
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantification of 71 detected elements from Li to U for aqueous samples by simultaneous-inductively coupled plasma-mass spectrometry†
Melanie Bäuchle a,
Tina Lüdeckeab,
Sasan Rabieha,
Khemet Calneka and
Timothy G. Bromage*acd
a,
Tina Lüdeckeab,
Sasan Rabieha,
Khemet Calneka and
Timothy G. Bromage*acd
aDepartment of Biomaterials, New York University College of Dentistry, 345 East 24th Street, New York, NY 10010, USA. E-mail: tim.bromage@nyu.edu
bSenckenberg Biodiversity and Climate Research Center, Senckenberganlage 25, 60325 Frankfurt, Germany
cDepartment of Paleoanthropology, Senckenberg Research Institute, National History Museum, Senckenberganlage 25, 60325 Frankfurt, Germany
dDepartment of Basic Science and Craniofacial Biology, New York University College of Dentistry, 345 East 24th Street, New York, NY 10010, USA
First published on 19th November 2018
Abstract
Quantitative analysis of multi-element concentrations in aqueous solutions, such as water, beverages and biofluids, has long been performed by sequential inductively coupled plasma-mass spectrometry. Recently, a fully simultaneous mass spectrum monitoring ICP-MS instrument that fits a compact Mattauch–Herzog geometry (MH-ICP-MS) with a permanent magnet and a large, spatially resolving semiconductor ion detector has been introduced. This technology allows coverage of the complete inorganic relevant mass range from 6Li to 238U in a single measurement, which helps to mitigate the restriction on the number of inorganic elements whose concentrations may be routinely measured from one sample, thus reducing operational assay times and aqueous sample volumes for evaluations across the breadth of the periodic table. We report here on a detailed method for utilizing MH-ICP-MS to detect all elements of the relevant inorganic spectrum in aqueous samples; 7 types of water, 4 types of beverage, and 4 biofluid biological samples. With this method 71 elements can be routinely detected simultaneously in seconds and in as little as 1–4 mL sample, when using a specific set of calibration and internal standards. Quantitative results reveal distinct element patterns between each sample and within types of samples, suggesting that different types of aqueous solutions can be recognized and distinguished by their elemental patterns. The method has implications for understanding elemental distribution and concentration for many fields, including nutrition, studies of the biosphere, ecological stoichiometry, and environmental health fields, among others, where broad elemental information is actually required.
Introduction
Pure water is almost non-existent in nature. What generally is called water is actually an accumulation of dissolved elements in the form of organic (typically carbon-hydrogen-based) and inorganic (non-organic) molecules. Given the importance of water to all life, it came to our attention that aqueous samples are not routinely measured for all element concentrations across the breadth of the chemical periodic table, despite the potential for some technologies to do so. This dearth of research is not for the lack of want for knowledge, but because technologies and methods were not specifically designed to accommodate such breadth on a routine basis. Hence, in this communication we report on a detailed method for the instantaneous detection of 71 inorganic element concentrations from 7Li to 238U from aqueous samples using MH-ICP-MS technology designed to provide this multi-element capability.There are several techniques and methods presently in use to detect and quantify inorganic elements in fluids and water in particular, such as graphite furnace atomic absorption spectroscopy (GFAAS), inductively coupled plasma optical emission spectrometry (ICP-OES), and most prominently, quadrupole, sector field, and time of flight inductively coupled plasma mass spectrometry (Q-ICP-MS, SF-ICP-MS, ICP-TOF-MS respectively).1–14 These technologies were designed for targeted analyses of single or moderate numbers of elements. In particular, forms of ICP-MS have been successfully used for nearly three decades to determine concentrations of some multiple number of elements, mostly trace elements or rare earth elements, in various water samples,3,6,8,15–17 wine,2,12,13 milk and formula,5,18,19 saliva, blood and urine,1,3,20–34 liquefied tomatoes,35 as well as in sediments and rocks.6,36,37 Hence, ICP-MS technology is the technique issued by national and international guidelines to monitor, among others, inorganic water quality.6,16,38,39 However, ICP-MS studies rarely exceed measurement of 40 elements due to the duty cycle and scanning characteristics of popular quadrupole-based mass analysers.
Until recently, most ICP-MS instruments have been so-called “sequential” ICP-MS, in which elements are analysed consecutively, one by one, including the ‘almost simultaneous’ ICP-TOF-MS.40 However, the employ of ICP-MS requires that sample volumes be adjusted upward to accommodate measurements of increasingly comprehensive multi-element composition, increasing also the time and consumables required to operate such instruments. Thus while it is possible, it is somewhat impractical to evaluate the entire inorganic spectrum with ICP-MS because this technology was not purposefully designed to perform such analyses.
MH-ICP-MS for simultaneous detection was developed to permit evaluation across the complete inorganic relevant spectrum from small sample volumes (1–4 mL) in seconds and potentially lower consumables costs. It uses a Mattauch–Herzog geometry in which the configuration of electrostatic and magnetic fields spreads the ions over the mass range and distributes them along a flat plane.41 This technology differs from multicollector-ICP-MS in that it incorporates a single detector with 4800 pixel elements, the dimension of which permits simultaneous detection of isotopes over the full relevant inorganic mass spectrum from 6Li to 238U (SPECTRO MS, SPECTRO Analytical Instruments GmbH, Kleve, Germany); technical specifications can be found elsewhere.40 With this technology, targeted evaluations may be performed on any number of elements, while also permitting discovery-based research that would be impractical using sequential ICP-MS. However, the method has been lacking to quantitatively evaluate the complete spectrum using MH-ICP-MS, which we present here.
The need for such a method, especially for water, is justified by the lack of comprehensive data regarding element distributions and concentrations for most elements across the breadth of the inorganic spectrum.24,42 For example, the European Union (EU) and the United States Environmental Protection Agency (EPA) are monitoring and have issued maximum concentration limits for a number of elements in drinking water considered to be health risks,43,44 yet many elements commonly known as harmful to human health, such as lithium or tin, are neither monitored nor regulated in drinking water. Hence, we don't know for the majority of elements which concentrations are normal, below, or above average.
The elemental content of water typically mirrors its natural environmental exposure; e.g. its mineral content reflects the geochemical environment through which water runs.3,42 Aqueous solutions employ water as a medium, and as such, in the case of, say, beverages, the elemental content will additionally include the effects of manufacturing processes. Aqueous biofluids primarily reflect biological functions and consumption and environmental exposure. Some aqueous solutions such as commercial milk, will include elemental signatures that reflect beverage manufacturing as well as those of biofluids. In all, aqueous solutions are expected to “fingerprint” their origin. Here we present an MH-ICP-MS calibration method to quantify the total elemental composition of aqueous samples by simultaneously measuring 71 elements from 7Li to 238U in a variety of water, beverage, and biofluid samples with the advantage of full spectrum monitoring in which no elemental information is lost and that is essential in several research areas.
