
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn efficient and atom-economical route to N-aryl amino alcohols from primary amines†
Zhen Xiaoa,
Juanjuan Lia,
Qiang Yuea,
Qian Zhanga and
Dong Li *ab
*ab
aSchool of Materials and Chemical Engineering, Hubei University of Technology, Wuhan 430068, China. E-mail: dongli@mail.hbut.edu.cn
bHubei Key Laboratory of Drug Synthesis and Optimization, Jingchu University of Technology, Jingmen 448000, China
First published on 5th October 2018
Abstract
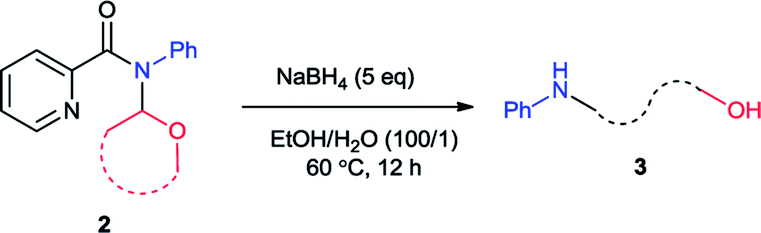
In this paper we reported a novel method for generation of N-aryl amino alcohols from N,N-disubstituted picolinamides through reduction/ring-opening reaction with NaBH4. The N,N-disubstituted picolinamides can be easily obtained from primary amines after convenient condensation with picolinic acid and coupling with cyclic ethers. The whole route proceeded under simple and mild conditions with high efficiency. Picolinic acid can be recovered in the form of piconol after reaction. It indicated an efficient and atom-economical route for the preparation of N-aryl amino alcohols from primary amines.
Introduction
Amino alcohols, which contain both an amine and an alcohol functional groups, are an important class of organic compounds. They are extensively used as solvents, ligands, plasticizers, agrochemicals, pharmaceuticals and also organic synthetic intermediates.1 Among them, the N-substituted linear alkanolamines have attracted much attention for a long time. The copper-catalyzed Ullmann type C–N coupling between aryl halides and primary alkanolamines served as a typical route for preparation of N-aryl amino alcohols.2 These methods required the used of amino alcohols as substrates which are normally not readily available. The catalytic hydrogenation of N-aryl cyclic amides provided an atom-economical route for the access of N-aryl amino alcohols. In 2011, Bergens and Ikariya groups independently reported efficient ruthenium-catalyzed hydrogenation of N-aryl pyrrolidinone and piperidinone which generate N-aryl-4-amino-1-butanol and pentanol respectively (Scheme 1a).3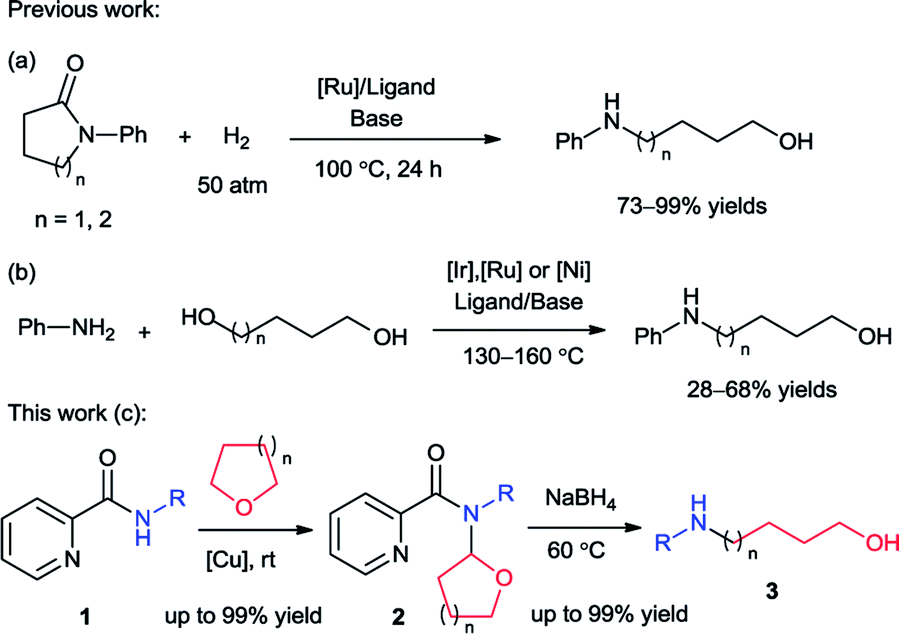 | ||
| Scheme 1 Preparation of N-arylamino-1-butanol/pentanol. | ||
However the requirement of expensive ruthenium catalysts, complicated ligands and harsh reaction conditions (high pressure and elevated temperature) might limit its synthetic applications. Another reaction to synthesize N-aryl-4-amino-1-butanol was reported in the same year which revealed an iridium-catalyzed alkylation of primary amines with 1,4-butanediol under microwave-assisted conditions.4 Recently, ruthenium and nickel catalysts were also developed for this reaction. With 1,5-pentanediol it can provide N-aryl-5-amino-1-pentanol as well (Scheme 1b).5 In these methods, additional ligand and base, high reaction temperature (130–160 °C) were still necessary and the reaction efficiency was unsatisfactory (28–68% yields).
