properties are another fundamental feature that determines the upper limit of the application temperature in developing high-performance phthalonitrile resin. The dynamic mechanical properties of DPPH resins cured with TPPA-Ph (P4) were investigated to evaluate changes in the resin modulus as a function of temperature and to determine the glass transition temperature of DPPH resins. The dynamic mechanical behavior of resins, including storage modulus (G′) and damping factor (tanδ) at a fixed frequency of 1 Hz in single cantilever mode, are shown in Fig. 10. All cured resins exhibited a high storage modulus of more than 3.0 GPa at 50 °C, which decreased as the temperature gradually increased. This high modulus should be due to heteroaromatic structures and the high degree of crosslinking. Furthermore, the declining modulus with increasing temperature was attributed to stress relaxation of the cured resins. The tand vs. temperature curves for DPPH cured resins are shown in Fig. 10b, showing that the tanδ values were below 0.03 when heated to 380 °C with no obvious relaxation transition, suggesting that the glass transition temperature of DPPH resins were all above 380 °C within the testing range. Some small transitions were ascribed to secondary relaxation of the phthalonitrile structures. As determined above, DPPH resins cured with various TPPA-Ph (P4) contents possessed excellent thermal stability. For DPPH cured with DDS, no Tg was detected within the tested temperature range and the storage modulus was much higher than that using the TPPA-Ph system due to the rigid structure of DDS.
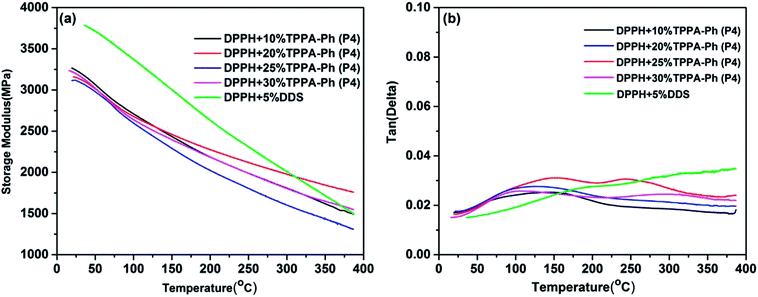 |
| | Fig. 10 Dynamic mechanical properties of DPPH resins cured with DDS and different contents of TPPA-Ph (P4): (a) storage modulus vs. temperature; (b) tanδ vs. temperature. | |
Another important factor in the use of resins under real conditions is water absorption caused by the cyano and phenolic hydroxyl groups. The water absorption was investigated by immersing the DPPH resins cured with TPPA-Ph (P4) in boiling deionized water for 100 h. Data, including the weight increase at 100 h, are summarized in Fig. 11. However, the water absorption of all resins with different TPPA-Ph (P4) contents was less than 4%, which was lower than that of the DDS curing system, guaranteeing the effective use of DPPH resin in practical production.
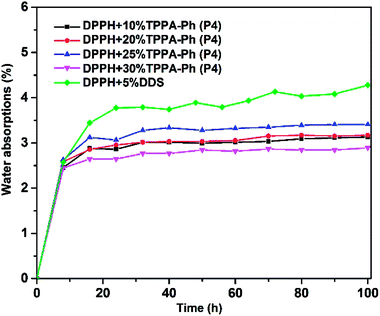 |
| | Fig. 11 Moisture absorption curves for DPPH phthalonitrile resins cured with DDS and different TPPA-Ph (P4) contents. | |
Conclusions
In this research, a novel multifunctional self-curing triphenol A-based phthalonitrile resin precursor was successfully synthesized from TPPA and NPh. The reaction conditions afforded a mixture of five TPPA-Ph compounds containing different amounts of underivatized phenolic hydroxyl groups, which act as curing centers to facilitate curing of the TPPA-based phthalonitrile resin in the absence of an additional curing reagent. The melting point of self-curing TPPA-Ph precursors was less than 100 °C, and the thermal properties and processing window of self-cured TPPA-Ph resins was regulated by adjusting the phenolic hydroxyl content in TPPA-Ph precursors. Furthermore, TPPA-Ph precursor systems contained phenolic hydroxyl groups and flexible structures that can act as curing agents and flexibilizers to optimize other phthalonitrile resins. For example, resorcinol-based phthalonitrile resin (DPPH) cured with various TPPA-Ph (P4) contents possessed excellent thermal stability and thermo-oxidative stability, and a wide processing window. Therefore, this study provides a strategy for constructing a multifunctional phthalonitrile resin precursor that can improve the curing process without sacrificing other properties, such as thermal stability and thermo-oxidative stability.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Nature Science Foundation of China (Nos. 51673033, U1663226 and 51873027) and the Fundamental Research Funds for the Central Universities (DUT17LK39).
References
- A. Rawal, P. V. Kameswara, S. Russell and A. Jeganathan, J. Appl. Polym. Sci., 2010, 116, 2668–2673 CrossRef.
- S. B. Sastri, J. P. Armistead and T. M. Keller, Polym. Compos., 1996, 17, 816–822 CrossRef.
- S. B. Sastri, J. P. Armistead and T. M. Keller, Polym. Compos., 1997, 18, 48–54 CrossRef.
