DOI:
10.1039/C8RA07539E
(Paper)
RSC Adv., 2018,
8, 39957-39966
Targeted delivery of paclitaxel by functionalized selenium nanoparticles for anticancer therapy through ROS-mediated signaling pathways
Received
10th September 2018
, Accepted 7th November 2018
First published on 30th November 2018
Abstract
As a therapeutic anticancer agent, the clinical use of paclitaxel (PTX) is limited by its poor water solubility and serious adverse side effects. The targeted-specific intracellular delivery of an anticancer drug as a new therapeutic modality is promising for cancer treatment. The anticancer activity of selenium nanoparticles (SeNPs) with low toxicity and excellent activity has attracted increasing attention for use in biomedical intervention in recent years. In this study, β-cyclodextrin (β-CD)-folate (FA)-modified selenium nanoparticles (SeNPs) loaded with paclitaxel (PTX) (Se@β-CD-FA@PTX) were successfully fabricated through a layer-by-layer method. The nanosystem is able to enter cancer cells through FA receptor-mediated endocytosis to achieve targeted-specific intracellular delivery. Se@β-CD-FA@PTX was found to increase the selectivity between normal and cancer cells. The viability in MCF-7 cells was remarkably lower than in MCF 10A cells, which may promote the specific targeted delivery of Se@β-CD-FA@PTX into MCF-7 cells. Moreover, Se@β-CD-FA@PTX was found to enhance the cytotoxic effect on MCF-7 cells via the induction of apoptosis activation of ROS-mediated p53 and AKT signaling pathways. The results demonstrate that Se@β-CD-FA@PTX nanoparticles provide a strategy for the design of cancer-targeted nanosystems for use in cancer therapy.
1 Introduction
According to the World Health Organization (WHO), breast cancer remains the leading diagnosed cancer that causes death in women worldwide.1 It has been reported that millions of new cases are detected and over 450![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 women die from breast cancer each year.2 A multidisciplinary approach is used to treat breast cancer, combining radiation, surgical and medical oncology. Surgery combined with chemotherapy and radiation is used to deal with the early stages of breast cancer.3,4 Of these, chemotherapy is widely used and has been established as a vital treatment for women with breast cancer.5 Paclitaxel is a microtubule-stabilizing agent that inhibits cancer cell proliferation and induces apoptosis6 and has been used in the treatment of several types of solid tumors, such as those in breast, colon, ovarian, head, lung, neck and liver cancers.7–12 Compared to conventional anticancer drugs, paclitaxel disrupts the tubulin–microtubule equilibrium to induce cancer cell apoptosis.13 However, the major problem for the clinical efficacy of paclitaxel is that it is limited by its poor water solubility and serious adverse side effects.14 Resistance is the greatest obstacle concerned with using paclitaxel to treat breast cancer.15
000 women die from breast cancer each year.2 A multidisciplinary approach is used to treat breast cancer, combining radiation, surgical and medical oncology. Surgery combined with chemotherapy and radiation is used to deal with the early stages of breast cancer.3,4 Of these, chemotherapy is widely used and has been established as a vital treatment for women with breast cancer.5 Paclitaxel is a microtubule-stabilizing agent that inhibits cancer cell proliferation and induces apoptosis6 and has been used in the treatment of several types of solid tumors, such as those in breast, colon, ovarian, head, lung, neck and liver cancers.7–12 Compared to conventional anticancer drugs, paclitaxel disrupts the tubulin–microtubule equilibrium to induce cancer cell apoptosis.13 However, the major problem for the clinical efficacy of paclitaxel is that it is limited by its poor water solubility and serious adverse side effects.14 Resistance is the greatest obstacle concerned with using paclitaxel to treat breast cancer.15
To overcome multidrug resistance, nanotechnology plays an important role in cancer therapy.16 It improves the delivery of drugs and strengthens the targeting in anticancer applications.17 Nanomaterials have peculiar properties, they are smart, highly stabile, have controllable morphologies and can be surface fictionalized. For these reasons, they are considered to be a promising solution to deal with resistance.18,19 As a special selenium species that operates as a drug carrier, selenium nanoparticles have attracted much attention in drug development.20 Furthermore, SeNPs have gained wide attention due to their potent activity, including antibacterial, antiviral, antiangiogenic, antifungal and anti-inflammatory properties.21–24 β-Cyclodextrin is a widely used compound in medicine due to its ready availability and suitable cavity size25 and its ability to complex a wide range of drugs.26,27 As a targeting ligand, folate has a greater affinity toward folate receptors, which are overexpressed in many cancer cell lines.28–31 To obtain selective antitumor activity, we developed β-CD-FA-modified selenium nanoparticles loaded with paclitaxel (Se@β-CD-FA@PTX) and used them to induce breast cancer cell apoptosis.
Reactive oxygen species (ROS) are one of the most important factors in cell apoptosis.32,33 ROS are generated in several cellular systems, such as peroxisomes, cytosol, plasma membrane, the membranes of mitochondria and in endoplasmic reticulum.34 The imbalance between ROS generation and antioxidant system could trigger cancer.35 Therefore, we hypothesized that Se@β-CD-FA@PTX could act as a novel chemotherapeutic agent to achieve cancer-targeted therapy. The anticancer mechanisms of Se@β-CD-FA@PTX involving ROS were also explored.
2 Experimental
2.1 Materials
The MCF-7 cells (ATCC® HTB-22™) and MCF 10A cells (ATCC® CRL-10317™) were obtained from ATCC (Manassas, VA, US). MCF 10A was used as a normal cell model. Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco. β-cyclodextrin (β-CD), folate (FA), paclitaxel (PTX), and Na2SeO3 were obtained from Sigma-Aldrich. Capase-3, P53, P-AKT, T-AKT, and β-actin antibodies were obtained from CST (Boston, MA, USA). Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and caspase-3 assay kits were acquired from the Beyotime Institute of Biotechnology (Shanghai, People's Republic of China).
2.2 Preparation and characterization of Se@β-CD-FA@PTX
Briefly, 3.405 g of β-CD and 1.323 g of FA were dissolved in 30 ml of dried DMSO, and then 0.14 g of DCC and 0.12 g of NHS were added to the reaction mixture at room temperature, which was then stirred for 24 h. The product was isolated by filtration and was then freeze-dried. β-CD-FA was obtained as the product. SeNPs were prepared as follows: briefly, 2 ml of a vitamin C solution (50 mM) was added dropwise into 0.25 ml of Na2SeO3 (0.1 M) under magnetic stirring for 2 h. Then, SeNPs were mixed in 3.792 mg of β-CD-FA and 0.32 ml of 6 mg ml−1 PTX with Milli-Q water to make up a final volume of 25 ml. The Se@β-CD-FA@PTX complex was purified by dialysis overnight. The Se@β-CD-FA@PTX nanoparticles were sonicated and filtered through a 0.2 μm pore size filter. The SeNPs, Se@β-CD-FA, and Se@β-CD-FA@PTX were characterized using a transmission electron microscope (TEM, H-7650 Hitachi) and a Zetasizer Nano ZS (Malvern Instruments Limited) particle analyzer was used to measure the particle zeta potential and size distribution.
2.3 Cell culture and viability assay
The MCF-7 and MCF 10A cells were maintained in DMEM blended with an antibiotic, fetal bovine serum (10%), penicillin (100 units per ml) and streptomycin (50 units per ml) at 37 °C in 5% CO2. The proliferative inhibition of MCF-7 cells by Se@β-CD-FA@PTX nanoparticles was measured through MTT assay, as previously described.36 Briefly, the cells were treated with a control, SeNPs, Se@β-CD-FA, PTX and Se@β-CD-FA@PTX at a density of 4 × 104 cells per well. Then, to each well, 20 μl of MTT solution was added and then the cells were incubated for 5 h. The cell viability was read as percentages of MTT reduction relative to the 570 nm absorbance by a microplate reader. The loading capacity of PTX was 60%. PTX = 0.32 ml × 6 mg ml−1 = 1.92 mg. 60% of 1.92 mg = 1.152 mg. The concentration of Se@β-CD-FA@PTX was 50 μg ml−1 and PTX was 8 μg ml−1. Se@β-CD-FA@PTX conjugated at a PTX dose of 8 μg ml−1. One mg of Se@β-CD-FA@PTX contains 0.16 mg PTX (8 μg ml−1 × 1 mg/50 μg ml−1 = 0.16 mg). The total yielded product of Se@β-CD-FA@PTX was 7.2 mg (1.152 × 1 mg/0.16 mg = 7.2 mg).
2.4 Localization and cellular uptake of Se@β-CD-FA@PTX
The localization of Se@β-CD-FA@PTX by 6-coumarin labeled MCF-7 cells was monitored by DAPI (4',6-diamidino-2-phenyindole) and LysoTracker Red, as previously described.37 The most important cellular uptake pathway for extracellular nanoparticles is endocytosis. Se@β-CD-FA@PTX was located in MCF-7 cells and marked by DAPI (cell nucleus) and LysoTracker (lysosome). MCF-7 cells were incubated with 6-coumarin-loaded Se@β-CD-FA@PTX for various periods of time, cultured on a cover glass in 6-well plates and then observed under a fluorescence microscope. Endocytosis inhibitors were used to investigate the mechanisms of the cellular uptake of Se@β-CD-FA@PTX in MCF-7 cells.
2.5 Mitochondrial membrane potential (ΔΨm)
The change in the mitochondrial membrane potential in MCF-7 cells exposed to Se@β-CD-FA@PTX was detected by JC-1, as previously reported.38,39 The cells were harvested by centrifugation, resuspended in PBS and analyzed by flow cytometry. The percentage of the green fluorescence from the JC-1 monomers was used to represent the cells that lost ΔΨm.
