
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbohydrate-conjugated 4-(1,3,2-dithiarsolan-2-yl)aniline as a cytotoxic agent against colorectal cancer
Boqiao Fu *a,
Xiaolin Wangb,
Yingjie Lib,
Jingying Huc,
Dai Lud,
Wei Lia,
Kewang Zhenga and
Caiqin Qin*a
*a,
Xiaolin Wangb,
Yingjie Lib,
Jingying Huc,
Dai Lud,
Wei Lia,
Kewang Zhenga and
Caiqin Qin*a
aHubei Provincial Collaborative Innovation Center of Biomass Resources Transformation and Utilization, College of Chemistry and Materials Science, Hubei Engineering University, Hubei 432000, P. R. China. E-mail: fuboqiao@126.com
bGuangdong Institute of Gastroenterology, Guangdong Provincial Key Laboratory of Colorectal and Pelvic Floor Diseases, The Sixth Affiliated Hospital, Sun Yat-sen University, Guangzhou, Guangdong 510655, China
cState Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Renji Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, 200032, China
dDepartment of Pharmaceutical Sciences, Rangel College of Pharmacy, Texas A&M University, Texas 78363, USA
First published on 5th December 2018
Abstract
Arsenic trioxide (As2O3) has been approved for the treatment of acute promyelocytic leukemia (APL); however, its use in the treatment of solid tumors is limited due to its pharmacokinetic properties. Organic arsenic compounds provide better options for pharmaceutical optimization. p-Aminophenyl arsenoxide (p-APAO), an organic arsenic compound, was found to interact with the promyelocytic leukemia–retinoic acid receptor alpha (PML–RARα) fusion protein in a similar manner to arsenic trioxide. Analogs of p-APAO such as 4-(1,3,2-dithiarsolan-2-yl)aniline (p-APDTAs) were recently found to show improved cytotoxicity toward several solid tumor cell lines with lower toxicity to normal cells. Here, we synthesized a carbohydrate-conjugated 4-(1,3,2-dithiarsolan-2-yl)aniline (p-APDTAs) and showed that it exhibited reduced cytotoxicity to normal cells, suggesting a feasible approach to improve the therapeutic index of arsenic-containing compounds as chemotherapeutic agents.
Arsenic trioxide (As2O3) has been used for various purposes in ancient China and Greece for more than 2400 years.1,2 Use of arsenic-containing compounds to treat hematologic disorders such as leukemia, Hodgkin's disease, and pernicious anemia has been reported since the 1700s.3 In the 1970s, a group of physicians in China reported that a formulation of arsenic trioxide induced complete remission in a small group of patients with acute promyelocytic leukemia (APL).1 Later on, the follow-up clinical trials by American investigators confirmed the clinical efficacy of arsenic trioxide in the management of APL. Subsequently, arsenic trioxide (Trisenox™) was approved by the U.S. Food and Drug Administration (FDA) in 2000 for the treatment of relapsed or refractory APL.4
Investigations to decipher the mechanism of action of arsenic trioxide revealed that the drug is able to promote the catabolic degradation of an oncogenic fusion protein that derives the proliferation of APL cells and has been found in over 98% of cases of human APL.5 The oncogenic fusion protein is the result of chromosomal rearrangements that juxtapose the promyelocytic leukemia (PML) gene and the retinoic acid receptor alpha (RARα) gene.6 Arsenic trioxide is capable of covalently modifying a cysteine-rich region of the PML moiety of the PML–RARα fusion protein, leading to proteasome-dependent degradation of the oncogenic PML–RARα fusion protein.7,8 Further studies have shown that arsenic trioxide can induce apoptosis of hematologic cancer cells and solid tumor cells via a myriad of mechanisms such as disruption of mitochondrial functions and cellular redox processes, activation of different caspases, and downregulation of Bcl-2 expression.9,10
Despite the remarkable success of arsenic trioxide in the treatment of APL, the inorganic arsenic compound has certain limitations as a chemotherapeutic such as the systemic toxicity associated with a high amount of arsenic compounds in the blood, and poor pharmacokinetic properties.2,11 Although arsenic trioxide has shown efficacy for solid tumors in many preclinical studies, the therapeutic effects of low-dose arsenic oxide for solid tumors have not been clinically proven yet. This may be attributed to the rapid renal clearance of arsenic trioxide metabolites. This likely results in an insufficient amount of arsenic oxide at the tumor sites.2