Experimental
Instrumentation
All measurements were performed with a MH-ICP-MS SPECTRO MS (SPECTRO Analytical Instruments GmbH, Kleve, Germany). The SPECTRO MS was launched in 2010, and it fits a compact Mattauch–Herzog geometry with a permanent magnet and a large, spatially resolving semiconductor ion detector covering the complete inorganic relevant mass range from 6Li to 238U in a single measurement. Fluid samples were introduced via a Teledyne ASX-560 autosampler (Teledyne CETAC Technologies, Omaha, NE, USA) by pneumatic nebulization with ultrahigh purity argon as carrier gas (Airgas, Randor, PA, USA), using a SeaSpray nebulizer (Glass Expansion, Pocasset, MA, USA) and nickel sampler and skimmer cones (ICPMS Cones Limited, Tarvin Nr. Chester, Cheshire, United Kingdom). Typical operating parameters are given in ESI Table S1.†Optimization and calibration
Prior to making measurements, the SPECTRO MS is optimized by a routine to provide maximum sensitivity over the entire mass/ratio range with reasonably low levels of oxide ion formation (BaO+/Ba+) and for peak shape.30 Ba++ ranges from −0.007 to 0.081 counts per second (cps), and BaO from 0.003 to 0.592 cps, and background standard deviation (SD) values from 27 to 86 cps (119 cps in alcoholic samples and 3673 cps in biofluids), depending of the samples analysed. The optimization routine includes a detector calibration (DC) and an ICALisation for adjusting, if necessary, peak positions according to set references. It is carried out using SPECTRO MS software, Mass Analyzer Vision (v. 1.32.1405) and employ of the MERCK VI multi-element standard (Merck Multi element standard VI, Merck Darmstadt, Germany) solution at the concentration of 20 ppb. When alcoholic beverages were to be analysed, we added 0.9% (v/v) dehydrated to match the physicochemical properties of the alcoholic samples.Consumables
Standards preparation and procedures
We achieve high pipetting precision and repeatability at room temperature from pipetting volumes of 10 μL to 10 mL using the pipetting robot ANDREW (Andrew Alliance S.A., Geneva, Switzerland).Internal standards
To account for day-to-day variability in instrument conditions (instrument drift and physical interferences) during the time of measurement,15 we chose isotopes as internal standards that are in close proximity (in terms of their atomic mass number) to the measured and calibrated elements, but that also cover the mass range (7Li, to 238U) in a most uniform way.To represent the entire mass spectrum, we found it best to use 6Li, 103Rh,1,6,12,17,21,23,32,35 and 232Th as internal standards; thus 6Li, 103Rh, and 232Th cannot be measured in test samples. We used internal standard stock solution concentrations of 6Li (10 ppm), 103Rh (2 ppm), and 232Th (2 ppm) (LiRhTh) in order to overwhelm any traces of these elements/isotopes that might conceivably be found in a sample. The internal stock solution is made of single element standards of Li, Rh, and Th (Inorganic Ventures, Christiansburg, Virginia, USA). 6Li was used for elements in the m/z range of 7 (Li) to 54 (Fe), 103Rh for the m/z range 55 (Mn) to 159 (Tb), and 232Th for elements within m/z range of 161 (Dy) to 238 (U). We also achieved good results using 89Y instead of 103Rh, and/or 159Tb instead of 232Th.
Blank standard
The blank standard is made of ultrapure water, 2% (v/v) ultrapure HNO3 to facilitate element release from sample tube surfaces,40 and the internal standards LiRhTh.Calibration standards
Up to 12 calibration standards (ESI Table S2†) were designed to calibrate all relevant elements between 7Li and 238U (Li, Be, B, Na, Mg, Al, Si, P, S, Cl, K, Ca, Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ga, Ge, As, Se, Br, Rb, Sr, Y, Zr, Nb, Mo, Ru, Pd, Ag, Cd, In, Sn, Sb, Te, I, Cs, Ba, La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu, Hf, Ta, W, Re, Os, Ir, Pt, Au, Hg, Tl, Pb, Bi, and U). Each calibration standard contains ultrapure water, 2% ultrapure HNO3, and the internal standards LiRhTh, and 0.9% (v/v) dehydrated ethanol for alcoholic beverages. In the case of Mix2 and Os calibration standards (see below), 0.5% ultrapure HCl (Merck, Darmstadt, Germany) is added to ensure element stability. We grouped the 71 measurable elements into six standard groups: (1) the ICAL-Group (ICAL): ICAL is a 30 element multi standard in 5–10% (v/v) HNO3 (Certipur® Certified Reference Material ICP multi-element standard VI: Merck KGaA, Germany)8,42 and contains Li, Be, B, Na, Mg, Al, K, Ca, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ga, Ge, As, Se, Rb, Sr, Mo, Ag, Cd, Te, Ba, Tl, Pb, Bi and U in various concentrations. This ICAL multi standard is also used to calibrate the MH-ICP-MS each operational day; (2) the Mix1-Group (Mix1): elemental composition is very similar to the ICAL-group, with the additional elements S, P, Si, In, Cs, but without Mo. These elements are mixed as Periodic Table Mix 1 for ICP (Trace cert® Sigma-Aldrich Production GmbH, Buchs, Switzerland) and are stored in 10% (w/w) HNO3; (3) the Mix2-Group (Mix2): its elemental composition of Ti, Ge, Zr, Nb, Mo, Ru, Rh, Pd, Sn, Sb, Hf, Ta, W, Re, Ir, Pt, and Au needs special storage conditions (5% (w/w) HNO3 and 1% (w/w) HF) and are available through as Periodic Table Mix 2 for ICP (Trace cert® Sigma-Aldrich Production GmbH, Buchs, Switzerland); (4) the rare Earths- or Mix3-Group (Mix3): this group consists of elements belonging to the lanthanide series (La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, and Lu), as well as scandium (Sc) and yttrium (Y). They are contained in the Periodic Table Mix 3 for ICP (Trace cert® Sigma-Aldrich Production GmbH, Buchs, Switzerland), in 5% (w/w) HNO3; (5) the single element-group: these standards are all single elements and are either not present in any of the above groups 1–4, or they are required in very different concentrations. Elements that belong to this group are: Cl and Br (in H2O), K (in 0.1% v/v HNO3), In and Ti (in 2% v/v HNO3), Cs (in 7% v/v HNO3), Hg (in 10% v/v HNO3), Os (in 10% v/v HCl), and I (in 1% TEA) (all acquired from: Inorganic Ventures, Christiansburg, Virginia, USAA). These elements are used as single element standards; (6) the multi-group (Multi): elements within this group are P and S (in H2O), Na, Mg, Ca, and K (in 0.1% v/v HNO3), and Si (in tr. HNO3 and tr. HF) (all produced by Inorganic Ventures Christiansburg, Virginia, USA). The elements in this group are mixed into one solution in our laboratory, to account for the need of higher concentrations than found in the ICAL or Mix1 group. All elements, whether in mixes or as single element standards, are certified reference materials (CRM) that are directly traceable to the corresponding NIST SRM®. The six groups result in 11 (12 for biofluids) calibration standards, with at least two concentrations for each element, or a larger range of concentrations when needed. Concentrations of the calibration standards were chosen, with regard to the expected concentrations in test samples (see ESI Table S2†). Standards with elemental concentrations of less than 20 ppb have to be prepared on the day of measurement to avoid degradation. Ideally, Mix2, Os and Cl calibrations standards should be prepared immediately prior to measurement.Reference material and controls
To assure accuracy and reproducibility, we use the Standard Reference Material (SRM) NIST 1640a, as well as certified reference materials ICAL (20 ppb and 100 ppb for wine), Mix 2 and Mix 3 (10 ppb), and the lower concentrated Multi standard. To furthermore account for the accuracy of measurements, ICAL 20 ppb was measured at least every 5 to 10 samples.Sample preparation and procedures
