
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCancer cell membrane-modified biodegradable mesoporous silica nanocarriers for berberine therapy of liver cancer†
Juan Yuea,
Zheng Wang*b,
Dan Shaob,
Zhimin Changb,
Rui Huc,
Li Li b,
Shi-zhong Luo*a and
Wen-fei Dong*b
b,
Shi-zhong Luo*a and
Wen-fei Dong*b
aCollege of Chemistry and Materials Science, The Key Laboratory of Functional Molecular Solids, Ministry of Education, Anhui Laboratory of Molecular-Based Materials, Center for Nano Science and Technology, Anhui Normal University, No. 1, Beijing East Road, Wuhu 241000, PR China. E-mail: shzhluo@ahnu.edu.cn
bCAS Key Laboratory of Bio-Medical Diagnostics, Suzhou Institute of Biomedical Engineering and Technology, Chinese Academy of Sciences, Suzhou 215163, China. E-mail: wangz@sibet.ac.cn; wenfeidong@sibet.ac.cn
cDepartment of Radiation Oncology, Affiliated Suzhou Hospital of Nanjing Medical University, China
First published on 4th December 2018
Abstract
Berberine (Ber) is regarded as a new, active and natural anti-cancer product; however, its clinical application has been limited due to its low aqueous solubility, poor gastrointestinal absorption, short residence time and poor targeting abilities. Hence, we reported a biomimetic nanoparticle as a drug delivery system to surmount these obstacles. We fabricated disulfide (S–S)-bridged mesoporous organosilica nanoparticles (ss-MONs) for Ber loading, which possessed uniform morphology, controllable mesoporous properties, highly-efficient drug loading capacity and superior biocompatibility. More interestingly, ss-MONs exhibited effective biodegradability under glutathione conditions through the breakage of the disulfide bond in ss-MONs, which promoted the Ber release. After coating human liver cancer HepG2 cell membranes (CM) on the surface of ss-MONs, the obtained CM-ss-MONs-Ber enhanced accumulation in liver cancer tissue through homologous targeting and effectively avoiding rapid blood clearance. Our findings indicate that CM-ss-MONs might be desirable drug carriers to promote the clinical use of Ber against liver cancer.
1 Introduction
Liver cancer is one of the most common tumors with an extreme mortality rate of about 600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people per year globally according to the World Health Organization.1–4 Clinically, chemotherapy is commonly used as the standard treatment for patients after surgery and some patients with unresectable liver cancer. However, traditional chemotherapeutic drugs possess high cytotoxicity to normal tissues and entail some limitations, including drug resistance and lack of specificity.5–8 Hence, it is urgent to seek novel chemotherapeutic agents and develop new strategies for highly effective and safe chemotherapy for liver cancer.9,10
000 people per year globally according to the World Health Organization.1–4 Clinically, chemotherapy is commonly used as the standard treatment for patients after surgery and some patients with unresectable liver cancer. However, traditional chemotherapeutic drugs possess high cytotoxicity to normal tissues and entail some limitations, including drug resistance and lack of specificity.5–8 Hence, it is urgent to seek novel chemotherapeutic agents and develop new strategies for highly effective and safe chemotherapy for liver cancer.9,10
Berberine (Ber), a traditional Chinese medicine, which is an isoquinoline alkaloid in the Ber species, has gained increasing attention due to its extensive pharmacological activity.11–13 Recently, its anti-liver-cancer activity has been demonstrated as Ber can significantly promote AIF-mediated apoptosis in liver cells by suppressing the protein expression levels of cytosolic phospholipase A2 (c PLA2) and cyclooxygenase (COX)-2 and elevating the content ratio of AA to prostaglandin E2 (PGE2).14–16 More interestingly, during liver cancer treatment, Ber showed low cytotoxicity to normal tissues. Hence, Ber is regarded as a promising candidate for liver cancer therapy. However, its further clinical application is limited due to low local concentrations, short residence time and poor intestinal absorption, resulting in reduced bioavailability.17,18
Recently, the rapid advancement of nanomedicine made it possible to solve the above-mentioned dilemmas.19–21 Among them, mesoporous silica nanomaterials exhibited excellent potential as drug delivery vehicles owing to their superb biocompatibility, large surface areas and easy functionalization.22–24 Compared with traditional inorganic mesoporous silica nanoparticles, mesoporous organosilica nanoparticles (MONs), with two or more alkoxysilanes, not only inherit the advantages of MSNs in preloading drugs but also achieve biodegradable properties,25–27 which facilitate drug release and promote the in vivo metabolism of nanomaterials.28,29 In our previous study, we designed MONs containing disulfide bridges (ss-MONs) and utilized them in the preloading of the model drug doxorubicin. We provided a detailed comparison of ss-MONs with MSNs in terms of biocompatibility, drug loading capacity, drug release fashion and therapeutic efficacy and discovered better efficiency of ss-MONs in liver cancer treatment owing to the pH/glutathione-dual responsive drug release property.30 Therefore, we believe that ss-MONs might be an ideal drug vehicle for the Ber treatment of liver cancer.
To improve the specificity of Ber to liver cancer and avoid rapid blood clearance, adopting optimal strategies to functionalize ss-MONs are imperative. However, the existing modification methods of ss-MONs such as ligands, aptamers, and antibodies are unsatisfactory due to their poor cancer specificity, weak binding affinity and the relative residence time.31,32 Recently, biomimetic cell membrane-coated nanocarriers have attracted increasing attention owing to their homologous target property and immune escape capacity.33,34 Hence, nanocarriers with liver cancer cell membrane modifications can significantly increase drug accumulation in liver cancer tissues and avoid the clearance of macrophages. In this system (Scheme 1), liver cancer cell membrane-coating ss-MONs (CM-ss-MONs) were designed and utilized to load Ber (CM-ss-MONs-Ber). The CM-ss-MONs-Ber exhibited uniform morphology, excellent monodispersity, superb drug loading capacity and superior biodegradability. More importantly, amounts of Ber entered into the cytoplasms of liver cancer cells rather than normal cells, which resulted from the targeted endocytic ability of CM-ss-MONs in the liver cancer cells and the tumor-microenvironment responsive Ber-release property. Additionally, in vitro and in vivo experiments were carried out to demonstrate the high therapeutic efficiency and excellent safety of CM-ss-MONs-Ber. In summary, our results revealed that CM-ss-MONs-Ber might be a potential chemotherapeutic agent for liver cancer treatment.
