DOI:
10.1039/C8RA07876A
(Paper)
RSC Adv., 2018,
8, 38013-38021
A novel resource utilization method using wet magnesia flue gas desulfurization residue for simultaneous removal of ammonium nitrogen and heavy metal pollutants from vanadium containing industrial wastewater
Received
21st September 2018
, Accepted 26th October 2018
First published on 13th November 2018
Abstract
In the present study, a novel resource utilization method using wet magnesia flue gas desulfurization (FGD) residue for the simultaneous removal of ammonium nitrogen (NH4–N) and heavy metal pollutants from vanadium (V) industrial wastewater was proven to be viable and effective. In this process, the wet magnesia FGD residue could not only act as a reductant of hexavalent chromium [Cr(VI)] and pentavalent vanadium [V(V)], but also offered plenty of low cost magnesium ions to remove NH4–N using struvite crystallization. The optimum experimental conditions for Cr(VI) and V(V) reduction are as follows: the reduction pH = 2.5, the wet magnesia FGD residue dose is 42.5 g L−1, t = 15.0 min. The optimum experimental conditions for NH4–N and heavy metal pollutants removal are as follows: the precipitate pH = 9.5, the n(Mg2+)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n(NH4+):n(PO43−) = 0.3:1.0:1.0, t = 20.0 min. Finally the NH4–N, V and Cr were separated from the vanadium containing industrial wastewater by forming the difficult to obtain, soluble coprecipitate containing struvite and heavy metal hydroxides. The residual pollutant concentrations in the wastewater were as follows: Cr(VI) was 0.047 mg L−1, total Cr was 0.1 mg L−1, V was 0.14 mg L−1, NH4–N was 176.2 mg L−1 (removal efficiency was about 94.5%) and phosphorus was 14.7 mg L−1.
:n(NH4+):n(PO43−) = 0.3:1.0:1.0, t = 20.0 min. Finally the NH4–N, V and Cr were separated from the vanadium containing industrial wastewater by forming the difficult to obtain, soluble coprecipitate containing struvite and heavy metal hydroxides. The residual pollutant concentrations in the wastewater were as follows: Cr(VI) was 0.047 mg L−1, total Cr was 0.1 mg L−1, V was 0.14 mg L−1, NH4–N was 176.2 mg L−1 (removal efficiency was about 94.5%) and phosphorus was 14.7 mg L−1.
Introduction
Sulfur dioxide (SO2) is one of the primary atmospheric pollutants attributed to the burning of fossil fuels, and the emission of SO2 leads to acid precipitation, vegetation destruction, and airborne particulate matter.1,2 Out of all the emission sources of oxysulfide in China, thermal power plants are primarily responsible, and the flue gas desulfurization (FGD) system is the main method used for reducing SO2 because it is a mature method and is widely used by the majority of the thermal power plants.1–4 The FGD technologies can be roughly divided into three methods: wet processes, semi-dry processes and dry processes. Of all these methods, the take-up rate of use of wet FGD processes was about 85% for worldwide use of FGD technology because of its high efficiency and wide applicability. In the past few decades, the wet limestone–gypsum process was the mainstream technology, which had a take-up rate of about 90% for the reduction of SO2 emissions in China.4–6 It is worth noting that the wet magnesia FGD technology has been gradually applied to thermal power plants in China. Compared with the conventional wet limestone–gypsum process, wet magnesia FGD technology has many merits, such as high and stable desulfurization efficiency, simple and compact flow sheet, low investment and energy consumption. In particular, China has the most abundant reserves of the magnesite mineral, which has encouraged the development of wet magnesia FGD technology.7–9
The mechanism of the wet magnesia FGD process is similar to the wet lime–gypsum method. In the wet magnesia FGD process, magnesium hydroxide slurry is used as the SO2 absorbent, which is prepared using an aging treatment of a mixture of magnesium oxide and water at a particular ratio. After removing the dust, the flue gas entered the absorption tower, and the SO2 in the flue gas was subsequently absorbed by small magnesium hydroxide droplets from the spraying system. Therefore, the SO2 in the flue gas could be removed and fixed in the desulfurization slurry because of the formation of low solubility magnesium sulfite.2,7–11 After the slurry has reached the saturation absorption, the desulfurization product can be separated from the spent slurry by filtration to form the FGD residue, and the main composition of the FGD residue is magnesium sulfite hydrate.
At present, the main resource utilization method of the wet magnesia FGD residue was recovery of magnesium sulfate using forced catalytic oxidation and crystallization. The current research focuses on the development of a low cost and highly efficient catalyst.2,12–14 As well as the regeneration of activated magnesia for FGD and regeneration of SO2 for acid making using thermal decomposition of wet magnesia, FGD residue was also an attractive approach. But the oxidation of the magnesium sulfite should be prevented by adding an oxidation inhibitor during the FGD process because of the higher thermal decomposition temperature of magnesium sulfate.8,9,15,16 Nowadays, the vast majority of the wet magnesia FGD residue is casually stacked without further treatment or discarded after forced aeration oxidation, and this is attributed to the high investment associated with the equipment for the treatment process and the low economic benefit of recycled products.7
The wastewater discharged from the sodium roasting vanadium extraction technology is complex, hard to degrade biochemically, has a high pollutant gas concentration, and is a highly hazardous acidic wastewater containing high concentrations of vanadium (V), hexavalent chromium [Cr(VI)] and ammonium nitrogen (NH4–N).17–20 The pollution emission concentration ranges of Cr(VI), total chromium, vanadium and NH4–N in the wastewater were approximately 2500–4500 mg L−1, 3000–5000 mg L−1, 30–150 mg L−1 and 2000–3500 mg L−1, respectively. According to the national discharge standard of pollutants for the vanadium industry (GB 26452-2011), the residual concentration of Cr(VI), total Cr, V and NH4–N in the wastewater must be below 0.5 mg L−1, 1.5 mg L−1, 1.0 mg L−1 and, 40 mg L−1, respectively. In reality, the heavy metal pollutants of the vanadium containing industrial wastewater were treated using chemical reduction precipitation, where the NH4–N was treated using air stripping or struvite precipitation. In the traditional treatment progress, a lot of reagents were consumed, such as the reductants for the heavy metal removal, and the magnesium salts for the NH4–N removal.5,7
In this study, a novel resource utilization of the wet magnesia FGD residue for simultaneous removal of NH4–N and heavy metal pollutants from the vanadium containing industrial wastewater is presented. In this process, the wet magnesia FGD residue could not only reduce the Cr(VI) and V to Cr(III) and V(IV) which were low in toxicity and easy to separate under neutral conditions, but also offered plenty of low cost magnesium ions for removing ammonium using struvite crystallization. Therefore, the wet magnesia FGD residue could simultaneously remove V, Cr and NH4–N from wastewater by forming the difficult to obtain, soluble coprecipitate containing struvite and heavy metal hydroxides. Compared with the conventional methods, this technology not only reduced the treatment cost of vanadium containing industrial wastewater and wet magnesia FGD residue, but also achieved the excellent objective of “waste treated by waste”.
