
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsight into trifluoromethylation – experimental electron density for Togni reagent I†‡
R. Wang a,
I. Kalfa and
U. Englert*b
a,
I. Kalfa and
U. Englert*b
aInstitute of Inorganic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany. E-mail: ullrich.englert@ac.rwth-aachen.de
bInstitute of Molecular Science, Shanxi University, Taiyuan, Shanxi 030006, P. R. China
First published on 5th October 2018
Abstract
3,3-Dimethyl-1-(trifluoromethyl)-1,3-dihydro-1-λ3,2-benziodoxole represents a popular reagent for trifluoromethylation. The σ hole on the hypervalent iodine atom in this “Togni reagent” is crucial for adduct formation between the reagent and a nucleophilic substrate. The electronic situation may be probed by high resolution X-ray diffraction: the experimental charge density thus derived shows that the short intermolecular contact of 3.0 Å between the iodine and a neighbouring oxygen atom is associated with a local charge depletion on the heavy halogen in the direction of the nucleophile and visible polarization of the O valence shell towards the iodine atom. In agreement with the expectation for λ3-iodanes, the intermolecular O⋯I–Caryl halogen bond deviates significantly from linearity.
A “halogen bond” denotes a short contact between a nucleophile acting as electron density donor and a (mostly heavy) halogen atom as electrophile;1,2 halogen bonds are a special of σ hole interactions.3–5 Such interactions do not only play an important role in crystal engineering;6–11 rather, the concept of a nucleophile approaching the σ hole of a neighbouring atom may also prove helpful for understanding chemical reactivity.
The title compound provides an example for such a σ hole based reactivity: 3,3-dimethyl-1-(trifluoromethyl)-1,3-dihydro-1-λ3,2-benziodoxole, 1, (Scheme 1) commonly known as “Togni reagent I”, is used for the electrophilic transfer of a trifluoromethyl group by reductive elimination. The original articles in which the application of 112 and other closely related “Togni reagents”13 were communicated have been and still are highly cited. Trifluoromethylation is not the only application for hypervalent iodine compounds; they have also been used as alkynylating14 or azide transfer reagents.15,16 The syntheses of hypervalent iodanes and their application in organofluorine chemistry have been reviewed,17 and a special issues of the Journal of Organic Chemistry has been dedicated to Hypervalent Iodine Catalysis and Reagents.18 Recently, Pietrasiak and Togni have expanded the concept of hypervalent reagents to tellurium.19