Along with the inorganic arsenic trioxide, organic arsenic compounds such as p-aminophenylarsine oxide (p-APAO)5,12,13 and others3,14–16 were investigated either as preclinical and clinical experimental drugs or as molecular probes in cancer cells. Arsenic sulfide and its derivatives were reported with potent antitumor activities to solid tumor cell lines such as HCT 116.17,18 In comparison with inorganic arsenic compounds, organic arsenic compounds offer certain advantages. They are generally more chemically stable than inorganic arsenic compounds and are easier for structural optimization to improve drug-like properties such as pharmacokinetic properties, therapeutic efficacy, and target selectivity etc. Some of the organic arsenic compounds, such as melarsoprol (Fig. 1D) and ZIO-101 (Fig. 1E) are more efficacious than arsenic trioxide to induce apoptosis in cancer cells.3 Interestingly, a biotin-conjugated p-APAO was found capable to bind to PML–RARα in a manner similar to arsenic trioxide (As2O3).5,13 These suggested that compounds containing a p-APAO moiety may possess anticancer activities As2O3. However, p-APAO is a substituted phenyl arsenoxide, which can be easily oxidized to pentavalent arsenic (AsV) compound and has higher tendency to form polymers.15,19 Recently, it was reported that p-APAO analogues 4-(1,3,2-dithiarsolan-2-yl)aniline (p-APDTAs, Fig. 1C) obtained by protecting the arsenoxide moiety of p-APAO with dithiols are more efficacious than p-APAO in inducing apoptosis in several cancer cell lines. Furthermore, the p-APDTAs showed less cytotoxicity toward normal cells than p-APAO.20 Collectively, these triggered our interests to employ p-APDTAs (Fig. 1C) rather than the p-APAO (Fig. 1B) as the arsenic-containing moiety for developing novel arsenic-containing compounds as anticancer agents.
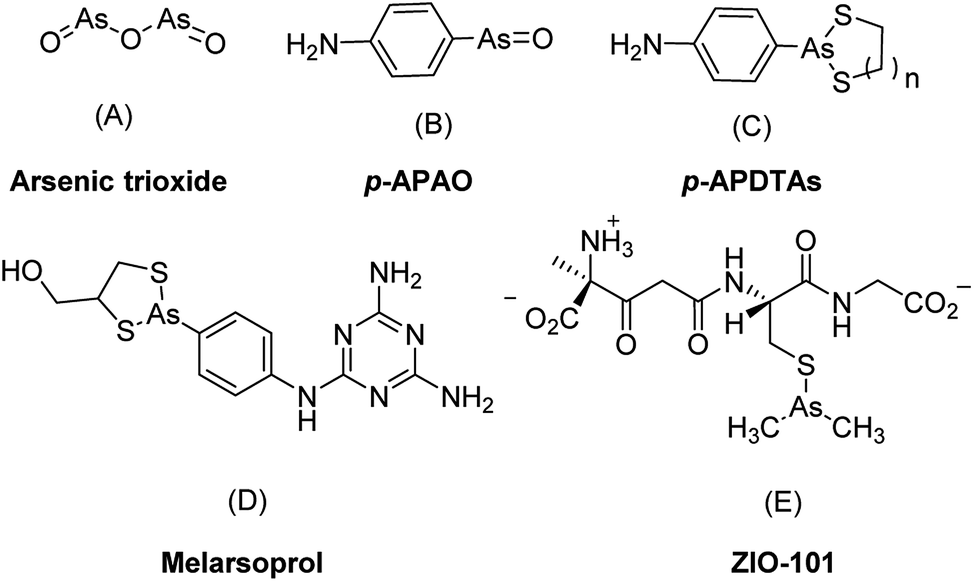 | ||
| Fig. 1 Representative arsenic-containing compounds showing anticancer activities. | ||
In our earlier investigation of detoxification of p-APAO with various carbohydrates, we found that 1,3,4,6-tetra-O-acetyl-β-D-glucosamine (OADG) is capable of reducing the toxicity of p-APAO toward the normal organelle modeled by S. cerevisiae.21 The natural product Jusbetonin is an indolo[3,2-b]quinoline alkaloid glycoside containing a β-D-glucose moiety. In a structural optimization, the replacement of its β-D-glucose with OADG was shown to enhance the cytotoxicity against breast cancer cell line MDA-231.22 Based on these results, we hypothesized that introducing OADG into organic arsenic compounds such as p-APDTAs may preserve the anticancer effects of organic arsenic compounds while the cytotoxicity toward normal cells can be reduced. Hence, we designed and synthesized OADG-conjugated p-APAO analogue (8) and p-APDTAs analogue (9). We assessed the cytotoxicity of the synthesized conjugates toward the solid tumor cells from three colorectal cancer cell lines HCT116, DLD1, and RKO and the cytotoxicity toward normal cells using epithelial NCM460 cells as the model.23
Click chemistry has been frequently applied in conjugation of bioactive molecules with various pharmaceutical agents and biomolecules for preclinical and clinical applications.24 Its typical process involves azide–alkyne [3 + 2] dipolar cycloaddition to form a 1,4-regioselectivity 1,2,3-triazole-based linker under the catalysis of CuI. The products from click chemistry has been proven to be superior in satisfying many criteria in drug development and biomedical research such as excellent biocompatibility, selectivity, yield, and stereospecificity.24 Many known 1,2,3-triazoles have various biological activities such as anti-HIV, anticancer, and antibacterial activities.25–27 In our study, the target compound was synthesized through the click reaction between a carbohydrate (i.e., OADG) moiety functionalized with an azido group and a p-APDTAs moiety functionalized by a propargyl group. The synthetic chemistry employed in this work is illustrated in Scheme 1 and 2.