We tested the method on 15 different types of aqueous sample, which include environmental water (tap, well, rain, lake, river, sea, and snow), beverages (bottled water, wine, beer and milk) and human biofluid samples (blood plasma, whole blood, saliva and urine) (Table 1). With the exception of the plasma and blood samples, all samples were collected directly into the prepared HDPE containers (Fischer Scientific, USA), without any intermediate container. The blood samples were collected into commercially available EDTA blood collection vials (BD Vacutainer® K2EDTA Tubes, Becton Dickinson, Franklin Lakes, NY, USA) and transferred into HDPE containers within 10 min of collection.| Samples | Location | GPS Coordinates |
|---|---|---|
| Environmental water | ||
| Tap water (0 and 5 min) | Yonkers, NY, USA | Private home |
| Well water | ||
| Well Karachi | Karachi, Pakistan | 24.8427554, 67.061033299 |
| Well Mecca | Mecca, Saudi Arabia | 21.4226, 39.8265000 |
| Snow | ||
| Sljeme Mountain | Zagreb, Croatia | 45.9142956, 15.9745177 |
| Otzi the Iceman | Italy | 46.77891, 10.839854 |
| Rain water | ||
| Porto Allegre | Porto Allegre, RS, Brazil | −30.107778, −51.209444 |
| Karachi | Karachi, Pakistan | 24.8427554, 67.06103329 |
| Lake water | ||
| Lake Zurich | Zürich, Switzerland | 47.358207, 8.547451 |
| Lake TrećeJezero | Zagreb, Croatia | 45.824564, 16.01882815 |
| River water | ||
| Medveščak feeder | Zagreb, Croatia | 45.890557, 15.97427 |
| Oswegatchie river | Wegatchie, NY, USA | 44.304508, −75.587172 |
| Sea water | ||
| Fukuoka | Fukuoka, Japan | 33.596823, 130.359027 |
| Copacabana Beach | Rio de Janeiro, RJ, Brazil | −22.977854, −43.187257 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Beverage samples | ||
| Bottled water | ||
| VOSS, Natural Artesian Water | Vatnestrøm, Norway | 58.412781, 8.084090 |
| Poland Spring, Natural Spring | Maine, USA | N/A |
| Beer | ||
| Budweiser (glass, #8071041B1310) | Anheuser-Busch, St. Louis, MO, USA | N/A |
| Heineken (can, #8163528A0559) | White Plains, NY, USA | N/A |
| Wine | ||
| ‘Old Soul’ Petite Sirah, Oak Ridge Winery, Lodi 2015 | Lodi, CA, USA | 38.136526, −121.2415530 |
| Familia Montana, Tempranillo Reserva, Rioja DOCa 2012 | Fuenmayor, La Rioja, Spain | 42.465949, −2.5599039999 |
| Milk | ||
| Elmhurst Dairy (#36-2107) | Worcester Crys DBA Mountainside Farm, Roxbury, NY, USA | 42.306469, −74.549747 |
| Farmland Fresh Dairies (#42-169) | Dairy, Pottsville Pike, Reading, PA, USA | 40.381500, −75.935056 |
|
||
| Biofluid biological samples | ||
| Saliva | ||
| Female | New York City, NY, USA | 40.737957, −73.978213 |
| Male | New York City, NY, USA | 40.737957, −73.978213 |
| Urine | ||
| Female | New York City, NY, USA | 40.737957, −73.978213 |
| Male | New York City, NY, USA | 40.737957, −73.978213 |
| Whole blood | ||
| Female | New York City, NY, USA | 40.737957, −73.978213 |
| Male | New York City, NY, USA | 40.737957, −73.978213 |
| Blood plasma | ||
| Female | New York City, NY, USA | 40.737957, −73.978213 |
| Male | New York City, NY, USA | 40.737957, −73.978213 |
Upon arrival in the laboratory, all samples were acidified. Water samples were stored in a cool dark place, beverages and biofluids were refrigerated at 4 °C.45 Further preparation and analysis were carried out at room temperature.
When dilution was required, samples were diluted with ultrapure water. For analysis, all (diluted) samples contain 2% (v/v) HNO3 and internal standards were added (1:100).
It has been reported that Hg is likely to be volatilized and adsorbed onto the inner walls of HDPE containers16 and that therefore chloride should be added to the sample to prevent the loss of Hg.47 To avoid high percentages of total dissolved solids, in assessing the method for this communication we elected to test only the supernatants of milk, saliva, whole blood and plasma samples, without the employ of certified reference materials specifically associated with these sample types. Such samples as these generally should be acid digested in order to include the complete sample with its solids. Our future in depth MH-ICP-MS research on such categories of samples will be performed using acid digestion preparation protocols combined with the appropriate certified reference material standards.
:20 and 1:40 (v/v)8 to enable quantification of elements in low and high concentrations.:10 (v/v)2,12,13 because of the high content of dissolved solids. The beer samples were shaken multiple times, then heated in a water bath for 20 min at 90 °C to eliminate carbon dioxide and subsequently diluted 1:5 and 1:10 (v/v). The milk samples were diluted 1:25 (v/v), as suggested by,31 because of the high content of dissolved solids. The diluted milk samples were vortexed for 2 min and then let stand to settle the solids. The supernatant was used for analysis. Milk and beer were collected from local supermarkets, and the wine was purchased in liquor retail stores in the U.S. Bottled water samples were not diluted.In contrast to ref. 20, 27 and 31 who used a saliva collecting device, we collected the saliva directly into a prepared sample tube to avoid contamination from the saliva collecting device itself and its containing buffers.
The saliva was collected prior to eating, rinsing, and tooth brushing. In previous studies, saliva has been prepared by various procedures e.g. microwave48 or heat-acid22 digestion, using ultrasound,49,50 or without any digestion.20,27,32 Here, we acidified the saliva samples (1 to 2% HNO3 (ref. 7, 32, 33 and 49–51)) and centrifuged33,49–51 them at 3500 rpm for 40 min. The supernatant was then diluted 1:10 (v/v). The midstream urine samples were directly collected into 50 mL HDPE containers. The samples were acidified with 2% (v/v) HNO3. For analysis, the urine samples were diluted 1:2 (v/v) in contrast to published dilutions of 1:5 or 1:10,23,24 to ensure the recovery of low-concentration elements.
A fasting, venous blood sample was collected into two K2EDTA blood collection vials via a pyrogen free blood collection set. The first collection vial was discharged to minimize contamination from the blood collection set.42 Blood from the second collection vial was used to generate two subsamples: whole blood and plasma. For the whole blood samples, 2 mm steel metal beads, washed overnight in ultrapure 2% (v/v) HNO3, were added to the samples and the whole blood samples vortexed at about 2000 rpm for 30 s in 4–6 repeating cycles. The beads were removed and then the samples diluted 1:100. For the plasma samples, the blood was centrifuged for 20 min at 10500 rpm. The supernatant plasma was diluted 1:10 (v/v) (male) and 1:20 (v/v) (female) with 2% (v/v) HNO3.33
Measurement procedure
To achieve sound values for elements in each concentration for the blank, each calibration standard, controls and the samples, the respective solution is flushed into the MH-ICP-MS for at least 30 s after reaching the plasma, known as “preflush”. Three aliquots are measured and a mean value for each measured isotope is established (isotopes monitored are given in ESI Table S3†). A total solution volume of 4 mL or greater is required at the minimum sample aspiration rate (0.5 mL min−1) when using an autosampler. When introducing samples, controls etc. manually, the volumes needed for analyses can be as low as 1 mL.Between the introduction of the blank, each calibration standard, controls and samples, the instrument is rinsed with an ultrapure 2% (v/v) HNO3 for 2 min (ref. 15) (for water based samples) to 4 min (milk and blood samples), or longer if needed. The overall time per one sample analyses is about 5 min.
Generally, the MH-ICP-MS produces stable data over several hours and we measure up to 60 samples after the set of calibration standards, resulting in about 7 h per run. This time can vary based on preflush and rinse time or the number of aliquots analyzed.