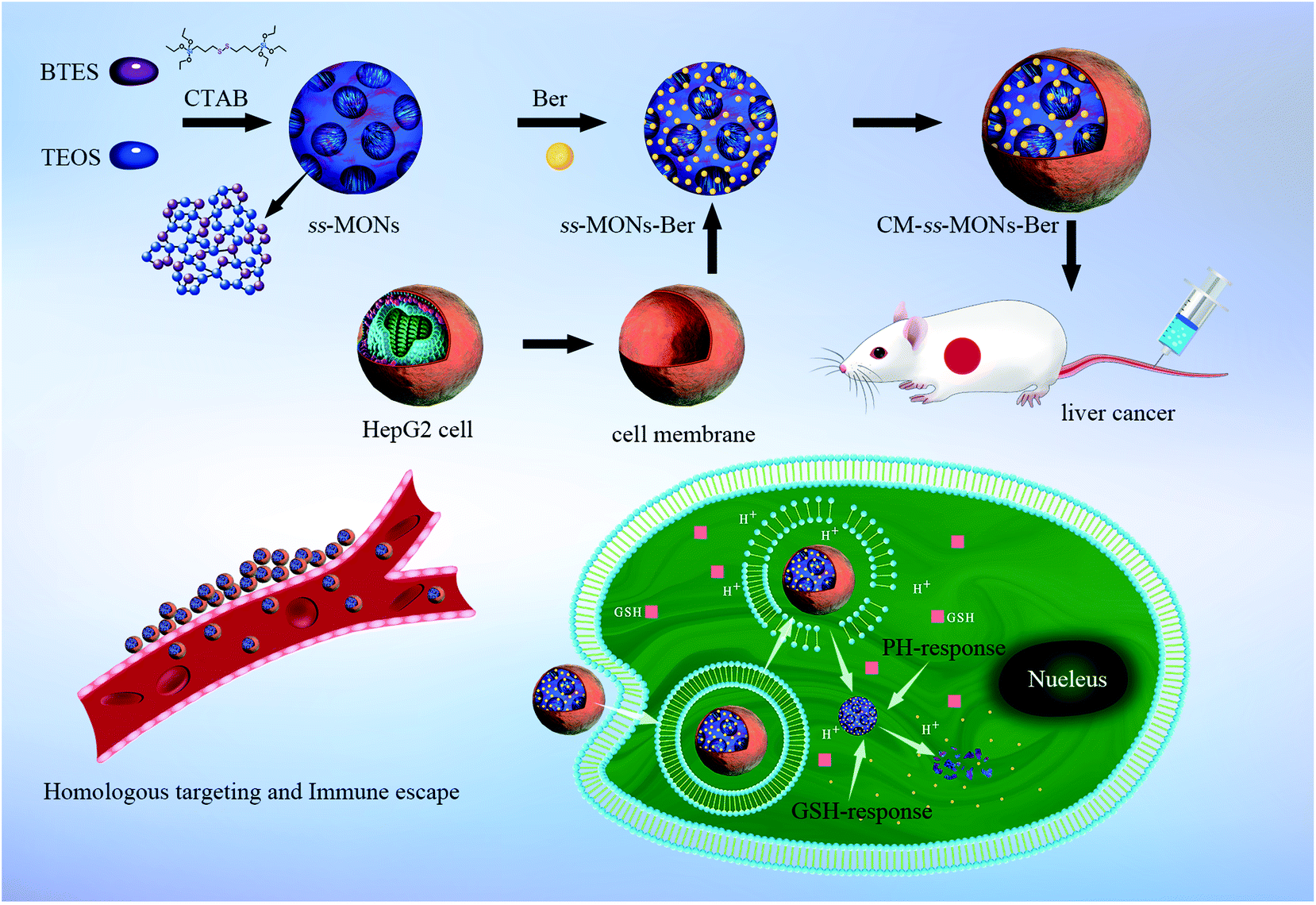 | ||
| Scheme 1 Schematic of the HepG2 cell membrane-coated biodegradable ss-MONs with dual-responsive berberine release for the homotypic targeting therapy for liver cancer. | ||
2 Experimental
2.1 Preparation of ss-MONs
Biodegradable ss-MONs were synthesized through a modified sol–gel method using cetyltrimethylammonium bromide (CTAB) as a template, tetraethoxysilane (TEOS) and bis[3-(triethoxysilyl)propyl]tetrasulfide (BTES) with disulfide-bridged groups as co-silica sources.35ss-MONs were synthesized by the following procedure: 0.6 g CTAB, 0.15 g triethanolamine (TEA) and 50 mL deionized water were stirred in a three-necked flask at 80 °C for 30 min. Then, a solution of 900 μL TEOS and 100 μL BTES was added dropwise to the surfactant solution. The resulting mixture was stirred at 80 °C for another 4 h with a stirring speed of 1000 rpm. The products were collected by centrifugation, washed three times with ethanol, and then refluxed in an ethanol solution of HCl (2% w/v) for 12 h. Finally, the as-synthesized ss-MONs were collected, washed and dried for subsequent experiments.
2.2 Amination and carboxylation of ss-MONs
The post-grafting method was used according to the method described in the previous report, with modification.36,37 Briefly, 50 mg of ss-MONs and 1.0 mL of APS were added to 40 mL water with ultrasound. We stirred the solution at room temperature for 24 h. The reaction was heated to 100 °C and reacted for 4 h with reflux. Then, we cooled the reaction mixture to room temperature. The ss-MONs modified through amination (ss-MONs-NH2) were obtained and collected by centrifugation. Carboxylation was introduced by suspending succinic anhydride in DMF. Briefly, the as-synthesized ss-MONs-NH2 and 50 mg of succinic anhydride were dispersed in 40 mL of DMF and stirred for 12 h at room temperature. Ethanol was used to centrifuge and wash the carboxylated ss-MONs (ss-MONs-COOH).2.3 Preparation of CM-ss-MONs-Ber
First, we prepared the ss-MONs-COOH with Ber (ss-MONs-Ber). Briefly, 10 mg of ss-MONs-COOH was dispersed in 10 mL of PBS buffer. Then, 10 mL of ethyl alcohol containing Ber (0.5 mg mL−1) was mixed with 10 mL of ss-MONs-COOH solution (1 mg mL−1), and the mixture was stirred at room temperature in the dark for 24 h. The ss-MONs-COOH with Ber (ss-MONs-Ber) was obtained through centrifugation at 5000 × g. Second, CM-ss-MONs-Ber was prepared by the classical extrusion approach. HepG2 cell membrane vesicles (CM) were then prepared. Briefly, hypertonic tris buffer (pH = 7.4) was used to treat the HepG2 cells for 1 h at 4 °C and centrifuged for 10 min at 5000 × g, so that the intracellular contents were removed. Supernatants were centrifuged at 10000 × g for 10 min and 100000 × g for 1 h; the cell membranes were obtained and an ultrasonicator was used to sonicate the cell membranes for 5 s, which was extruded through 400 nm and 200 nm polycarbonate membranes. Finally, the ss-MONs-Ber (0.1 mg mL−1) were mixed with the HepG2 cell membranes (0.05 mg mL−1), which extruded the mixture through 400 nm, 200 nm and 100 nm polycarbonate membranes. Then, the residual free cell membrane was removed by centrifuging and washing the mixture with PBS to obtain CM-ss-MONs-Ber. We also prepared CM-ss-MONs without Ber in the same way.
The optical densities of the CM-ss-MONs-Ber and ss-MONs-Ber were measured by a UV-visible spectrophotometer to calculate the efficiency and content of drug loading through eqn (1) and (2):38,39
| efficiency of loading (%) = mass of drug loaded by nanoparticles/initial mass of drug | (1) |
| content of loaded drug (%) = mass of drug loaded by nanoparticles/(mass of nanoparticles + mass of drug loaded by nanoparticles) | (2) |
2.4 Biodegradation of CM-ss-MONs
10 mg of CM-ss-MONs and ss-MONs were added to 20 mL PBS buffer solution (pH 7.4) with glutathione (10 mM), respectively, and stirred at 37 °C. The suspensions collected after 1 day, 3 days and 5 days were analyzed by TEM.2.5 Characterization of CM-ss-MONs
The size and morphology of ss-MONs were analyzed via transmission electron microscopy (TEM) using a transmission electron microscope (JEOL Ltd, Japan) with an accelerating voltage of 200 kV. A Zetasizer Nano-ZS-90 (Malvern Instruments, UK) was adopted to measure the hydrodynamic size distribution and zeta potential of ss-MONs. The pore size distribution and surface area of ss-MONs were measured using the Barret–Joyner–Halenda method and the Brunauer–Emmett–Teller (BET) method.2.6 Endocytosis and immune escape
5 × 104 human normal liver cell line HL-7702 cells per well and 5 × 104 human hepatocellular carcinoma cell lines HepG2 (the HepG2 cell line, HL-7702 cell line and RAW264.7 cells were obtained from the cell bank of Shanghai, China) per well were cultured in 24-well plates through DMEM with 10% fetal bovine serum (FBS) to measure the ss-MONs uptake. After 12 hours, the medium was replaced with fresh medium containing FITC-labelled ss-MONs or CM-ss-MONs at concentrations of 12.5 μg mL−1. The cells were incubated for 3 hours, washed with PBS three times and treated through a 50 nM LysoTracker Red DND-99 (Invitrogen) for 1 hour. Subsequently, the cells were washed twice and stained for 5 min with Hoechst 33258 (5 mg mL−1). PBS was used to wash the cells twice. The fluorescence in the cells was detected through confocal scanning microscopy (CLSM) by an Olympus FV1000 microscope which was equipped with a 30 mW laser and a 405 nm laser class 3D laser, and the distribution of intracellular nanoparticles was observed. Flow cytometry analysis was used to quantify the uptake of 12.5 μg mL−1 FITC-labelled ss-MONs or CM-ss-MONs in HepG2 cells or HL-7702 cells.
The mouse macrophage RAW264.7 cells were used to evaluate the uptake of nanoparticles. The cells were treated with the CM-ssMONs and ss-MONs for 1 h. Subsequently, the cells were washed with PBS and fixed with 4% paraformaldehyde for 30 min before imaging using the CLSM microscope.
2.7 Dual-responsive drug release
Firstly, we simulated the tumor tissue and normal tissue microenvironment and measured the Ber cumulative release at different times. Briefly, 10 mg of ss-MONs-Ber and CM-ss-MONs-Ber were encapsulated respectively to a dialysis bag with or without glutathione (GSH) (10 mM) to investigate the release of the drug of ss-MONs-Ber for varied pH values (pH = 7.4 or 5.5). The solution was stored in the dark and shaken at 37 °C. UV-visible spectroscopy was used to detect the Ber release at specific intervals.In order to analyze the intracellular Ber release process, we cultured 5 × 104 HL-7702 cells per well and 5 × 104 HepG2 cells per well in 24-well plates through free Ber or ss-MONs-Ber or CM-ss-MONs-Ber (10 μg mL−1) under a Ber concentration of 2.5 μg mL−1 for 3 hours. PBS was used to remove and wash the medium twice. The nuclei were stained for 5 min through Hoechst 33258 (5 μg mL−1). Finally, flow cytometry was used to detect the fluorescence of these two kinds of cells.