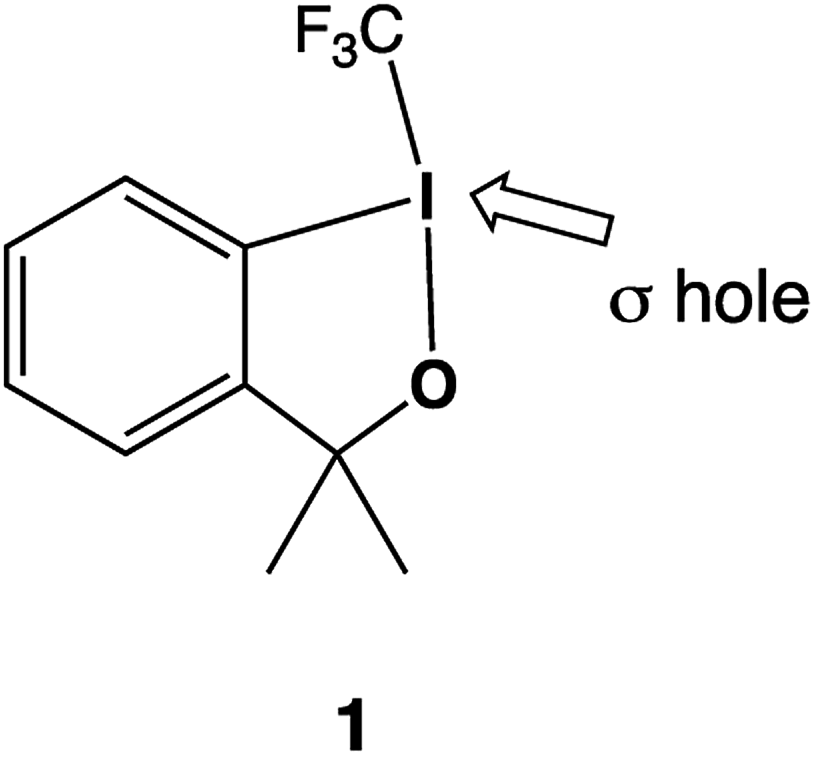 | ||
| Scheme 1 Lewis structure of the Togni reagent, 1. | ||
Results from theory link σ hole interactions and chemical properties and indicate that the electron density distribution associated with the hypervalent iodine atom in 1 is essential for the reactivity of the molecule in trifluoromethylation.20 Lüthi and coworkers have studied solvent effects and shown that activation entropy and volume play relevant roles for assigning the correct reaction mechanism to trifluoromethylation via 1. These authors have confirmed the dominant role of reductive elimination and hence the relevance of the σ hole interaction for the reactivity of 1 in solution by ab initio molecular dynamics (AIMD) simulations.21,22 An experimental approach to the electron density may complement theoretical calculations: low temperature X-ray diffraction data of sufficient resolution allow to obtain the experimental charge density and associate it with intra- and inter-molecular interactions.23–25 Such advanced structure models based on aspherical scattering factors have also been applied in the study of halogen bonds.26–30 In this contribution, we provide direct experimental information for the electronic situation in Togni reagent I, 1; in particular, we analyze the charge distribution around the hypervalent iodine atom.
Excellent single crystals of the title compound were grown by sublimation.§ The so-called independent atom model (IAM), i.e. the structure model based on conventional spherical scattering factors for neutral atoms, confirms the solid state structure reported by the original authors,12 albeit with increased accuracy. As depicted in Fig. 1, two molecules of 1 interact via a crystallographic center of inversion. The pair of short intermolecular O⋯I contacts thus generated can be perceived as red areas on the interaction-sensitive Hirshfeld surface.31
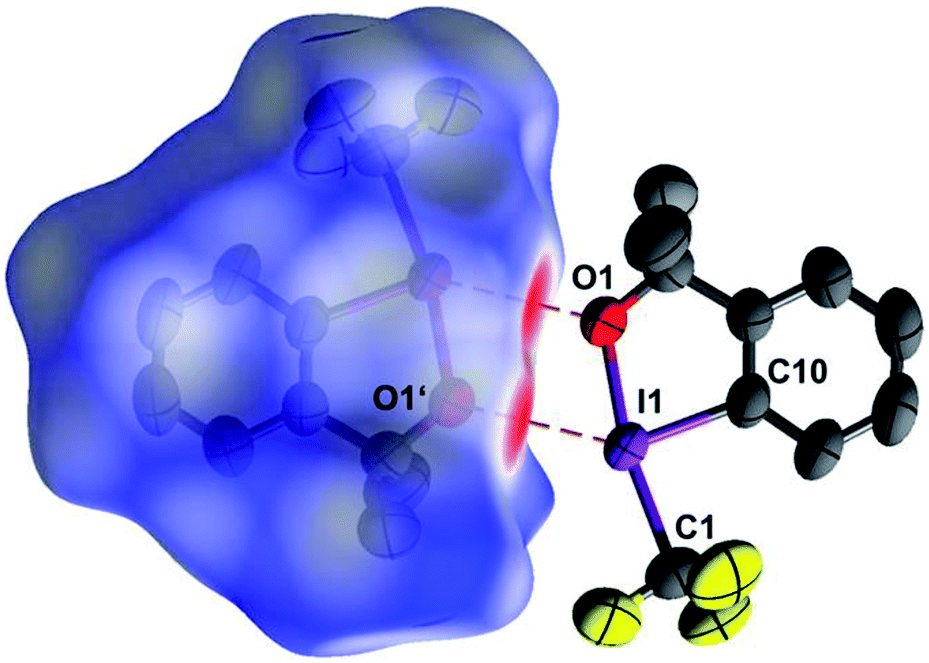 | ||
| Fig. 1 Two neighbouring molecules of 1, related by a crystallographic center of inversion. The short intermolecular I⋯O contacts show up in red on the Hirshfeld surface32 enclosing the left molecule. (90% probability ellipsoids, H atoms omitted, symmetry operator 1 − x, 1 − y, 1 − z). | ||
The high resolution of our diffraction data ¶ for 1 allowed an atom-centered multipole refinement33,34 and thus an improved model for the experimental electron density which takes features of chemical bonding and lone pairs into account.