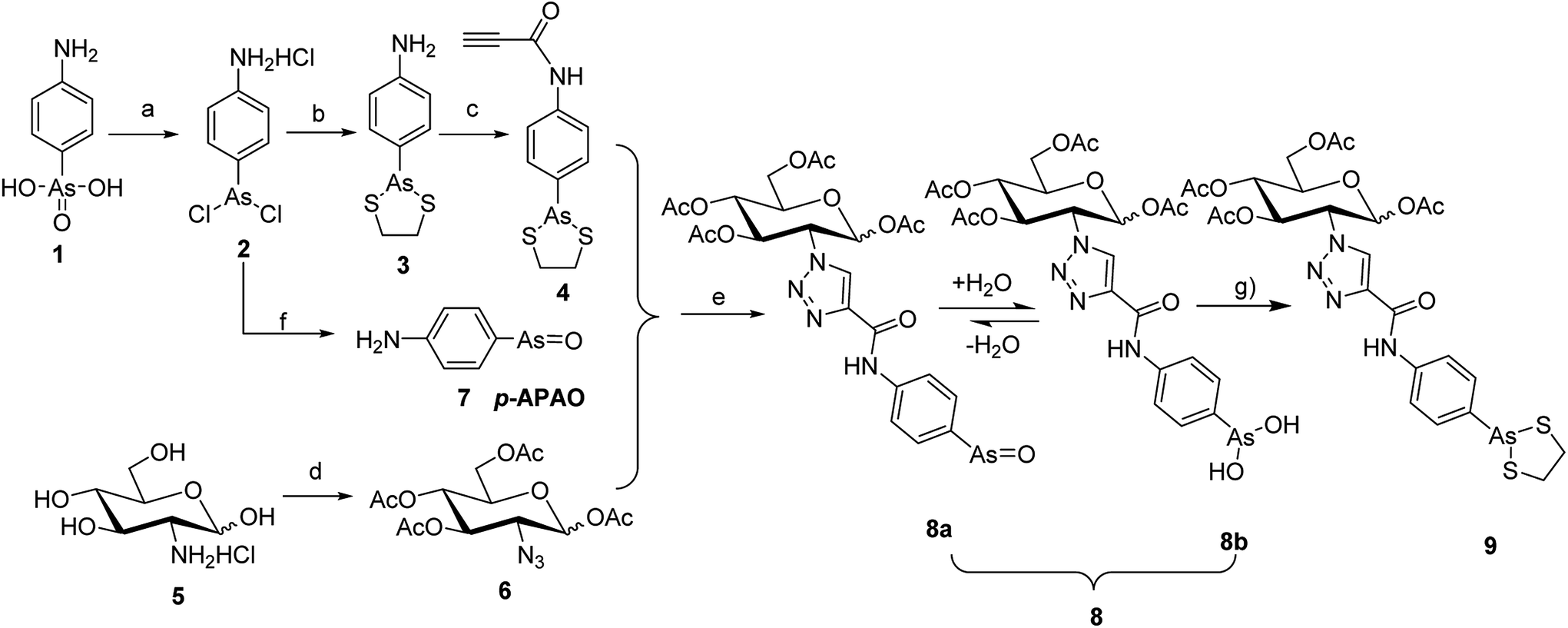 | ||
| Scheme 1 Synthesis routes of compounds 7 and 9. Reaction conditions: (a) SO2, KI, HCl, MeOH; (b) ethane-1,2-dithiol, NaHCO3, MeOH, 82%; (c) propiolic acid, dicyclohexyl carbodiimide, CH2Cl2, r.t, 64%; (d) (i) TfN3, CuSO4·5H2O, CH3CN; (ii) Ac2O, pyridine; (e) sodium L-ascorbate, CuSO4·5H2O, microwave, 100 °C, 38.3%; (f) NH3·H2O, N2, 53.9%; (g) ethane-1,2-dithiol, MeOH, 69%. | ||
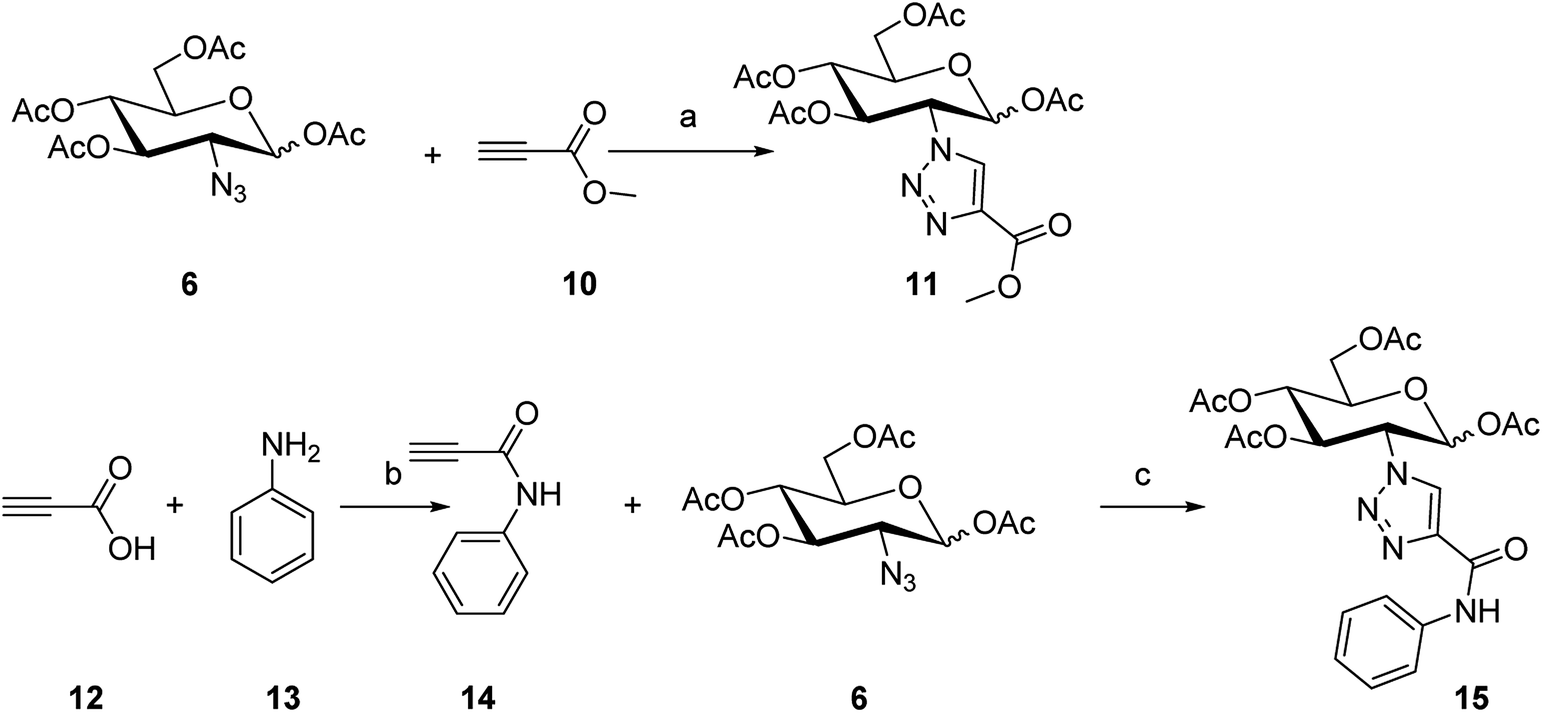 | ||
| Scheme 2 Synthesis routes of compounds 11 and 15. Reaction conditions: (a) sodium L-ascorbate, CuSO4·5H2O, microwave, 100 °C, 61.1%; (b) propiolic acid, dicyclohexyl carbodiimide, CH2Cl2, r.t, 70%; (c) sodium L-ascorbate, CuSO4·5H2O, microwave, 100 °C, 32.5%. | ||
To synthesize the alkyn-containing reactant (4) for click reaction, commercially available p-arsanilic acid 1 was first converted into 4-aminophenyldichloroarsine (2) according to reported protocols.20,28,29 The product 2 was easily transformed into the intermediate 3 through reaction with 1,2-ethandithiol under aqueous sodium carbonate. Compound 3 reacted with propiolic acid in the presence of DCC to provide the click chemistry reactant 4. To synthesize the other reactant of the click reaction (i.e., azide 6), which bears the 1,3,4,6-tetra-O-acetyl-β-D-glucosamine (OADG) moiety, the amino group of commercially available 2-amino-2-deoxy-D-glucose (5) was converted to azido functionality by reaction with trifluoromethanesulfonyl azide, followed by acetylation of its 1,3,4,6 hydroxyl groups to yield the key building block 6.30,31 Coupling of 4 with 6 under typical conditions of click reaction produced the desired conjugate 8, which exists as a mixture of equilibrium of arsenic oxide 8a and its hydrated analog arsonous acid 8b. The presence of the arsenic oxide 8a and arsonous acid 8b was confirmed by NMR and mass spectrometry analysis.32 To obtain the p-APDTAs analogue (9), the mixture of arsenic compounds 