2.8 Cytotoxicity of CM-ss-MONs-Ber
The cytotoxicity of different samples in comparison with that of ss-MONs, CM-ss-MONs, Ber, ss-MONs-Ber and CM-ss-MONs-Ber were evaluated through MTT experiment.33 HepG2 cells and HL-7702 cells were seeded in 96-well plates, and the density was 2 × 104 cells per well. Then, ss-MONs, ss-MONs-Ber and CM-ss-MONs-Ber at different concentrations were co-incubated with HL-7702 cells and HepG2 cells for 24 hours. The supernatant was removed and the cells were washed through PBS. Thereafter, 20 μL of a PBS solution of MTT (5 mg mL−1) was added to each well slowly, which was incubated for 4 h. A microplate reader was used to read the absorbance to calculate the cell viability of treatment groups.2.9 Anti-tumor efficacy in vivo
This study was performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH publication no. 85-23 rev. 1985) and was approved by the Ethics Committee for the Use of Experimental Animals of the Suzhou Institute of Biomedical Engineering and Technology, Chinese Academy of Sciences (Suzhou, Jiangsu, China).The HepG2 tumors on the right shoulder of BALB/c nude mice were divided into five groups randomly (PBS, ss-MONs, Ber, ss-MONs-Ber and CM-ss-MONs-Ber). At a tumor volume of 80 mm3, PBS, Ber, ss-MONs-Ber and CM-ss-MONs-Ber (a dose of 5 mg Ber kg−1) solutions were injected into the mice intravenously every day. The volumes of tumors in each group were measured. The volume of each tumor was calculated through the formula: V = (length of tumor) × (width of tumor)2/2. Moreover, the weight of each mouse was also measured.
2.10 In vivo biosafety assessment
After 22 days from the first injection, whole blood and serum were collected, and the mice were sacrificed. The heart, liver, spleen, lung and kidney were then harvested from each. The blood samples were used to detect alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN) and creatinine (CRE) parameters for hematological and biochemical analysis. The obtained tissues were fixed in formalin for the next paraffin sectioning and H&E assays to evaluate the histopathologic toxicity of the tissues.3 Results and discussion
3.1 Preparation and characterization
ss-MONs was synthesized by a classical sol–gel method. Transmission electron microscopy (TEM) was used to characterize its morphology and structure. TEM analysis results indicated that ss-MONs possessed a uniform spherical structure, and its diameter was approximate 35 nm (Fig. 1A). Then, we assessed its mesoporous property. The N2 adsorption–desorption isotherm curve of ss-MONs showed IV-type curves (Fig. 1B). The pore volume and surface area were 1.19 cm3 g−1 and 902.1 m2 g−1, respectively. The average pore diameter was 3.1 nm as determined by the Barrett–Joyner–Halenda method in Fig. S1A.† After post-grafting modification, these parameters of mesoporosity were correspondingly decreased. The as-synthesized ss-MONs-NH2 showed surface area, pore volume and pore diameter values as small as 430.5 m2 g−1, 0.78 cm3 g−1 and 2.87 nm, respectively, and the values of surface area, pore volume and pore diameter for ss-MONs-COOH were 301 m2 g−1, 0.56 cm3 g−1 and 2.63 nm, respectively. This reducing tendency should be attributed to the covalent fixation of organic groups on the ss-MONs (Fig. S1B and 1C†).40 The superb mesoporous property of ss-MONs suggested the possibility of Ber loading. We then functionalized carboxyl groups on the mesoporous surface of ss-MONs to payload Ber by the electrostatic interaction between –COOH groups and –NH2 groups. The drug efficiency and drug content of ss-MONs were 50.5% and 20.1%, respectively, as determined by UV-visible spectrophotometry.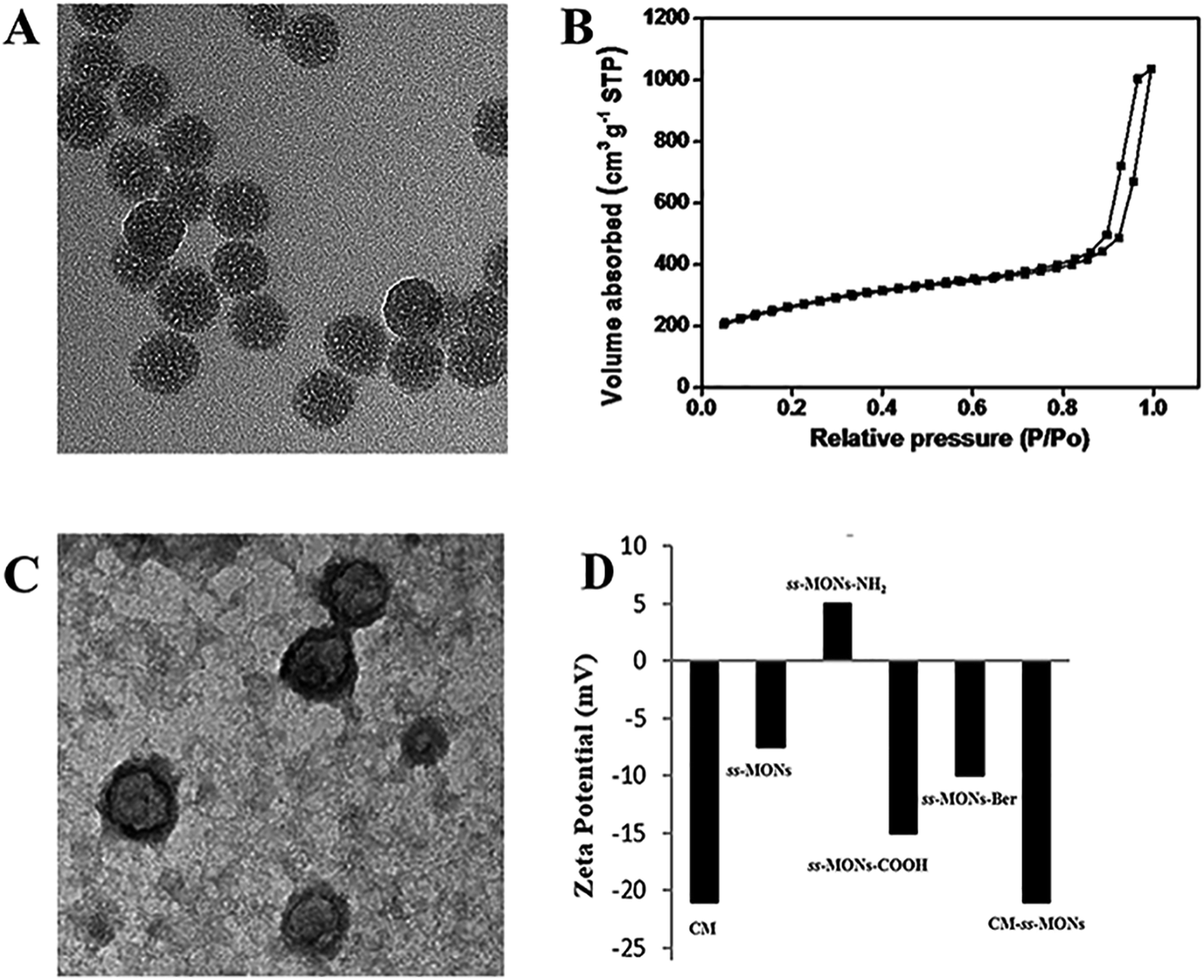 | ||
| Fig. 1 Characteristics of ss-MONs and CM-ss-MONs. (A) TEM image of ss-MONs, bar = 50 nm. (B) N2 sorption isotherms and pore size distribution of ss-MONs. (C) TEM image of CM-ss-MONs, bar = 50 nm. (D) Zeta potentials of the CM, ss-MONs, ss-MONs-NH2 and CM-ss-MONs. | ||
To endow the ss-MONs-Ber with liver cancer targeting and immune escape properties, cell membranes (CM) derived from human liver cancer HepG2 cells were modified on the surface of ss-MONs (CM-ss-MONs-Ber). As illustrated in Fig. 1C, the CM-ss-MONs-Ber showed uniform morphology and similarly spherical structures with a diameter of approximately 53 nm and a lipid shell of ∼8 nm in thickness, which was consistent with the reported cell membranes of 5–10 nm in thickness. Fig. S2† demonstrated that both ss-MONs-Ber and CM-ss-MONs-Ber possessed a narrow size distribution, which indicated their excellent monodispersity. Besides, the CM-ss-MONs-Ber displayed an averaged hydrodynamic diameter of 57 nm, which was larger than that of the ss-MONs. The zeta potential of CM, ss-MONs, ss-MONs-NH2, ss-MONs-COOH, ss-MONs-Ber and CM-ss-MONs-Ber were −21 mV, −7.5 mV, 5 mV, −15 mV, −10 mV and −21 mV, repectively (Fig. 1D). The changed potentials indicated the successful process of carboxyl groups functionalization, Ber loading and HepG2 cell membranes modification.