Fig. 2 shows the deformation density, i.e. the difference electron density between this advanced multipole model and the IAM in the same orientation as Fig. 1.|| The orientation of an oxygen lone pair (blue arrow) pointing towards the σ hole of the heavy halogen in the inversion-related molecule and the region of positive charge at this iodine atom (red arrow) are clearly visible. Single-bonded terminal halides are associated with one σ hole opposite to the only σ bond, thus resulting in a linear arrangement about the halogen atom. Different geometries and potentially more than a single σ hole are to be expected for λ3-iodanes such as our target molecule, and as a tendency, the resulting halogen bonds are expected to be weaker than those subtended by single-bonded iodine atoms.35 In agreement with these theoretical considerations, the closest I⋯O contacts in 1 amount to 2.9822(9) Å. This distance is significantly shorter than the sum of the van-der-Waals radii (I, 1.98 Å; O, 1.52 Å (ref. 36)) but cannot compete with the shortest halogen bonds between iodine and oxygen37,38 or iodine and nitrogen.39–41 Fig. 1 and 2 show that the Caryl–I⋯O contacts are not linear; they subtend an angle C10–I1⋯O1′ of 141.23(3)° at the iodine atom. On the basis of theoretical calculations, Kirshenboim and Kozuch35 have suggested that the split σ holes should be situated in the plane of the three substituents of the hypervalent atom and that halogen and covalent bonds should be coplanar. Fig. 3 shows that the Caryl–I⋯O interaction in 1 closely matches this expectation, with the next oxygen neighbour O1′ only 0.47 Å out of the least-squares plane through the heavy halogen I1 and its three covalently bonded partners C1, O1 and C10.
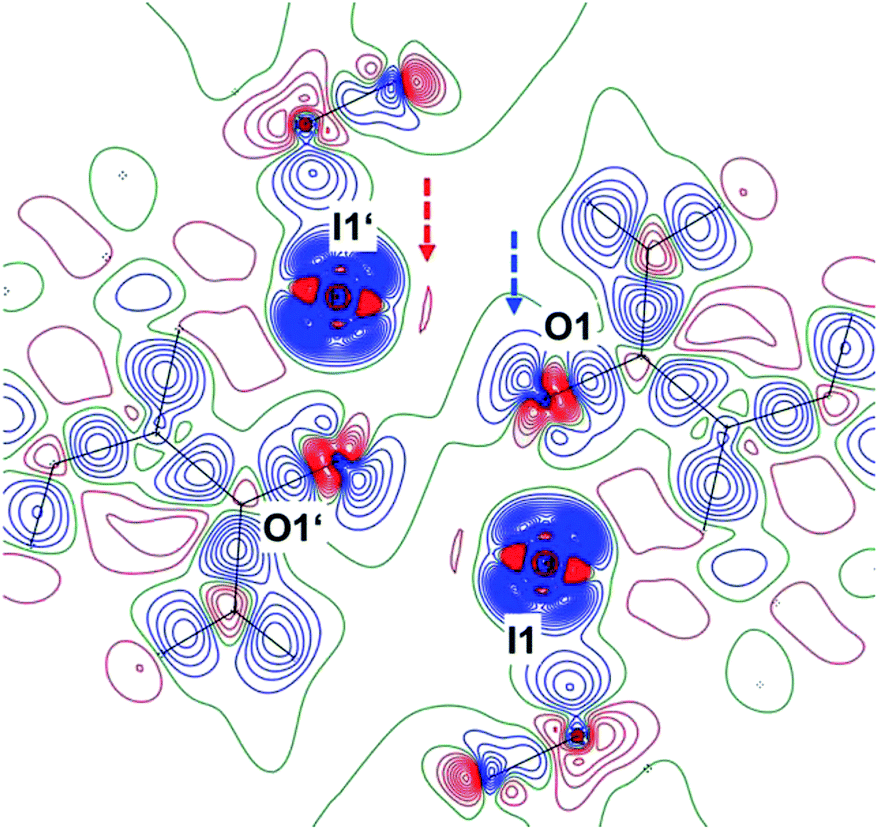 | ||
| Fig. 2 Deformation density for the pair of neighbouring molecules in 1; the dashed blue and red arrows indicate regions of opposite charge. (Contour interval 0.10e Å−3; blue lines positive, red lines negative, green lines zero contours, symmetry operator 1 − x, 1 − y, 1 − z). | ||
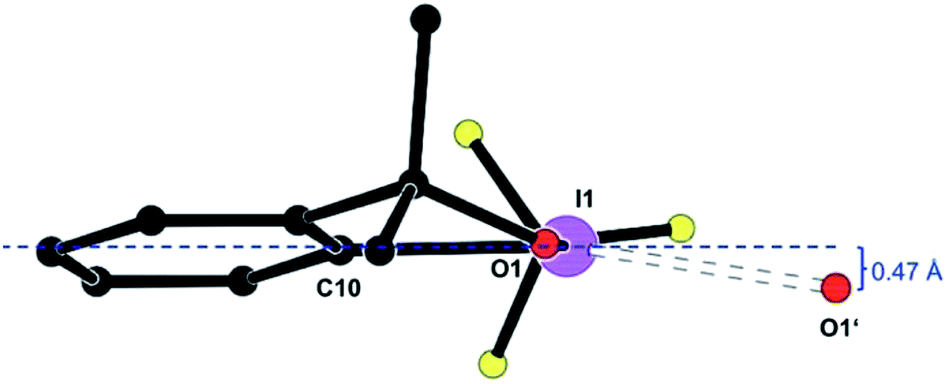 | ||
| Fig. 3 A molecule of 1, shown [platon] along O1⋯C1, and its halogen-bonded neighbour O1i. Symmetry operator 1 − x, 1 − y, 1 − z. | ||