Recently, our group developed an efficient copper-catalyzed C–N cross dehydrogenative coupling between picolinamides (2) and simple ethers which provided the N,N-disubstituted picolinamides (2).6 During the study of the coupling product, it was found that this compound converted to N-phenyl-4-amino-1-butanol (3) solely with sodium borohydride (NaBH4) (Scheme 1c). Sodium borohydride is a very common and useful reductant which be widely used for reducing many organic carbonyls, both in the laboratory and on a technical scale. Normally it is efficient for reduction of acyl chlorides, anhydrides, ketones, aldehydes and imines. Esters and carboxylic acids react slowly and inefficiently while amides are generally not reduced at all.7 Furthermore, typical reduction of tertiary amides commonly provide tertiary or secondary amines.8 This unexpected result propelled us to further exploration. After extensive study, herein we reported the synthesis of a series of N-aryl amino alcohols from the reduction/ring-opening of N,N-disubstituted picolinamide which can be easily obtained from coupling between N-aryl picolinamides and cyclic ethers. The N-aryl picolinamides were prepared from condensation of picolinic acid and primary amines. The whole route proceeded under simple and mild conditions with high efficiency. Picolinic acid can be recovered in the form of piconol after reaction which enabled the method meet the requirement of atom-economy.9 It indicated an efficient and atom-economical route for the preparation of N-aryl amino alcohols from primary amines.
Results and discussion
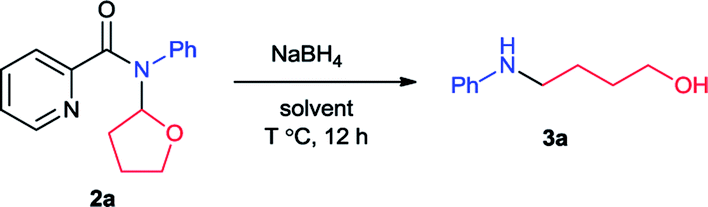
Initially we carried out the reaction of N-(2-tetrahydrofuranyl)-picolinamide (2a) with 2 equiv. of NaBH4 in 2 mL EtOH at room temperature (25 °C). After 12 hours, the desired product 3a was isolated in only 32% yield (Table 1, entry 1). Thus we increased the amount of NaBH4 to pursue higher conversion. With 3, 4 and 5 equiv. of NaBH4, the product yields became 36, 56 and 73% respectively (entries 2–4). Further increasing of the NaBH4 amount did not produce higher yield (entry 5). With 5 equiv. of NaBH4, we modulated the reaction temperature next. The product yield was improved to 75% under 40 °C (entry 6). Further elevation of the reaction temperature provided the product in 78% yield under 60 °C and 81% under 80 °C (entries 7 and 8). Subsequently we examined the effect of reaction concentration. To our delight, the product yields were generally promoted with higher concentration which was 76, 83 and 82% at 40, 60 and 80 °C respectively (entries 9–11). Among them, the reaction at 60 °C exhibited the best result. A surprising result was found that small amount of H2O could facilitate the reaction effectively. With 0.1 mL H2O, the reaction yield was improved to 94% (entry 12). Alteration of the H2O amount to 0.01 mL led to completely full conversion (entry 13). We also conducted the reaction at room temperature and 84% yield of product was still generated (entries 14). From the above, we decided the best reaction conditions as shown in entry 13.