Materials and methods
Wet magnesia flue gas desulfurization residue
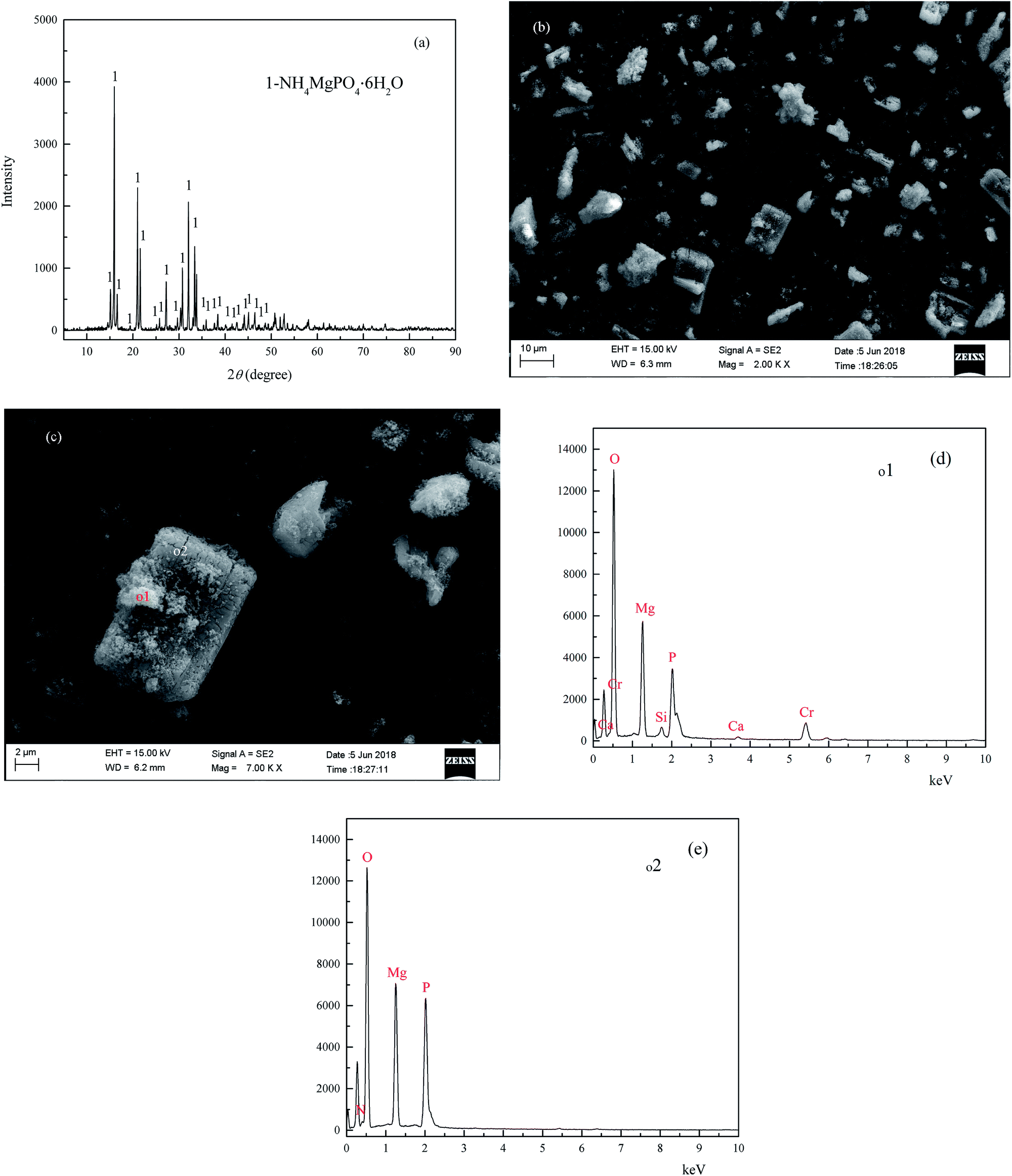
The wet magnesia FGD residue used in this work was provided by the Datang Lubei thermal power plant in the Shandong province of China. Because the residue was stacked at a waste landfill site for about three months under natural conditions, the FGD residue could be crushed directly and stored in a plastic valve bag. The chemical composition of the wet magnesia FGD residue was as follows (wt%): magnesium oxide (MgO): 25.9, sulfite (SO32−): 23.75, sulfate (SO42−): 12.81, silicon dioxide (SiO2): 3.35, calcium oxide (CaO): 1.56, chloride (Cl): 0.72, carbonate (CO32−): 6.75, TFe: 0.23. The results of the X-ray diffraction (XRD) analysis of the wet magnesia FGD residue are shown in Fig. 1(a), and the peaks of the wet magnesia FGD residue were consistent with those of magnesium sulfate trihydrate (MgSO3·3H2O, JCPDS no. 00-024-0738), magnesium carbonate (MgCO3, JCPDS no. 00-008-0479), magnesium sulfate hexahydrate (MgSO4·7H2O, JCPDS no. 00-036-0419), and magnesium sulfate tetrahydrate (MgSO4·4H2O, JCPDS no. 00-024-0720). As is shown in Fig. 1(b) and (c), wet magnesia FGD residue consists of irregular particles and its size is about 1–30 μm, the chemical element composition of the wet magnesia FGD residue contained oxygen, magnesium, sulfur and a trace of silicon, calcium, ferric iron. The main mineral phases of the wet magnesia flue gas desulfurization residue were MgSO3·3H2O and MgCO3 with minor amounts of MgSO4·7H2O and MgSO4·4H2O.
 |
| | Fig. 1 The XRD (a) and SEM (b) EDS (c) of wet magnesia flue gas desulfurization residue. | |
Vanadium containing industrial wastewater
The wastewater discharged from the sodium roasting vanadium extraction plant was provided by Jianlong Steel Co., Ltd. The concentrations of the main pollutants in the wastewater were as follows: Cr(VI) was 3800 mg L−1, total Cr was 4320 mg L−1, V was 75 mg L−1, NH4–N was 3200 mg L−1 and the pH = 2.14.
Chemical reagents
Potassium dichromate (K2Cr2O7), ammonium metavanadate (NH4VO3), ammonium chloride (NH4Cl), sulfuric acid (H2SO4), diphenylcarbazide (C13H14N4O), magnesium chloride (MgCl2), sodium hydroxide (NaOH), sodium phosphate (Na3PO4), acetone (CH3COCH3), Nessler's reagent and sodium potassium tartrate tetrahydrate (KNaC4H4O6·4H2O) were purchased from Tianjin Kemiou Chemical Reagent Co., Ltd.
Analysis and detection methods
The acidity was determined using pH detectors (PHS-3E). The 1,5-diphenylcarbazide spectrophotometric method was used to determine the Cr(VI) concentration. The Nessler's reagent spectrophotometry method was used to determine the NH4–N concentration. Inductively-coupled plasma-optical emission spectrometry (ICP-OES, Optima 8300DV, PerkinElmer) was used to determine the content of total Cr ions, V ions, phosphorus ions and magnesium ions.
The XRD (PW3040/60, Philips) with Cu Kα radiation (λ = 1.54056 Å) was used to identify the crystalline structure of the wet magnesia FGD residue and the coprecipitate at the sweep rate of 6° min−1 in the range from 5° to 90°. The scanning electron microscopy-energy dispersive X-ray spectrometry (SEM-EDS, Sigma 500, Zeiss) was used to study the morphology and elemental composition of the wet magnesia FGD residue and the coprecipitate.
Experimental procedure
Batch experiments were performed in a 250 mL glass beaker on a magnetic heated stirrer at room temperature. The diluted NaOH solution and diluted H2SO4 solution were used to control the acidity of the reaction solutions during the course of the experiment. Firstly, a certain amount of wet magnesia flue gas desulfurization residue was added into 200 mL of wastewater in a glass beaker with continuous stirring, and the pH of wastewater was controlled at an acidic level until the reduction reaction was completed. Subsequently, a given amount of soluble magnesium and phosphate was added separately into the wastewater to give the desired molar ratio of Mg2+, NH4+ and PO43− with constant stirring, and the pH of the wastewater was then adjusted to an alkaline level. Finally, the vanadium, chromium and ammonium in the vanadium containing industrial wastewater were removed synchronously by forming the difficult to obtain soluble coprecipitate, and the coprecipitate was then dried at 60 °C before further analysis.