The Laplacian, the scalar derivative of the gradient vector field of the electron density, emphasizes local charge accumulations and depletions and it allows to assess the character of intra- and inter-molecular interactions. A detailed analysis of all bonds in 1 according to Bader's Atoms In Molecules theory42 is provided in the ESI.‡ We here only mention that the electron density in the bond critical point (bcp) of the short intermolecular I⋯O contact amounts to 0.102(5)e Å−3; we are not aware of charge density studies on λ3-iodanes, but both the electron density and its small positive Laplacian match values experimentally observed for halogen bonds involving O and terminal I in the same distance range.43
The crystal structure of 1 necessarily implies additional contacts beyond the short halogen bond shown in Fig. 1 and 2. The shortest among these secondary interactions is depicted in Fig. 4: it involved a non-classical C–H⋯F contact with a H⋯F distance of 2.55 Å.
 | ||
| Fig. 4 C–H⋯F contact in 1; additional information has been compiled in the ESI.‡ Symmetry operators i = 1 − x, 1 − y, 1 − z; ii = 1 − x, 1 − y, −z. | ||
The topological analysis of the experimental charge density reveals that this non-classical C–H⋯F hydrogen bond and all other secondary contacts are only associated with very small electron densities in the bcps. Table S8 in the ESI‡ provides a summary of this analysis and confirms that the I⋯O halogen bond discussed in Fig. 1 and 2 represents by far the most relevant intermolecular interaction.
The relevance of this halogen bond extends beyond the crystal structure of 1: Insight into the spatial disposition of electrophilic and nucleophilic regions and hence into the expected reactivity of a molecule may be gained from another electron-density derived property, the electrostatic potential (ESP). The ESP for the pair of interacting molecules in 1 is depicted in Fig. 5.
 | ||
| Fig. 5 Electrostatic potential for a pair of molecules in 1 mapped on an electron density isosurface (ρ = 0.5e Å−3; program MoleCoolQt44,45). | ||
Fig. 5 underlines the complementary electrostatic interactions between the positively charged iodine and the negatively charged oxygen atoms. One can easily imagine to “replace” the inversion-related partner molecule in crystalline 1 by an incoming nucleophile.