8a and 8b was treated with 1,2-ethandithiol in anhydrous methanol to provide the target compound. The reference compound p-APAO (7) was prepared as previously described,28 and used to compare with other derivatives of dithiarsolanes in the cytotoxicity assays. Because of formation of a triazole moiety during the linkage by click reaction, we synthesized the carbohydrate-triazole-containing compounds 11 and 15 to study the biological influence of the triazole moiety in the structure of 9 (Scheme 2).
It was reported that derivatives of arsenic trioxide (As2O3) slightly inhibited the proliferation of human colon cancer cell line HCT116.17,34 To determine the antitumor activities of the synthesized compounds in colorectal cancer, three common colorectal cancer cell lines (i.e., HCT116, DLD1, and RKO)33 were selected for cytotoxicity assessment of the synthesized compounds (3, 4, p-APAO, 8, 9, 11, 15) and the standard chemotherapeutic for colorectal cancer 5-FU was used as a positive control drug.35 Their cytotoxicity in intestinal epithelial cell line NCM460, which is a model of normal cells, were also evaluated. The half maximal inhibitory concentrations (IC50) of the synthesized compounds are shown in Table 1 and the dose dependent inhibition of proliferation by compound 9 and 5-FU are illustrated in Fig. 2.
| IC50 (μM) ± SD (μM) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compounds | 5-FU | p-APAO | 3 | 4 | 8 | 9 | 11 | 15 |
| a NCM460 cell is used as the model of normal cell.b ND = not determined.c Selectivity index: the IC50 against HCT116 over the IC50 against NC460. | ||||||||
| NCM460a | 44.57 ± 4.69 | 3.67 ± 0.10 | 11.11 ± 0.82 | 3.20 ± 0.78 | 16.15 ± 2.21 | 50.75 ± 0.97 | NDb | NDb |
| HCT-116 | 3.62 ± 1.22 | 5.03 ± 0.66 | 1.86 ± 0.34 | 0.53 ± 0.20 | 17.69 ± 1.44 | 1.29 ± 0.21 | >50 | 43.39 ± 4.17 |
| DLD1 | >50 | 2.17 ± 0.21 | 6.90 ± 1.81 | 1.41 ± 0.59 | 14.96 ± 2.72 | 13.96 ± 2.05 | >50 | 20.28 ± 2.30 |