|
||||
|---|---|---|---|---|
| Entry | NaBH4 (eq.) | Solvent (mL) | T (°C) | Yieldb (%) |
| a Reactions were performed using 1a (0.2 mmol) in solvent with NaBH4 for 12 h.b Isolated yield. | ||||
| 1 | 2 | EtOH (2) | rt | 32 |
| 2 | 3 | EtOH (2) | rt | 36 |
| 3 | 4 | EtOH (2) | rt | 56 |
| 4 | 5 | EtOH (2) | rt | 73 |
| 5 | 6 | EtOH (2) | rt | 73 |
| 6 | 5 | EtOH (2) | 40 | 75 |
| 7 | 5 | EtOH (2) | 60 | 78 |
| 8 | 5 | EtOH (2) | 80 | 81 |
| 9 | 5 | EtOH (1) | 40 | 76 |
| 10 | 5 | EtOH (1) | 60 | 83 |
| 11 | 5 | EtOH (1) | 80 | 82 |
| 12 | 5 | EtOH/H2O:10/1 (1) | 60 | 94 |
| 13 | 5 | EtOH/H2O:100/1 (1) | 60 | 99 |
| 14 | 5 | EtOH/H2O:100/1 (1) | rt | 84 |
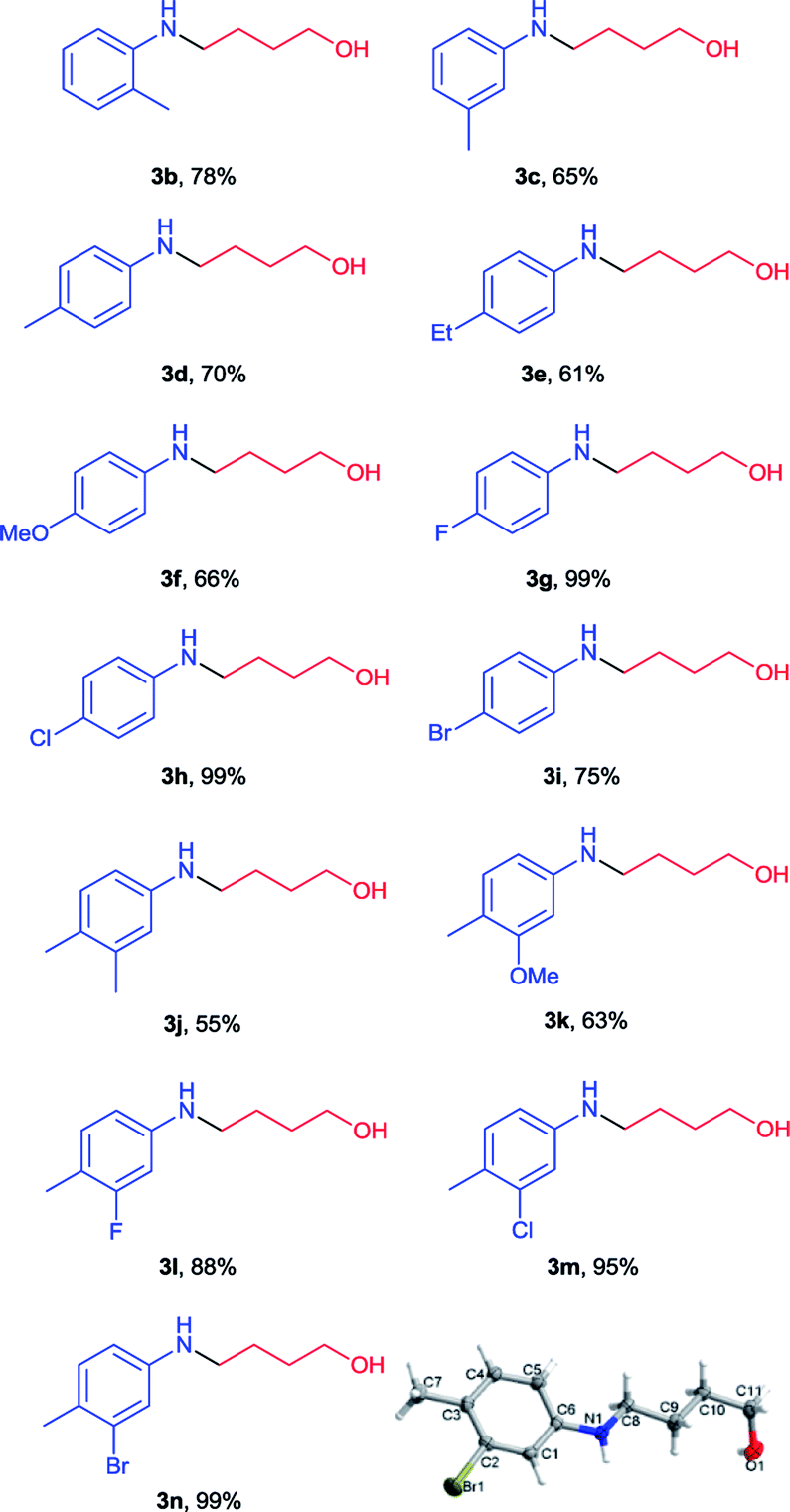
After optimization of reaction conditions, we applied this method for a series of N-(2-tetrahydrofuranyl)-picolinamides (2) which were obtained in our previous work. As shown in Table 2, all the desired N-aryl-4-amino-1-butanols were generated in good to excellent yields (61–99%) (3b–3n). Functional groups such as OMe, F, Cl and Br were tolerated on the benzene ring. There was no obvious steric effect observed as the 4-(o-tolylamino)-1-butanol (3b), 4-(m-tolylamino)-1-butanol (3c) and 4-(p-tolylamino)-1-butanol (3d) were all isolated in roughly similar yields. But electron-withdrawing group substituted substrates (3g–3i) provided better results than those with electron-donating substituents (3d–3f). Among them the 4-(4-fluorophenyl)amino-1-butanol (3g) and 4-(4-chlorophenyl)amino-1-butanol (3h) were obtained in almost quantitative amount. Multi-substituted substrates were also examined. With different combination of electron-withdrawing and electron-donating substituents, the corresponding products were all generated in high yields (3j–3n). 