2.6 TUNEL – DAPI staining
DNA fragmentation in MCF-7 induced by Se@β-CD-FA@PTX was detected using a TUNEL apoptosis detection kit.40 Briefly, MCF-7 cells were fixed with formaldehyde (3.7%) and then incubated with 0.1% Triton X-100 in PBS. The MCF-7 cells were blended with TUNEL for 1 h. Then, cells were incubated with 1 μg ml−1 of DAPI for nuclear staining at 37 °C. After 30 min, the MCF-7 cells were rinsed with PBS and detected under a fluorescence microscope (Nikon Eclipse 80i).
2.7 Caspase-3 activity
The caspase-3 activity was determined using a fluorometric method, as previous reported.41 Harvested cells were suspended in a cell lysis buffer on ice for 1 h, followed by centrifugation for 10 min at 10000g. The caspase activity was determined from the fluorescence intensity using caspase-3 substrates with a 380 nm excitation wavelength and 460 nm emission wavelength, respectively.
2.8 ROS generation
ROS accumulation by Se@β-CD-FA@PTX treated MCF-7 cells was detected by staining the cells by DCF fluorescence assay.42,43 Briefly, after treatment with Se@β-CD-FA@PTX, the MCF-7 cells were suspended in PBS containing 10 μM of 2′,7′-dichlorofluorescein diacetate. ROS generation was measured using a microplate reader with an excitation wavelength of 488 nm and emission wavelength of 525 nm, respectively.
2.9 Western blot analysis
Western blotting was used to examine the expression of proteins by Se@β-CD-FA@PTX in MCF-7 cells, as previously reported.44,45 Briefly, the total cellular proteins were obtained from MCF-7 cells treated by Se@β-CD-FA@PTX with a lysis buffer. A BCA kit was used to detect the amount of protein. The proteins were observed by ECL chemiluminescence in solution and examined on X-ray film.
2.10 MCF-7 cell xenograft mice model
32 female nude mice were injected in the right armpit region with MCF-7 cells. After that, the tumor-bearing mice were randomly divided into a control group and three treatment groups. Se@β-CD-FA@PTX was injected into the tumor-bearing mice at dosages of 4 mg kg−1, 8 mg kg−1 and 12 mg kg−1 through intravenous injection every 2 days and this continued for 16 days. Then, the tumor-bearing mice were sacrificed, and the tumor and body weights were measured. All mice experiments were approved and guided by the ethics committee of Guangzhou Medical University and Experimental Animal Center of Guangzhou Medical University.
2.11 Statistical analysis
Experiments were performed at least three times. All data were processed using the SPSS 19.0 software (IBM Corporation, Armonk, NY, USA). A probability of P < 0.05 (*) or P < 0.01 (**) indicates statistically significant values.
3 Results and discussion
3.1 Preparation and characterization of Se@β-CD-FA@PTX
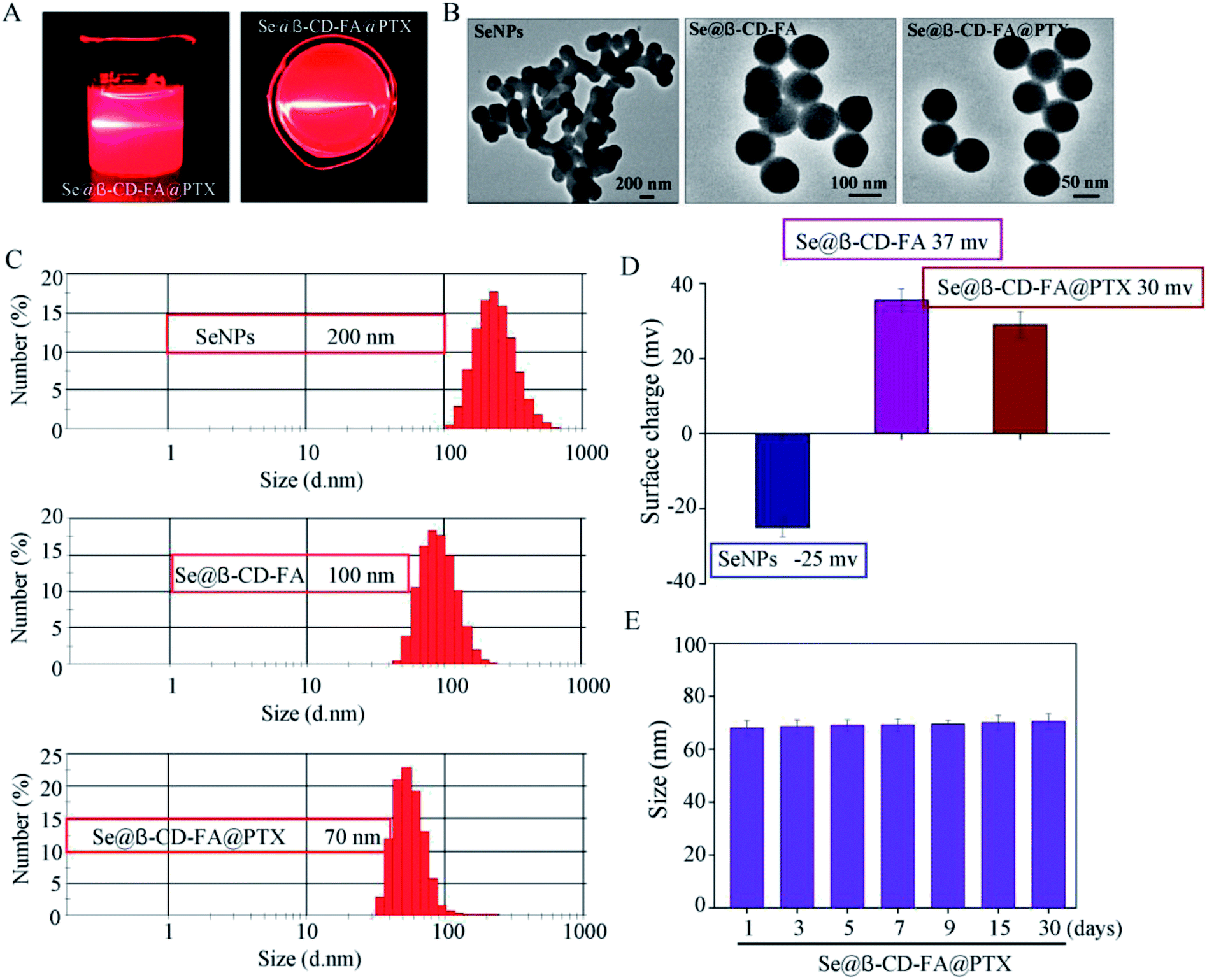
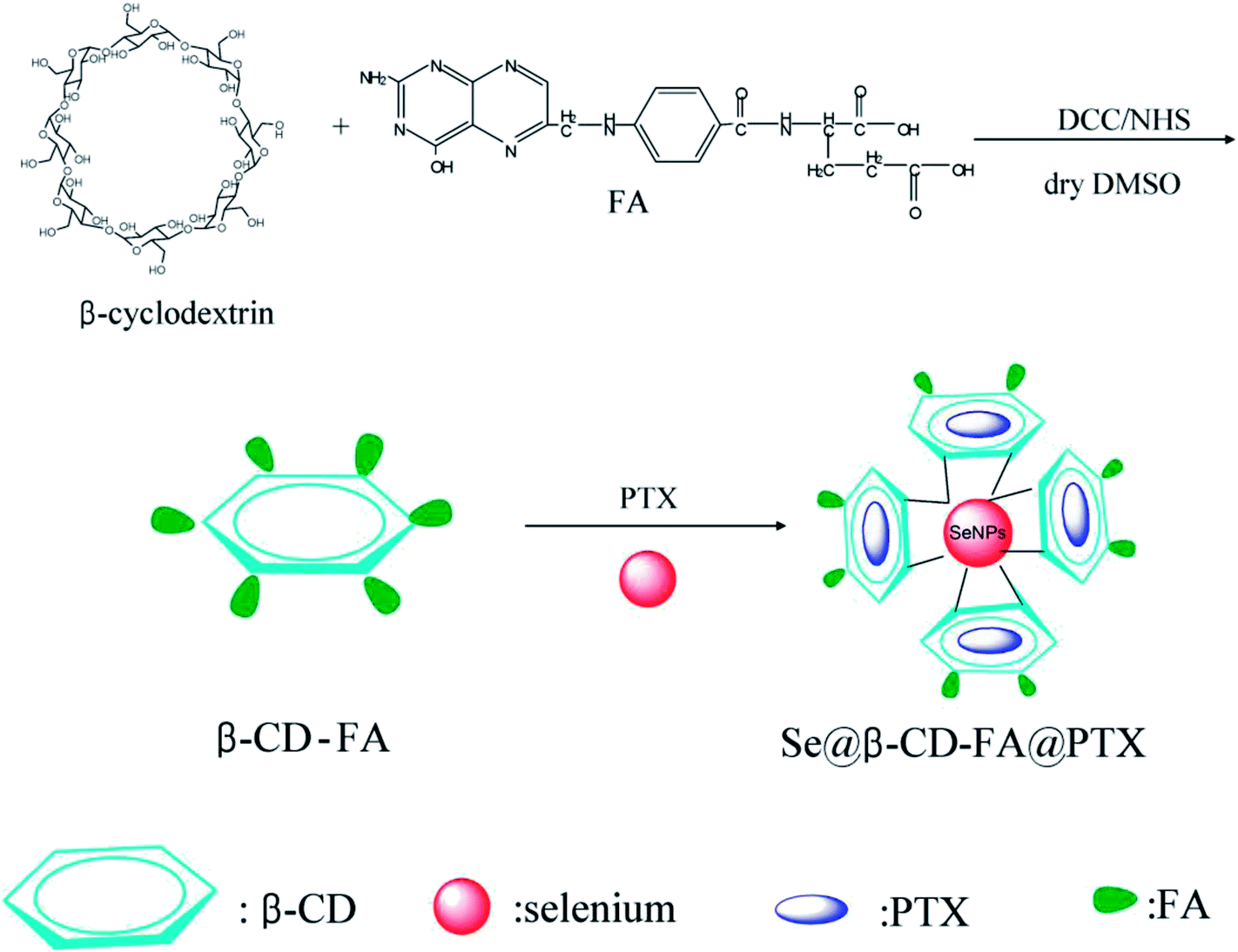
The morphology and stability of Se@β-CD-FA@PTX was determined and analyzed using various methods. As shown in Fig. 1A, the Tyndall effect of Se@β-CD-FA@PTX indicated that Se@β-CD-FA@PTX nanoparticles were synthesized. The TEM images show that Se@β-CD-FA@PTX is made up of monodisperse and spherical particles (Fig. 1B). The Se@β-CD-FA@PTX nanoparticles have an average diameter size of 70 nm (Fig. 1C). The zeta potential of the SeNPs was −25 mV and increased to 29 mV after capping with β-CD-FA and PTX (Fig. 1D), indicating that the Se@β-CD-FA@PTX nanoparticles are positively charge, which indicates that the nanoparticles can easily cross the cell membrane. The size distribution of the Se@β-CD-FA@PTX indicated that the decorated SeNPs were stable in aqueous solution for at least 30 days (Fig. 1E). Overall, with the properties of being smart, stable and potentially positively charged, Se@β-CD-FA@PTX can easily enter into cells (Scheme 1).