3.2 Endocytosis and immune escape
Encouraged by the uniform morphology and excellent Ber loading capacity of CM-ss-MONs, we further explored the uptake behaviour of CM-ss-MONs and ss-MONs in HepG2 cells and human liver normal HL-7702 cells. As shown in Fig. 2A and B, obviously green fluorescent signs were observed to accumulate and colocalize with the LysoTracker after 3 h incubation with FITC-labeled ss-MONs or CM-ss-MONs in both HepG2 cell lines and HL-7702 cell lines, which indicated that these nanoparticles could enter the cytoplasm through lysosomes. The green fluorescence of CM-ss-MONs and ss-MONs in HepG2 cells were both higher than that in HL-7702 cells, which indicated that these nanoparticles possessed better cellular internalization behaviour in cancer cells than in normal cells. Furthermore, CM-ss-MONs exhibited higher cellular uptake in comparison with the nontargeted ss-MONs in HepG2 cells, whereas the increase in the uptake of CM-ss-MONs in the HL-7702 cells wasn't obvious compared to that of ss-MONs, which was in line with flow cytometry analysis (Fig. 2C and D). These results demonstrated the selective cellular uptake behaviour and the improved liver cancer targeting after CM modification.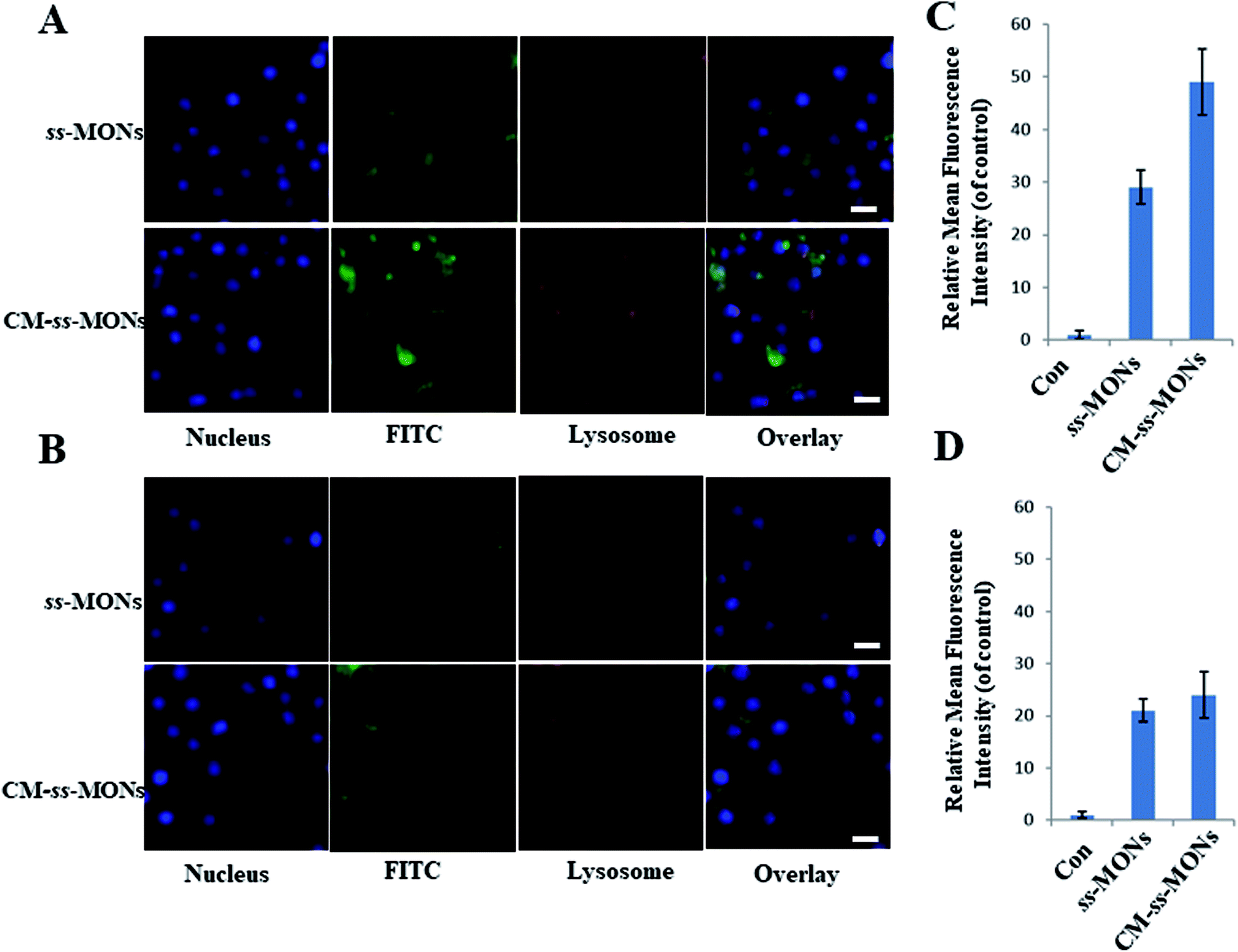 | ||
| Fig. 2 CLSM images of HepG2 cells (A) and HL-7702 cells (B) after treatment with FITC-labeled ss-MONs or FITC-labelled CM-ss-MONs for 3 h. Scale bars, 10 μm. Flow cytometry analysis of the uptake of 12.5 μg mL−1 FITC-labeled ss-MONs in HepG2 cells (C) and HL-7702 cells (D) after 3 h of exposure. | ||
We also explored the immune-invasion ability of CM-ss-MONs in RAW264.7 murine macrophages cells. As shown in Fig. S3,† large amounts of ss-MONs were endocytosed by macrophages. However, significant decreasing internalization was observed in the macrophages after CM modification. This was because protein CD47 expression on the cell membrane inhibited the phagocytic uptake. The excellent liver cancer targeting and immune escape ability suggested that CM-ss-MONs were ideal nanocarriers, which could effectively deliver Ber to the liver cancer tissue.