4-(3-Bromo-4-methylphenyl)amino-1-butanol (3n) was also received in perfect yield and its structure was unambiguously confirmed by X-ray crystallography.10 Unfortunately N-alkyl picolinamides were not applicable in this method and the results were not shown.
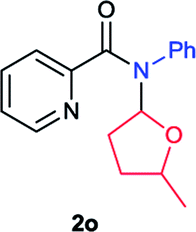
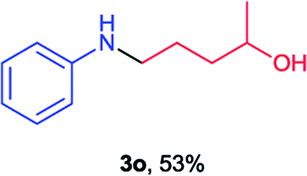
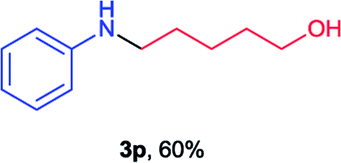
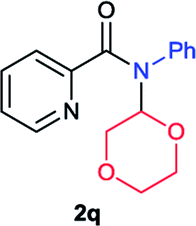
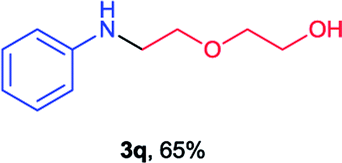
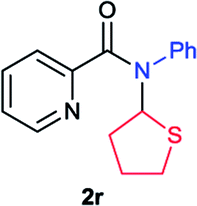
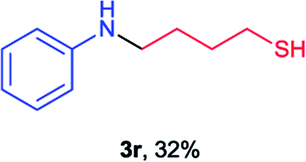
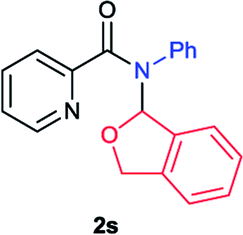
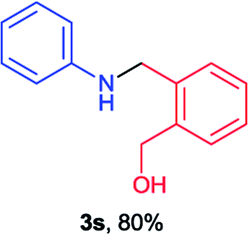
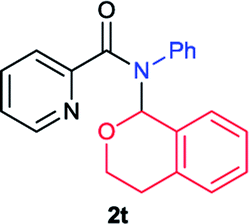
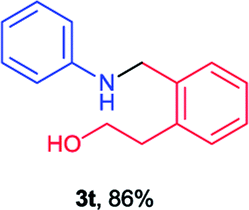
Next the N,N-disubstituted picolinamides containing other cyclic ether moieties were examined for this reaction and the results were showed in Table 3. The 2-methyltetrahydrofuran ring in compound 2o also cleaved during reaction to give the branched 5-phenylamino-2-pentanol (3o) in 53% yield. Compounds containing six-membered tetrahydropyrane and 1,6-dioxane ring (2p and 2q) proceeded to form corresponding 5-phenylamino-1-pentanol (3p) and 2-(2-(phenylamino)ethoxy)ethanol (3q) in good yields respectively. Cyclic thioether such as tetrahydrothiophene derived substrate (2r) was also applicable which provided the amino thiol product (3r). Notably, substrates prepared from benzo-cyclic ethers such as phthalan (2s) and isochroman (2t) were successfully converted to 2-phenylaminomethyl phenyl alcohols (3s and 3t) in high yields, which possessed application potential for various relative substrates and could undergo further transformations.