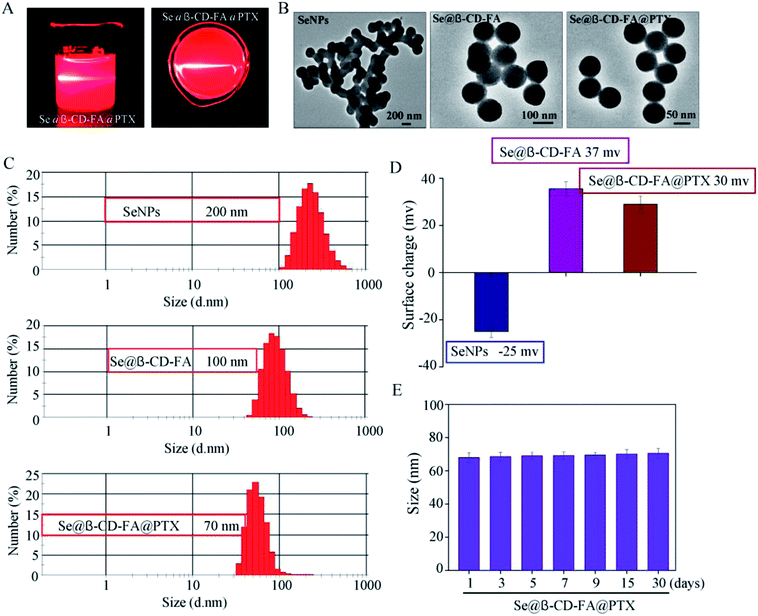 |
| | Fig. 1 Characterization of the SeNPs, Se@β-CD-FA and Se@β-CD-FA@PTX. (A) The Tyndall effect of Se@β-CD-FA@PTX. (B) TEM images of the SeNPs, Se@β-CD-FA and Se@β-CD-FA@PTX. (C and D) Size distribution and zeta potentials of the SeNPs, Se@β-CD-FA and Se@β-CD-FA@PTX. (E) Stability of Se@β-CD-FA@PTX in aqueous solution. | |
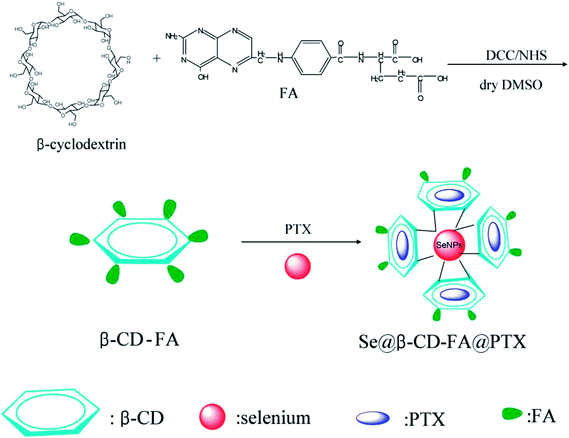 |
| | Scheme 1 Synthetic route of Se@β-CD-FA@PTX. | |
3.2 In vitro cytotoxicity of Se@β-CD-FA@PTX
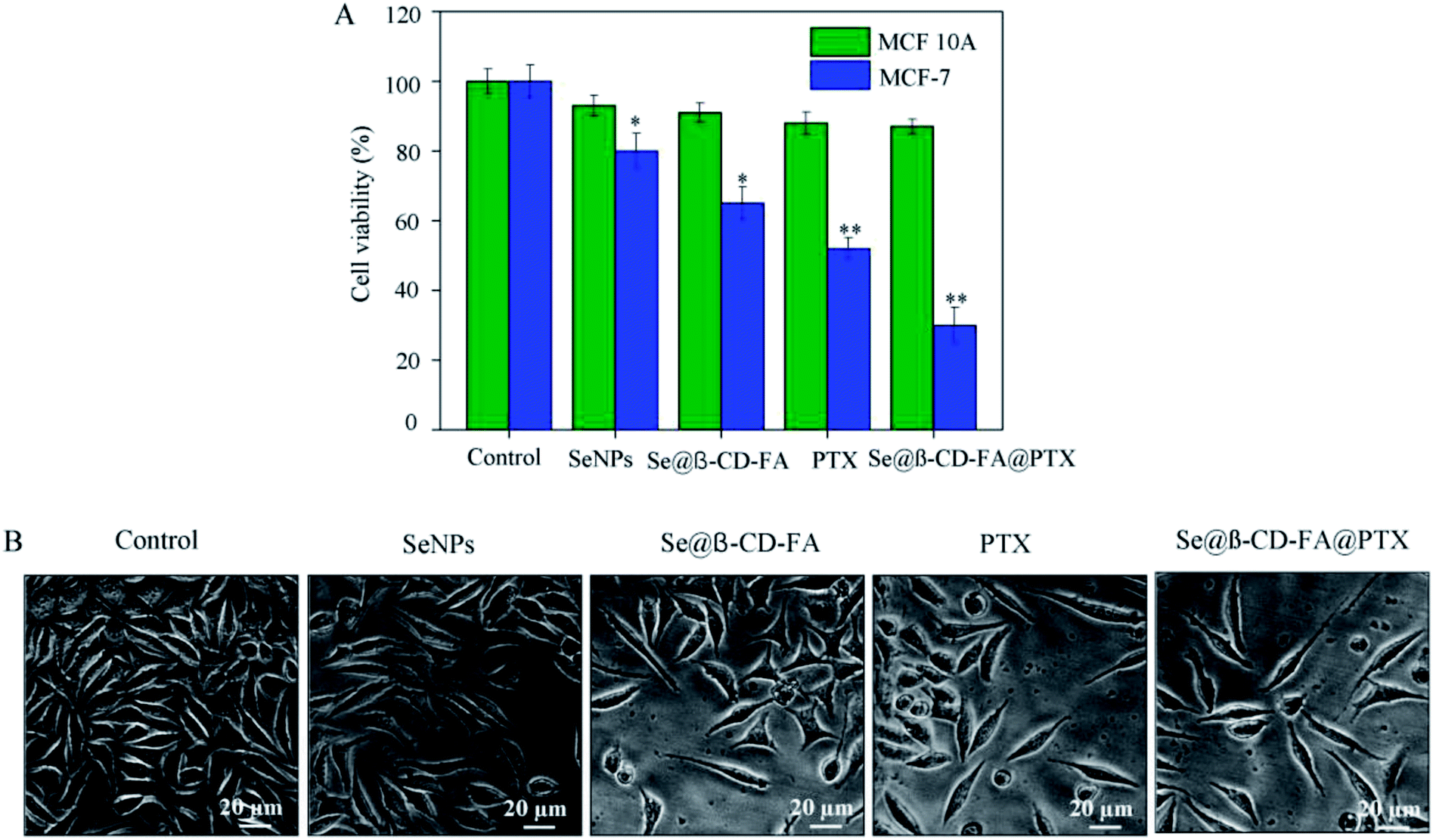
The anticancer activity of the SeNPs, Se@β-CD-FA, PTX and Se@β-CD-FA@PTX was detected by MTT assay. Fig. 2A shows that the cell viability of MCF-7 was remarkably lower than MCF 10A cells when treated with SeNPs (80% vs. 93%), Se@β-CD-FA (65% vs. 91%), PTX (52% vs. 88%) and Se@β-CD-FA@PTX (30% vs. 87%). Compared with the SeNPs, the PTX and Se@β-CD-FA, Se@β-CD-FA@PTX obviously inhibited the proliferation of MCF-7 cells and exhibited low cytotoxicity toward the MCF 10A cells. The effects of the SeNPs, Se@β-CD-FA, PTX and Se@β-CD-FA@PTX on the growth of the MCF-7 cells were further confirmed, as shown in Fig. 2B. After being treated with Se@β-CD-FA@PTX, the MCF-7 cell numbers reduced the cytoplasm shrinkage. The results suggested that Se@β-CD-FA@PTX effectively inhibited the proliferation of MCF-7 cells and induced cancer cell death.