Analysis procedure
Measured isotope peaks and backgrounds of the calibration standards and samples are manually defined.In general, the SPECTRO MS software Mass Analyzer Vision (v. 1.32.1405) factors in the influence of interferers on a target isotope by adjusting the calibration regression of the target isotope providing a mathematically corrected elemental concentration. When an interfering element (e.g. its isotopes) is present in a different calibration standard than the target isotope, the SPECTRO MS software calculates the respective interferer-factor. We tested the interferences of elements (e.g. their isotopes) that influence isotopes of elements within the same calibration standards separately, for each matrix. These elements are the rare earth elements Ce to Lu, as well as Na, Al, Si, S, Ca, Fe, Mn, Sr, Nb and I for non-alcoholic samples, and rare earth elements La to Lu and Na, Al, Si, S, K, Ca, Cr, Mn, Fe, Co, Cu, Sr, Nb, Mo, Cd, and I for alcoholic samples. The respective interference factors were then used in measurements within one matrix group (e.g. all water sample, or samples containing alcohol). We also tested potential interferences in all sample types resulting from our internal standards Li, Rh, and Th, with negative results. A list of all interferer factors for all target isotopes in all measurement runs can be found in ESI Table S3.†
To achieve a regression line with a correlation coefficient above, or equal to R2 = 0.996 (higher than suggested by (ref. 23)), readings of the blank and at least two calibrations standards are plotted, and the influence of interferers factored in. Elements that did not achieve the set correlation coefficient in the samples tested here were 27Al (R2 = 0.975), 34S (R2 = 0.963, 0.976), 42Ca (R2 = 0.995), and 127I (R2 = 0.994, 0.989) in the environmental waters, 34S (R2 = 0.992), 35Cl (R2 = 0.969), and 49Ti (R2 = 0.995) in bottled water, 39K (R2 = 0.991), 49Ti (R2 = 0.930), 62Ni (R2 = 0.994), 90Zr (R2 = 0.967), 178Hf (R2 = 0.975) for alcoholic beverages, 28Si (R2 = 0.994), 34S (R2 = 0.992), and 35Cl (R2 = 0.991) in milk, and 28Si (R2 = 0.995), 34S (R2 = 0.992), and 35Cl (R2 = 0.991) in body fluids. However, most isotopes had an R2 ≥ 0.999 and some achieved a R2 of 1 (environmental waters: 11B, 23Na, 24Mg, 44Ca, 90/91Zr, 99/101Ru, 108Pd, 109Ag, 111/112/114Cd, 118/120Sn, 140Ce, 143Nd, 166/167Er, 169Tm, 172/173Yb, 177/178/179Hf, 182W, 191Ir, 197Au, 207Pb, and 209Bi; bottled water: 24Mg, 39K, 140Ce, 162Dy, 166/167Er, 171/172Yb, 182/184W, and 187Re; alcoholic beverages: 9Be, 45Sc, 141Pr, 157Gd; milk: 23Na, 89Y, 133Cs, 139La, 141Pr, 166Er, 184W, 185Re, 192Os, 193Ir; body fluids: 23Na, 24Mg, 39K, 89Y, 133Cs,139La, 141Pr, 166Er, 184W, 193Ir, and 192Os).
When elemental concentration was measured on multiple isotopes (see ESI Table S3†), we selected the isotope with the lowest relative standard deviation using STOICHY software52 (OVAN GmbH, Berlin, Germany). Mean values of three consecutive measurements, relative standard deviation, and detection limits were recorded for each element.
Results
71 elements simultaneously detected and quantified
Mean elemental concentrations, relative standard deviations, and detection limits for each sample, as well as the control samples are reported in the ESI section.†Environmental Water Samples
Fig. 1A, ESI Tables S4.1–4.4.†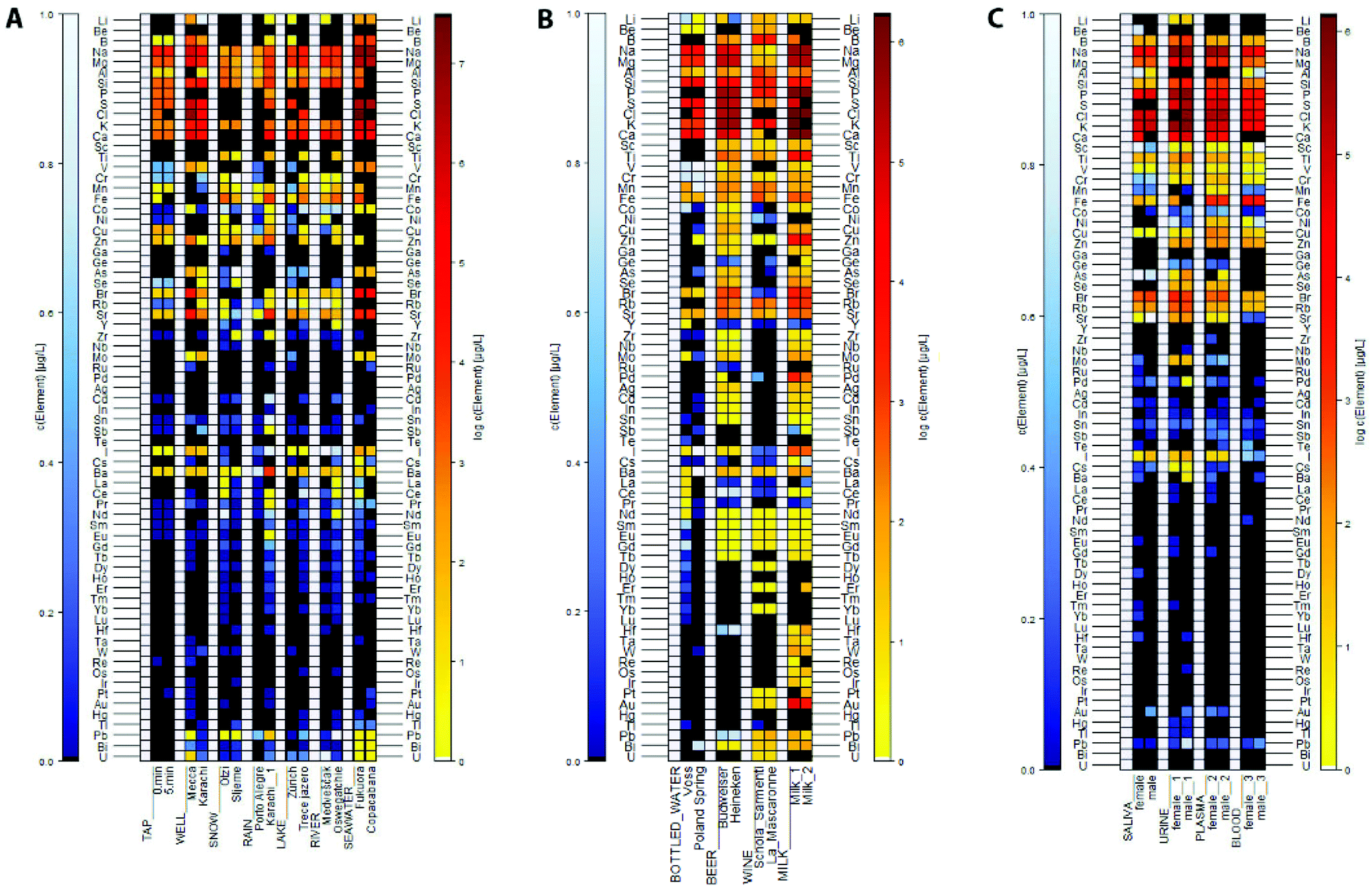 | ||
| Fig. 1 (A) Element concentration patterns in (environmental) water. Trace concentrations between ND (not detected, black) and 1 μg L−1 (white) are shown in shades of blue. Concentrations above 1 μg L−1 are represented by warm colours and given in a logarithmic scale and range from >1 μg L−1 (light yellow) to over 3600 mg L−1 (dark red); (B) element concentration patterns in beverages. Trace concentrations between ND (black) and 1 μg L−1 (white) are shown in shades of blue. Concentrations above 1 μg L−1 are represented by warm colours and given in a logarithmic scale and range from >1 μg L−1 (light yellow) to over 910 mg L−1 (dark red); (C) element concentration patterns in biofluids. Trace concentrations between ND (black) and 1 μg L−1 (white) are shown in shades of blue. Concentrations above 1 μg L−1 are represented by warm colours and given in a logarithmic scale and range from >1 μg L−1 (light yellow) to over 1300 mg L−1 (dark red). | ||
:40, concentrations of Na, Mg, S and Cl, still exceeded the set maximum calibration concentrations of 180 ppm (Na, Mg), 60 ppm (S) and 360 ppm (Cl). Therefore, data shown here refers to the 1:20 diluted sample. Seawater from Fukuora shows a high abundance of rare earth elements and higher concentrations of Co and Pb. In total, the Fukuora sample contains 41 elements, in comparison to the Copacabana sample (30).Elemental concentrations in environmental water were verified using ICAL, NIST 1640a CRM, Mix2, Mix3 and the mixture of single element reference materials Na, Mg, Si, P, S, and K (Multi). Twenty-two were covered by multiple controls, and with the exception of B and Fe, the controls were within the required ± 20% error margin. In samples of tap, well and rain waters, Lake Zurich and the Otzi snow sample, the low concentrations of iron may be slightly too high, as the lower concentrated control was measured at 125% of the reference concentration. Control concentrations of boron were at 67.6% only in one control and hence, the rivers, Treće Jezero and Sljeme samples might actually be higher, in contrast to iron, that has a control concentration of 168% above the anticipated concentration compared to NIST 1640a CRM (ESI Table S4.4†).