To examine the synthetic application of our method, we attempted to prepare a T-type calcium channel antagonist candidate 411 from 4-methoxyaniline on gram-scale (Scheme 2). Condensation of 4-methoxyaniline and picolinic acid generated the N-phenylpicolinamide 1f. Copper-catalyzed cross coupling of 1f with THF provided the N-(2-tetrahydrofuranyl)-picolinamides 2f, which was subsequently transferred to 4-(4-methoxyphenylamino)-1-butanol 3f through our method. So far all the atoms of 4-methoxyaniline and THF entered compound 3f except two hydrogens. Along with this transformation 70% of piconol was also recovered. Finally, an acid-catalyzed condensation of 3f and commercial available 4-chlorobenzhydrol afforded product 4 in 86% yield (68% overall yield). Thus, it demonstrated an efficient and atom-economic route for the preparation of amino alcohol/ether from primary amine.
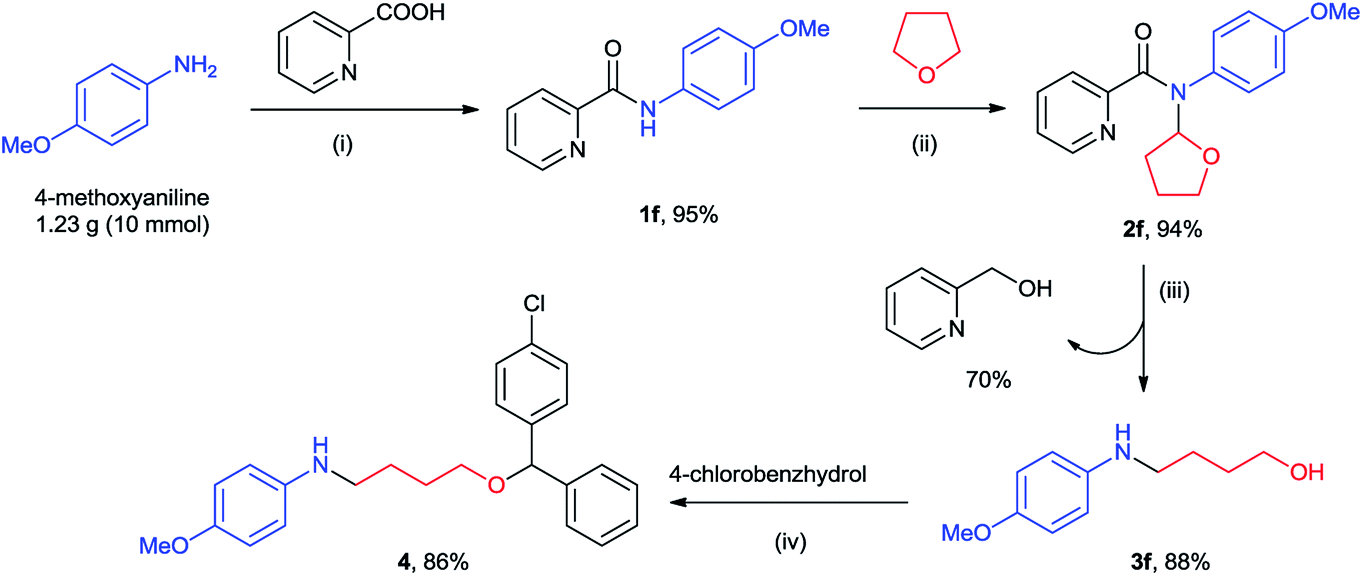 | ||
| Scheme 2 Gram-scale synthesis of a T-type calcium channel antagonist (4) from 4-methoxyaniline. Reaction conditions: (i) POCl3, Et3N, DCM, rt, 2 h; (ii) Cu(OAc)2/1,10-phenanthroline, TBHP, THF, rt, 8 h; (iii) NaBH4, EtOH/H2O, 60 °C, 12 h; (iv) p-TsOH, toluene, reflux. | ||
Although detailed reaction mechanism must await further investigation, based on the above results and literature survey12 we have suggested a plausible mechanism as shown in Scheme 3. With the activation of 2-tetrahydrofuranyl group, picolinamide (2) was first attacked by hydride to form intermediate A. Anion-induced isomerization of intermediate A led to the release of a pyridylaldehyde and cleavage of the tetrahydrofuran ring, generating intermediate B which would be protonated to imino alcohol C. Further reduction of pyridylaldehyde and C under reaction conditions gave piconol and amino alcohol 3 respectively. It is also explained the requirement of excess amount of NaBH4 in this reaction.