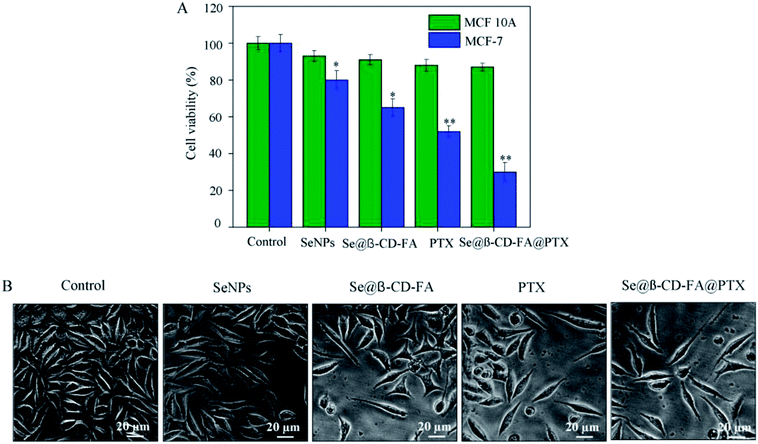 |
| | Fig. 2 Effects of the SeNPs, Se@β-CD-FA and Se@β-CD-FA@PTX on the growth of MCF-7 cells by MTT assay. (A) Antiviral activity of Se@β-CD-FA@PTX. The concentration of the SeNPs, Se@β-CD-FA and Se@β-CD-FA@PTX was 50 μg ml−1, PTX was 8 μg ml−1, and the Se@β-CD-FA@PTX conjugates were at a PTX dose of 8 μg ml−1. (B) Morphological changes in the MCF-7 cells observed by phase-contrast microscopy. The asterisks over the bars indicate the statistical difference at the *p < 0.05 or **p < 0.01 level. | |
3.3 Localization and cellular uptake of Se@β-CD-FA@PTX
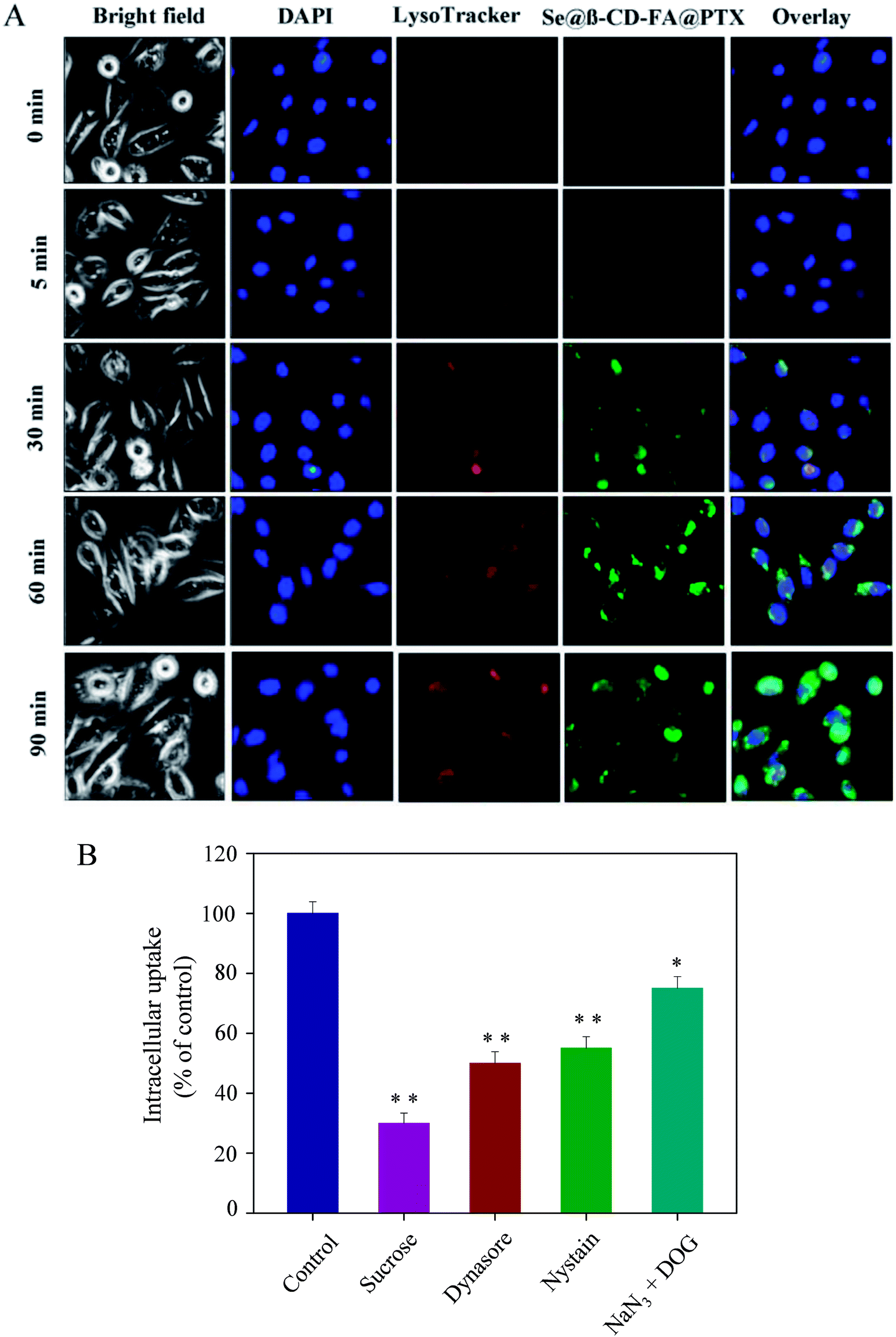
As shown in Fig. 3A, Se@β-CD-FA@PTX and lysosomes were found to be colocalized in the MCF-7 cells and gradually enhanced after then. Se@β-CD-FA@PTX escaped from the lysosome after 60 min and transported into the cytosol and distributed in cells after 90 min. To further detect the Se@β-CD-FA@PTX endocytosis mechanism, endocytosis inhibitors were used to treat the MCF-7 cells before the addition of Se@β-CD-FA@PTX. As shown in Fig. 3B, treatments with sucrose markedly inhibited the internalization of Se@β-CD-FA@PTX by 30% compared to that of the control. Dynasore and nystatin decreased the Se@β-CD-FA@PTX internalization to 50% and 55%, compared with the control. Moreover, NaN3 and DOG induced the uptake of Se@β-CD-FA@PTX to 75%. This result indicated that the lysosome was the target organelle of Se@β-CD-FA@PTX. Se@β-CD-FA@PTX showed time dependent cell-penetration for cancer therapy.
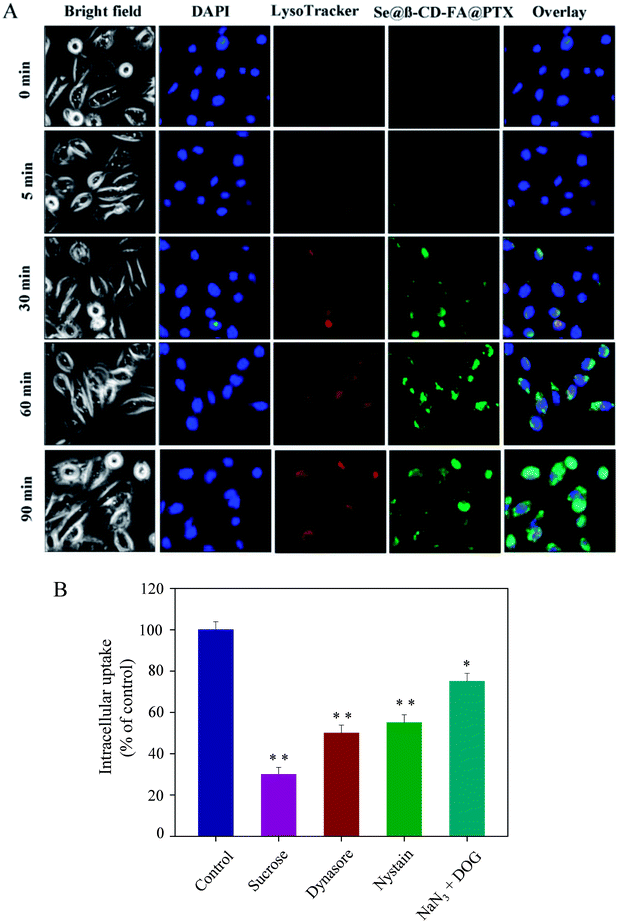 |
| | Fig. 3 Localization and uptake pathways of coumarin-6-loaded Se@β-CD-FA@PTX in MCF-7 cells. (A) The cells were treated with coumarin-6-loaded Se@β-CD-FA@PTX for different periods of time and were observed under a fluorescence microscope, stained with LysoTracker (red fluorescence, lysosome) and DAPI (blue fluorescence, nucleus) (magnification, 100×). (B) Intracellular uptake of Se@β-CD-FA@PTX in MCF-7 cells with different endocytosis-inhibited conditions. Significant difference between the treatment and control groups is indicated at the P < 0.05 (*) or P < 0.01 (**) level. | |
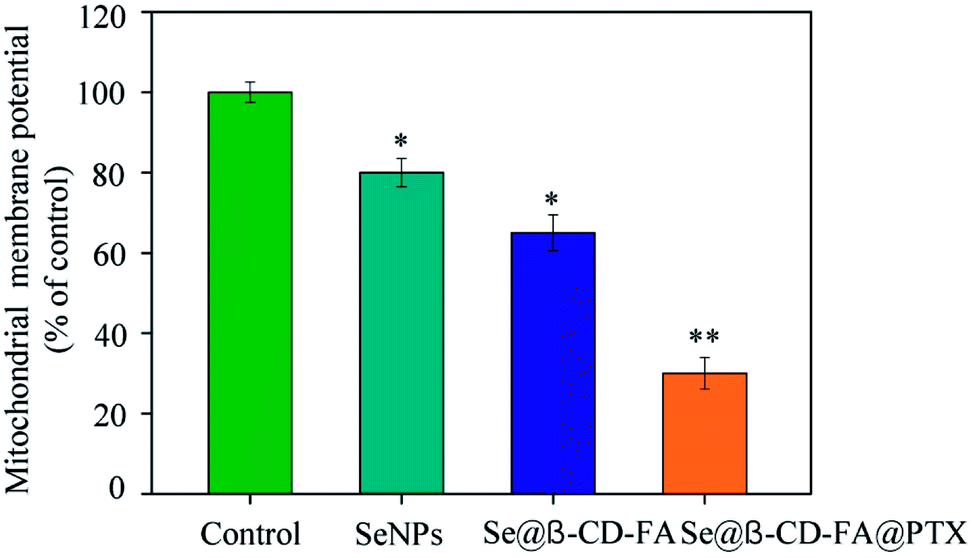
3.4 Mitochondrial membrane potential
The treatment of MCF-7 cells resulted in the elevation of mitochondria depolarization. As shown in Fig. 4, the mitochondrial membrane potential was reduced significantly to 80%, 65%, and 30% for the SeNPs, Se@β-CD-FA, and Se@β-CD-FA@PTX, respectively. These results indicated that Se@β-CD-FA@PTX triggered MCF-7 cell apoptosis through the induction of mitochondrial dysfunction.