Beverage samples
Fig. 1B, ESI Tables S5.1 and 5.2.†Aluminium concentrations in both milk samples are reported to be above 400 mg L−1, these values should be interpreted with caution, as the control resulted in values 41% above the correct concentration.
Biofluid biological samples
Fig. 1C, ESI Tables S6.1 and 6.2.†Blood samples in comparison. Overall the male sample has higher elemental concentration in the plasma sample, whereas the female shows higher concentrations in the whole blood sample. In both sexes, Al is present in whole blood samples, yet not in plasma. Contrary, are Ca, Ge, As (in the male), Se, Mo, and Au present in plasma, but not in the whole blood samples.
Biofluid samples in comparison. Elements are more abundant in the female samples, with the exception of the whole blood sample. Elements that are not present are Ga, Ag, Pr, Sm, Tb, Ho, Er, Lu, Ta, W, Os, Ir Pt, Bi, and U. Overall, concentrations of elements are slightly higher in urine and elements are least concentrated in whole blood samples.
Discussion
Results presented here illustrate the potential for using the MH-ICP-MS calibration method to detect and quantify all elements present in an aqueous sample from 7Li to 238U simultaneously and in as little as 1–4 mL of aqueous sample.Choice of internal standards
The EPA Method 200.8 (ref. 38) suggests to use five internal standards to cover the mass range from 7Li to 238U, with a minimum of three. We elect to use only three elements as internal standards, 6Li, 103Rh and 232Th, since each element used as an internal standard cannot be determined in unknown samples, therefore reducing the number of measurable elements. We did not find that the reduction of internal standards affected elemental quantification, as our controls were within their ±20% margin.We also decided to deviate from the suggested elements and use a highly concentrated 6Li internal standard, even though lithium is a bi-isotope element (6Li 7.5% and 7Li 92.5% natural abundances) to cover the lower mass ranges. Experimental data shows that the fraction of naturally occurring lithium in environmental water on mass six is so little (even with high lithium concentrations) that it does not interfere with the 100 ppb 6Li internal standard. Although it is not known whether Rh and Th are naturally occurring in water samples, it can be expected that their concentrations are very low, and the influence of potential sample concentrations are negligible compared to the artificially added, highly concentrated isotopes within the internal standards. Even if the samples would contain Rh and/or Th, the additional amounts would be measurable and could be accounted for in the process of data analysis; e.g., Rh has been widely used in body fluid analysis.23
Choice of calibration standards
The combination of calibration standards used in this study represents the minimum number of standards necessary, without risking inaccurate concentration measurements due to peculiarities of inorganic chemistry, such as (1) a conflation of too many isotopes leading to polyatomic complexes, (2) elemental stability: we found that some combinations of mixes and single element standards resulted in precipitation, and (3) interferences (such as isobaric interferences, isobaric polyatomic interferences, abundance sensitivity, physical interferences, and memory interferences).3,8,10,21,38,53 However, the multi-element calibration standards were not adequate to account for all interferences that occur across the isotopic spectrum, and hence additional single element standards are necessary (see below).Elemental limitations
One limitation of the MH-ICP-MS calibration method is that it prohibits detection of elements with mass to charge ratios of less than 5 and more than 243. In addition, noble gases and fluorine (F) cannot be detected by our MH-ICP-MS instrument. Carbon, nitrogen and oxygen are common contaminants in argon gas and samples, which prohibits us from analysing these elements as well. Radioactive elements such as Tc, Pm, Po, At, Rn, Fr, Ra, Ac, Pa, Np, Pu, and Am are not measured, as their standards are available commercially only with special permits.Interferences
SPECTRO MS software can account for interferences that are caused by elements that have been calibrated for, and hence we cannot account for interferences caused by H, C, N and O, other than avoiding isotopes that are affected by those interfering elements (e.g. 56Fe, which is interfered by 40Ar16O+). Especially in alcoholic and biofluid solutions, these elements can cause non-spectral interferences, which can be accounted for by making calibration standard matrices to imitate the sample matrix (e.g. by adding EtOH into the calibration standards). Therewith, interferences would occur at similar rates in the calibration standards as well as the samples and affects can be mathematically corrected for. In general, we also cannot control for interferences caused by elements of the internal standard: 6Li together with hydrogen interferes on 7Li, to some degree. The amount of formed 6Li1H is being recorded by the software in the blank and subtracted from the measured 7Li cps. Other interferences caused by the internal standards were not detected.Isobaric interferences were avoided by choosing isotopes that are not affected by those interferences (e.g. measuring 42Ca, 43Ca and 44Ca, instead of its most abundant isotope 40Ca, on which 40Ar is an isobaric interference). However, isobaric interferences still occur on fifteen isotopes: 54Fe/Cr, 58Ni/Fe, 82Se/Kr, 86Sr/Kr, 102Ru/Pd, 115In/Sn, 128Te/Xe, 138Ba/La/Ce, 144Nd/Sm, 152Sm/Gd, 158Gd/Dy, 162Dy/Er, 184W/Os, 187Re/Os, 192Os/Pt. As Kr and Xe are noble gases, and potential contaminants in the carrier gas, they would be present to the same degree in the calibration standards and samples, and not affect the measurements. Assuming a natural abundance of isotopes, the percentage of, e.g. chromium, that will occur on 54Fe does not change and is accounted for by the SPECTRO MS software when calculating the elemental concentration of iron on mass 54. The same principal applies to all other isobaric interferences.