The ESP tentatively obtained for a single molecule in the structure of 1 did not differ significantly from that derived for the inversion-related pair (Fig. 5), and even the results from theoretical calculations in the gas phase for an isolated molecule20 are in good qualitative agreement with our ESP derived from the crystal structure. In the absence of very short contacts, polarization by neighbouring molecules only has a minor influence on the ESP. The experimentally observed electron density matches the proven reactivity for the title compound, and we consider it rewarding to extend our charge density studies on related hypervalent reagents.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge support by Deutsche Forschungsgemeinschaft for their project EN309/10, “Charge density of halogen bonds and interhalogen contacts”.Notes and references
- O. Hassel, Science, 1970, 170, 497–502 CrossRef CAS PubMed.
- P. Metrangolo and G. Resnati, Chem.–Eur. J., 2001, 7, 2511–2519 CrossRef CAS.
- T. Brinck, J. S. Murray and P. Politzer, Int. J. Quantum Chem., 1992, 44(suppl 19), 57–64 CrossRef.
- T. Brinck, J. S. Murray and P. Politzer, Int. J. Quantum Chem., 1993, 48(suppl 20), 73–88 CrossRef CAS.
- P. Politzer, J. S. Murray, T. Clark and G. Resnati, Phys. Chem. Chem. Phys., 2017, 19, 32166–32178 RSC.
- G. R. Desiraju and R. Parthasarathy, J. Am. Chem. Soc., 1989, 111, 8725–8726 CrossRef CAS.
- F. Zordan, L. Brammer and P. Sherwood, J. Am. Chem. Soc., 2005, 127, 5979–5989 CrossRef CAS PubMed.
- F. Zordan, S. L. Purver, H. Adams and L. Brammer, CrystEngComm, 2005, 7, 350–354 RSC.
- F. C. Pigge, V. R. Vangala and D. C. Swenson, Chem. Commun., 2006, 2123–2125 RSC.
- D. S. Yufit, R. Zubatyuk, O. V. Shishkin and J. A. K. Howard, CrystEngComm, 2012, 14, 8222–8227 RSC.
- G. M. Espallargas, A. Recuenco, F. M. Romero, L. Brammer and S. Libri, CrystEngComm, 2012, 14, 6381–6383 RSC.
- I. Kieltsch, P. Eisenberger and A. Togni, Angew. Chem., Int. Ed., 2007, 46, 754–757 CrossRef CAS PubMed.
- P. Eisenberger, S. Gischig and A. Togni, Chem.–Eur. J., 2006, 12, 2579–2586 CrossRef CAS PubMed.
- J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165–4179 RSC.
- V. V. Zhdankin, C. J. Kuehl, A. P. Krasutsky, M. S. Formaneck and J. T. Bolz, Tetrahedron Lett., 1994, 35, 9677–9680 CrossRef CAS.
- M. Tiffner, L. Stockhammer, J. Schörgenhumer, K. Röser and M. Waser, Molecules, 2018, 23, 1142, DOI:10.3390/molecules23051142.
- J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650–682 CrossRef CAS PubMed.
- V. V. Zhdankin and K. Muñiz, J. Org. Chem., 2017, 82, 11667–11668 CrossRef CAS PubMed.
- E. Pietrasiak and A. Togni, Organometallics, 2017, 36, 3750–3757 CrossRef CAS.
- H. Pinto De Magalhães, A. Togni and H. P. Lüthi, J. Org. Chem., 2017, 82, 11799–11805 CrossRef PubMed.
- O. Sala, H. P. Lüthi and A. Togni, J. Comput. Chem., 2014, 35, 2122–2131 CrossRef CAS PubMed.
- O. Sala, H. P. Lüthi, A. Togni, M. Iannuzzi and J. Hutter, J. Comput. Chem., 2015, 36, 785–794 CrossRef CAS PubMed.