3.3 Dual-responsive drug release
To investigate the Ber release behaviour of CM-ss-MONs-Ber, we simulated the tumor tissue and normal tissue microenvironment and measured the Ber cumulative release at different times. As shown in Fig. 3B, CM-ss-MONs-Ber showed a pH-responsive release property. The cumulative Ber release amount was less than 5% at pH 7.4 PBS solution without glutathione (GSH), whereas as much as 55% Ber was released from CM-ss-MONs-Ber at pH 5.5 without GSH after 96 h. The pH-responsive release behaviour facilitated Ber release in liver cancer tissues rather than normal tissues, since the extracellular environment of the tumor tissue and intracellular lysosomes and endosomes were acidic. Furthermore, the amount of GSH in the cancer cells was obviously higher than that in normal cells. The high level of GSH could degrade CM-ss-MONs-Ber by disrupting the disulfide bonds of CM-ss-MONs through reduction reactions, which is favorable for drug release. To verify our hypothesis, we firstly evaluated the biodegradability of CM-ss-MONs in the presence of the PBS solution of GSH (10 mM), which were intracellular equivalents of GSH. As shown in Fig. 3B, irregular particles existed around CM-ss-MONs after 24 h, and then the spherical shape of CM-ss-MONs disappeared at 72 h. Finally, CM-ss-MONs were degraded completely into fragments after 120 h. From the above results, it can be inferred that CM-ss-MONs and ss-MONs have excellent biodegradability in the presence of GSH. Then, we measured the Ber cumulative release with the aid of GSH. As shown in Fig. 3A, the content of releasing Ber from CM-ss-MONs-Ber was up to 80% at pH 5.5 after 96 h. The pH and GSH dual-responsive drug property would contribute to Ber accumulation in cancer cells and decrease the concentration of the intracellular drug in normal cells.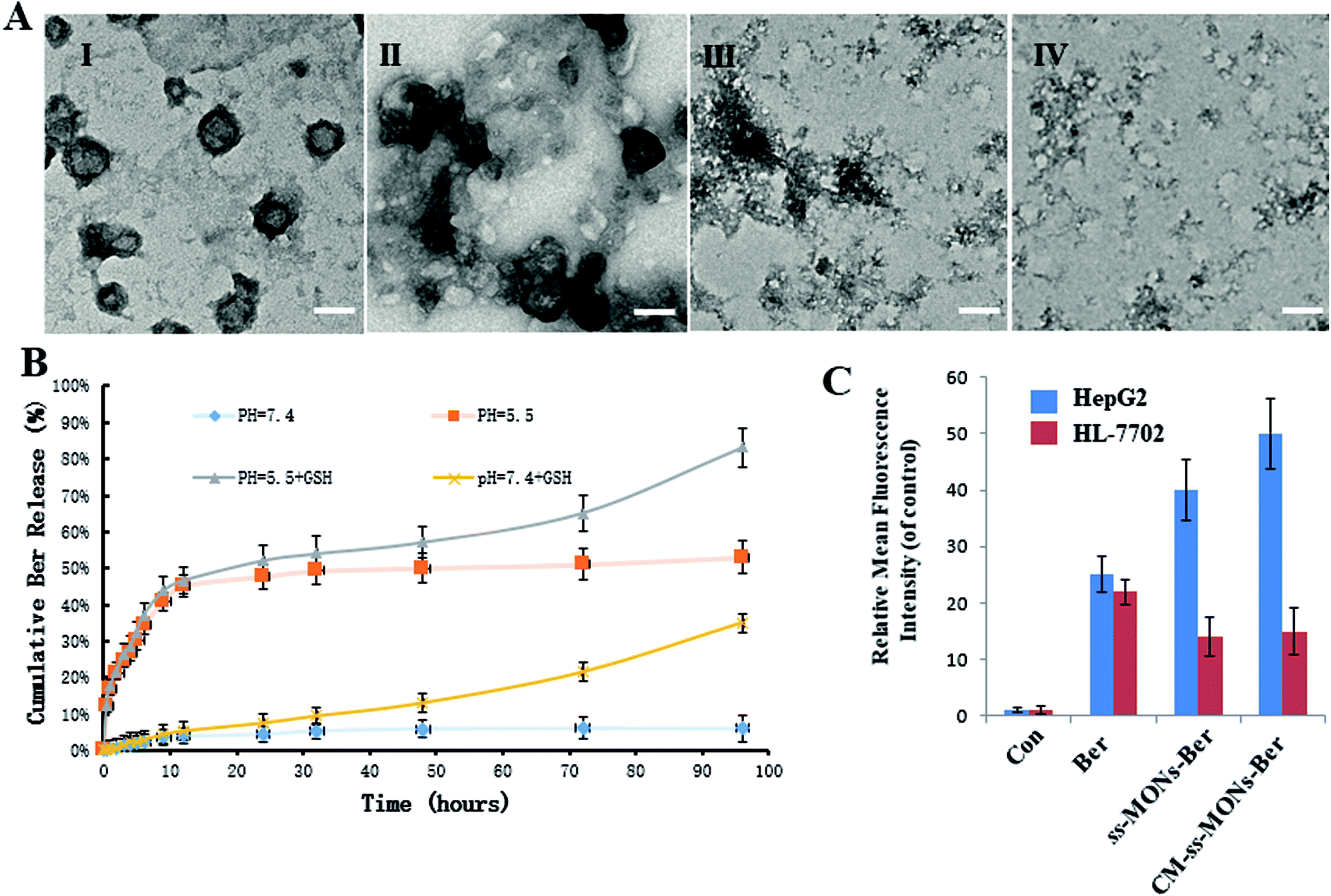 | ||
| Fig. 3 Dual-responsive release behaviour of CM-ss-MONs-Ber. (A) TEM image of CM-ss-MONs treated with glutathione (GSH) after 0 days (I), 1 day (II), 3 days (III) and 5 days (IV). (B) Ber release from ss-MONs-Ber at different pH values (pH 7.4 and 5.5) with or without GSH at the designed intervals (C) the fluorescence intensity of Ber in HepG2 cells and HL-7702 cells after treatment with ss-MONs-Ber or CM-ss-MONs-Ber for 24 h. | ||
To further investigate the drug release behaviours of Ber by CM-ss-MONs-Ber in vitro, the intracellular Ber release in HepG2 cells and HL-7702 cells were detected using flow cytometry. As shown in Fig. 3C, in the HepG2 cells, the fluorescence intensity of Ber in the ss-MONs-Ber-treated group was higher than that in the Ber-treated group. Particularly, the fluorescence intensity was the highest in CM-ss-MONs-Ber-treated group. This was because of the excellent targeting endocytosis and highly efficient drug release of CM-ss-MONs-Ber in cancer cells. In contrast, lower fluorescence was detected in HL-7702 cells after incubation with ss-MONs-Ber or with CM-ss-MONs-Ber in comparison to the Ber-treated group, which was due to the weaker release of Ber in normal cells. The selective Ber release behaviour would enhance the therapeutic effect in cancer cells while reducing the adverse impact on normal cells.