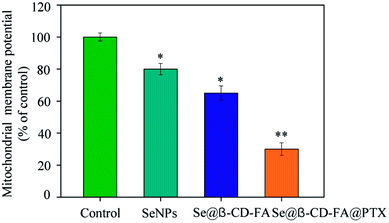 |
| | Fig. 4 Depletion of the mitochondrial membrane potential induced by the SeNPs, Se@β-CD-FA and Se@β-CD-FA@PTX on the growth of the MCF-7 cells. The asterisks over the bars indicate the statistical difference at the *p < 0.05 or **p < 0.01 level. | |
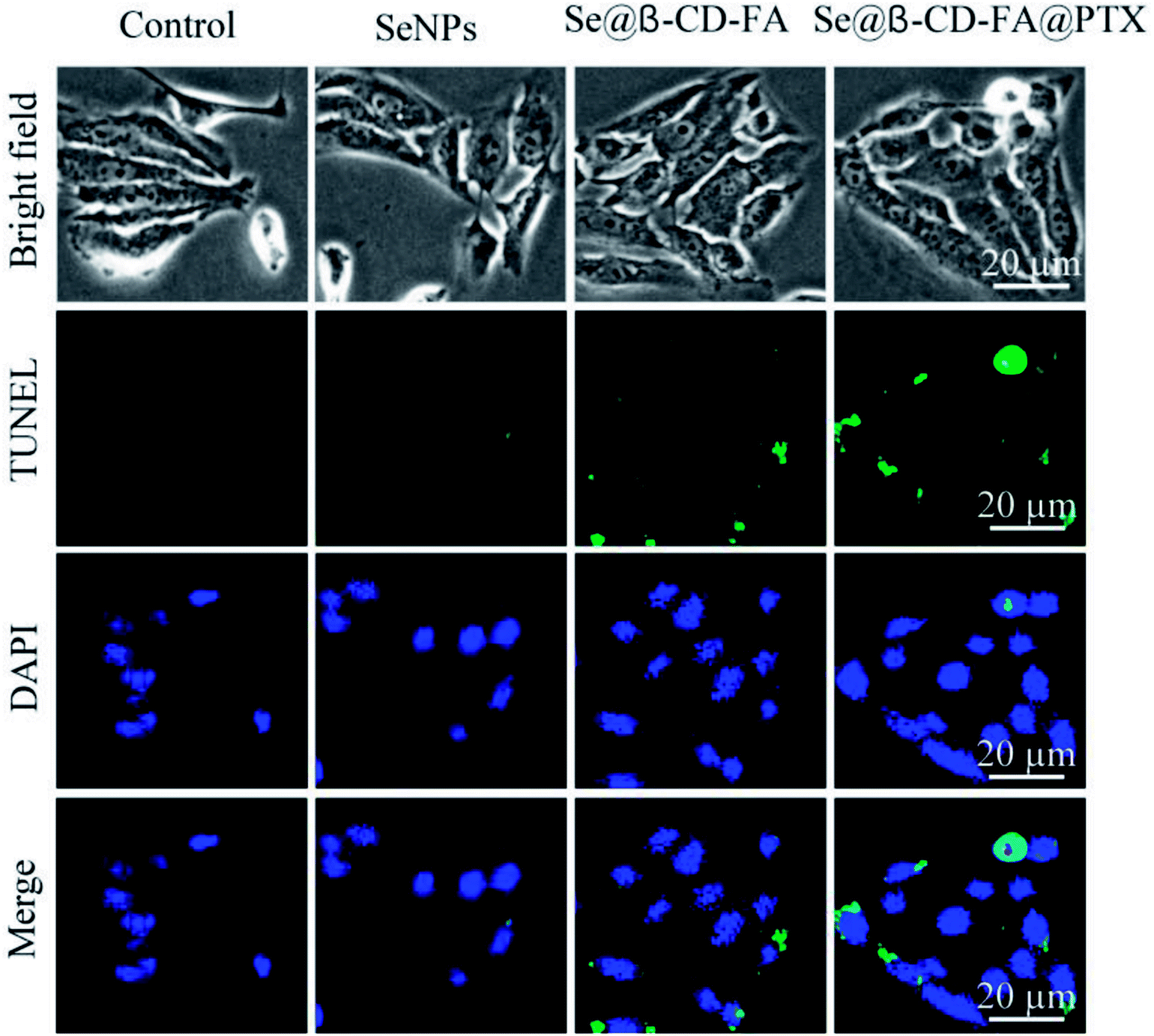
3.5 Se@β-CD-FA@PTX induced MCF-7 cell apoptosis
TUNEL enzyme labeling and DAPI co-staining was used to confirm MCF-7 cell apoptosis, which is one of the most crucial mechanisms for anticancer action. As shown in Fig. 5, compared to the control, SeNPs and Se@β-CD-FA, the MCF-7 cells exhibited typical apoptotic features with Se@β-CD-FA@PTX, such as DNA fragmentation and nuclear condensation. These results demonstrated that Se@β-CD-FA@PTX induced MCF-7 cell apoptosis.
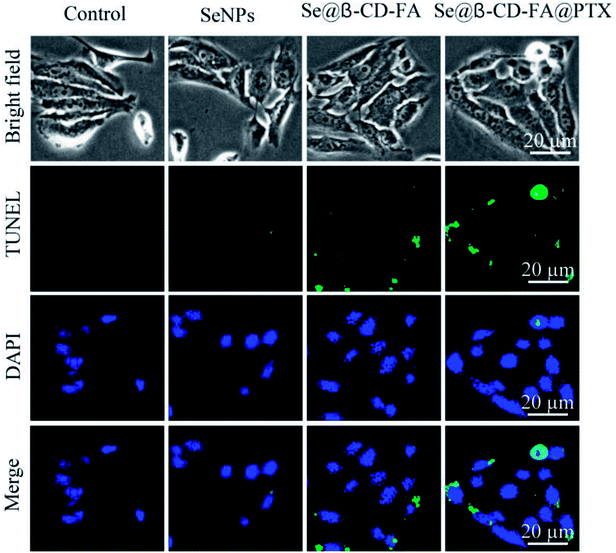 |
| | Fig. 5 Se@β-CD-FA@PTX induced apoptosis in the MCF-7 cells. Representative photomicrographs of DNA fragmentation and nuclear condensation as detected by a TUNEL-DAPI co-staining assay. | |
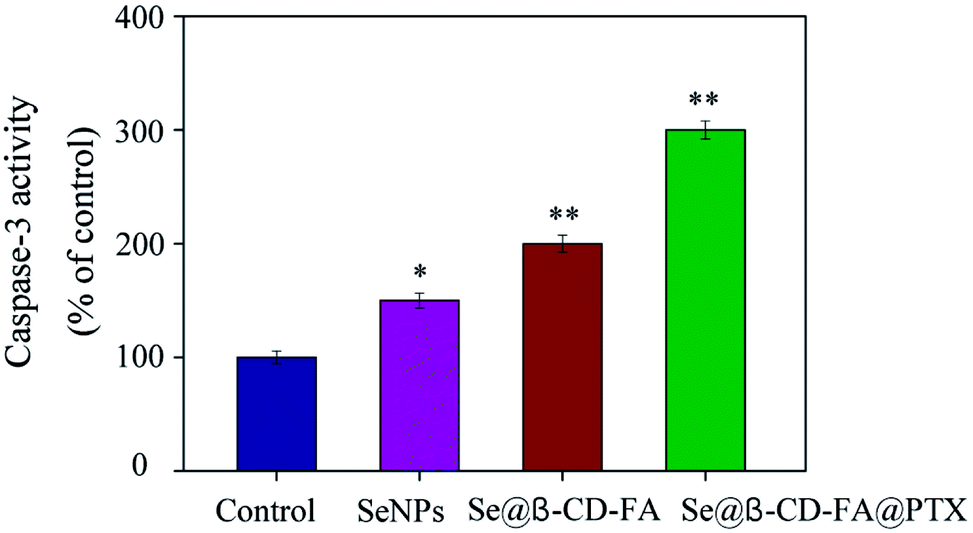
3.6 Caspase-3 activation by Se@β-CD-FA@PTX
Caspase-3 has been considered as the key mediator of apoptosis, as it is responsible for the cleavage of many proteins in programmed cell death. Western blotting was used to evaluate the caspase-3 activity in terms of its involvement and contribution to cell apoptosis. As shown in Fig. 6, compared with the control group (100%), SeNPs (150%) and Se@β-CD-FA (200%), treatments of the MCF-7 cells with Se@β-CD-FA@PTX (300%) significantly increased the activity of caspase-3. The results show that caspase-3 activity was significantly strengthened by Se@β-CD-FA@PTX. Overall, the results exhibited that the nanosystem inhibits cancer cell growth by inducing MCF-7 cell apoptosis.