Polyatomic interferences are abundant and cannot be avoided, but they can be accounted for. When elements that interfere with a target isotope are calibrated in a differing calibration standard than the target isotope, the SPECTRO MS software calculates the respective interferer-factor. The factors caused by interfering isotopes are then used to adjust the calibration regression of the target isotope providing a mathematically corrected elemental concentration. To keep the number of calibration standards to a minimum we used thirty single element standards to investigate the possible impact of interferences that cannot arise from the multi-element calibration standards for each sample type. With that, we assume that the factors with which interferences influence the target isotopes do not change significantly between runs of the same sample types, given that instrument conditions do not change significantly either (e.g. in oxide molecular yields). We successfully achieved and maintain instrument stability by profound maintenance and optimization of the instrument.
The formation of doubly charged ions is minimized by our optimization procedure10,15,21,40 and monitored by the formation of BaO+/Ba+. Nevertheless, we still detected double charged strontium ions (on 44Ca) and some rare earth element (Ce, Nd, Sm, Gd, Dy, Er, Yb, and Lu) ions between 71Ga and 88Sr. These interferences were accounted for by establishing interference-factors, as described for polyatomic interferences.
Analytical limitations
For environmental water samples, bottled water, wine and beer, as well as saliva, urine and plasma, we assumed to meet the requirements of matrix similarities in calibration standards, blanks, controls and samples, as all samples have a water content of 95% or higher, especially given their dilutions. We neglected certified reference materials when analysing seawater, milk and whole blood samples, since our goal in this communication was predominantly to show the wide range of applications of the described method. When pursuing research on significant numbers of seawater, milk and whole blood analysis, we strongly recommend matching the matrices of calibration standards, blank and respective controls with the respective samples, as suggested.8,20,24,27,31,32,45,46,49,54–61 However, we did use controls for all of our sample types, but for alcoholic beverages we adjusted the control matrix to match alcohol content. Overall, the controls established were within the required ±20% margin of error. Interestingly, the aluminium control is measured higher than its actual concentration (in all runs but the environmental water run) and so is sulphur in biofluids and bottled water, the control concentration, however, being at 36% only.“Simultaneous” ICP-MS
No existing quadrupole-based se-ICP-MS is providing simultaneous detection and recording of multiple elements. There is thus some confusion in the literature regarding the use of the term “simultaneous”. Many articles use this term in their titles, yet they employ a conventional quadrupole mass spectrometer in their experimental design.5,9,11,13,14,21,35,62 What these investigators mean by the term simultaneous is storage and readout of multi-element data from the mass spectrometer using a multichannel analyser. Multi-element detection is still classical sequential ICP-MS, measuring one element at a time. For this reason, quantitative multi-element measurements on sequential ICP-MS instruments are somewhat constrained in the breadth of the inorganic spectrum that can be measured with any one or two multi-element calibration standards.2,4,6,8,10,12,15–19,22,23,31,42,45 The MH-ICP-MS technology and the method advanced in this study, in which six grouped standards are iteratively dispensed to calibrate the instrument, make simultaneous detection and measurement from 7Li to 238U possible.Samples
In respect of the aqueous samples chosen as exemplars in this study, patterns of elemental concentrations throughout the periodic table are clearly distinguishable between the various categories of water, beverage, and biofluid. The numbers of samples are small in this communication, as in this communication we only mean to apply the method to a variety of sample types. Our future investigations will meet statistical standards and generate more robust interpretations of the results.Environmental waters
Beverage samples
Concentrations among the beverages tested reveal considerable variability.Biofluid samples
Results show that some elements can only be detected in certain body fluids, e.g. rare earth elements are found in saliva and urine, but not in blood samples; Be only present in saliva and Li is in urine only. Investigating differences of elemental composition in different body fluids can help determine which body fluid would be best for detecting and measuring certain elements in the future. For example, elemental concentrations are reasonably high and common in saliva and urine, and hence these may be good alternatives over invasive blood sampling for having a comprehensive look at the inorganic elemental content in humans.Whether females, in general, possess higher amounts of elements in their bodily fluids when compared to males as shown here, needs further investigation. Differences found here may be consequences of age, lifestyle (diet, medication and exercise) or permanent (geographical) residence, rather than sex.
Due to their different components, we measured blood twice: the whole blood and the plasma. We would have expected higher concentrations, or at least similarly high concentrations of elements in the whole blood sample compared to the plasma sample, as plasma is an integral part of the whole blood sample. However, we only found this to be true for Fe, which is significantly higher in the whole blood samples than in plasma, as anticipated, since it is mainly present in erythrocytes. The lack of higher/similar concentrations of elements in the whole blood sample might be due to inadequate method design: The matrices of the calibration standards, the blank, and controls were not adjusted to the respective sample matrices (plasma, whole blood), as suggested elsewhere,31,32,45 and hence interferences might have caused inaccurate values.77 Furthermore, sample preparation might have been insufficient: others suggested to use Triton-X 100 as a detergent.25,31,45,46,77 We decided not to use any detergent to keep the samples as pure as possible, and to prevent contamination from additional chemicals. An alternative might be heated acid digestion using ultrapure HNO3.32 Furthermore, we noticed residue on the instrument after inserting the whole blood samples, which may have been prevented if using a detergent, or a higher dilution factor. This leads us to the assumption that some parts of the sample may have not been analysed. Adjustment of the calibration standards, etc. is probably of lesser importance for the plasma samples, as plasma consists of 95% water. Here, the matrices are much more alike, compared to the whole blood sample (with 44% cellular components) and the calibration standards, etc.
Although the water content is high in body fluids (saliva: 98%, urine: 91–96% and plasma: up to 95%), we suggest adapting the calibration standards matrices to the respective body fluid.31,32,45 Some studies use diluted body fluids as matrix in calibration standards and blanks.31,45 However, when aiming to quantify all elements between 7Li and 238U, in our opinion this approach is rather counterproductive. The NIST 1640 CRM has been used for blood samples,27 and we encourage the use of specific certified reference materials for blood,20,27,31,45,46 saliva,27,31,49, and urine24 in addition to the broad set of controls. Nevertheless, we have shown that it is possible to detect the entire range of elements from 7Li and 238U in blood samples.
Applicability of a Water Fingerprinting method
The MH-ICP-MS calibration method is essentially a “Water Fingerprinting” method, which is applicable to all aqueous samples and can be used to advance personal health in general, and public health in particular, because the monitoring of 71 elements and their patterns in water we consume and in biofluids is relatively easy, quick, and cost efficient. The method can be used to understand common concentration level ranges of each relevant inorganic element between 7Li and 238U, as well as element patterns in drinking water, beverages, and human biofluids, exceeding by far the number of commonly monitored elements. The mapping of elemental patterns in human bodily fluids might also be beneficial for the understanding of diseases, their origin, emergences, or manner of transmission. In combination with (tap) water analysis, the method may find its application in forensics: unique total elemental patterns in environmental water might be matched to unique total elemental blood, plasma, saliva or urine patterns. The digestion of hair and tissue samples or the extension of this method to solids by a developed laser ablation-MH-ICP-MS method, will further aid the identification of specific elemental concentration patterns in human tissues.The elemental mapping of concentration levels in bottled water and tap water will help to increase our understanding of “normal” concentration levels of all elements in water. Many elements are known to affect human health negatively, yet only 19 elements are monitored by the EPA.
Besides the method's applicability to (public) health, it may also find an application in “forensic” tracing of the origins of (bottled) water, wine, milk, beer, and other foods. Knowing the elemental composition of beverages and foods will increase our understanding of elemental distributions among consumed items (especially macro- and micronutrients) and be used to determine their origin.