| RKO | 2.09 ± 0.87 | 2.99 ± 0.04 | 3.74 ± 0.88 | 0.79 ± 0.05 | 4.57 ± 0.17 | 12.55 ± 1.90 | 20.03 ± 3.38 | >50 |
| Selectivity indexc | 12.31 | 0.73 | 5.97 | 6.04 | 0.91 | 39.34 | — | — |
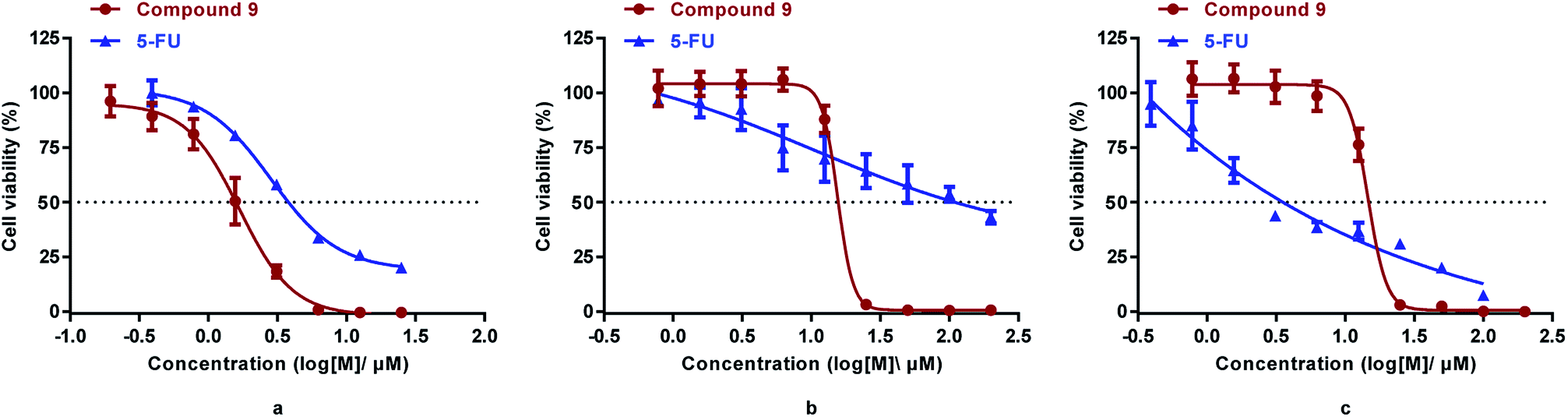 | ||
| Fig. 2 Cytotoxicity of compound 9 and 5-FU in HCT-116 (a), DLD1(b), and RKO (c) cells measured by the MTS assay (mean ± SD). | ||
Similar to 5-FU, arsenic containing compounds 3, 4, 8, 9 and p-APAO all exhibited anti-proliferation activity in the three colorectal cancer cell lines. Particularly, arsenic compound 4 showed significantly enhanced activity cross the three colorectal cancer cell lines in comparison with 5-FU. However, these compounds showed higher cytotoxicity in normal cells except the p-APDTAs analogue (9), which exhibited potent activity against HCT116 cells with IC50 value of 1.29 ± 0.21 μM. It also showed strong activities against DLD1 and RKO cell lines with IC50 values of 13.96 ± 2.05 μM and 12.55 ± 1.90 μM, respectively. Compound 9 showed stronger cytotoxicity than 5-FU in HCT-116 and DLD1 cells, but not in RKO cells. Compare to 5-FU, compound 9 displayed similar cytotoxicity towards normal colorectal endothelial cells NCM460, but with over 1.8 fold increase of cytotoxicity to colorectal cancer HCT-116 cells.
The results from compounds 11 and 15 indicated that the p-APDTAs moiety is critical for the anticancer effects of compound 9. The absence of the arsolane group significantly reduced antitumor activity as observed from compound 15. Similarly, the triazole-containing compound 11, which is derived from 1,3,4,6-tetra-O-acetyl-β-D-glucosamine (OADG), was not responsible for the anticancer effects of compound 9. The compound 8 is the OADG-conjugated derivative of p-APAO and the compound 9 is the OADG-conjugated derivative of p-APDTAs. In comparison with their non-conjugated count part (i.e. 8 vs. p-APAO, and 9 vs. 3), the OADG-conjugated organic arsenic compounds 8 and 9 showed significantly