- F. Zuo and X. B. Liu, J. Appl. Polym. Sci., 2010, 117, 1469–1475 Search PubMed.
- T. M. Keller, SAMPE Symp. Proc., 1986, 31, 528–532 Search PubMed.
- T. M. Keller, CHEMTECH, 1988, 18, 635–639 Search PubMed.
- J. F. Giuliani and T. M. Keller, Sens. Mater., 1989, 1, 22–27 Search PubMed.
- A. Badshah, M. R. Kessler, Z. Heng, J. H. Zaidi, S. Hameed and A. Hasan, Polym. Chem., 2013, 4, 3617–3622 RSC.
- W. H. Zhu and G. S. Wu, J. Phys. Chem. A, 2001, 105, 9568–9574 CrossRef.
- T. M. Keller and D. D. Dominguez, Polymer, 2007, 48, 91–97 CrossRef.
- S. B. Sastri and T. M. Keller, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1885–1890 CrossRef.
- T. M. Keller, Polym. Prepr., 1992, 33, 422–423 Search PubMed.
- T. M. Keller and T. R. Price, J. Macromol. Sci., Chem., 1982, 18, 931–937 CrossRef.
- P. J. Burchill, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1–8 CrossRef.
- T. R. Walton, J. R. Griffith and J. G. O'Rear, J. Adhes. Sci. Technol., 1975, 9, 665–676 Search PubMed.
- L. Y. Sun and Z. T. Huang, Chin. J. Polym. Sci., 1990, 8, 177–182 Search PubMed.
- S. B. Sastri and T. M. Keller, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1885–1890 CrossRef.
- T. M. Keller, Chem. Mater., 1994, 6, 302–305 CrossRef.
- T. M. Keller, Polymer, 1993, 34, 952–955 CrossRef.
- T. M. Keller and T. R. Price, Polym. Commun., 1984, 25, 42–44 Search PubMed.
- T. M. Keller and D. D. Dominguez, Polymer, 2005, 46, 4614–4618 CrossRef.
- X. Fu, G. P. Yu, C. Liu, J. Y. Wang, C. Y. Pan and X. G. Jian, High Perform. Polym., 2014, 26, 540–549 CrossRef.
- X. Y. Yu, K. Naito, C. Kang, X. W. Qu and Q. X. Zhang, Macromol. Chem. Phys., 2013, 214, 361–369 CrossRef.
- T. M. Robert, D. Augustine, S. Chandran, D. Mathew and C. P. R. Nair, RSC Adv., 2015, 5, 1198–1204 RSC.
- H. T. Sheng, X. G. Peng, H. Guo, X. Y. Yu, K. Naito and X. W. Qu, Thermochim. Acta, 2014, 577, 17–24 CrossRef.
- M. Laskoski, A. Neal, T. M. Keller, D. D. Dominguez, C. A. Klug and A. P. Saab, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1662–1668 CrossRef.
- G. P. Yu, C. Liu, X. P. Li, J. Y. Wang, X. G. Jian and C. Y. Pan, Polym. Chem., 2012, 3, 1024–1032 RSC.
- Z. Q. Wu, J. H. Han, N. Li, Z. H. Weng, J. Y. Wang and X. G. Jian, Polym. Int., 2017, 66, 876–881 CrossRef.
- Z. H. Weng, J. Y. Fu, L. S. Zong, C. Liu, J. Y. Wang and X. G. Jian, RSC Adv., 2015, 5, 92055–92060 RSC.
- S. Vyazovkin, Thermochim. Acta, 2000, 355, 155–163 CrossRef.
- C. F. Dickinson and G. R. Heal, Thermochim. Acta, 2009, 494, 15–25 CrossRef.
- S. Vyazovkin, A. K. Burnham, J. M. Criado, L. A. Perez-Maqueda, C. Popescu and N. Sbirrazzuoli, Thermochim. Acta, 2011, 520, 1–19 CrossRef.
- K. Zeng, K. Zhou, S. H. Zhou, H. B. Hong, H. F. Zhou and Y. P. Wang, Eur. Polym. J., 2009, 45, 1328–1335 CrossRef.
- M. Z. Xu, M. D. Liu, S. H. Dong and X. B. Liu, J. Mater. Sci., 2013, 48, 8108–8116 CrossRef.
- Z. Lei and H. Xiao, Polymer, 2010, 51, 3814–3820 CrossRef.
- C. Jubsilp, S. Damrongsakkul, T. Takeichi and S. Rimdusit, Thermochim. Acta, 2006, 447, 131–140 CrossRef.
- T. Zhang, J. Wang, M. Derradju, N. Ramdani, H. Wang and Z. W. Lin, Thermochim. Acta, 2015, 602, 22–29 CrossRef.
- M. Z. Xu, Y. S. Luo, Y. X. Lei and X. B. Liu, Polym. Test., 2016, 55, 38–43 CrossRef.
- G. M. Roudsari, A. K. Mohanty and M. Misra, ACS Sustainable Chem. Eng., 2014, 2, 2111–2116 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06926c |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Yu Qi,
Jinyan Wang
*,
Yu Qi,
Jinyan Wang