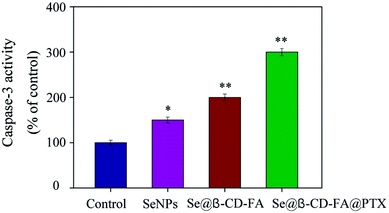 |
| | Fig. 6 Caspase-3 mediated apoptosis induced by Se@β-CD-FA@PTX in MCF-7 cells. MCF-7 cells treated with Se@β-CD-FA@PTX and caspase-3 activity were analyzed using a synthetic fluorogenic substrate. | |
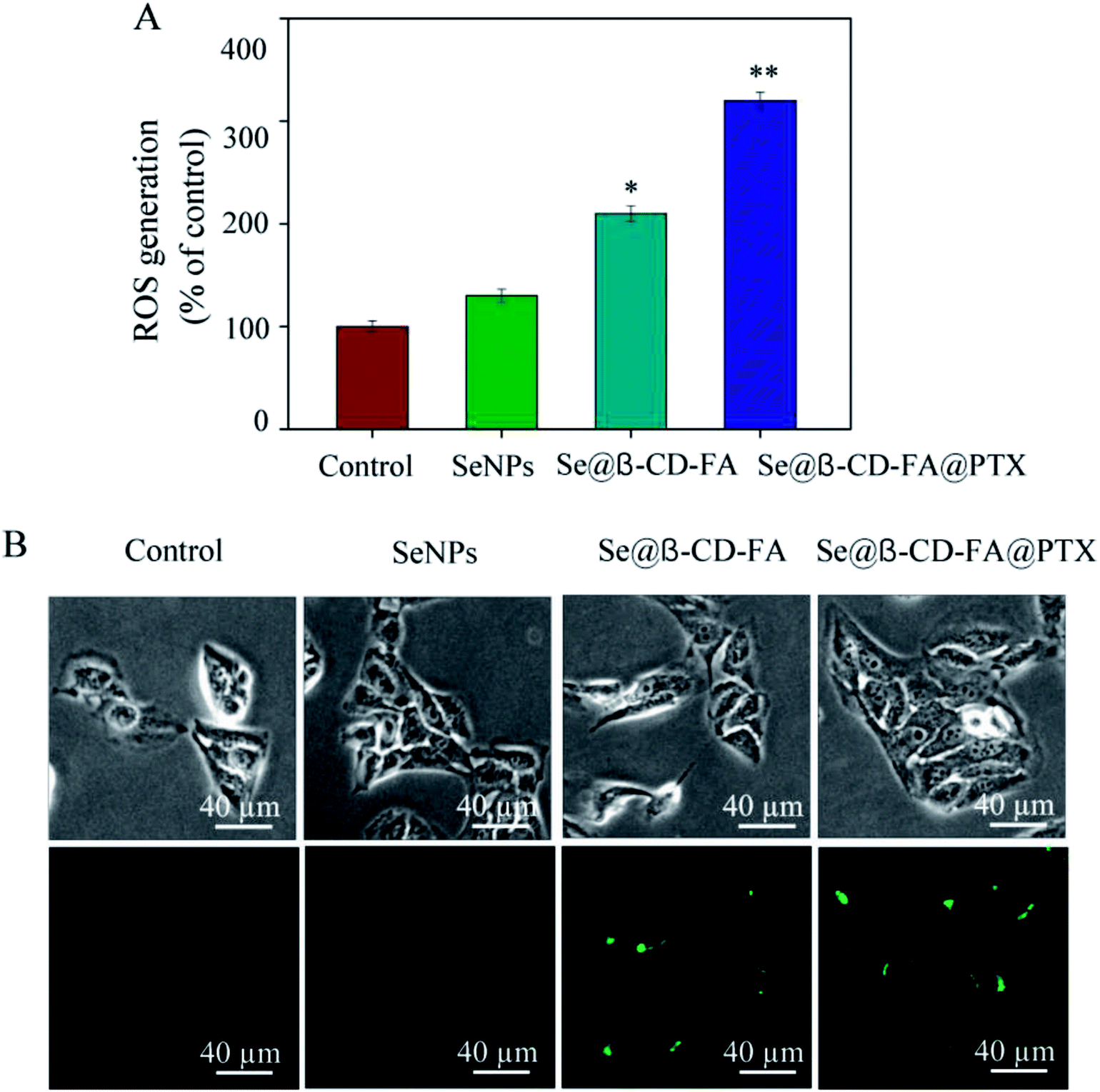
3.7 Induction of ROS generation by Se@β-CD-FA@PTX
To investigate whether Se@β-CD-FA@PTX could trigger ROS-mediated apoptosis, the intracellular ROS level was detected by DCF fluorescence. In Fig. 7A, compared with the control group (100%), SeNPs (130%), and Se@β-CD-FA (210%), the ROS generation of MCF-7 cells increased significantly after treatment with Se@β-CD-FA@PTX (320%). As shown in Fig. 7B, the fluorescence intensity of DCF in MCF-7 cell exposure to Se@β-CD-FA@PTX was the strongest among the groups. The results demonstrate that ROS generation was induced in the anticancer activity of Se@β-CD-FA@PTX.
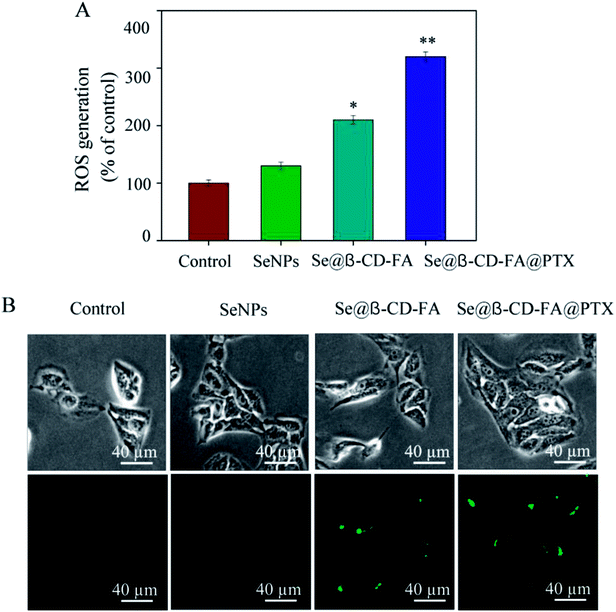 |
| | Fig. 7 ROS overproduction induced by Se@β-CD-FA@PTX in MCF-7 cells. (A) Changes in intracellular ROS generation detected by measuring the DCF fluorescence intensity. (B) MCF-7 cells were incubated with 10 μM DCF-DA in PBS for 30 min and then treated with SeNPs, Se@β-CD-FA and Se@β-CD-FA@PTX. The asterisks over the bars indicate the statistical difference at the *p < 0.05 or **p < 0.01 level. | |
3.8 In vivo anticancer activity of Se@β-CD-FA@PTX
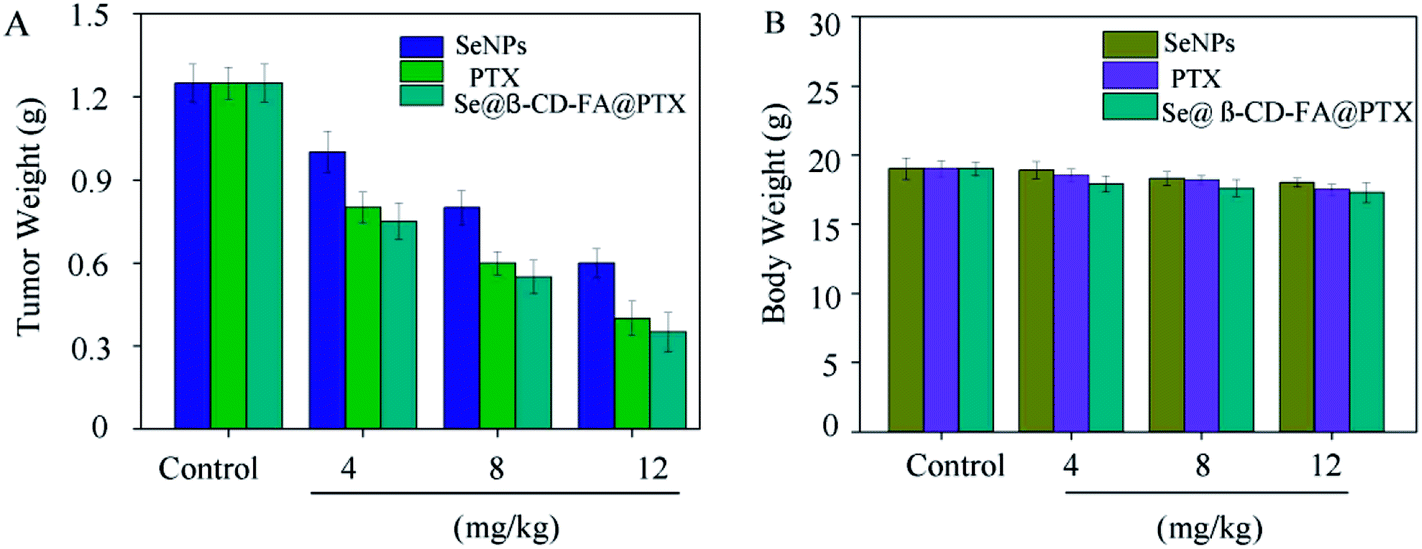
To further confirm the potential anticancer of Se@β-CD-FA@PTX, we assessed the in vivo therapeutic efficacy of this nanosystem with MCF xenograft nude mice. As shown in Fig. 8A, compared to SeNPs and PTX, Se@β-CD-FA@PTX significantly inhibited MCF-7 tumor growth, as evidenced by the decrease in the tumor weight. The MCF-7 xenograft nude mice were treated with Se@β-CD-FA@PTX at a dose of 4, 8, 12 mg kg−1 for a period of 16 days, and the tumor weights after last treatment were 0.75 g, 0.55 g and 0.35 g, respectively. Meanwhile, after treatment with different concentrations of Se@β-CD-FA@PTX (4, 8, 12 mg kg−1), compared to the control group (Fig. 8B), no significant reduction was observed in the body weight of the nude mice, suggesting a reduction in the side effects of the SeNPs brought about by surface decoration with β-CD-FA@PTX. Taken together, the data suggested that Se@β-CD-FA@PTX significantly suppressed the proliferation of the tumors.