- P. Coppens, X-ray Charge Densities and Chemical Bonding, Oxford University Press, 1997 Search PubMed.
- P. Luger, Org. Biomol. Chem., 2007, 5, 2529–2540 RSC.
- D. Stalke, Chem.–Eur. J., 2011, 17, 9264–9278 CrossRef CAS PubMed.
- R. Bianchi, A. Forni and T. Pilati, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 559–568 CrossRef PubMed.
- R. H. Venkatesha and T. N. Guru Row, J. Phys. Chem. A, 2010, 114, 13434–13441 CrossRef PubMed.
- R. Wang, T. S. Dols, C. W. Lehmann and U. Englert, Chem. Commun., 2012, 48, 6830–6832 RSC.
- R. Wang, T. S. Dols, C. W. Lehmann and U. Englert, Z. Anorg. Allg. Chem., 2013, 639, 1933–1939 CrossRef CAS.
- A. Wang, R. Wang, I. Kalf, A. Dreier, C. W. Lehmann and U. Englert, Cryst. Growth Des., 2017, 17, 2357–2364 CrossRef CAS.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129 CrossRef CAS.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University of Western Australia, 2017 Search PubMed.
- N. K. Hansen and P. Coppens, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1978, 34, 909–921 CrossRef.
- A. Volkov, P. Macchi, L. J. Farrugia, C. Gatti, P. R. Mallinson, T. Richter and T. Koritsanszky, XD2006, University of New York at Buffalo, USA, 2006 Search PubMed.
- O. Kirshenboim and S. Kozuch, J. Phys. Chem. A, 2016, 120, 9431–9445 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- R. Bianchi, A. Forni and T. Pilati, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 559–568 CrossRef PubMed.
- J. N. Moorthy, K. Senapati and K. N. Parida, J. Org. Chem., 2010, 75, 8416–8421 CrossRef CAS PubMed.
- R. Bianchi, A. Forni and T. Pilati, Chem.–Eur. J., 2003, 9, 1631–1638 CrossRef CAS PubMed.
- L. C. Roper, C. Präsang, V. N. Kozhevnikov, A. C. Whitwood, P. B. Karadakov and D. W. Bruce, Cryst. Growth Des., 2010, 10, 3710–3720 CrossRef CAS.
- R. Wang, D. Hartnick and U. Englert, Z. Kristallogr. - Cryst. Mater., 233 DOI:10.1515/zkri-2018-2069.
- R. F. W. Bader, Atoms in Molecules - a Quantum Theory, Clarendon Press, 1990 Search PubMed.
- C. Merkens, F. Pan and U. Englert, CrystEngComm, 2013, 15, 8153–8158 RSC.
- C. B. Hübschle and P. Luger, J. Appl. Crystallogr., 2006, 39, 901–904 CrossRef.
- C. B. Hübschle and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 238–240 CrossRef PubMed.
Footnotes |
| † Dedicated to Prof. Dietmar Stalke on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Quality indicators associated with the experimental charge density study, topological analysis of the electron density, structural information in CIF format for both IAM and MM structure models. CCDC 1862413 and 1862414. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra07187j |
| § In an oil bath, ca. 10 mg of 1 were heated in a Schlenk tube to 75 °C under a slow stream of dry N2. Transparent crystals in the shape of elongated blocks formed at the glass wall immediately above the heated zone. |
¶ Key quality criteria for the diffraction experiment at 100 K: resolution sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θmax/λ = 1.10 Å−1, 151509 reflections, 12366 independent reflections, R = 0.022, wR2 = 0.055 for the IAM; R = 0.019, wR2 = 0.051 for the MM. θmax/λ = 1.10 Å−1, 151509 reflections, 12366 independent reflections, R = 0.022, wR2 = 0.055 for the IAM; R = 0.019, wR2 = 0.051 for the MM. |
| || CCDC-1862413 and 1862414 contain the crystallographic data for the IAM and the MM structure model, respectively. |
| This journal is © The Royal Society of Chemistry 2018 |