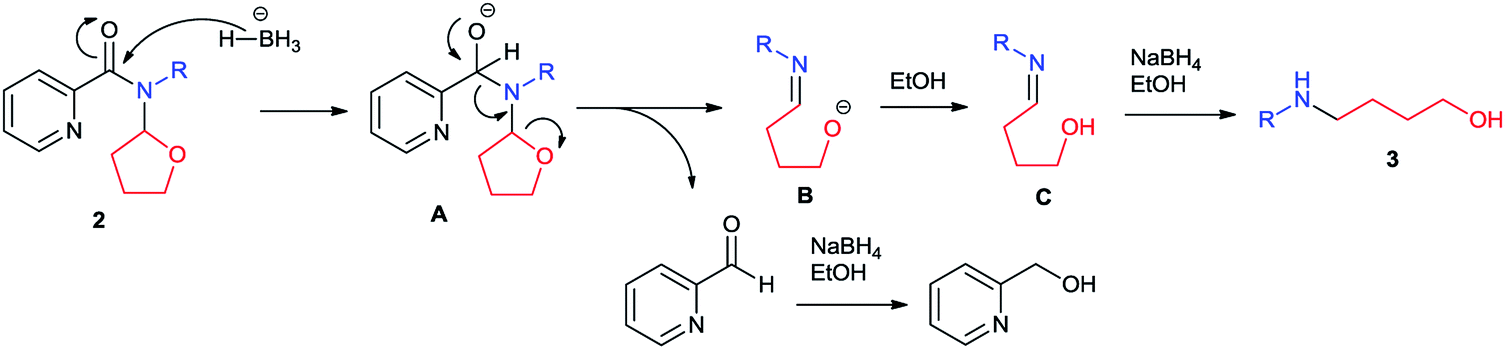 | ||
| Scheme 3 Plausible mechanism. | ||
Conclusions
In summary, we have developed a novel method for generation of N-aryl amino alcohols from N,N-disubstituted picolinamides through reduction/ring-opening reaction with NaBH4. It can also be treat as an efficient and atom-economical route for the preparation of N-aryl amino alcohols from primary amines, as the N,N-disubstituted picolinamides can be easily obtained from primary amines after convenient condensation with picolinic acid and coupling with cyclic ethers. Each step showed high yields and the picolinic acid can be recovered in the form of piconol after reaction. Thus this method exhibited excellent efficiency and atom-economy, which suggest that it should be useful in complex molecule synthesis and related synthetic applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (NSFC, No. 21702054) and the open project program of Hubei Key Laboratory of Drug Synthesis and Optimization, Jingchu University of Technology (OPP2016ZD01) for financial support.Notes and references
- (a) M. Frauenkron, J.-P. Melder, G. Ruider, R. Rossbacher and H. Höke, Ethanolamines and Propanolamines, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2002 Search PubMed; (b) J. Takehara, S. Hashiguchi, A. Fujii, S. Inoue, T. Ikariya and R. Noyori, Chem. Commun., 1996, 233 RSC; (c) C. Palomo, M. Oiarbide and A. Laso, Angew. Chem., Int. Ed., 2005, 44, 3881 CrossRef CAS PubMed; (d) R. W. Marquis, A. M. Lago, J. F. Callahan, R. E. L. Trout, M. Gowen, E. G. DelMar, B. C. V. Wagenen, S. Logan, S. Shimizu, J. Fox, E. F. Nemeth, Z. Yang, T. Roethke, B. R. Smith, K. W. Ward, J. Lee, R. M. Keenan and P. Bhatnagar, J. Med. Chem., 2009, 52, 3982 CrossRef CAS PubMed.