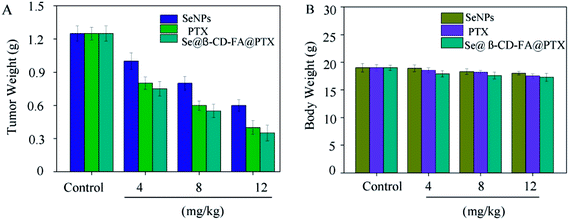 |
| | Fig. 8 Anticancer effects of Se@β-CD-FA@PTX in vivo. (A) Tumor weights of xenograft MCF-7 tumor nude mice after treatment with different concentrations of SeNPs, PTX and Se@β-CD-FA@PTX. (B) Body weights of xenograft MCF-7 tumor nude mice after treatment with different concentration of SeNPs, PTX and Se@β-CD-FA@PTX. The asterisks over the bars indicate the statistical difference at the *p < 0.05 or **p < 0.01 level. | |
3.9 Activation of ROS-mediated signaling pathways by Se@β-CD-FA@PTX
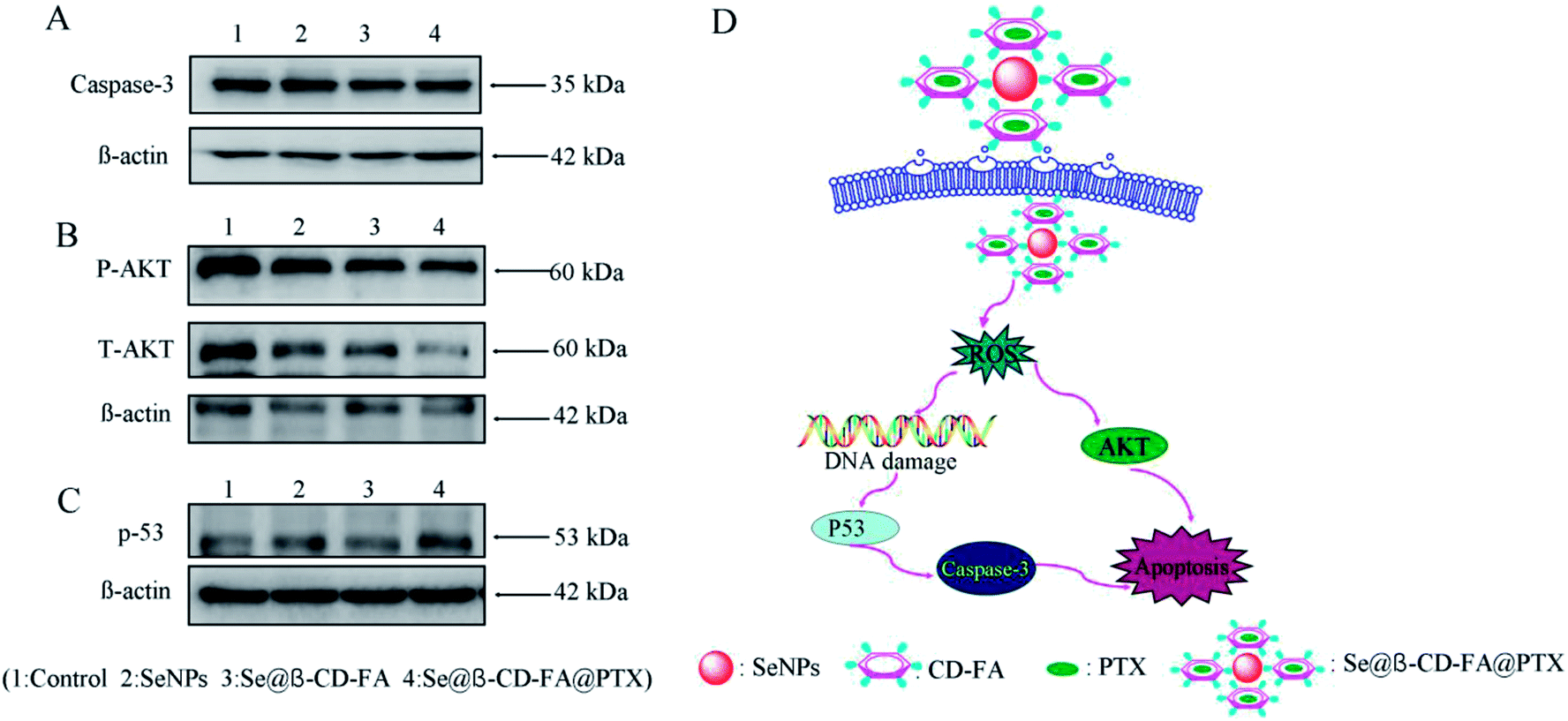
The mechanism of apoptosis by Se@β-CD-FA@PTX induced in the MCF-7 cells was further investigated in this study. Western blotting was used to detect the effects on the ROS-mediated signaling pathway. As shown in Fig. 9A, compared with the SeNPs and Se@β-CD-FA, the expression of caspase-3 was downregulated after treatment with Se@β-CD-FA@PTX. Meanwhile, as shown in Fig. 9B, MCF-7 cells treated with Se@β-CD-FA@PTX effectively increased the expression of total AKT in the MCF-7 cells. For the p53 signaling pathway, Se@β-CD-FA@PTX significantly increased the expression levels of total p53 (Fig. 9C). The results indicated that Se@β-CD-FA@PTX induces MCF-7 cell apoptosis through the regulation of the ROS-mediated AKT and p53 signaling pathways (Fig. 9D).
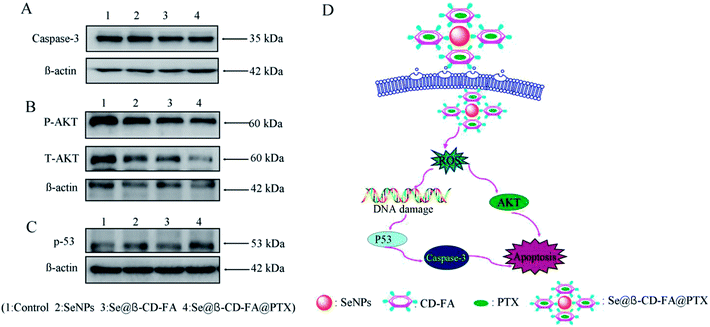 |
| | Fig. 9 Activation of ROS-mediated apoptosis signal pathways by Se@β-CD-FA@PTX in MCF-7 cells. (A) Activation of caspase-3. (B) and (C) Activation of the AKT and p53 signal pathways. (D) The main signal pathway of ROS-mediated AKT, the p53 pathway. | |
4 Conclusions
In summary, β-CD-FA modified SeNPs loaded with PTX were successfully fabricated in the present study. Se@β-CD-FA@PTX showed dramatic cytotoxic effects towards MCF-7 cells rather than normal cells, which improved its anticancer drug sensitivity and induced apoptosis. The molecular mechanisms demonstrated that Se@β-CD-FA@PTX activated caspase-3-mediated MCF-7 cell apoptosis via ROS generation. Furthermore, the results revealed the apoptotic signaling pathway through ROS triggered by the Se@β-CD-FA@PTX in the MCF-7 cells, including AKT and p53. In conclusion, this study highlights the possibility of using a cancer-targeted nanosystem to induce MCF-7 cell apoptosis and offers a new strategy for potential cancer therapy.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Technology Planning Project of Guangzhou (201804010183), the Medical Scientific Research Foundation of Guangdong Province (A2018306), and the Pediatrics Institute Foundation of Guangzhou Women and Children's Medical Centre (YIP-2018-036).
Notes and references
- C. M. Rodriguez-Razon, I. Yanez-Sanchez, V. O. Ramos-Santillan, C. Velasquez-Ordonez, S. A. Gutierrez-Rubio, M. R. Garcia-Garcia, R. I. Lopez-Roa, P. E. Sanchez-Hernandez, A. Daneri-Navarro and T. Garcia-Iglesias, Int. J. Nanomed., 2018, 13, 1081–1095 CrossRef CAS PubMed.
- J. Ferlay, I. Soerjomataram, R. Dikshit, S. Eser, C. Mathers, M. Rebelo, D. M. Parkin, D. Forman and F. Bray, Int. J. Cancer, 2015, 136, E359–E386 CrossRef CAS PubMed.
- H. S. Rugo, N. Engl. J. Med., 2014, 371, 175–176 CrossRef CAS PubMed.
- M. Ahmed, A. D. Purushotham and M. Douek, Lancet Oncol., 2014, 15, E351–E362 CrossRef PubMed.