reduced toxicity towards normal colorectal cells while their anti-proliferation activities were generally maintained. We also found that deacetylation of compound 9 with sodium methoxide and then neutralization with acid resin couldn't give deacetylation compound. Actually dithiol protective group on the arsenic compound 9 was also deprotected under acid resin.36,37 When we used dithiol to protect arsenic acid of deacetylation carbohydrate-conjugated arsenic compound again, all the naked hydroxyl groups could be protected. So it is very difficult to get the deacetyl compound from 9. The usefulness of 1,3,4,6-tetra-O-acetyl-β-D-glucosamine (OADG) in anticancer molecules were also demonstrated by others. The acetyl protective groups were deemed beneficial in comparison with the unprotected β-D-glucosamine bearing analogues.22
Conclusions
In this investigation, it was found that conjugation of cytotoxic arsenic-containing compound p-APDTAs with β-D-glucose 9 derived 1, 3, 4, 6-O-acetyl-β-D-glucoamine (OADG) can significantly reduce the toxicity to normal colorectal endothelial cells while the cytotoxicity towards colorectal cancer cells can be maintained. The conjugation linker introduced by click chemistry did not abolish the anticancer activity of the resultant compound, which showed anticancer activity comparable to the standard chemotherapeutic 5-flurouraicil (5-FU) in colorectal cancer models. These results suggested a new approach for the discovery and development of arsenic-containing compounds as novel chemotherapeutics for the treatment of solid tumors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Natural Science Foundation of Hubei Province, China (2014CFB570), the National Natural Science Foundation of China (Grant No. 31371750 and 81502022) and the Fundamental Research Fund for the Sun Yat-sen University (Grant No. 16ykpy35).References
- S. Waxman and K. C. Anderson, Oncologist, 2001, 6, 3–10 CrossRef CAS PubMed.