- (a) H. Yin, M. Jin, W. Chen, C. Chen, L. Zheng, P. Wei and S. Han, Tetrahedron Lett., 2012, 53, 1265 CrossRef CAS; (b) D. Chen, K. Yang, H. Xiang and S. Jiang, Tetrahedron Lett., 2012, 53, 7121 CrossRef CAS.
- (a) M. Ito, T. Ootsuka, R. Watari, A. Shiibashi, A. Himizu and T. Ikariya, J. Am. Chem. Soc., 2011, 133, 4240 CrossRef CAS PubMed; (b) J. M. John and S. H. Bergens, Angew. Chem., Int. Ed., 2011, 50, 10377 CrossRef CAS PubMed.
- W. Zhang, X. Dong and W. Zhao, Org. Lett., 2011, 13, 5386 CrossRef CAS PubMed.
- (a) F.-L. Yang, Y.-H. Wang, Y.-F. Ni, X. Gao, B. Song, X. Zhu and X.-Q. Hao, Eur. J. Org. Chem., 2017, 3481 CrossRef CAS; (b) M. Vellakkaran, K. Singh and D. Banerjee, ACS Catal., 2017, 7, 8152 CrossRef CAS.
- Q. Yue, Z. Xiao, Z. Kuang, Z. Su, Q. Zhang and D. Li, Adv. Synth. Catal., 2018, 360, 1193 CrossRef CAS.
- L. Banfi, E. Narisano, R. Riva, N. Stiasni, M. Hiersemann, T. Yamada and T. Tsubo, Sodium Borohydride, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd., 2014 Search PubMed.
- For recent examples: (a) D. H. O'Donovan, C. D. Fusco and D. R. Spring, Tetrahedron Lett., 2016, 57, 2962 CrossRef; (b) D. Mukherjee, S. Shirase, K. Mashima and J. Okuda, Angew. Chem., Int. Ed., 2016, 55, 13326 CrossRef CAS PubMed; (c) R. C. Chadwick, V. Kardelis, P. Lim and A. Adronov, J. Org. Chem., 2014, 79, 7728 CrossRef CAS PubMed; (d) W. Xie, M. Zhao and C. Cui, Organometallics, 2013, 32, 7440 CrossRef CAS; (e) J. A. Fernández-Salas, S. Manzini and S. P. Nolan, Chem. Commun., 2013, 49, 9758 RSC.
- (a) B. M. Trost, Science, 1991, 254, 1471 CrossRef CAS PubMed; (b) B. M. Trost, Angew. Chem., Int. Ed., 1995, 34, 259 CrossRef CAS.
- CCDC-1858880..
- W. F. McCalmont, J. R. Patterson, M. A. Lindenmuth, T. N. Heady, D. M. Haverstick, L. S. Grayb and T. L. Macdonald, Bioorg. Med. Chem., 2005, 13, 3821 CrossRef CAS PubMed.
- (a) M. Periasamy and M. Thirumalaikumar, J. Organomet. Chem., 2000, 609, 137 CrossRef CAS; (b) M. V. N. de Souza and T. R. A. Vasconcelos, Appl. Organomet. Chem., 2006, 20, 798 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and analytical data, and copies of NMR spectra. CCDC 1858880. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra07355d |
| This journal is © The Royal Society of Chemistry 2018 |