3.4 Cytotoxicity of CM-ss-MONs-Ber
To assess the therapeutic effect of the CM-ss-MONs-Ber in vitro, the cell viability of HepG2 cells and HL-7702 cells were tested after various treatments for 24 h at different concentrations by the MTT assay. As shown in Fig. 4A and B, there was no significant decrease in cell viability after treatment with CM-ss-MONs or ss-MONs in neither the HepG2 cells nor the HL-7702 cells even at very high concentrations (100 μg mL−1), which demonstrated that both CM-ss-MONs and ss-MONs were non-toxic. The inhibition of the three other treated groups (CM-ss-MONs-Ber, ss-MONs-Ber and free Ber) on both the HepG2 cells and HL-7702 cells showed dose-dependent manners. ss-MONs-Ber killed more HepG2 cells than free Ber, and CM-ss-MONs-Ber exhibited the best anti-tumor effect of all the groups (Fig. 4A). Conversely, both CM-ss-MONs-Ber and ss-MONs-Ber caused lower amounts of deaths in normal HL-7702 cells than an equal dose of free Ber after 24 h and 48 h (Fig. 4B). The above results indicated that ss-MONs-Ber and CM-ss-MONs-Ber possessed a selective killing effect on cancer cells rather than normal cells, and CM-ss-MONs-Ber possessed a higher anti-cancer efficacy than ss-MONs-Ber because of their superb homotypic targeting ability.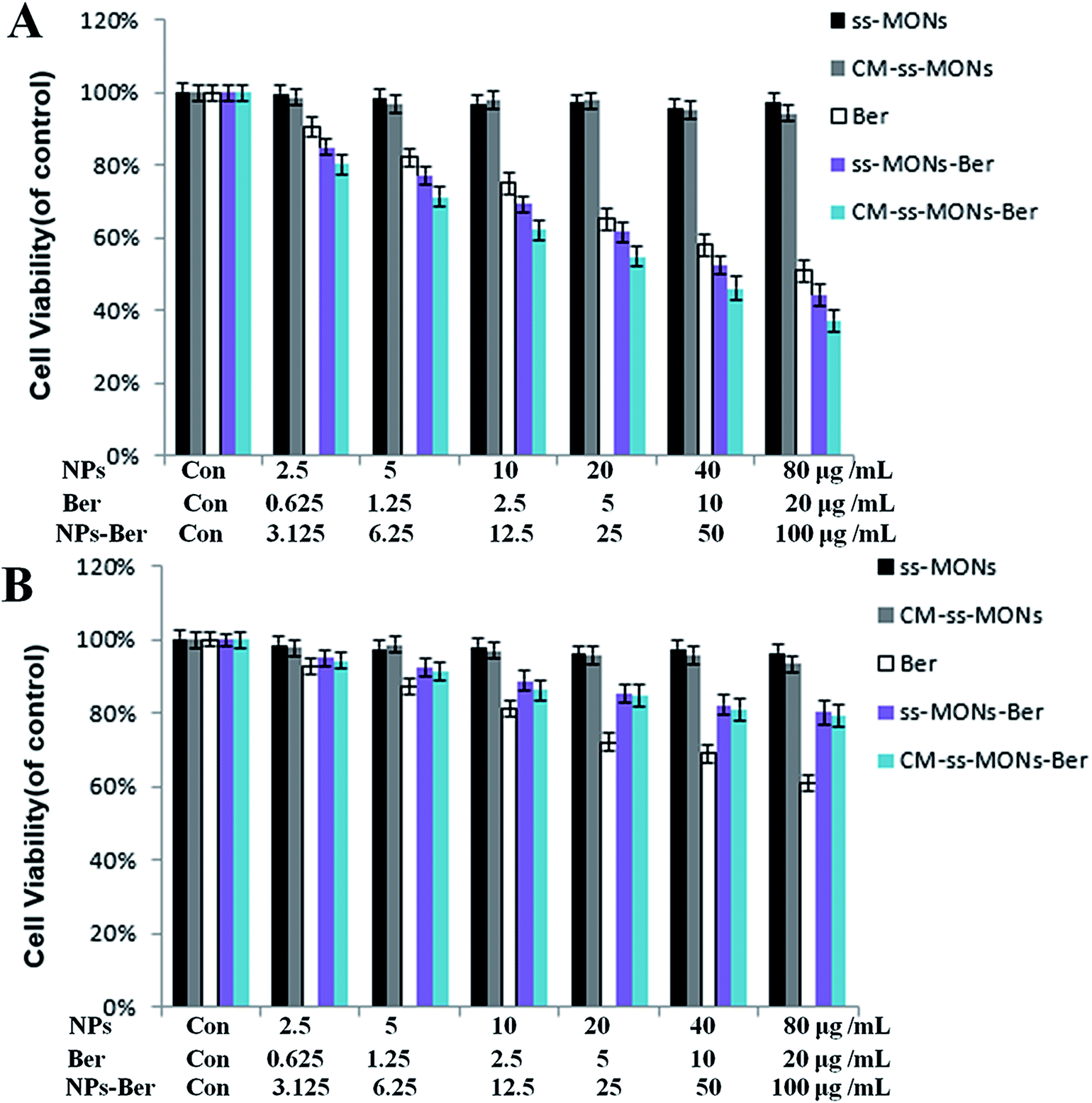 | ||
| Fig. 4 Cytotoxicity of ss-MONs, CM-ss-MONs, Ber, ss-MONs-Ber, CM-ss-MONs-Ber on (A) HepG2 and (B) HL-7702 cells. | ||
3.5 Anti-tumor efficacy in vivo
To further investigate their anti-tumor efficacy in vivo, we intravenously injected PBS, CM-ss-MONs, Ber, ss-MONs-Ber and CM-ss-MONs-Ber into nude mice bearing HepG2 tumor xenografts. As shown in Fig. 5A–C, when treated with PBS, the tumors grew rapidly during the test. The mice treated with ss-MONs showed a similar growth behaviour as the PBS group, which indicated that the ss-MONs had no anti-tumor efficacy. Negligible inhibition efficacy was found in the free Ber, which was due to the poor bioavailability of free Ber. ss-MONs-Ber showed obvious antitumor growth efficiency, resulting from how the ss-MONs-Ber could enhance the bioavailability of Ber and promote the accumulation of Ber in tumor tissue. It is noteworthy that the group treated with CM-ss-MONs-Ber exhibited dramatically suppressed efficiency compared to all the other groups, which could be attributed to the cell membrane mediating the homotypic target and immune escape capacity. Hence, the above results demonstrated that the CM-ss-MONs were excellent nanocarriers for the highly efficient Ber treatment of liver cancer.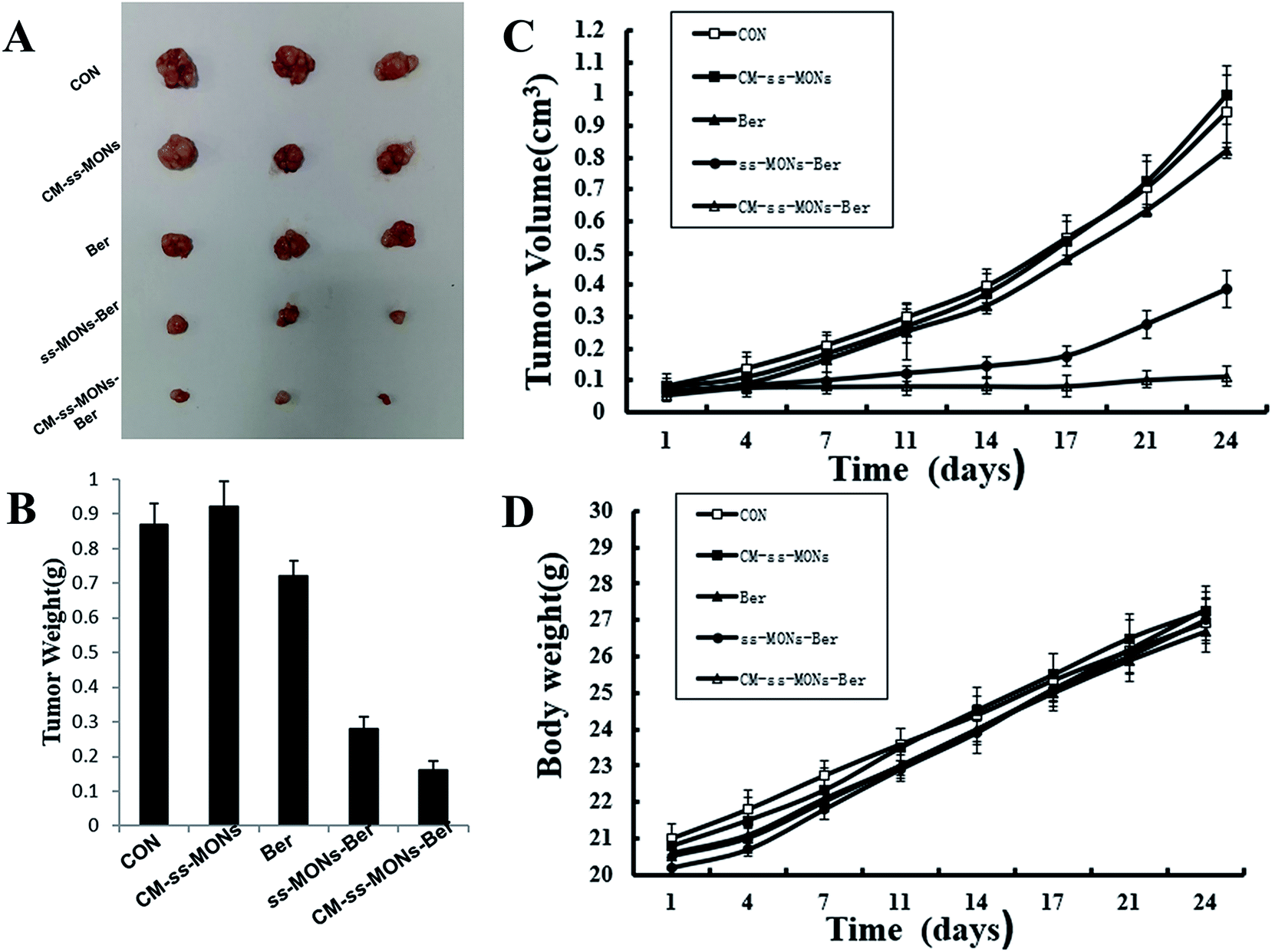 | ||
| Fig. 5 In vivo antitumor effects of CM-ss-MONs-Ber in HepG2 tumor-bearing nude mice: (A) tumor photographs, (B) tumor volume, (C) tumor weight and (D) body weight. | ||
3.6 In vivo biosafety assessment
Moreover, the therapeutic biosafety of these nanoparticles cannot be ignored. Thus, we detected systemic toxicity using mice weight tests, blood chemistry and routine blood tests as well as histopathological analysis. As shown in Fig. 5D, no significant weight loss could be found in any of the treated groups compared to the PBS treated-group. Additionally, there was no obvious change in the level of ALT, AST, BUN and CRE in Fig. 6A–D, suggesting negligible impact of the treatments on liver or kidney function. Furthermore, H&E staining analysis showed no significant pathological changes in the heart, liver, spleen, lung or kidney sections of any of the groups (Fig. 7). In all, CM-ss-MONs-Ber possessed excellent biosafety not only because of the lower toxicity of Ber compared with clinical chemotherapeutic drugs but also due to the fact that silica materials are superb biocompatible elements approved by the FDA for clinical use.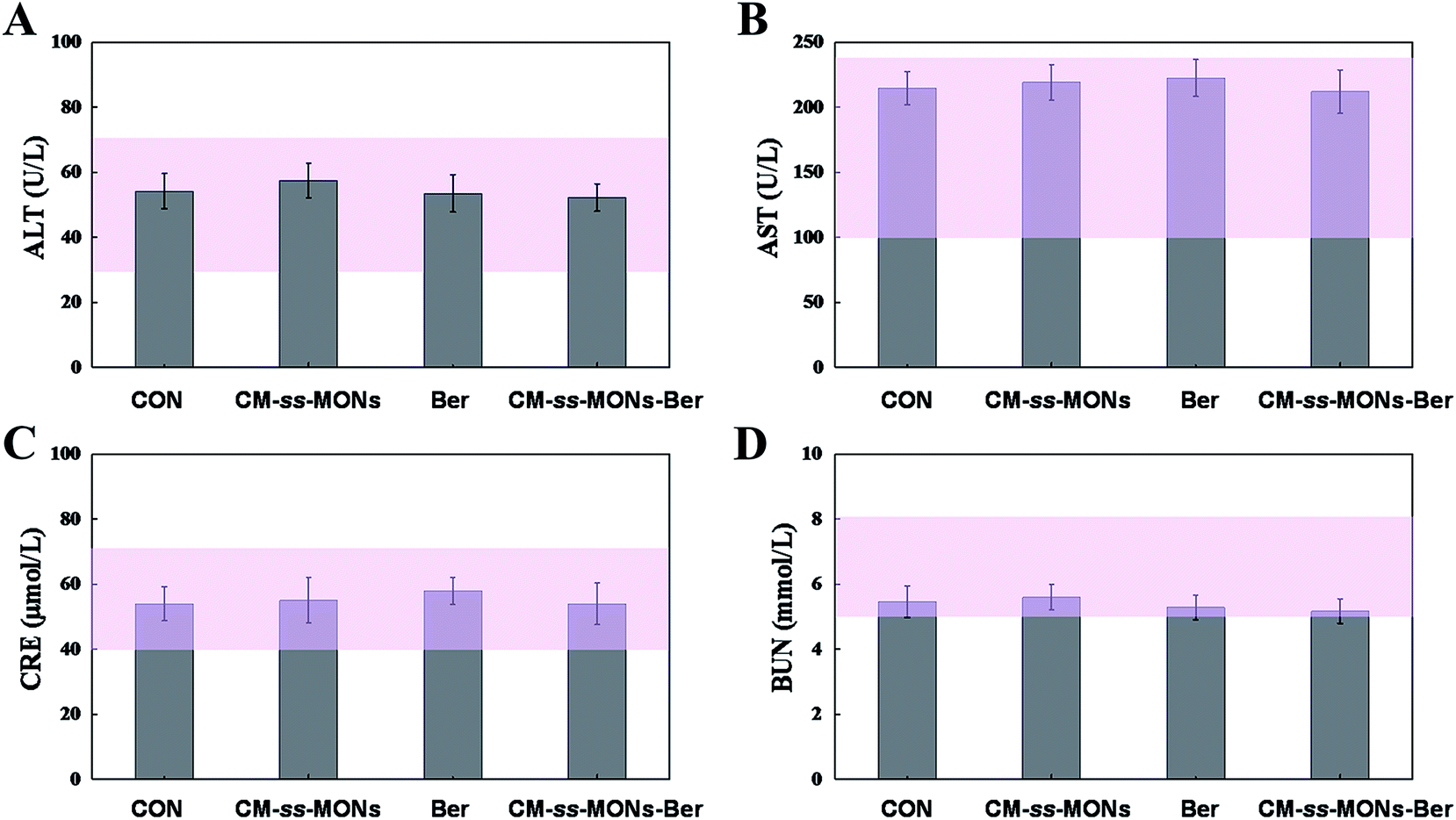 | ||
| Fig. 6 Blood chemistry and routine blood test for each group on the last day of the 21 day treatment. (A) ALT, (B) AST, (C) CRE and (D) BUN for mice from each group. The pink bars represented the range of values obtained from healthy nude mice. | ||
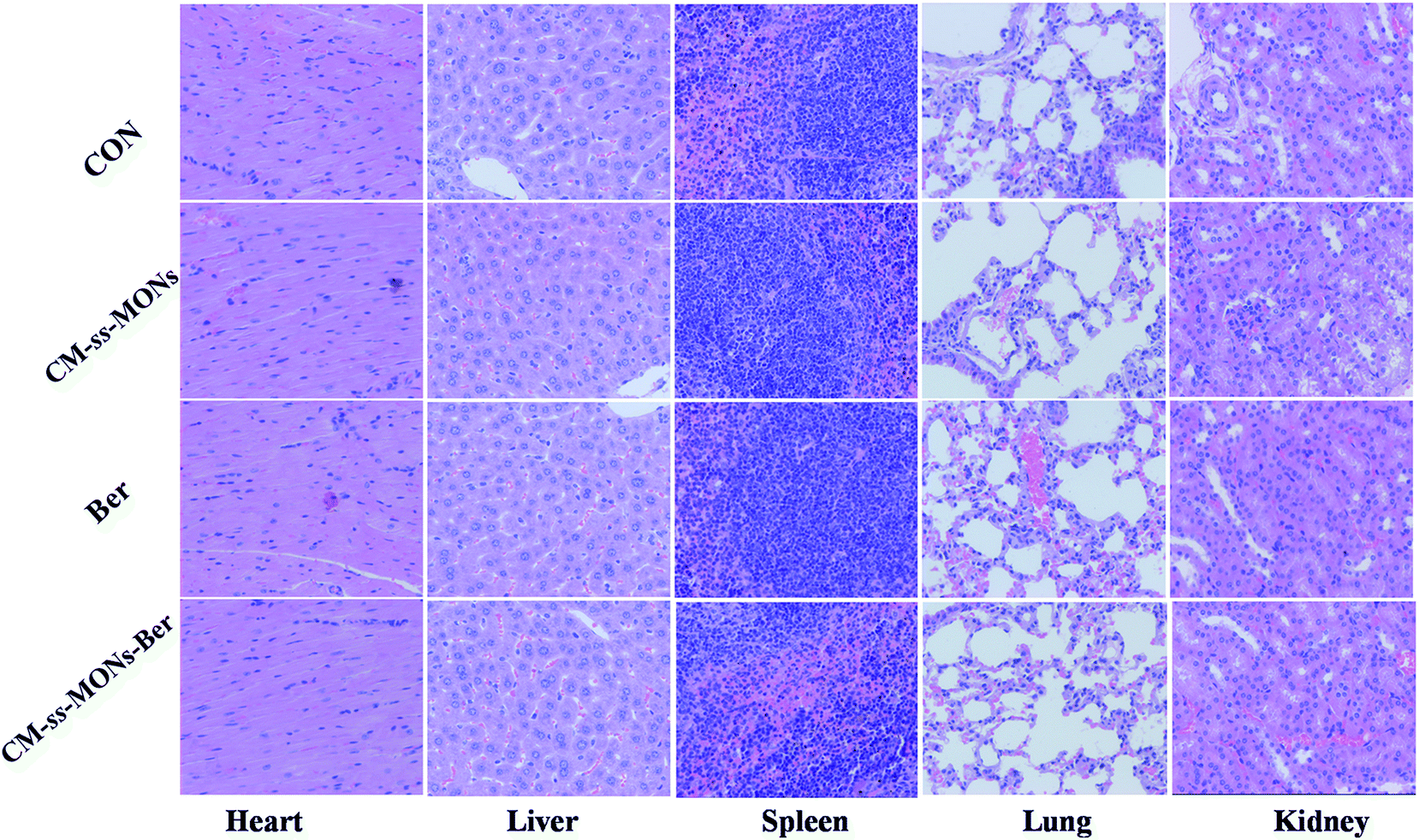 | ||
| Fig. 7 H&E staining images of the heart, liver, spleen, lung and kidney of a mouse from each group. The scale bars represent 100 mm. | ||
4 Conclusions
In summary, we prepared biodegradable ss-MONs with the HepG2 cell membrane coating as a biomimetic nanodrug delivery system for the Ber treatment of liver cancer. The ss-MONs-Ber exhibited uniform morphology, superb monodispersity and excellent Ber loading capacity. After HepG2 cell membrane modification, ss-MONs-Ber obtained superior liver cancer cell targeting efficiency and immune escape ability. More importantly, the pH and GSH dual-responsive drug release properties further enhanced Ber accumulation in tumor tissues rather than in normal tissues. Thereby, during in vitro and in vivo experiments, CM-ss-MONs-Ber exhibited an enhanced Ber therapeutic effect and excellent biosafety. We believe that CM-ss-MONs-Ber are promising agents for the Ber treatment of liver cancer in clinical practice.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (Grant No. 2017YFF0108600, 2017YFC0211900 and 2016YFF0103800), the National Natural Science Foundation of China (Grant No. 81771982, 61535010 and 8160071152), the Key Research Program of the Chinese Academy of Sciences (No. KFZD-SW-210), the Natural Science Foundation of Jiangsu Province (No. BE2015601) and the Science and Technology Department of Suzhou City (No. SS201539 and ZXY201434).Notes and references
- S. Bloom, W. Kemp, A. Nicoll, S. Roberts, K. Gow, P. Dev, A. Bell, S. Sood, S. Kronborg, I. Knight, V. Lewis and D. Lubel, J. Hepatol., 2018, 0168–8278 Search PubMed.
- W. K. Park, A. W. Maxwell, V. E. Frank, M. P. Primmer, S. A. Collins, G. L. Baird and D. E. Dupuy, Theranostics, 2017, 7, 1026–1035 CrossRef CAS PubMed.
- A. Venook, C. Papandreou and J. Laura Ladrón de Guevara, Oncologist, 2010, 15, 5–13 CrossRef PubMed.
- M. Maluccio and A. Covey, Ca-Cancer J. Clin., 2012, 62, 394 CrossRef PubMed.
- U. Asghar and T. Meyer, J. Hepatol., 2012, 56, 686 CrossRef PubMed.
- V. W. Lam, C. Spiro, J. M. Laurence, E. Johnston, M. J. Hollands, H. C. Pleass and A. J. Richardson, Ann. Surg. Oncol., 2012, 19, 1292 CrossRef PubMed.
- M. M. Lu, J. Bai, D. Shao, J. Qiu, M. Li, X. Zheng, Y. Xiao, Z. Wang, Z. M. Chang, L. Chen, W. F. Dong and C. B. Tang, Int. J. Nanomed., 2018, 13, 5849–5863 CrossRef PubMed.
- M. M. Lu, Q. J. Wang, Z. M. Chang, Z. Wang, X. Zheng, D. Shao, W. F. Dong and Y. M. Zhou, Int. J. Nanomed., 2017, 12, 3577–3589 CrossRef CAS PubMed.
- L. Wu, D. Leng, D. Cun, C. Foged and M. Yang, J. Controlled Release, 2017, 260, 78–91 CrossRef CAS PubMed.
- E. Polo, M. Collado, B. Pelaz and P. Del Pino, ACS Nano, 2017, 11, 2397–2402 CrossRef CAS.
- J. Du, Y. Sun, Y. Y. Lu, E. Lau, M. Zhao, Q. M. Zhou and S. B. Su, Anticancer Res., 2017, 37, 6141–6151 Search PubMed.
- J. Liu, X. Zhao, D. Pei, G. Sun, Y. Li, C. Zhu, C. Qiang, J. Sun, J. Shi, Y. Dong, J. Gou, S. Wang and A. Li, Sci. Rep., 2018, 8, 2828 CrossRef.
- M. Tillhon, L. M. G. Ortiz, P. Lombardi and A. I. Scovassi, Biochem. Pharmacol., 2012, 84, 1260 CrossRef CAS PubMed.
- J. Li, O. Li, M. Kan, M. Zhang, D. Shao, Y. Pan, H. Zheng, X. Zhang, L. Chen and S. Liu, Mol. Med. Rep., 2015, 12, 4572 CrossRef CAS PubMed.
- Y. Q. Shan, Y. P. Zhu, J. Pang, Y. X. Wang, D. Q. Song, W. J. Kong and J. D. Jiang, Biol. Pharm. Bull., 2013, 36, 1562 CrossRef CAS.
- C. Ma, K. L. Tang, Q. Liu, R. X. Zhu and Z. W. Cao, Chem. Biol. Drug Des., 2013, 81, 775 CrossRef CAS.
- C. S. Liu, Y. R. Zheng, Y. F. Zhang and X. Y. Long, Fitoterapia, 2016, 109, 274 CrossRef CAS PubMed.
- Z. Wang, Y. S. Wang, Z. M. Chang, L. Li, Y. Zhang, M. M. Lu, X. Zheng, M. Li, D. Shao, J. Li, L. Chen and W. F. Dong, Chem. Biol. Drug Des., 2017, 89, 464–469 CrossRef CAS PubMed.
- L. Yu, Y. Chen, H. Lin, W. Du, H. Chen and J. Shi, Biomaterials, 2018, 161, 292–305 CrossRef CAS PubMed.
- M. Dang, W. Li, Y. Y. Zheng, X. D. Su, X. B. Ma, Y. L. Zhang, Q. Q. Ni, J. Tao, J. J. Zhang, G. G. Lu, Z. G. Teng and L. H. Wang, J. Mater. Chem. B, 2017, 5, 2625–2634 RSC.
- E. P. F. Nhavene, G. Ferreira Andrade, J. A. C. Pinese, J. Q. Lin, U. Milbreta, M. Q. Li, Y. C. Wang, K. W. Leong and S. Y. Chew, Acta Biomater., 2018, 76, 164–177 CrossRef PubMed.
- Q. Arantes Faria, D. Assis Gomes and E. Martins Barros de Sousa, ChemEngineering, 2018, 2(2), 24 CrossRef.
- S. P. H. Moghaddama, M. Yazdimamaghania and H. Ghandeharia, J. Controlled Release, 2018, 282, 62–75 CrossRef PubMed.
- C. Pinese, J. Q. Lin, U. Milbreta, M. Q. Li, Y. C. Wang, K. W. Leong and S. Y. Chew, Acta Biomater., 2018, 76, 164–177 CrossRef CAS PubMed.
- P. Y. Huang, H. Chen, L. Lin, L. Yu, L. Zhang, Y. Wang and J. Zhu, Biomaterials, 2017, 125, 125 CrossRef PubMed.
- J. G. Croissant, Y. Fatieiev, K. Julfakyan, J. Lu, A.-H. Emwas, D. H. Anjum, H. Omar, F. Tamanoi, J. I. Zink and N. M. Khashab, Chem.–Eur. J., 2016, 22, 14806 CrossRef CAS PubMed.
- J. G. Croissant, Y. Fatieiev and N. M. Khashab, Adv. Mater., 2017, 29, 9 Search PubMed.
- J. Lu, M. Liong, Z. Li, J. I. Zink and F. Tamanoi, Small, 2010, 6, 1794 CrossRef CAS.
- J. Yue, S. Z. Luo, M. M. Lu, D. Shao, Z. Wang and W. F. Dong, Chem. Biol. Drug Des., 2018, 92, 2 CrossRef.
- M. Zhou, X. Du, W. Li, X. Li, H. Huang, Q. Liao, B. Shi, X. Zhang and M. Zhang, J. Mater. Chem. B, 2017, 5, 4455 RSC.
- W. Gao, C. M. Hu, R. H. Fang, B. T. Luk, J. Su and L. Zhang, Adv. Mater., 2013, 25, 3549 CrossRef CAS.
- H. P. Sun, J. H. Su, Q. S. Meng, Q. Yin, L. L. Chen, W. G. Gu, Z. W. Zhang, H. J. Yu, P. C. Zhang, S. L. Wang and Y. P. Li, Adv. Funct. Mater., 2017, 27, 1604300 CrossRef.
- D. Shao, M. Li, Z. Wang, X. Zheng, Y. H. Lao, Z. Chang, F. Zhang, M. Lu, J. Yue, H. Hu, H. Yan, L. Chen, W. F. Dong and K. W. Leong, Adv. Mater., 2018, 1801198 CrossRef.
- L. Rao, B. Cai, L. L. Bu, Q. Q. Liao, S. S. Guo, X. Z. Zhao, W. F. Dong and W. Liu, ACS Nano, 2017, 11(4), 3496–3505 CrossRef CAS PubMed.
- N. E. T. Furman, Y. Lupu-Haber, T. Bronshtein, L. Canetti, N. Letko, E. Weinstein, L. Baruch and M. Machluf, Nano Lett., 2013, 13, 3248 CrossRef.
- Z. Wang, D. Shao, Z. Chang, M. Lu, Y. Wang, J. Yue, D. Yang, M. Li, Q. Xu and W. F. Dong, ACS Nano, 2017, 11, 12732–12741 CrossRef CAS PubMed.
- B. Chang, J. Guo, C. Liu, J. Qian and W. Yang, J. Mater. Chem., 2010, 20, 9941 RSC.
- J. Croissant, X. Cattoen, M. W. Man, A. Gallud, L. Raehm, P. Trens, M. Maynadier and J. O. Durand, Adv. Mater., 2014, 26, 6174–6180 CrossRef CAS PubMed.
- D. Shao, J. Li, X. Zheng, Y. Pan, Z. Wang, M. Zhang, Q.-X. Chen, W.-F. Dong and L. Chen, Biomaterials, 2016, 100, 118 CrossRef CAS PubMed.
- B. S. Chang, J. Guo, C. Y. Lu, J. Qian and W. L. Yang, J. Mater. Chem., 2010, 20(44), 9941 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07574c |
| This journal is © The Royal Society of Chemistry 2018 |