Given that each water sample, may it come from rain, snow, a puddle, a river, stream, lake or the sea, has its own unique elemental concentration pattern, the Water Fingerprinting method enables global ecological stoichiometry, to map the world according to its elemental distributions and concentrations, hence revealing element flows through the environment.
Conclusions
In summary, the method facilitates the simultaneous capability of MH-ICP-MS technology for the quantification of 71 elements in aqueous solutions without the loss of elemental data by a single analytical run. The described method may be used to explore and understand the distribution of inorganic elements in aqueous solutions in the environment, those that we consume, and in biofluids beyond stereotypical inorganic analyses or organic analytics of carbon, nitrogen, and oxygen. With the MH-ICP-MS method it is possible to comprehensively engage the fields of ecological stoichiometry and metabolic ecology and contribute to many fields relating to states of health and the environment. Additionally, once elemental distribution and concentration patterns for specific environments have been established, the method can and should be applied to answer pending and vexing questions in fields that relate the present to the past, such as would relate to the study of the paleoenvironment and climate change.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Johannes Pfleging for obtaining the Otzi snow sample and Maham Khan for obtaining the Mecca water and Karachi rain samples. We thank Nick Michelson for help in creating the heat maps. The technical support of George Cisneros Jr and Emily Xu is also gratefully acknowledged, as is the method applications support of Willi Barger. Reviewer appreciation for the scope of the work is acknowledged, and critique was also greatly appreciated, which we used to significantly improve the paper. Research support was provided by the Max Planck Prize to TGB, endowed by the German Federal Ministry of Education and Research to the Max Planck Society and the Alexander von Humboldt Foundation. Further financial support came from the Human Microassay, Inc., Collierville, TN, USA to MB.References
- C. Ding, C. Zhu, D. Liu, M. Dong, A. Zhang, Y. Pan and H. Yan, Zhonghua Yufang Yixue Zazhi, 2012, 46, 745–749 CAS.
- A. Gonzalvez, S. Armenta, A. Pastor and M. De La Guardia, J. Agric. Food Chem., 2008, 56, 4943–4954 CrossRef CAS PubMed.
- J. M. Henshaw, E. M. Heithmar and T. A. Hinners, Anal. Chem., 1989, 61, 335–342 CrossRef CAS.
- M. Jabłońska-Czapla, S. Szopa, K. Grygoyć, A. Łyko and R. Michalski, Talanta, 2014, 120, 475–483 CrossRef PubMed.
- N. Khan, I. S. Jeong, I. M. Hwang, J. S. Kim, S. H. Choi, E. Y. Nho, J. Y. Choi, B.-M. Kwak, J.-H. Ahn and T. Yoon, Food Chem., 2013, 141, 3566–3570 CrossRef CAS PubMed.
- M. Krachler and W. Shotyk, Sci. Total Environ., 2009, 407, 1089–1096 CrossRef CAS PubMed.
- J. Kubová, V. Nevoral and V. Streško, J. Anal. At. Spectrom., 1994, 9, 241–243 RSC.
- P. Leonhard, R. Pepelnik, A. Prange, N. Yamada and T. Yamada, J. Anal. At. Spectrom., 2002, 17, 189–196 RSC.
- G. R. Loope, L. R. Kump and M. A. Arthur, Chem. Geol., 2013, 351, 195–208 CrossRef CAS.
- D. Pröfrock and A. Prange, Appl. Spectrosc., 2012, 66, 843–868 CrossRef PubMed.
- R. Rahil-Khazen, H. Henriksen, B. Bolann and R. Ulvik, Scand. J. Clin. Lab. Invest., 2000, 60, 677–686 CrossRef CAS PubMed.
- V. S. Šelih, M. Šala and V. Drgan, Food Chem., 2014, 153, 414–423 CrossRef PubMed.
- V. F. Taylor, H. P. Longerich and J. D. Greenough, J. Agric. Food Chem., 2003, 51, 856–860 CrossRef CAS PubMed.
- D. Yeghicheyan, C. Bossy, M. Bouhnik Le Coz, C. Douchet, G. Granier, A. Heimburger, F. Lacan, A. Lanzanova, T. C. Rousseau and J. L. Seidel, Geostand. Geoanal. Res., 2013, 37, 449–467 CrossRef CAS.
- J. De Boer, W. Verweij, T. Van Der Velde-Koerts and W. Mennes, Water Res., 1996, 30, 190–198 CrossRef CAS.
- H. Louie, C. Wong, Y. J. Huang and S. Fredrickson, Anal. Methods, 2012, 4, 522–529 RSC.
- K. J. Stetzenbach, M. Amano, D. K. Kreamer and V. F. Hodge, Groundwater, 1994, 32, 976–985 CrossRef CAS.
- N. Khan, I. S. Jeong, I. M. Hwang, J. S. Kim, S. H. Choi, E. Y. Nho, J. Y. Choi, K. S. Park and K. S. Kim, Food Chem., 2014, 147, 220–224 CrossRef CAS PubMed.
- N. Mohd-Taufek, D. Cartwright, M. Davies, A. K. Hewavitharana, P. Koorts, P. N. Shaw, R. Sumner, E. Lee and K. Whitfield, Food Anal. Methods, 2016, 9, 2068–2075 CrossRef.
- F. Barbosa, M. H. C. Rodrigues, M. R. Buzalaf, F. J. Krug, R. F. Gerlach and J. E. Tanus-Santos, Arch. Toxicol., 2006, 80, 633–637 CrossRef CAS PubMed.
- S. D'Ilio, N. Violante, M. Di Gregorio, O. Senofonte and F. Petrucci, Anal. Chim. Acta, 2006, 579, 202–208 CrossRef PubMed.
- R. Forrer, K. Gautschi and H. Lutz, Biol. Trace Elem. Res., 2001, 80, 77 CrossRef CAS PubMed.
- J.-P. Goullé, L. Mahieu, J. Castermant, N. Neveu, L. Bonneau, G. Lainé, D. Bouige and C. Lacroix, Forensic Sci. Int., 2005, 153, 39–44 CrossRef PubMed.
- P. Heitland and H. D. Köster, Clin. Chim. Acta, 2006, 365, 310–318 CrossRef CAS PubMed.
- D. Koh, V. Ng, L. Chua, Y. Yang, H. Ong and S. Chia, Occup. Environ. Med., 2003, 60, 696–698 CrossRef CAS PubMed.
- T. D. Lyon, G. S. Fell, R. C. Hutton and A. N. Eaton, J. Anal. At. Spectrom., 1988, 3, 265–271 RSC.
- J. Nriagu, B. Burt, A. Linder, A. Ismail and W. Sohn, Int. J. Hyg. Environ. Health, 2006, 209, 109–121 CrossRef CAS PubMed.
- I. Rodushkin and F. Ödman, J. Trace Elem. Med. Biol., 2001, 14, 241–247 CrossRef CAS.
- I. Rodushkin, F. Ödman and S. Branth, Fresenius J. Anal. Chem., 1999, 364, 338–346 CrossRef CAS.
- I. Rodushkin, F. Ödman, R. Olofsson and M. D. Axelsson, J. Anal. At. Spectrom., 2000, 15, 937–944 RSC.
- J. F. Staff, A.-H. Harding, J. Morton, K. Jones, E. A. Guice and T. McCormick, Toxicol. Lett., 2014, 231, 270–276 CrossRef CAS PubMed.
- M. Vaughan, A. Baines and D. Templeton, Clin. Chem., 1991, 37, 210–215 CAS.
- D. Wang, X. Du and W. Zheng, Toxicol. Lett., 2008, 176, 40–47 CrossRef CAS PubMed.