Results and discussion
The main mechanism of the pollutant removal process
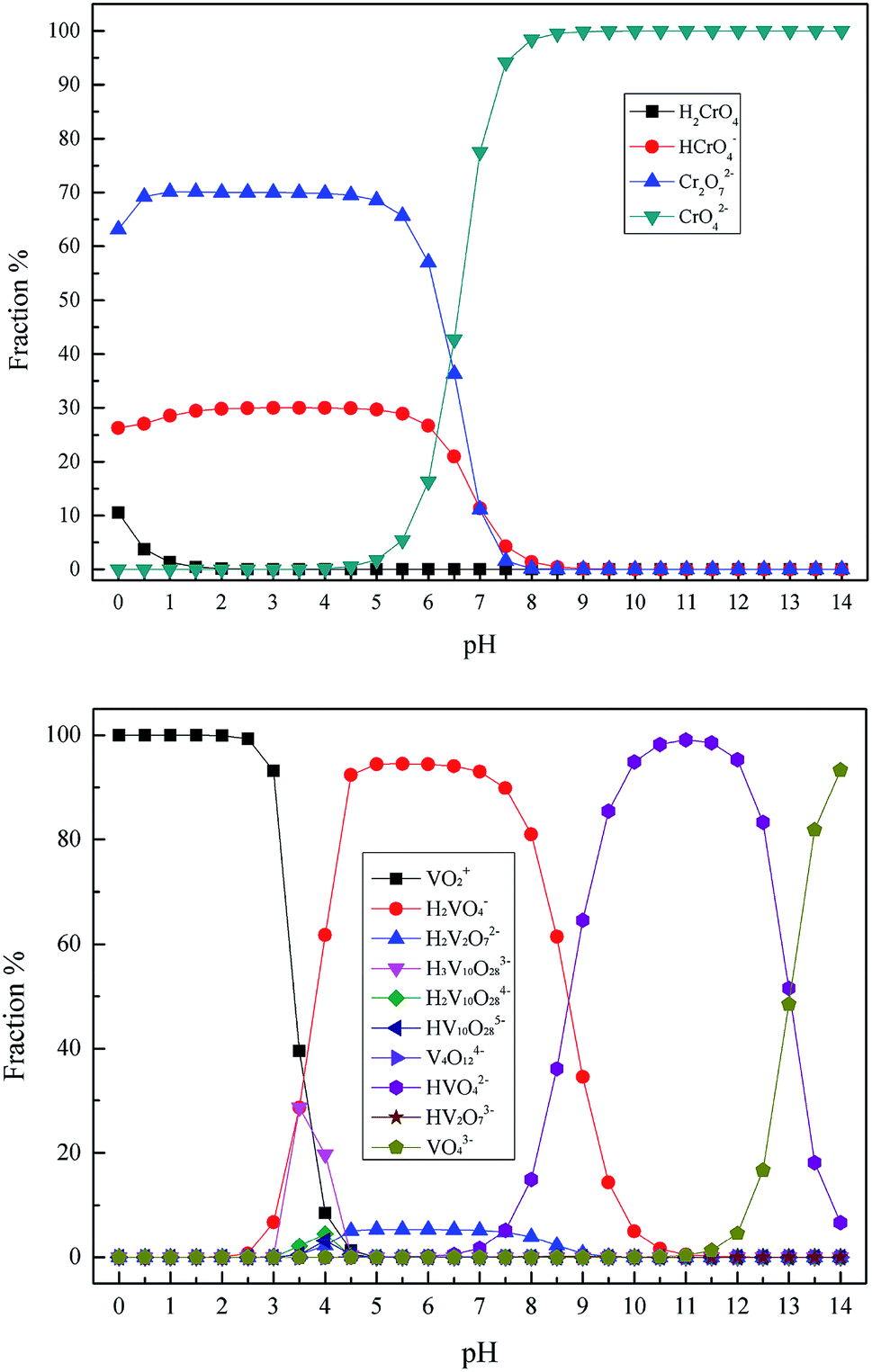
In the conventional sodium salt (Na2CO3 or NaCl) roasting technique, both vanadium and chromium in vanadium bearing slag were oxidized to a high valence state and then transformed into the soluble vanadate and chromate. The roasted products were washed repeatedly in hot water, and the vanadium raw material was obtained from the high concentration vanadium extraction solution using ammonium polyorthovanadate precipitation in acid conditions.7,21–25 Therefore, the main existing valences of vanadium and chromium in the wastewater were Cr(VI) and pentavalent vanadium [V(V)], respectively. As is already known, the existing forms of Cr(VI) and V(V) are closely related to their concentration and pH of a solution. Therefore, the existing forms of the Cr(VI) and V(V) were calculated and determined using the Visual MINTEQ aqueous chemical equilibria software based on the actual heavy metal pollutant's concentration in the wastewater. The speciation distribution of Cr(VI) and V(V) were calculated over the range of pH = 0–14.0, as shown in Fig. 2. Consequently, it was found that the main existing forms of heavy metal pollutants in vanadium containing industrial wastewater were Cr2O72− and VO2+.26–28
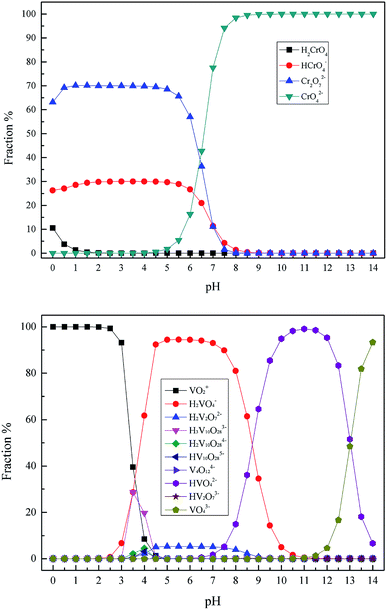 |
| | Fig. 2 The species distribution of Cr(VI) and V(V) in aqueous solutions. | |
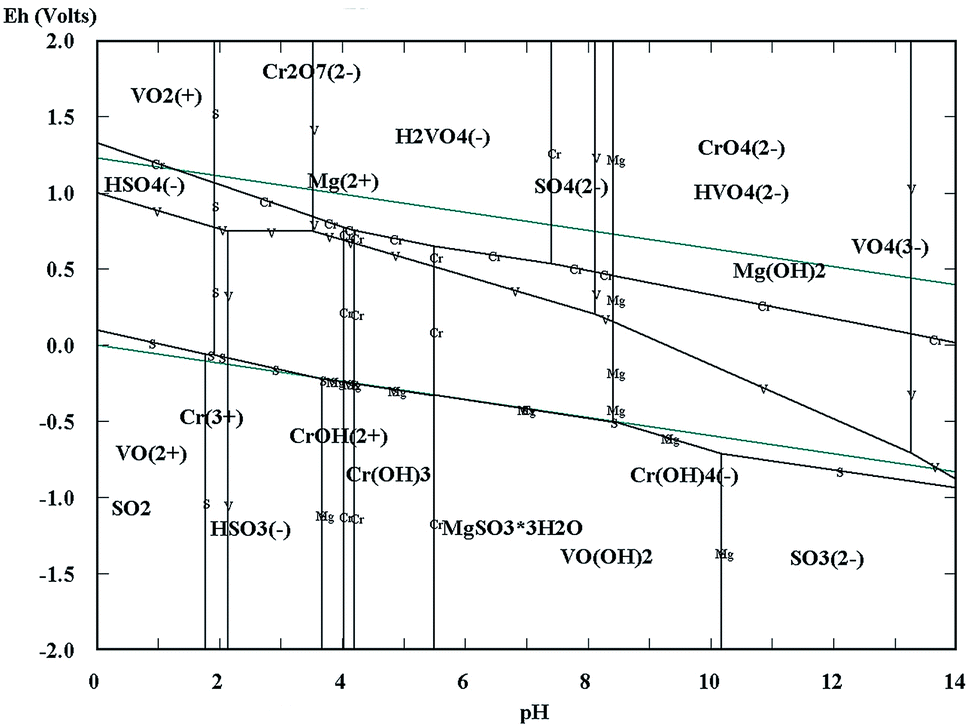
The Eh–pH diagrams of Cr–V–S–Mg–H2O are shown in Fig. 3. Depending on the acidity of the vanadium extraction wastewater (pH = 2.14), the redox potentials of VO2+ and Cr2O72− were higher than that of HSO3−.5,7,26–31 Consequently, the VO2+ and Cr2O72− could be reduced to VO2+ and Cr3+ by HSO3− which is the acidolysis product of wet magnesia FGD residual (main composition is MgSO3·3H2O), as shown in eqn (1)–(3). In addition, the appropriate low pH condition was conducive to oxidation–reduction reactions because the H+ also took part in the reaction. At the same time, a large quantity of Mg2+ was obtained by the dissolution of MgSO3·3H2O, MgCO3 [as shown in eqn (1) and (4)] and MgSO4·nH2O in the acidic conditions which would be regarded as the low cost magnesium ion source of struvite (MgNH4PO4·6H2O) crystallization.32–35 Following the completion of the reductive reaction, the given amount of soluble magnesium and phosphate were added separately into the wastewater. Then, the pH of the wastewater was adjusted to alkalinity. Because the VO2+, Cr3+ and NH4+ could be separated synchronously from the wastewater by forming an insoluble sediment including vanadium and chromium hydroxides and struvite in alkaline conditions, as shown in eqn (5)–(7). In conclusion, the simultaneous removal of NH4–N and heavy metal pollution from vanadium containing industrial wastewater could be realized using wet magnesia FGD residue.