- C. Q. Pan, Y. Q. Liu, M. Y. Zhou, W. S. Wang, M. Shi, M. Xing and W. J. Liao, Int. J. Nanomed., 2018, 13, 1119–1137 CrossRef CAS PubMed.
- L. Zhu, J. Wang, W. M. Zuo, R. Lin, T. T. Lin, Y. Lei, B. S. Ren, J. Lu, H. Y. Dong, L. J. Lin, L. H. Huang, Q. H. Wang, Y. J. Ma, H. Lyu, B. L. Liu, J. M. Tan and S. L. Wang, Cancer Res., 2016, 76, 1538 CrossRef PubMed.
- J. Ye, X. J. Xia, W. J. Dong, H. Z. Hao, L. H. Meng, Y. F. Yang, R. Y. Wang, Y. F. Lyu and Y. L. Liu, Int. J. Nanomed., 2016, 11, 4125–4140 CrossRef CAS PubMed.
- T. Yue, X. D. Zheng, Y. L. Dou, X. H. Zheng, R. Sun, Z. G. Tian and H. M. Wei, BMC Cancer, 2016, 16, 665 CrossRef PubMed.
- T. Voloshin, M. Munster, R. Blatt, A. Shteingauz, P. C. Roberts, E. M. Schmelz, M. Giladi, R. S. Schneiderman, E. Zeevi, Y. Porat, Z. Bomzon, N. Urman, A. Itzhaki, S. Cahal, E. D. Kirson, U. Weinberg and Y. Palti, Int. J. Cancer, 2016, 139, 2850–2858 CrossRef CAS PubMed.
- H. E. Barker, R. Patel, M. McLaughlin, U. Schick, S. Zaidi, C. M. Nutting, K. L. Newbold, S. Bhide and K. J. Harrington, Mol. Cancer Ther., 2016, 15, 2042–2054 CrossRef CAS PubMed.
- L. Simon-Gracia, H. Hunt, P. Scodeller, J. Gaitzsch, V. R. Kotamraju, K. N. Sugahara, O. Tammik, E. Ruoslahti, G. Battaglia and T. Teesalu, Biomaterials, 2016, 104, 247–257 CrossRef CAS PubMed.
- T. Tanei, F. Leonard, X. W. Liu, J. F. Alexander, Y. Saito, M. Ferrari, B. Godin and K. Yokoi, Cancer Res., 2016, 76, 429–439 CrossRef CAS PubMed.
- J. H. Xu, S. L. Hu, G. D. Shen and G. Shen, Cancer Cell Int., 2016, 16, 13 CrossRef PubMed.
- Z. J. He, X. M. Wan, A. Schulz, H. Bludau, M. A. Dobrovolskaia, S. T. Stern, S. A. Montgomery, H. Yuan, Z. B. Li, D. Alakhova, M. Sokolsky, D. B. Darr, C. M. Perou, R. Jordan, R. Luxenhofer and A. V. Kabanov, Biomaterials, 2016, 101, 296–309 CrossRef CAS PubMed.
- C. Holohan, S. Van Schaeybroeck, D. B. Longley and P. G. Johnston, Nat. Rev. Cancer, 2013, 13, 714–726 CrossRef CAS PubMed.
- Y. H. Li, M. Guo, Z. F. Lin, M. Q. Zhao, M. S. Xiao, C. B. Wang, T. T. Xu, T. F. Chen and B. Zhu, Int. J. Nanomed., 2016, 11, 6693–6702 CrossRef CAS PubMed.
- M. Guo, Y. H. Li, Z. F. Lin, M. Q. Zhao, M. S. Xiao, C. B. Wang, T. T. Xu, Y. Xia and B. Zhu, RSC Adv., 2017, 7, 52456–52464 RSC.
- R. Wahab, M. A. Siddiqui, Q. Saquib, S. Dwivedi, J. Ahmad, J. Musarrat, A. A. Al-Khedhairy and H. S. Shin, Colloids Surf., B, 2014, 117, 267–276 CrossRef CAS PubMed.
- Y. H. Li, Z. F. Lin, M. Q. Zhao, T. T. Xu, C. B. Wang, H. M. Xia, H. Z. Wang and B. Zhu, Int. J. Nanomed., 2016, 11, 3065–3076 CrossRef CAS PubMed.
- Y. H. Li, Z. F. Lin, M. Guo, M. Q. Zhao, Y. Xia, C. B. Wang, T. T. Xu and B. Zhu, Int. J. Nanomed., 2018, 13, 2005–2016 CrossRef CAS PubMed.
- X. Fang, C. Li, L. Zheng, F. Yang and T. Chen, Chem.–Asian J., 2018, 13, 996–1004 CrossRef CAS PubMed.
- Z. N. Zhao, P. Gao, Y. Y. You and T. F. Chen, Chem.–Eur. J., 2018, 24, 3289–3298 CrossRef CAS PubMed.
- L. Chan, L. Z. He, B. W. Zhou, S. H. Guan, M. J. Bo, Y. H. Yang, Y. Liu, X. Liu, Y. Y. Zhang, Q. Xie and T. F. Chen, Chem.–Asian J., 2017, 12, 3053–3060 CrossRef CAS PubMed.
- Y. H. Li, Z. F. Lin, M. Q. Zhao, T. T. Xu, C. B. Wang, L. Hua, H. Z. Wang, H. M. Xia and B. Zhu, ACS Appl. Mater. Interfaces, 2016, 8, 24385–24393 CrossRef CAS PubMed.
- S. V. Kurkov and T. Loftsson, Int. J. Pharm., 2013, 453, 167–180 CrossRef CAS PubMed.
- B. Gidwani and A. Vyas, Colloids Surf., B, 2014, 114, 130–137 CrossRef CAS PubMed.
- M. J. O'Neill, A. M. O'Mahony, C. Byrne, R. Darcy and C. M. O'Driscoll, Int. J. Pharm., 2013, 456, 390–399 CrossRef PubMed.
- K. Kameyama, K. Motoyama, N. Tanaka, Y. Yamashita, T. Higashi and H. Arima, Int. J. Nanomed., 2017, 12, 3433–3446 CrossRef CAS PubMed.
- K. M. Elamin, K. Motoyama, T. Higashi, Y. Yamashita, A. Tokuda and H. Arima, Int. J. Biol. Macromol., 2018, 113, 386–394 CrossRef CAS PubMed.
- A. Kasprzak, I. P. Grudzinski, M. Bamburowicz-Klimkowska, A. Parzonko, M. Gawlak and M. Poplawska, Macromol. Biosci., 2018, 18(2), 1–7 CrossRef PubMed.
- D. J. Zhang, C. Zhou, P. Lv, Y. L. Zhao, J. Liang, X. L. Liao and B. Yang, Mater. Sci. Eng., C, 2018, 86, 48–55 CrossRef CAS PubMed.
- Y. H. Li, X. L. Li, W. J. Zheng, C. D. Fan, Y. B. Zhang and T. F. Chen, J. Mater. Chem. B, 2013, 1, 6365–6372 RSC.
- Y. H. Li, Z. F. Lin, M. Q. Zhao, M. Guo, T. T. Xu, C. B. Wang, H. M. Xia and B. Zhu, RSC Adv., 2016, 6, 89679–89686 RSC.
- Z. F. Lin, Y. H. Li, M. Guo, T. T. Xu, C. B. Wang, M. Q. Zhao, H. Z. Wang, T. F. Chen and B. Zhu, RSC Adv., 2017, 7, 742–750 RSC.
- B. Zhu, Y. H. Li, Z. F. Lin, M. Q. Zhao, T. T. Xu, C. B. Wang and N. Deng, Nanoscale Res. Lett., 2016, 11, 1–8 CrossRef CAS PubMed.
- Z. F. Lin, Y. H. Li, M. Guo, M. S. Xiao, C. B. Wang, M. Q. Zhao, T. T. Xu, Y. Xia and B. Zhu, RSC Adv., 2017, 7, 35290–35296 RSC.
- Y. Y. Huang, Y. Luo, W. J. Zheng and T. F. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 19217–19228 CrossRef CAS PubMed.
- A. Johannisson, M. I. Figueiredo, Z. Al-Kass and J. M. Morrell, Anim. Reprod. Sci., 2018, 192, 290–297 CrossRef CAS PubMed.
- A. T. Mbaveng, B. L. Ndontsa, V. Kuete, Y. M. M. Nguekeu, I. Celik, R. Mbouangouere, P. Tane and T. Efferth, Phytomedicine, 2018, 43, 78–85 CrossRef CAS PubMed.
- X. Y. Fu, Y. H. Yang, X. L. Li, H. Q. Lai, Y. Y. Huang, L. Z. He, W. J. Zheng and T. F. Chen, Nanomedicine, 2016, 12, 1627–1639 CrossRef CAS PubMed.
- X. L. Li, L. J. Ma, W. J. Zheng and T. F. Chen, Biomaterials, 2014, 35, 8596–8604 CrossRef CAS PubMed.
- H. Zhu, B. W. Zhou, L. Chan, Y. X. Du and T. F. Chen, Int. J. Nanomed., 2017, 12, 5023–5038 CrossRef CAS PubMed.
- P. Wu, S. Y. Liu, J. Y. Su, J. P. Chen, L. Li, R. G. Zhang and T. F. Chen, Food Funct., 2017, 8, 3707–3722 RSC.
- Y. Xia, T. T. Xu, C. B. Wang, Y. H. Li, Z. F. Lin, M. Q. Zhao and B. Zhu, Int. J. Nanomed., 2018, 13, 143–159 CrossRef CAS PubMed.
- B. Ma, L. Z. He, Y. Y. You, J. B. Mo and T. F. Chen, Drug Delivery, 2018, 25, 293–306 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c
*c