- E. P. Swindell, P. L. Hankins, H. Chen, Đ. U. Miodragović and T. V. O'Halloran, Inorg. Chem., 2013, 52, 12292–12304 CrossRef CAS PubMed.
- P. J. Dilda and P. J. Hogg, Cancer Treat. Rev., 2007, 33, 542–564 CrossRef CAS PubMed.
- K. H. Antman, Oncologist, 2001, 6, 1–2 CrossRef CAS PubMed.
- X.-W. Zhang, X.-J. Yan, Z.-R. Zhou, F.-F. Yang, Z.-Y. Wu, H.-B. Sun, W.-X. Liang, A.-X. Song, V. Lallemand-Breitenbach and M. Jeanne, Science, 2010, 328, 240–243 CrossRef CAS PubMed.
- G.-B. Zhou, J. Zhang, Z.-Y. Wang, S.-J. Chen and Z. Chen, Philos. Trans. R. Soc., B, 2007, 362, 959–971 CrossRef CAS PubMed.
- A. Emadi and S. D. Gore, Blood Rev., 2010, 24, 191–199 CrossRef CAS PubMed.
- J. Zhu and V. Lallemand-Breitenbach, Oncogene, 2001, 20, 7257 CrossRef CAS PubMed.
- A. M. Evens, M. S. Tallman and R. B. Gartenhaus, Leuk. Res., 2004, 28, 891–900 CrossRef CAS PubMed.
- K. Alimoghaddam, Int. J. Hematol. Oncol. Stem Cell Res., 2014, 8, 44 Search PubMed.
- I. Khairul, Q. Q. Wang, Y. H. Jiang, C. Wang and H. Naranmandura, Oncotarget, 2017, 8, 23905 CrossRef PubMed.
- H.-N. Zhang, L. Yang, J.-Y. Ling, D. M. Czajkowsky, J.-F. Wang, X.-W. Zhang, Y.-M. Zhou, F. Ge, M.-K. Yang and Q. Xiong, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15084–15089 CrossRef CAS PubMed.
- X. Zhang, F. Yang, J.-Y. Shim, K. L. Kirk, D. E. Anderson and X. Chen, Cancer Lett., 2007, 255, 95–106 CrossRef CAS PubMed.
- E. Hikita, M. Arai, S. Tanaka, K. Onda, H. Utsumi, B. Yuan, H. Toyoda and T. Hirano, Anticancer Res., 2011, 31, 4169–4178 CAS.
- N. Wimmer, J. A. Robinson, N. Gopisetty-Venkata, S. J. Roberts-Thomson, G. R. Monteith and I. Toth, Med. Chem., 2006, 2, 79–87 CrossRef CAS.
- X.-Y. Fan, X.-Y. Chen, Y.-J. Liu, H.-M. Zhong, F.-L. Jiang and Y. Liu, Sci. Rep., 2016, 6, 29865 CrossRef CAS.
- L. Zhang, Y. Tong, X. Zhang, M. Pan and S. Chen, Drug Des., Dev. Ther., 2015, 9, 5851–5862 CAS.
- W. Ding, Y. Tong, X. Zhang, M. Pan and S. Chen, Sci. Rep., 2016, 6, 19793 CrossRef CAS PubMed.