| | |
MgSO3·3H2O + H+ → Mg2+ + HSO4− + 3H2O
| (1) |
| | |
2VO2+ + HSO3− + H+ → 2VO2+ + SO42− + H2O
| (2) |
| | |
Cr2O72− + 3HSO3− + 5H+ → 2Cr3+ + 3SO42− + 4H2O
| (3) |
| | |
MgCO3 + 2H+ → Mg2+ + CO2↑+ H2O
| (4) |
| | |
Cr3+ + 3OH− → Cr(OH)3↓
| (5) |
| | |
VO2+ + 2OH− → VO(OH)2↓
| (6) |
| | |
Mg2+ + NH4+ + HPO42− + 6H2O → MgNH4PO4·6H2O↓ + H+
| (7) |
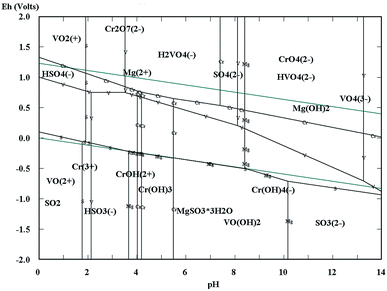 |
| | Fig. 3 The Eh–pH diagrams of the Cr–V–S–Mg–H2O system. | |
The optimum pH of the Cr(VI) and V(V) reduction
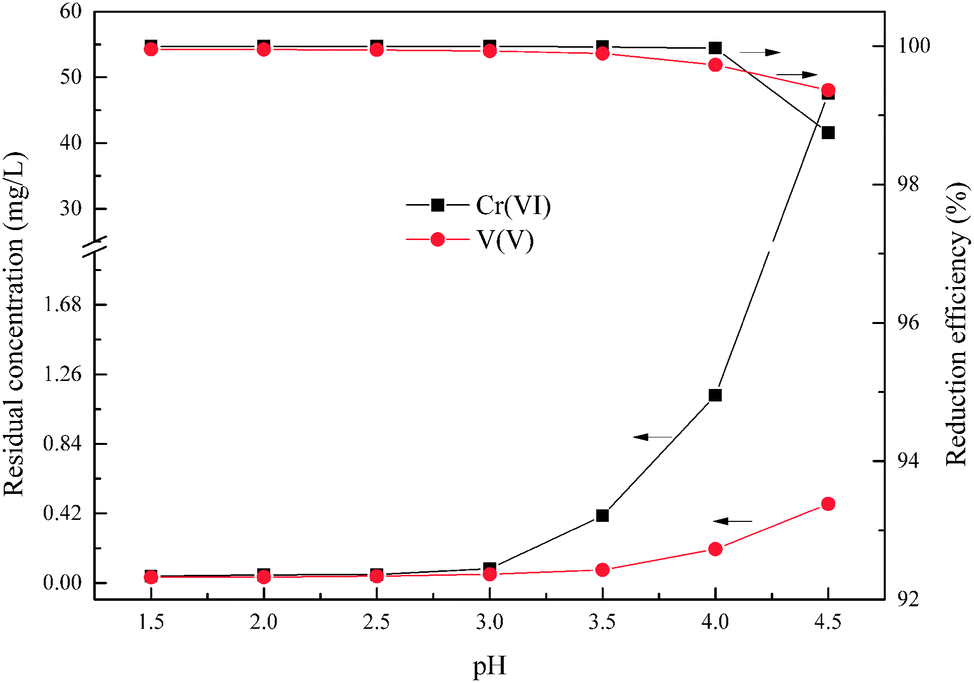
According to the reductive mechanism of Cr(VI) and V(V), the reductive reaction pH is an important factor to ensure that the Cr(VI) and V(V) could be reduced adequately and rapidly. This experiment was used to determine the optimum pH within the range from 1.5 to 4.5, and the wet magnesia FGD residue dosage was 8.5 g, and reaction time was 15 min.
As shown in Fig. 4, the degree of reduction of Cr(VI) and V(V) is closely related to the pH. Together with an increase of pH from 1.5 to 4.5, the reduction efficiencies of Cr(VI) and V(V) reduced from 99.99% and 99.95% to 98.74% and 99.36%, respectively, and the residual Cr(VI) and V(V) increased from 0.041 mg L−1 and 0.035 mg L−1 to 47.56 mg L−1 and 0.478 mg L−1, respectively. It is worth noting that the main reduction product of Cr(VI) was Cr3+ in the pH range from 2.5 to 4.0, as shown in eqn (3). Whereas the main reduction products of Cr(VI) were Cr(OH)2+ or Cr(OH)3 in the pH range from 4.0 to 4.5, as shown in eqn (8) and (9). The reaction rate of eqn (8) and (9) was slower than that of eqn (3) which resulted in the significant reduction of Cr(VI) reduction efficiency. In the other words, the low pH condition was conducive to the reduction of Cr(VI) and V(V) using wet magnesia FGD residue as a reductant. As required by national Standards, the hazardous heavy metal pollutants' emission concentration limits of hexavalent chromium and total vanadium were 0.5 mg L−1 and 1.0 mg L−1, respectively. In conclusion, the optimum pH of the Cr(VI) and V(V) reduction process was pH = 2.5.
| | |
Cr2O72− + 3MgSO3·3H2O + 6H → 2Cr(OH)2+ + 3SO42− + 3Mg2+ + 11H2O
| (8) |
| | |
Cr2O72− + 3MgSO3·3H2O + 2H+ → 2Cr(OH)3 + 3SO42− + 3Mg2+ + 7H2O
| (9) |
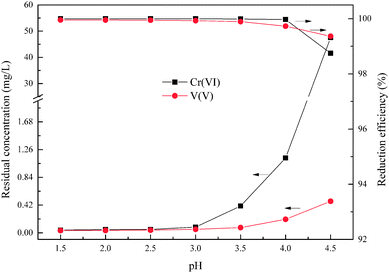 |
| | Fig. 4 Effect of pH on Cr(VI) and V(V) reduction. The wet magnesia FGD residue dosage was 8.5 g, and t = 15.0 min. | |
The optimum wet magnesia FGD residue dose for Cr(VI) and V(V) reduction
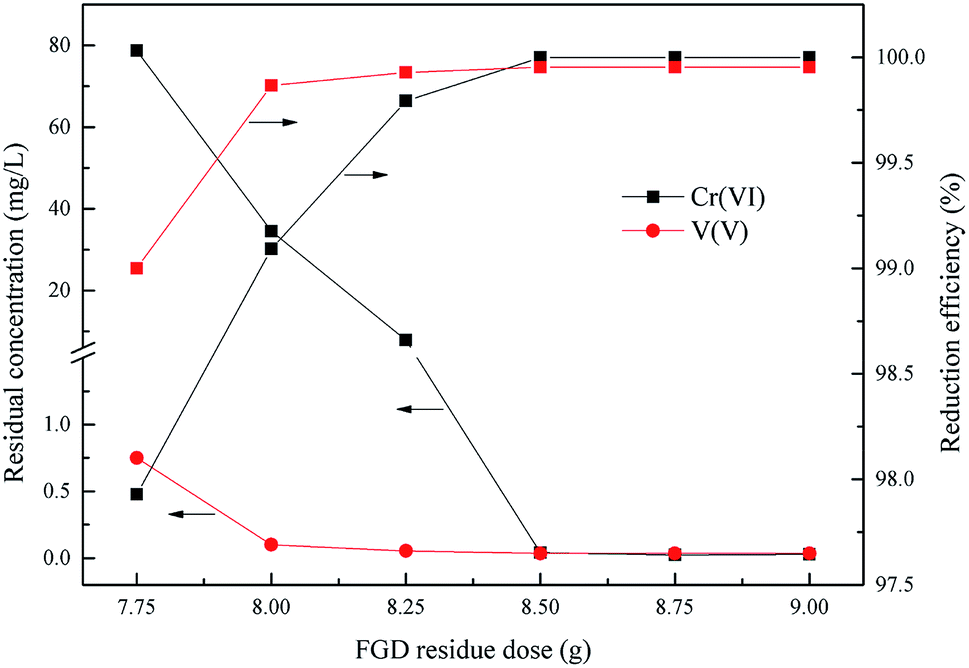
As is already known, the dose of the reducing agent is a key factor of the redox reaction between Cr(VI), V(V) and the wet magnesia FGD residue. This experiment was done to determine the wet magnesia FGD residue dosage within the range from 7.75 g to 9.0 g in 200 mL of wastewater, the optimum reduction pH was 2.5, and the reduction reaction time was 15 min.
As shown in Fig. 5, the degree of reduction of Cr(VI) and V(V) was closely related to the dose of wet magnesia FGD residue. With an increase of the wet magnesia FGD residue dose from 7.75 g to 9.0 g, the reduction efficiency of Cr(VI) and V(V) also increased from 97.92% and 99.0% to 99.99% and 99.95%, respectively, and the residual Cr(VI) and V(V) reduced from 78.7 mg L−1 and 0.75 mg L−1 to 0.029 mg L−1 and 0.035 mg L−1, respectively. Once the dose of wet magnesia FGD residue reached 8.5 g, the reduction efficiency of Cr(VI) and V(V) was steady and the residual pollutant concentration was far below the emission concentration limits. Consequently, the optimum wet magnesia FGD residue dose for Cr(VI) and V(V) reduction was 8.5 g (42.5 g L−1).
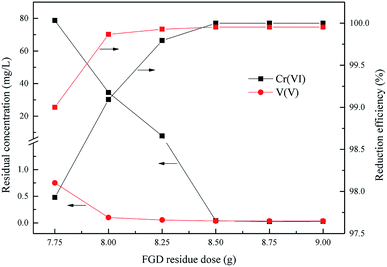 |
| | Fig. 5 Effect of wet magnesia FGD dose on Cr(VI) and V(V) reduction. The optimum pH = 2.5, and t = 15.0 min. | |
The optimum time for Cr(VI) and V(V) reduction
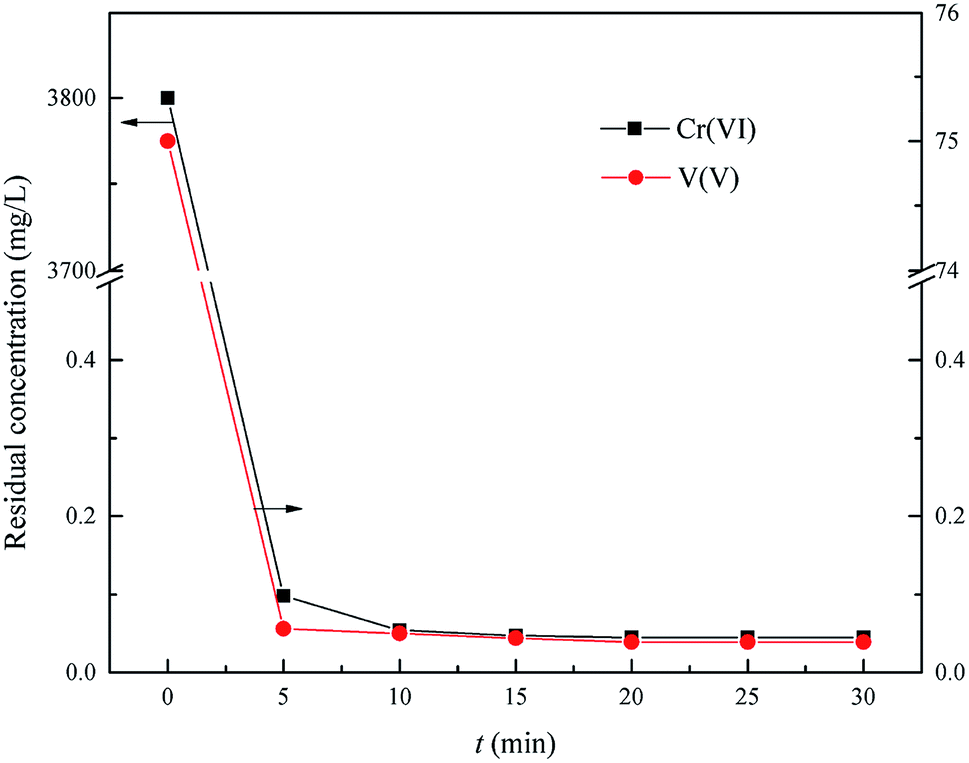
This experiment was carried out to determine the optimum time of Cr(VI) and V(V) reduction within the range of reaction time from 5.0 min to 30.0 min, the optimum reduction pH = 2.5, the optimum wet magnesia FGD residue dose was 8.5 g in 200 mL of wastewater.
As shown in Fig. 6, the residual Cr(VI) and V(V) reduced significantly from the initial 3800 mg L−1 and 75 mg L−1 to 0.045 mg L−1 and 0.039 mg L−1, respectively, with an increase of reduction time from 0 min to 30.0 min, respectively. Therefore, the Cr(VI) and V(V) could be reduced quickly to Cr(III) and V(IV) by using the wet magnesia FGD residue as a reductant within 15.0 min. So, the wet magnesia FGD residue could be regarded as a low cost and environmentally effective substitute for conventional reductants, such as sodium metabisulfite (Na2S2O5) or iron(II) sulfate (FeSO4). Consequently, the optimum reduction time was 15.0 min, and the concentration of the by-product, magnesium ions was about 5517 mg L−1.
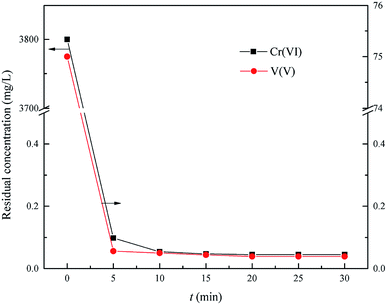 |
| | Fig. 6 Effect of reaction time on Cr(VI) and V(V) reduction. The optimum pH is 2.5, and the wet magnesia FGD residue dose is 8.5 g. | |
The optimum pH for ammonium nitrogen and heavy metal pollutants removal
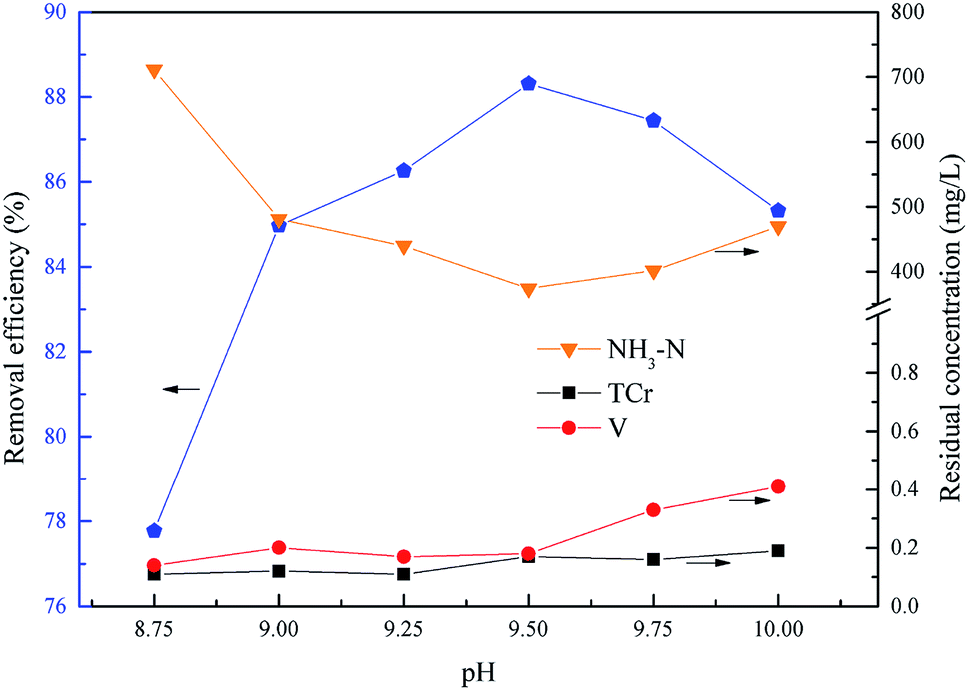
The pH was a key factor for struvite crystallization and the formation of chromium and vanadium hydroxides. This experiment was performed to determine the optimum pH of total chromium, vanadium and ammonium removal within the pH range 8.75 to 10.0, with the molar ratio of n(Mg2+):n(NH4+):n(PO43−) = 1.0:1.0:1.0, and the reaction time was 15.0 min. The molar ratio of the residual Mg2+ (5517 mg L−1) and NH4+ was about 1.0:1.0, so the actual added molar ratio of n(Mg2+):n(NH4+):n(PO43−) was 0:1.0:1.0.
As shown in Fig. 7, the removal efficiency of ammonium increased first from 77.77% to 88.31% and then reduced to 85.32% with an increase of pH, and the ammonium removal efficiency reached a peak at pH = 9.5, and the residual concentrations of chromium and vanadium were always below the emission concentration limits. The main form of phosphate was HPO42− in alkaline conditions within the experimental pH range. As shown in eqn (7), the excess H+ would inhibit the formation of struvite. With the pH increasing, more PO43− was dissociated from HPO42− which lead to the struvite being replaced by magnesium phosphate [Mg3(PO4)2].34–36 Consequently, the optimum pH of total chromium, vanadium and ammonium removal was 9.5.
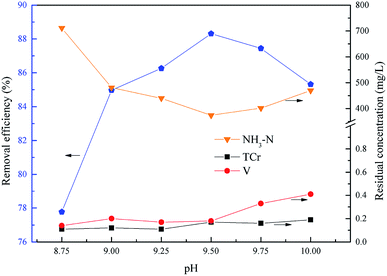 |
| | Fig. 7 Effect of pH on NH4–N and heavy metal pollutants removal. The molar ratio n(Mg2+):n(NH4+):n(PO43−) = 0:1.0:1.0, and t = 15.0 min. | |
The optimum molar ratio of n(Mg2+)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :n(NH4+):n(PO43−) for NH4–N removal
:n(NH4+):n(PO43−) for NH4–N removal
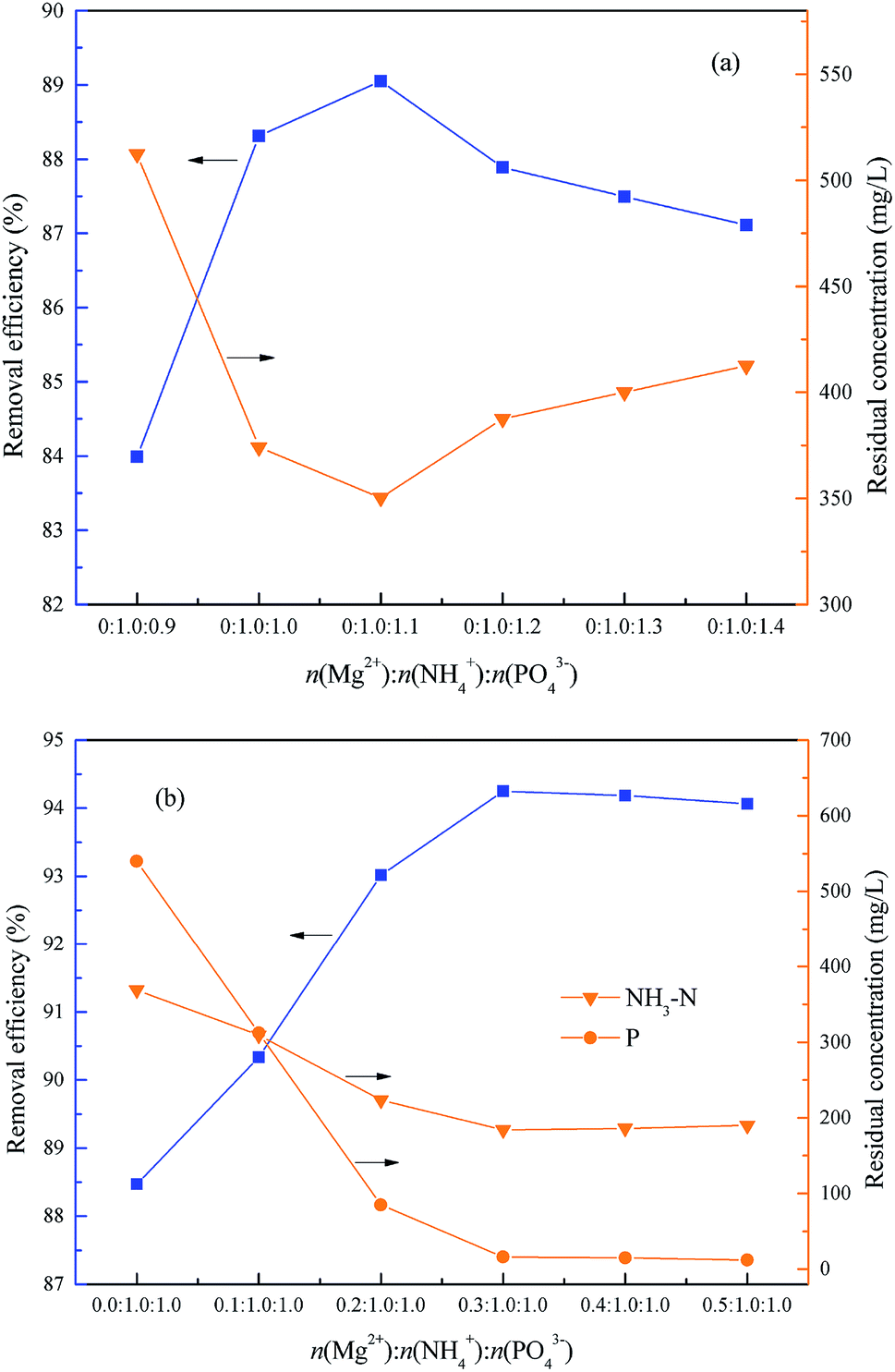
This experiment was used to determine the optimum molar ratio n(Mg2+):n(NH4+):n(PO43−) for NH4–N removal within 15.0 min. Firstly, the optimum molar ratio between NH4+ and PO43− was investigated within the range of n(Mg2+):n(NH4+):n(PO43−) = 0:1.0:0.9 to 0:1.0:1.4, and the optimum pH = 9.5. As shown in Fig. 8(a), the removal rate of NH4–N increased first from 83.99% to 89.05% and then reduced slightly to 87.10% together with an increase of the molar ratio n(Mg2+):n(NH4+):n(PO43−) from 0:1.0:0.9 to 0:1.0:1.4. Consequently, the optimum molar ratio between NH4+ and PO43− was 1.0:1.0 because the excess PO43− lead to a phosphorus residue with only a little increase of NH4–N removal.
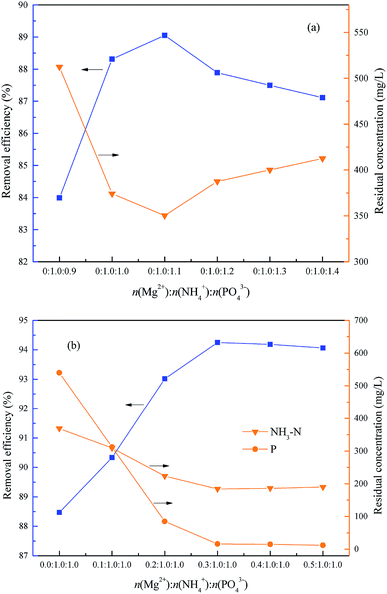 |
| | Fig. 8 Effect of the molar ratio n(Mg2+):n(NH4+):n(PO43−) on NH4–N removal. The optimum pH = 9.5 and t = 15 min. | |
Subsequently, the optimum molar ratio between Mg2+ and NH4+ was investigated within the range of n(Mg2+):n(NH4+):n(PO43−) = 0:1.0:1.0–0.5:1.0:1.0, and the optimum pH = 9.5. As shown in Fig. 8(b), with an increase of molar ratio of n(Mg2+):n(NH4+):n(PO43−) from 0:1.0:1.0 to 0.5:1.0:1.0, the NH4–N removal rate increased first from 88.46% to 94.25% and then reduced slightly to 94.06%, and the residual phosphorus was also reduced markedly from 540 mg L−1 to 12.0 mg L−1. Consequently, the optimum molar ratio of n(Mg2+):n(NH4+) was 0.3:1.0.
It is worth noting that the serious overdosing of Mg2+ or HPO42− would reduce the ammonium removal efficiency, because the concentration of PO43− would be increased together with the HPO42− increasing because of the dissociation of HPO42−, as shown in eqn (10). So, excessive PO43− could be consumed before more Mg2+ to produce Mg3(PO4)2 which would result in a shortage of Mg2+ to form struvite, as shown in eqn (11). Also, excessive Mg2+ would also consume more PO43− to produce Mg3(PO4)2 which would result in the shortage of HPO42− to form struvite.7,33,34 Consequently, the optimum molar ratio of n(Mg2+):n(NH4+):n(PO43−) = 0.3:1.0:1.0.
| | |
Mg2+ + PO43− → Mg3(PO4)2
| (11) |
The optimum reaction time for NH4–N and heavy metal pollutant removal
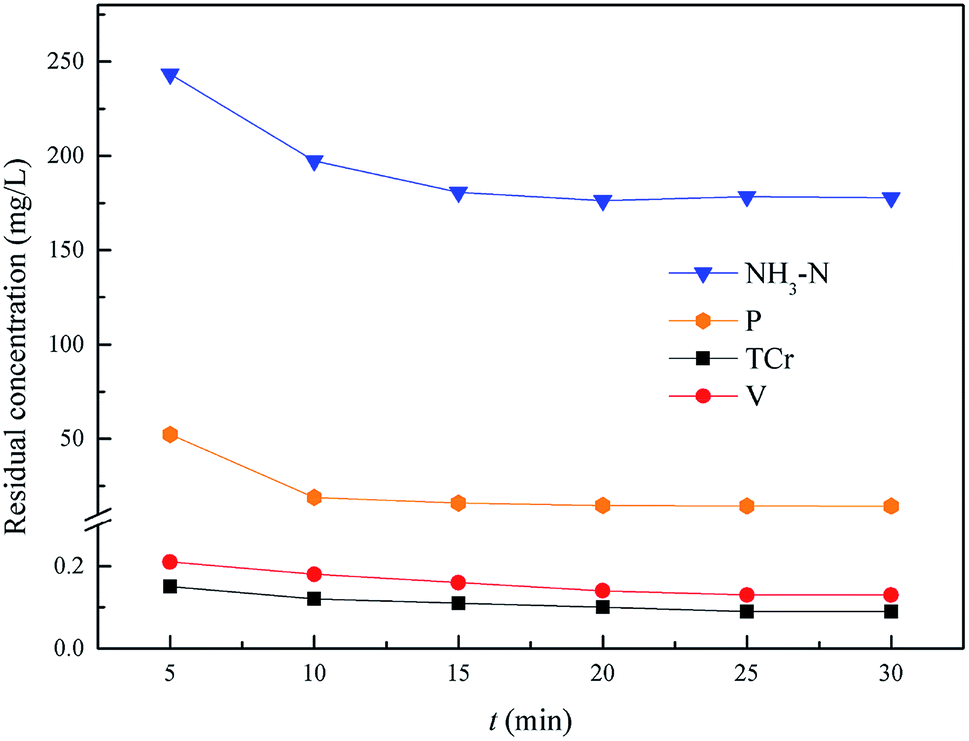
This experiment was used to determine the optimum reaction time of NH4–N and heavy metal pollutants from 5.0 min to 30.0 min, with an optimum pH = 9.5, and the optimum molar ratio n(Mg2+):n(NH4+):n(PO43−) = 0.3:1.0:1.0. As shown in Fig. 9, the residual NH4–N and heavy metal pollutants could reach an optimal state and remain stable within 20.0 min. So, the optimum reaction time of NH4–N and heavy metal pollutants removal was 20.0 min, and the residual pollutant levels were: Cr(VI): 0.047 mg L−1, total Cr: 0.1 mg L−1, V: 0.14 mg L−1, NH4–N: 176.2 mg L−1 (removal efficiency was 94.5%) and phosphorus: 14.7 mg L−1.
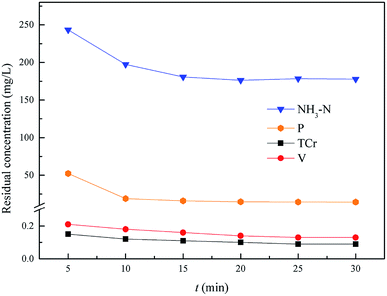 |
| | Fig. 9 Effect of NH4–N and heavy metal pollutant removal. The optimum pH = 9.5, and the molar ratio n(Mg2+):n(NH4+):n(PO43−) = 0.3:1.0:1.0. | |
Characterization analysis of the coprecipitate
The XRD analysis results of the coprecipitate are shown in Fig. 10(a), and the main peaks of the coprecipitate are entirely consistent with MgNH4PO4·6H2O (struvite JCPDS no. 01-077-2303). The micrographs of the coprecipitate are shown in Fig. 10(b) and (c), and show some irregular agglomerated particles attached to the surface of the bulk shape crystal. The EDS patterns of the coprecipitate are shown in Fig. 10(d) and (e), and the main chemical element constituent of the bulk shape crystal [o2, Fig. 10(e)] contained O, Mg, N, P, which is consistent with the element constituents of struvite. In comparison, the chemical element constituent of irregular agglomerated particles [o1, Fig. 10(d)] on the surface of the bulk shape crystal contained O, Mg, P, Cr, Ca and Si. The results of the quantitative analysis of the coprecipitate was (wt%): MgO: 17.45, P: 10.32, chromium(III) oxide (Cr2O3): 8.27, N: 4.17, Na: 0.29, S: 0.165, V: 0.14, CaO: 0.47, SiO2: 1.11, and TFe: 0.24. In conclusion, the heavy metal pollutants and NH4–N in the vanadium containing industrial wastewater were removed by forming the difficult to obtain, soluble coprecipitate containing struvite and chromium and vanadium hydroxide in alkaline conditions.
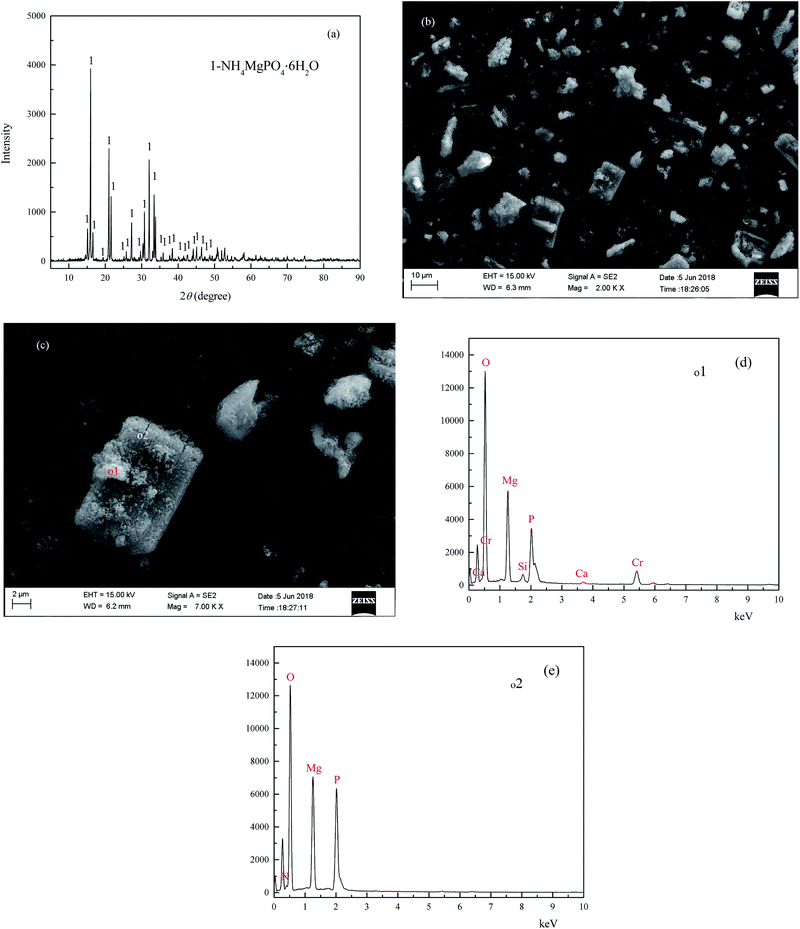 |
| | Fig. 10 XRD pattern (a), SEM (b and c) EDS patterns (d and e) of the coprecipitate. | |
Conclusions
In this paper, a novel resource utilization method of the wet magnesia flue gas desulfurization residue for simultaneously removing ammonium nitrogen and heavy metal pollutants from vanadium containing industrial wastewater was proven to be viable and effective. The main mineral phases of the wet magnesia flue gas desulfurization residue were MgSO3·3H2O and MgCO3 with a minor amount of MgSO4·7H2O and MgSO4·4H2O. The optimum experimental conditions of hexavalent chromium and pentavalent vanadium reduction were: the reduction pH = 2.5, wet magnesia flue gas desulfurization dose was 42.5 g L−1, and the reduction time was 15.0 min. The optimum experimental conditions of ammonium nitrogen and heavy metal pollutants removal were: the precipitate reaction pH = 9.5, the molar ratio n(Mg2+):n(NH4+):n(PO43−) = 0.3:1.0:1.0, and the reaction time was 20.0 min. The residual pollutant concentrations were: chromium(VI) was 0.047 mg L−1, total chromium was 0.1 mg L−1, vanadium was 0.14 mg L−1, ammonium nitrogen was 176.2 mg L−1 (removal efficiency was 94.5%) and phosphorus was 14.7 mg L−1. The main pollutants in the vanadium containing industrial wastewater could be removed by forming the difficult to obtain, soluble coprecipitate containing struvite, chromium and vanadium hydroxide.
In general, this research provided a relatively low cost, efficient and environmental treatment process of the vanadium containing industrial wastewater and wet magnesia FGD residue, which has realized the objective of “waste treated using waste”.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The authors are grateful for the support from the Natural Science Foundation of China (No. U1502273).
References
- H. Zhang, B. Zhang and J. Bi, Energy, 2015, 80, 1–9 CrossRef.
- T. Zhu, Y. Ma, H. Zhang, D. Li and L. Li, Catal. Today, 2015, 258, 70–74 CrossRef CAS.
- X. Liu, B. Lin and Y. Zhang, J. Cleaner Prod., 2016, 113, 133–143 CrossRef CAS.
- F. J. Gutiérrez Ortiz, Chem. Eng. J., 2010, 165, 426–439 CrossRef.
- D. Fang, X. Liao, X. Zhang, A. Teng and X. Xue, J. Hazard. Mater., 2017, 342, 436–445 CrossRef PubMed.
- D. Zheng, H. Lu, X. Sun, X. Liu, W. Han and L. Wang, Thermochim. Acta, 2013, 559, 23–31 CrossRef CAS.
- D. Fang, X. Zhang, M. Dong and X. Xue, J. Hazard. Mater., 2017, 336, 8–20 CrossRef CAS PubMed.
- Z. Shen, X. Chen, M. Tong, S. Guo, M. Ni and J. Lu, Fuel, 2013, 105, 578–584 CrossRef CAS.
- L. Wang, Y. Ma, W. Zhang, Q. Li, Y. Zhao and Z. Zhao, J. Hazard. Mater., 2013, 258–259, 61–69 CAS.
- R. Guo, W. Pan, X. Zhang, H. Xu and J. Ren, Fuel, 2011, 90, 7–10 CrossRef CAS.
- R. Valle-Zermeño, J. Formosa, J. Aparicio and J. Chimenos, Chem. Eng. J., 2014, 254, 63–72 CrossRef.
- L. Wang, S. Cui, Q. Li, J. Wang and S. Liu, Appl. Catal., A, 2016, 511, 16–22 CrossRef CAS.
- Q. Li, Y. Yang, L. Wang, P. Xu and Y. Han, Appl. Catal., B, 2017, 203, 851–858 CrossRef CAS.
- L. Guo, W. Liu, X. Tang, H. Wang, Q. Liu and Y. Zhu, Chem. Eng. J., 2017, 330, 870–879 CrossRef CAS.
- L. Yan, X. Lu, Q. Guo, Q. Wang and X. Ji, Adv. Powder Technol., 2014, 25, 1709–1714 CrossRef CAS.
- L. Yan, X. Lu, Q. Wang, Y. Kang, J. Xu and Y. Chen, Therm. Eng., 2014, 65, 487–494 CrossRef CAS.
- Y. Zhang, S. Bao, T. Liu, T. Chen and J. Huang, Hydrometallurgy, 2011, 109, 116–124 CrossRef CAS.
- R. R. Moskalyk and A. M. Alfantazi, Miner. Eng., 2003, 16, 793–805 CrossRef CAS.
- X. Zhang, F. Liu, X. Xue and T. Jiang, J. Alloys Compd., 2016, 686, 356–365 CrossRef CAS.
- M. R. Tavakoli, S. Dornian and D. B. Dreisinger, Hydrometallurgy, 2014, 141, 59–66 CrossRef CAS.
- H. Li, H. Fang, K. Wang, W. Zhou, Z. Yang, X. Yan, W. Ge, Q. Li and B. Xie, Hydrometallurgy, 2015, 156, 12–135 CrossRef.
- D. He, Q. Feng, G. Zhang, L. Ou and Y. Lu, Miner. Eng., 2007, 20, 1184–1186 CrossRef CAS.
- K. Yang, X. Zhang, X. Tian, Y. Yang and Y. Chen, Hydrometallurgy, 2010, 103, 7–11 CrossRef CAS.
- X. Li, B. Xie, G. Wang and X. Li, Trans. Nonferrous Met. Soc. China, 2011, 21, 1860–1867 CrossRef CAS.
- Y. Ji, S. Shen, J. Liu and Y. Xue, J. Cleaner Prod., 2017, 149, 1068–1078 CrossRef CAS.
- C. Barreradíaz, V. Lugolugo and B. Bilyeu, J. Hazard. Mater., 2012, 223–224, 1–12 CrossRef PubMed.
- S. Wilson and J. Weber, Chem. Geol., 1979, 26, 345–354 CrossRef CAS.
- B. Wehrli and W. Stumm, Geochim. Cosmochim. Acta, 1989, 53, 69–77 CrossRef CAS.
- J. A. Dean, Lange's Handbook of Chemistry, McGraw-Hill, New York, 1999 Search PubMed.
- S. Xu, S. Pan, Y. Xu, Y. Luo, Y. Zhang and G. Li, J. Hazard. Mater., 2015, 283, 7–13 CrossRef CAS PubMed.
- Y. Yuan, S. Yang, D. Zhou and F. Wu, J. Hazard. Mater., 2016, 307, 294–301 CrossRef CAS PubMed.
- D. Zhang, Y. Chen, G. Jilani, W. Wu, W. Liu and Z. Han, Bioresour. Technol., 2012, 116, 386–395 CrossRef CAS PubMed.
- T. Zhang, L. Ding, H. Ren and X. Xiong, Water Res., 2009, 43, 5209–5215 CrossRef CAS PubMed.
- T. Zhang, L. Ding and H. Ren, J. Hazard. Mater., 2009, 166, 911–915 CrossRef CAS PubMed.
- H. Huang, D. Xiao, Q. Zhang and L. Ding, J. Environ. Manage., 2014, 145, 191–198 CrossRef CAS PubMed.
- S. Zhou and Y. Wu, Environ. Sci. Pollut. Res. Int., 2012, 19, 347–360 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Xuefei Zhangab and
Xiangxin Xue*ab
ab,
Xuefei Zhangab and
Xiangxin Xue*ab