- S. Zhang, X. Zhuo and D. Ma, Fayixue Zazhi, 2012, 28, 456–460 CAS , 463.
- F. C. Bressy, G. B. Brito, I. S. Barbosa, L. S. Teixeira and M. G. A. Korn, Microchem. J., 2013, 109, 145–149 CrossRef CAS.
- C. D. Garbe-Schönberg, Geostand. Geoanal. Res., 1993, 17, 81–97 CrossRef.
- C. J. Kantipuly and A. D. Westland, Talanta, 1988, 35, 1–13 CrossRef CAS PubMed.
- S. Long and T. Martin, Determination of trace elements in waters and wastes by inductively coupled plasma-mass spectrometry: Method 200. 8. Version 4. 0, Environmental Protection Agency, Environmental Monitoring Systems Lab, Cincinnati, OH (USA), 1989 Search PubMed.
- WHO, Guidelines for drinking-water quality, Report 978-92-4-154815-1, 4th edn, 2011 Search PubMed.
- D. Ardelt, A. Polatajko, O. Primm and M. Reijnen, Anal. Bioanal.Chem., 2013, 405, 2987–2994 CrossRef CAS PubMed.
- G. D. Schilling, F. J. Andrade, J. H. Barnes, R. P. Sperline, M. B. Denton, C. J. Barinaga, D. W. Koppenaal and G. M. Hieftje, Anal. Chem., 2006, 78, 4319–4325 CrossRef CAS PubMed.
- J. Fernandez-Turiel, J. Llorens, F. López-Vera, C. Gómez-Artola, I. Morell and D. Gimeno, Fresenius J. Anal. Chem., 2000, 368, 601–606 CrossRef CAS PubMed.
- EPA, Drinking Water Standards and Health Advisories, Report EPA 822-S-12-001, U.S. Environmental Protection Agency, Washington, DC, 2012 Search PubMed.
- EU, Council Directive 98/83/EC on the quality of water intented for human consumption, Official Journal of the European Communities, Brussels, Belgium, 1998 Search PubMed.
- P. Heitland and H. D. Köster, J. Trace Elem. Med. Biol., 2006, 20, 253–262 CrossRef CAS PubMed.
- A. Schütz, I. A. Bergdahl, A. Ekholm and S. Skerfving, Occup. Environ. Med., 1996, 53, 736–740 CrossRef.
- C. Feldman, Anal. Chem., 1974, 46, 99–102 CrossRef CAS.
- Y. J. Kim, Y. K. Kim and H. S. Kho, Oral Dis., 2010, 16, 823–830 CrossRef PubMed.
- Z. T. Dame, F. Aziat, R. Mandal, R. Krishnamurthy, S. Bouatra, S. Borzouie, A. C. Guo, T. Sajed, L. Deng, H. Lin, P. Liu, E. Dong and D. S. Wishart, Metabolomics, 2015, 11, 1864–1883 CrossRef CAS.
- D. Wang, Y. Shimoda, S. Wang, Z. Wang, J. Liu, X. Liu, H. Jin, F. Gao, J. Tong and K. Yamanaka, Environ. Health Preventative Med., 2017, 22, 45 CrossRef PubMed.
- J. Morton, E. Leese, A.-H. Harding, K. Jones and O. Sepai, Biomonitoring, 2014, 1, 75–84 Search PubMed.
- OVAN, http://www.ovan.de, 2016.
- R. Thomas, Practical guide to ICP-MS: a tutorial for beginners, CRC Press, 2013 Search PubMed.
- A. Ataro, R. I. McCrindle, B. M. Botha, C. M. E. McCrindle and P. P. Ndibewu, Food Chem., 2008, 111, 243–248 CrossRef CAS.
- M. P. Field, J. T. Cullen and R. M. Sherrell, J. Anal. At. Spectrom., 1999, 14, 1425–1431 RSC.
- N. Herwig, K. Stephan, U. Panne, W. Pritzkow and J. Vogl, Food Chem., 2011, 124, 1223–1230 CrossRef CAS.
- F. F. Lopez, C. Cabrera, M. L. Lorenzo and M. C. Lopez, Sci. Total Environ., 1998, 220, 1–9 CrossRef CAS PubMed.
- F. A. R. Martino, M. a. L. F. Sánchez and A. Sanz-Medel, Anal. Chim. Acta, 2001, 442, 191–200 CrossRef CAS.
- A. Milne, W. Landing, M. Bizimis and P. Morton, Anal. Chim. Acta, 2010, 665, 200–207 CrossRef CAS PubMed.
- H. J. Reid, A. A. Bashammakh, P. S. Goodall, M. R. Landon, C. O'Connor and B. L. Sharp, Talanta, 2008, 75, 189–197 CAS.
- T. Sumida, T. Nakazato and H. Tao, Bunseki Kagaku, 2003, 52, 619–626 CrossRef.
- H. Peng, N. Zhang, M. He, B. Chen and B. Hu, Talanta, 2015, 131, 266–272 CrossRef CAS PubMed.
- M. Li, Z. Liu, Y. Chen and Y. Hai, Water Res., 2016, 106, 593–603 CrossRef CAS PubMed.
- V. V. Bhaskar and P. Rao, J. Atmos. Chem., 2017, 74, 23–53 CrossRef CAS.
- M. A. Arain, Z. Haque, N. Badar and N. Mughal, JPMA, J. Pak. Med. Assoc., 2009, 59, 270–274 Search PubMed.
- M. A. Jaleel, R. Noreen and A. Baseer, J. Ayub. Med. Coll. Abbottabad, 2001, 13, 12–15 CAS.
- M. A. M. Sharaf and A. M. Subyani, International Journal of Scientific and Engineering Research, 2011, 2, 1–14 Search PubMed.
- AMAP, Arctic Pollution 2002: Persistent Organic Pollutants, Heavy Metals, Radioactivity, Human Health, Changing Pathways, 2002 Search PubMed.
- E. Steinnes, T. Berg and H. T. Uggerud, Sci. Total Environ., 2011, 412, 351–358 CrossRef PubMed.
- L. Yuefang, L. Zhen, G. Cozzi, C. Turetta, C. Barbante, H. Ju and X. Longfei, Chemosphere, 2018 DOI:10.1016/j.chemosphere.2018.01.039.
- K. Budhavant, P. Rao, P. Safai, L. Granat and H. Rodhe, Atmos. Environ., 2014, 88, 59–65 CrossRef CAS.
- S. Tyagi, S. Tiwari, A. Mishra, A. Chatterjee and D. S. Bisht, Earth Science India, 2016, 9, 150–166 CrossRef.
- I. Anil, O. Alagha and F. Karaca, Air Qual. Atmos. Health, 2017, 10, 1193–1206 CrossRef CAS.
- B. M. Ghauri, M. I. Mirza, R. Richter, V. A. Dutkiewicz, A. Rusheed, A. R. Khan and L. Husain, Chemosphere: Global Change Sci., 2001, 3, 51–63 CrossRef CAS.
- D. Migliavacca, E. Teixeira, M. Pires and J. Fachel, Atmos. Environ., 2004, 38, 1641–1656 CrossRef CAS.
- D. Migliavacca, E. Teixeira, F. Wiegand, A. Machado and J. Sanchez, Atmos. Environ., 2005, 39, 1829–1844 CrossRef CAS.
- R. Chemnitzer, Analytik News, Direkte Bestimmung von Spurenelementen in Körperflüssigkeiten mittels ICP-Massenspektrometrie, Dr. Beyer Internet-Beratung, 2015 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07070a |
| This journal is © The Royal Society of Chemistry 2018 |