- C. Huang, Q. Yin, J. Meng, W. Zhu, Y. Yang, X. Qian and Y. Xu, Chem.–Eur. J., 2013, 19, 7739–7747 CrossRef CAS PubMed.
- Y. Liu, D. Duan, J. Yao, B. Zhang, S. Peng, H. Ma, Y. Song and J. Fang, J. Med. Chem., 2014, 57, 5203–5211 CrossRef CAS PubMed.
- B. Fu, L. Lv, C. Xia, K. Zheng, L. Zhu, W. Li, Y. Yan and C. Qin, J. Med. Chem. Toxicol, 2017, 2, 28–33 Search PubMed.
- S. Liu, J. Meng, W. Zhang, S. Wan and T. Jiang, J. Heterocycl. Chem., 2016, 53, 1194–1199 CrossRef CAS.
- S. L. Organ, J. Hai, N. Radulovich, C. B. Marshall, L. Leung, T. Sasazuki, S. Shirasawa, C. Zhu, R. Navab, M. Ikura and M. Tsao, PLoS One, 2014, 9, e86103 CrossRef PubMed.
- K. Nwe and M. W. Brechbiel, Cancer Biother.Radiopharm., 2009, 24, 289–302 CrossRef CAS PubMed.
- R. Alvarez, S. Velazquez, A. San-Felix, S. Aquaro, E. De Clercq, C.-F. Perno, A. Karlsson, J. Balzarini and M. J. Carmarasa, J. Med. Chem., 1994, 37, 4185–4194 CrossRef CAS PubMed.
- (a) S. Velaquez, R. Alvarez, C. Perez, F. Gago, C. De, J. Balzarini and M. J. Camarasa, Antiviral Chem. Chemother., 1998, 9, 481–489 CrossRef CAS PubMed; (b) M. J. Genin, D. A. Allwine, D. J. Anderson, M. R. Barbachyn, D. E. Emmert, S. A. Garmon, D. R. Graber, K. C. Grega, J. B. Hester, D. K. Hutchinson, J. Morris, R. J. Reischer, C. W. Ford, G. E. Zurenko, J. C. Hamel, R. D. Schaadt, D. Stapert and B. H. Yagi, J. Med. Chem., 2000, 43, 953–970 CrossRef CAS PubMed.
- J. Heredia-Moya and K. L. Kirk, Bioorg. Med. Chem., 2008, 16, 5743–5746 CrossRef CAS PubMed.
- K. J. Stevenson, G. Hale and R. N. Perham, Biochemistry, 1978, 17, 2189–2192 CrossRef CAS PubMed.
- J. Liu, M. M. Numa, H. Liu, S.-J. Huang, P. Sears, A. R. Shikhman and C.-H. Wong, J. Org. Chem., 2004, 69, 6273–6283 CrossRef CAS PubMed.
- Q.-H. Fan, N.-T. Ni, Q. Li, L.-H. Zhang and X.-S. Ye, Org. Lett., 2006, 8, 1007–1009 CrossRef CAS PubMed.
- B. Fu, C. Qin, C. Xia and X. Yang, Synthesis Method of arsenic-sugar with anticancer biological activity and its application, Chinese Patent, ZL201510437907.9, 2015.
- H. R. Lee, H.-J. Cheong, S.-J. Kim, N.-S. Lee, H.-S. Park and J.-H. Won, Oncol. Rep., 2008, 20, 41–47 CAS.
- G. Malich, B. Markovic and C. Winder, Toxicology, 1997, 124, 179–192 CrossRef CAS PubMed.
- D. U. Miodragović, J. A. Quentzel, J. W. K. C. L. Stern, R. W. Ahn, I. Kandela, A. Mazar and T. V. O'Halloran, Angew. Chem., Int. Ed., 2013, 52, 10749–10752 CrossRef PubMed.
- B. Pardini, R. Kumar, A. Naccarati, J. Novotny, R. B. Prasad, A. Forsti, K. Hemminki, P. Vodicka and J. Lorenzo Bermejo, Br. J. Clin. Pharmacol., 2011, 72, 162–163 CrossRef CAS PubMed.
- C. Lin, T. Sugai, R. L. Halcomb, Y. Ichikawa and C. Wong, J. Am. Chem. Soc., 1992, 114, 10138–10145 CrossRef CAS.
- S. D. Rychnovsky and R. C. Hoye, J. Am. Chem. Soc., 1994, 116, 1753–1765 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |