
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInhibition of FAD-dependent lysine-specific demethylases by chiral polyamine analogues†
Naoki Umezawa *a,
Kasumi Tsujia,
Shin Satob,
Masaki Kikuchib,
Hisami Watanabeb,
Yuhei Horaia,
Masashi Yamaguchia,
Yosuke Hisamatsua,
Takashi Umehara*b and
Tsunehiko Higuchi*a
*a,
Kasumi Tsujia,
Shin Satob,
Masaki Kikuchib,
Hisami Watanabeb,
Yuhei Horaia,
Masashi Yamaguchia,
Yosuke Hisamatsua,
Takashi Umehara*b and
Tsunehiko Higuchi*a
aGraduate School of Pharmaceutical Sciences, Nagoya City University, 3-1 Tanabe-dori, Mizuho-ku, Nagoya 467-8603, Japan. E-mail: umezawa@phar.nagoya-cu.ac.jp; higuchi@phar.nagoya-cu.ac.jp
bLaboratory for Epigenetics Drug Discovery, RIKEN Center for Biosystems Dynamics Research, 1-7-22 Suehiro-cho, Tsurumi-ku, Yokohama, Kanagawa 230-0045, Japan. E-mail: takashi.umehara@riken.jp
First published on 31st October 2018
Abstract
Lysine-specific demethylases 1 and 2 (LSD1 and LSD2) are flavoenzyme demethylases, and their inhibitors are considered as potential chemical tools and anticancer agents. Here we report polyamine-based inhibitors of LSD1 and LSD2. In the initial screening, partially constrained polyamine 2 which contains three trans-cyclopentane units with a total of six stereogenic centers, showed the most potent LSD1-inhibitory activity. We then prepared a set of optical isomers of 2 and evaluated their inhibitory activities toward LSD1, LSD2, monoamine oxidases A and B (MAO-A and MAO-B). Optical isomers of 2 showed LSD1-inhibitory activity with Ki values of 2.2 to 6.4 μM, and LSD2-inhibitory activity with Ki values of 4.4 to 39 μM; there was a general preference for LSD1 to LSD2. All of them showed weak to negligible inhibition of MAO-A and MAO-B. This selectivity seemed to reflect the differences in the size and shape of the catalytic cavity of target enzymes, and our strategy of employing a set of optical isomers appears to be an effective approach for exploring the structural features of this family of enzymes. Polyamine 9 showed most potent LSD1-inhibitory activity (Ki = 2.2 μM in vitro), and it also inhibited the proliferation of HL-60 cells (IC50 = 49 μM). On the other hand, 12 was the most potent inhibitors of LSD2 with in vitro Ki values of 4.4 μM.
Introduction
Polyamines are organic molecules containing multiple amino groups. Representative biogenic polyamines are the tetraamine spermine [H2N(CH2)3NH(CH2)4NH(CH2)3NH2], the triamine spermidine [H2N(CH2)4NH(CH2)3NH2], and the diamine putrescine [H2N(CH2)4NH2]. These polyamines are linear aliphatic nitrogenous bases, and amino groups in the polyamines are protonated and serve as cations under physiological conditions. These biogenic polyamines are ubiquitous in cells and are essential for cell growth and differentiation.1,2 The pathways of biosynthesis and interconversion of polyamines in mammals are well established.1,3 Polyamine catabolism is a recycling pathway that converts spermine to spermidine and spermidine to putrescine, with the production of aldehydes and hydrogen peroxide (H2O2). The polyamines are interconverted by oxidative degradation directly via spermine oxidase (SMO) or by polyamine oxidase (PAO) after acetylation of the primary amino group of spermine or spermidine via spermidine/spermine-N1-acetyltransferase (SSAT).3,4Posttranslational modifications (PTMs), such as acetylation, acylation, phosphorylation, and methylation, play an important role in regulating many protein functions. Among such PTMs, reversible methylation/demethylation of lysine residues is typically involved in regulation of the chromatin environment and eukaryotic gene expression.5 Mammals have two flavoenzyme demethylases: lysine-specific demethylase 1 (LSD1/KDM1A) and lysine-specific demethylase 2 (LSD2/KDM1B). LSD1 removes mono- and dimethyl groups from histone H3 at Lys4 (H3K4) or Lys9 (H3K9) in complexes with several chromatin-associated factors, such as the corepressor CoREST.6–10 LSD2 also demethylates histone H3, mainly at mono- and dimethylated Lys4, although its activity is weaker than that of LSD1 in vitro.11 LSD1 and LSD2 both belong to the amine oxidase family that utilizes O2 and generates H2O2 and formaldehyde as by-products.6,11,12 The active site structures of LSD1 and LSD2 have sequence homology to monoamine oxidases A and B (MAO-A and MAO-B) and also to SMO and PAO.6,13,14 Indeed, the small-molecular MAO inhibitor tranylcypromine also inhibits LSD1 and LSD2 in an irreversible and mechanism-based manner.11,13–17 Since LSD1 is involved in tumorigenesis and progression of various cancers, LSD1-specific inhibitors are considered as not only chemical tools for biomedical research, but also as potential anticancer agents.18,19 Most cell-permeable LSD1 inhibitors reported so far, including ours,17 are based on the tranylcypromine scaffold, and are irreversible, covalently reacting inhibitors.15,19 On the other hand, no LSD2-specific inhibitors have yet been developed, which hinders the study of the chemical biology of LSD2.
Since LSD1 is homologous to the FAD-dependent SMO and PAO,6,20 polyamine analogue-based LSD1 inhibitors have been developed based on known PAO inhibitors.21,22 For example, bisguanidine and biguanide noncovalently inhibit LSD1.23 Their bisurea and bisthiourea derivatives also inhibit LSD1 with an IC50 value of approximately 5 μM for the most potent compound.24 These inhibitors contained an alkyldiamine moiety. Also, long-chain polyamine analogues, which possess 10 secondary amino groups, inhibit LSD1.25 These reports led us to speculate that simple polyamine derivatives might inhibit LSDs, although the natural polyamines are not substrates of LSD1.6 We report herein the development of novel polyamine-based small molecules that inhibit both LSD1 and LSD2, though with a preference for LSD1.
Results and discussion
Initial screening of polyamine backbones for LSD1 inhibition
To test our hypothesis that relatively simple polyamine might inhibit lysine-specific demethylases LSD1 and LSD2, we first screened seven structurally diverse analogues to identify the preferred backbone structure of polyamine (Fig. 1). Polyamines 1–4 were synthesized by the solid-phase method, as reported previously.26,27 In brief, once the peptide was constructed on solid support using standard Fmoc method, the resin was treated with BH3-THF to reduce the backbone amide functionality. The resultant polyamine was then treated with piperidine while still attached to the solid support, followed by TFA cleavage. These polyamines all contain a (4-methoxyphenyl)propyl group, which shows strong UV absorbance and is helpful for purification by reversed-phase HPLC (RP-HPLC).26,27 We chose (4-methoxyphenyl)propyl group, since it is readily available, relatively small, and stable under the condition of amide reduction. Also, we envisioned that this structure might participate in the favorable hydrophobic interaction with LSDs, since phenyl group is frequently appeared in the small molecule LSD1 inhibitors.19 The solid-phase method is relatively simple and possible to synthesize various types of polyamines.27 Polyamines 5–7 were prepared according to the literature.28,29 Polyamines 1–6 contain five primary, secondary, tertiary, quaternary amino or ammonium groups, whereas 7 has only four amino groups.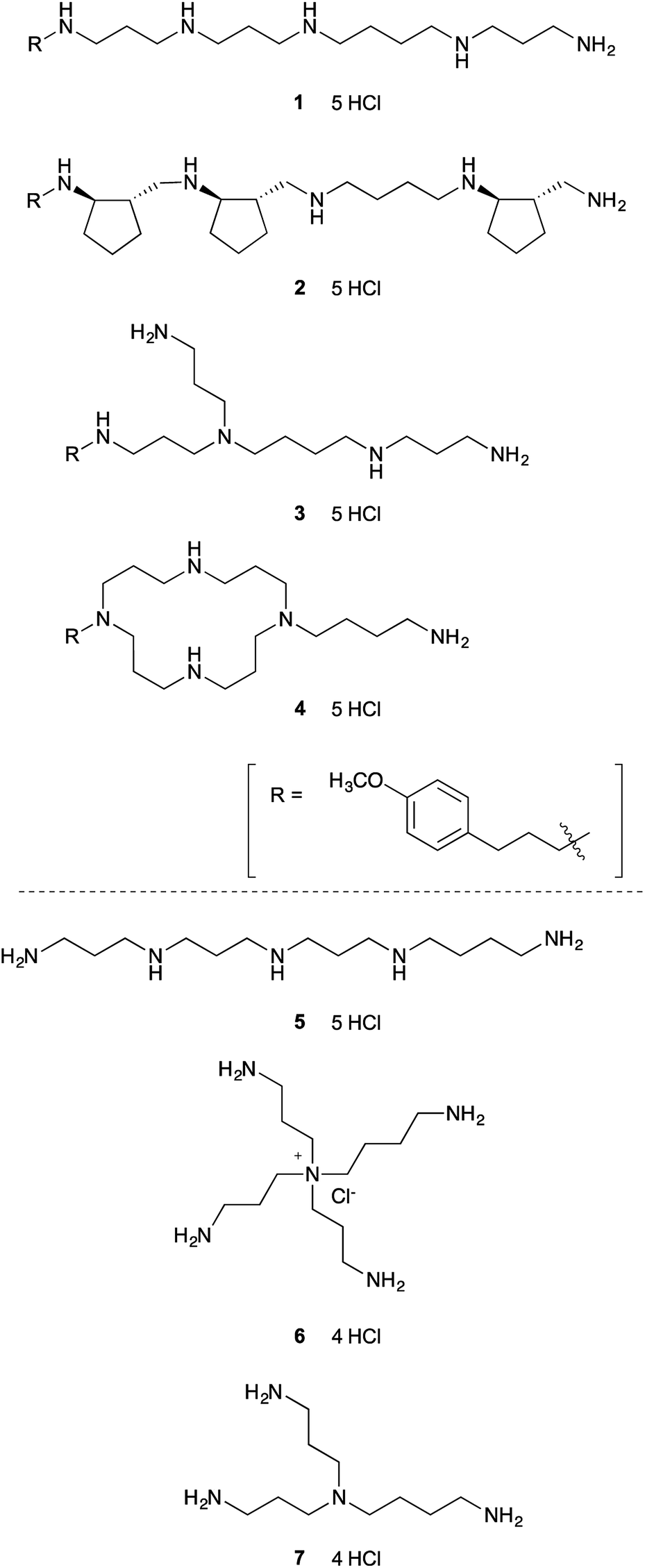 | ||
| Fig. 1 Structures of polyamines subjected to initial screening for LSD1-inhibitory activity. | ||
The LSD1-inhibitory activity of polyamines 1–7 (Fig. 1) was measured in vitro using a peroxidase-coupled assay system.30,31 The assay results (Table 1) showed that even the simple linear polyamine 5 exhibits modest LSD1-inhibitory activity (Ki = 13 μM). Addition of a (4-methoxyphenyl)propyl group (1) resulted in similar inhibitory potency (Ki = 14 μM). Branched polyamine 3 and cyclic polyamine 4 possessing a (4-methoxyphenyl)propyl group showed virtually no LSD1-inhibitory activity (Ki > 50 μM), suggesting that the structure of the polyamine itself is critical for the inhibitory activity. Huang et al. reported a series of conformationally restricted and saturated polyamine analogues, including pentamines, hexamines, octamines, and decamines, for their effect on LSD1 activity.25 Most compounds have four methylene (CH2) units between each amino groups, but in some cases, cyclopropane ring or double bond was incorporated for conformational restriction. Although conformational restriction sometimes worked positive, the LSD1 inhibitory activity generally depends on the number of amino groups; pentamine and hexamine analogues marginally affected LSD1 activity. In our initial screening, however, some of the pentamines exhibited considerable LSD1 inhibition. Among 1–7, the partially constrained polyamine 2 was the most potent LSD1 inhibitor (Ki = 6.3 μM), suggesting that partially constrained backbone, which contains three trans-cyclopentane units with a total of six stereogenic centers, is preferential for LSD1 inhibition.
Validation of LSD1–polyamine interaction site by X-ray crystal structure analysis
Next, we investigated the crystal structure of the complex of LSD1 with polyamine 2, with the aim of providing a rational basis for designing more potent and selective inhibitors. We obtained single crystals of LSD1·CoREST, and soaked them in a solution of polyamine 2 or histone H3(1–20)K4-dimethylated (K4me2) peptide. When LSD1·CoREST crystals were soaked in H3(1–20)K4me2 peptide solution, electron density corresponding to the H3(1–20)K4me2 peptide could not be detected in the catalytic cavity of LSD1 (Fig. S1A in ESI†). On the other hand, an Fo-Fc map of a LSD1·CoREST complex soaked with polyamine 2 revealed the presence of polyamine 2 in the catalytic cavity of LSD1 (Fig. S1B in ESI†). However, the observed electron density was insufficient to determine the orientation of 2. In order to uncover the binding mode, we synthesized 8, which is an asymmetrically brominated analogue of 2 (Fig. 2), anticipating that the strong anomalous signal of the bromine atom would be informative. We confirmed that polyamine 8 showed similar LSD1-inhibitory activity to 2 (Ki = 3.9 ± 0.32 μM). As expected, X-ray absorption fine structure (XAFS) spectroscopy of the LSD1·CoREST complex soaked with 8 showed anomalous dispersion from the bromine atom (Fig. S1C in ESI†), and the Fo-Fc map confirmed the presence of 8 in the catalytic cavity of LSD1 (Fig. S1D in ESI†). Unfortunately, though, the results were still insufficient to identify the binding mode of 8. A possible explanation of this result would be that 8 binds to LSD1 in several different modes.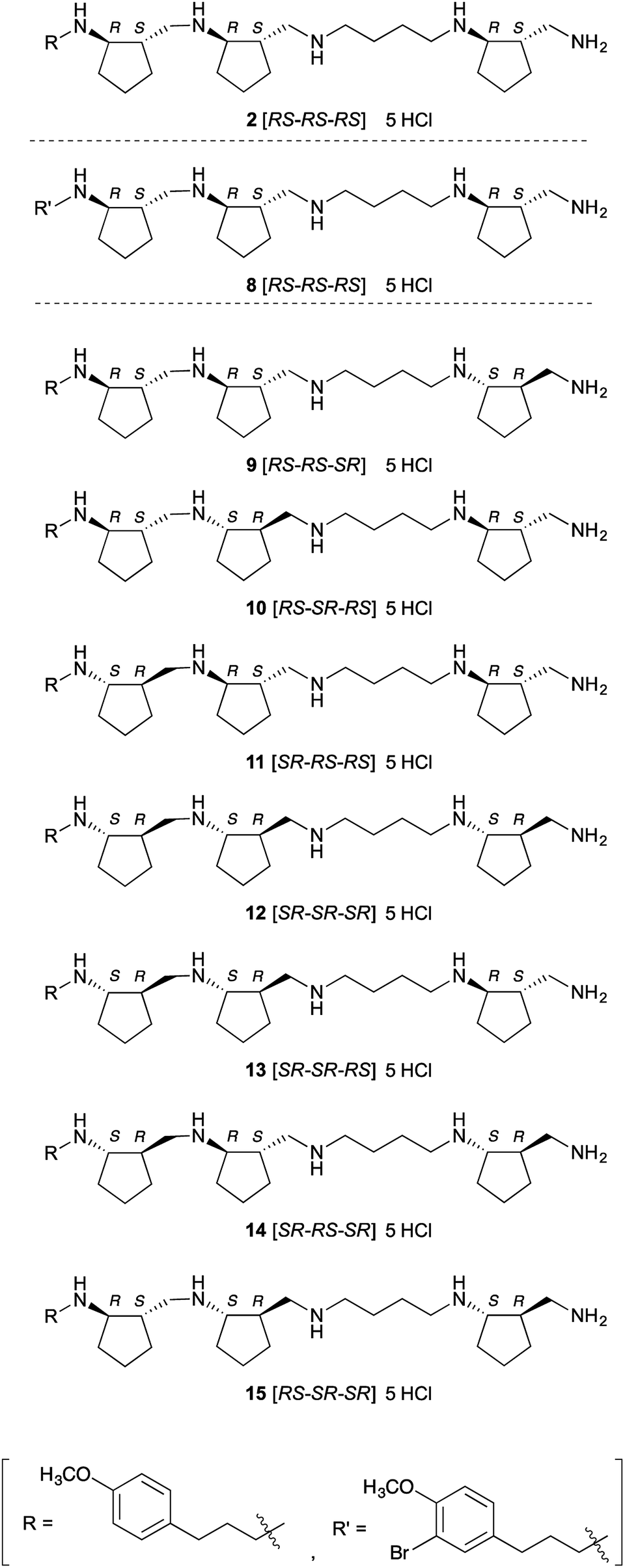 | ||
| Fig. 2 Structures of brominated analogue and optical isomers of polyamine 2. | ||
Since it was difficult to fully identify the structure of LSD1 in complex with polyamines, we carried out molecular modeling (docking) study to predict how polyamine 2 binds to LSD1. The docking program BIOVIA Discovery Studio software package was used for docking. The crystal structure of LSD1·CoREST complex soaked with 2 was used as a docking template, which was prepared by adding hydrogen atoms to the structure, removing the electron density of polyamine and water molecules, while keeping FAD as an integrated component of the protein. Polyamine 2 could be well docked into the active site of LSD1. Among the docking structures with lowest energies, several binding conformations in the active site of LSD1 were well overlapped to the observed electron density. Representative results were shown in Fig. S2 (ESI†). In general, polyamine 2 seemed to interact with LSD1 through electrostatic and hydrophobic interactions. The electrostatic interactions were observed between amino groups in polyamine 2 and negatively charged residues in the active site of LSD1. Cyclopentyl and (4-methoxyphenyl)propyl groups in polyamine 2 participated in the hydrophobic interactions with hydrophobic residues and FAD in LSD1. The binding conformations of 2 were divergent and the orientation of 2 could be opposite (Fig. S2 in ESI†). This result supports the above discussion that polyamine 2 or 8 binds to LSD1 in several different modes. Nonetheless, these results at least confirmed that 2 and 8 interact with the catalytic center of LSD1 in the crystal state, which is consistent with the finding that they inhibit the demethylation activity of LSD1 toward histone H3 tail peptide at single-digit micromolar concentrations in vitro.
Inhibition of LSDs and MAOs by optical isomers of 2
As a first step to the structural optimization of 2, we examined the chirality of backbone structure. The above results confirmed that 2 interacts with the catalytic cavity of LSD1 in the crystal, and since the catalytic cavity is a chiral environment, we envisioned that different optical isomers of polyamine 2 might exhibit distinct activities. Further, the amino acid residues in the catalytic cavity of LSD2 have bulkier side chains than the corresponding residues in LSD1, making the catalytic cavity of LSD2 smaller than that of LSD1,32,33 so we anticipated that some of the isomers might show isozyme selectivity. Also, from a different perspective, these compounds might serve as chemical probes to explore the structural features of this family of enzymes, such as differences of cavity size and shape. We synthesized a set of optical isomers of polyamine 2 with different combinations of six chiral centers (Fig. 2). A Lineweaver–Burk plot (Fig. S3 in ESI†) confirmed that one of these polyamines, 13, was a competitive inhibitor of LSD1. This result strongly suggests that all of these polyamine-based inhibitors are also competitive inhibitors. All the optical isomers showed relatively potent inhibition of both LSD1 and LSD2 (Table 2), but in all cases, the inhibitory activity towards LSD1 was greater than that towards LSD2: the Ki values for LSD1 inhibition were 2.2–6.4 μM, while the Ki values for LSD2 inhibition were 4.4–39 μM (the selectivity ratio ranged from 1.8 to 6.1). To the best of our knowledge, these compounds are the first examples of polyamine-based LSD2 inhibitors. Polyamine 9 [RS-RS-SR] was the most potent inhibitor of LSD1 (Ki = 2.2 μM), while 12 [SR-SR-SR] and 13 [SR-SR-RS] were comparably potent (Ki = 2.4 μM and 2.5 μM, respectively). On the other hand, polyamine 12 [SR-SR-SR] was the most potent inhibitors of LSD2 (Ki = 4.4 μM), while 13 [SR-SR-RS] was similarly potent (Ki = 4.5 μM). The overall preference to LSD1 could be explained by the differences in the size and shape of the catalytic cavity, since catalytic cavity of LSD2 is smaller than that of LSD1.32,33 The LSD1 inhibitory activities of these polyamines are fairly potent, since the Ki values are comparable to those of known LSD1 inhibitors. For example, Ki values of peptidic inhibitor SNAIL1 1–9,34 small-molecule irreversible inhibitors tranylcypromine14–16,35,36 and S2101 (ref. 17) were 1.9 μM,31 100 μM,17 and 0.61 μM,17 respectively in our assay system.| Polyamines | Ki for LSD1 inhibitionb (μM) | Ki for LSD2 inhibitionb (μM) | Selectivity [LSD1/LSD2] |
|---|---|---|---|
| a Kinetic plots are shown in Fig. S4 and S5 in ESI.b Mean ± SE (N = 3). | |||
| 2 [RS-RS-RS] | 6.3 ± 0.48 | 24 ± 2.1 | 3.8 |
| 9 [RS-RS-SR] | 2.2 ± 0.18 | 6.9 ± 0.56 | 3.1 |
| 10 [RS-SR-RS] | 6.4 ± 0.47 | 39 ± 3.1 | 6.1 |
| 11 [SR-RS-RS] | 4.2 ± 0.43 | 9.5 ± 0.54 | 2.3 |
| 12 [SR-SR-SR] | 2.4 ± 0.24 | 4.4 ± 0.39 | 1.8 |
| 13 [SR-SR-RS] | 2.5 ± 0.24 | 4.5 ± 0.42 | 1.8 |
| 14 [SR-RS-SR] | 3.3 ± 0.32 | 9.2 ± 0.45 | 2.8 |
| 15 [RS-SR-SR] | 3.8 ± 0.32 | 18 ± 0.93 | 4.7 |
In order to further evaluate the target selectivity, we also examined the inhibitory activity of these polyamines toward MAO-A and MAO-B, whose active site structures show sequence homology to those of LSDs. LSD1 is classified as a member of the PAO superfamily, and some small-molecular MAO inhibitors, such as pargyline, tranylcypromine, and phenelzine, also inhibit LSD1. As shown in Table 3, the polyamine analogues showed little or no inhibitory activity toward MAO-A or MAO-B; i.e., they are highly selective for LSDs over MAOs.
| Polyamines | Ki for MAO-A inhibitionb (μM) | Ki for MAO-B inhibitionb (μM) |
|---|---|---|
| a Kinetic plots are shown in Fig. S6 and S7 in ESI.b Mean ± SE (N = 3). | ||
| 2 [RS-RS-RS] | 380 ± 50 | >500 |
| 9 [RS-RS-SR] | 350 ± 49 | >500 |
| 10 [RS-SR-RS] | >500 | >500 |
| 11 [SR-RS-RS] | 330 ± 31 | >500 |
| 12 [SR-SR-SR] | 290 ± 59 | >500 |
| 13 [SR-SR-RS] | 310 ± 70 | >500 |
| 14 [SR-RS-SR] | 500 ± 35 | >500 |
| 15 [RS-SR-SR] | 190 ± 32 | >500 |
Given that these polyamine analogues bind within the catalytic cavity, the above results may reflect differences in the size of the catalytic cavity. It is reported that LSD1 has a wider catalytic cavity than LSD2,19 while MAO-A and MAO-B, which were negligibly inhibited by all of the polyamines, have even narrower cavities than LSD2.37 It is noteworthy that a cyclopentane substituent appears to reduce the inhibitory activity towards LSD2, presumably because of steric hindrance. Further modification of polyamines 12 or 13, such as systematic removal of cyclopentane rings and changing the linker length between amino groups, might yield LSD2-preferential inhibitors. On the other hand, the (4-methoxyphenyl)propyl group seemed to have little effect on the inhibition of LSD1, since 1 and 5 showed similar inhibition activity toward LSD1. Substitution of this group with other groups might further improve the activity. Overall, these results suggest that our strategy of employing a set of optical isomers is a useful approach for exploring the structural features of this family of enzymes, such as differences of cavity size and shape, and should be helpful in rationally designing more potent and selective inhibitors.
Finally, since 9 [RS-RS-SR] showed the strongest inhibitory activity toward LSD1 in vitro (i.e. Ki = 2.2 ± 0.18 μM) among the compounds tested, we assessed whether it can inhibit the proliferation of human acute promyelocytic leukemia cell line HL-60, which is sensitive to LSD1 inhibition.38 We found that 9 [RS-RS-SR] inhibited proliferation of HL-60 cells with IC50 = 49 ± 2.4 μM (n = 3). Since polyamines interact with various cellular targets, we cannot rule out the possibility that there may be unknown molecular target(s) other than LSD1. However, this result suggests that polyamine 9 is membrane-permeable, at least to some extent, and that the decrease of HL-60 cell proliferation is due at least in part to inhibition of LSD1. The difference between the in vitro (Ki = 2.2 μM) and in vivo (IC50 = 49 μM) effects might reflect limited ability of 9 to penetrate the cell membrane.
Conclusion
We prepared a series of polyamines with modified backbones and screened them for LSD-inhibitory activity. Polyamine 2 showed the most potent LSD1-inhibitory activity in the initial screening, suggesting that partially constrained backbone structure is preferential for LSD1 inhibition. We next synthesized a set of optical isomers of 2 in order to optimize the backbone structure. Among them, 9 was the most potent LSD1 inhibitor (Ki = 2.2 μM), while 12 was the most potent inhibitors of LSD2 (Ki = 4.4 μM) although 12 still showed higher inhibitory activities toward LSD1. To the best of our knowledge, these compounds are the first examples of polyamine-based LSD2 inhibitors. All of the polyamines negligibly inhibited MAO-A and MAO-B. Considering the structure of these enzymes, this selectivity would reflect differences in the size and shape of the catalytic cavity, suggesting that our strategy of employing a set of optical isomers appears to be an effective approach for exploring the structural features of this family of enzyme. Polyamine 9 also inhibited the proliferation of human acute promyelocytic leukemia cell line HL-60. These polyamines may be promising leads for the development of more potent and selective inhibitors of lysine-specific demethylases as potential chemical tools and pharmaceuticals. Since our solid-phase polyamine synthesis method is simple and well suited for library construction, further optimization would be relatively straight-forward. A large number of structurally and stereochemically diverse amino acid building blocks are easily accessible. Furthermore, since LSD1-inhibiting long-chain polyamines are synergistic with DNA methyltransferase inhibitors,25 our polyamines may also be useful in combination with DNA methyltransferase inhibitors for epigenetic therapy of cancer.Experimental
General
All regents and solvents were commercial products of the highest quality and were used without purification. Flash column chromatography was performed on silica gel (Fuji Silysia Chemical Ltd., BW-300) using forced flow. Thin layer chromatography (TLC) was performed on Merck pre-coated plates (Silica gel 60 F254, 0.25 mm) and bands were visualized by fluorescence quenching under UV light or by staining with potassium permanganate or ninhydrin. 1H-NMR and 13C-NMR spectra were recorded on a JEOL LNM-LA500 (JEOL Ltd., Tokyo, Japan) or a Varian VNMRS 500 at 500 MHz and 125 MHz, respectively. 13C-NMR spectra were also recorded on a Bruker Avance-600 (Bruker Biospin, Yokohama, Japan) at 150 MHz. Chemical shifts are expressed as parts per million (ppm) using tetramethylsilane, 1,4-dioxane (for 13C-NMR of polyamine), or solvent as an internal standard. Data are reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet; coupling constants in Hz. Infrared (IR) spectra were recorded on a JASCO FT/IR-680 Fourier-transform infrared spectrophotometer (JASCO Corporation, Tokyo, Japan). Electrospray ionization mass spectrometry (ESI-MS) was done with a JEOL JMS-T100LP4G (JEOL Ltd., Tokyo, Japan). Preparative RP-HPLC and analytical RP-HPLC were performed using a Shimadzu SPD-M10AVP variable-wavelength UV detector (Shimadzu Corporation, Kyoto, Japan). Inertsil ODS-3 (10 × 250 mm, GL Science Inc., Tokyo, Japan) and Inertsil ODS-3 (4.6 × 250 mm, GL Science Inc., Tokyo, Japan) columns were employed for chromatographic and analytical separations, respectively.Synthesis of polyamines
In vitro enzyme inhibition assays
LSD1 inhibition assay was performed using the peroxidase-coupled reaction method essentially as described previously.30,31 Human LSD1 (2.8 μM) was incubated with serial dilutions of each polyamine in 50 mM HEPES-Na (pH 7.5) buffer containing 400 μM 4-aminoantipyrine, modified Trinder's reagent TOOS (N-ethyl-N-(2-hydroxy-3-sulfopropyl)-3-methylaniline, sodium salt, dihydrate), and 40 μg mL−1 horseradish peroxidase at 25 °C for 10 min. The reaction mixture was subsequently incubated with 83 μM H3(1–20)K4-dimethylated (K4me2) peptide for 30 min. Inhibition assays for LSD2, MAO-A and MAO-B were performed in a similar manner to the LSD1 inhibition assay, using 167 μM H3(1–20)K4me2 peptide as the substrate for 2.7 μM LSD2, and 50 and 150 μM tyramine as the substrate for 1.4 and 2.8 μM MAO-A and MAO-B, respectively. Absorbance at 562 nm of the hydrogen peroxide by-products generated by H3(1–20)K4me2 demethylation or tyramine oxidation was measured with a 96-well microplate reader (Ultrospec Visible Plate Reader II 96; GE Healthcare). Steady-state reaction ranges from 0 to 10 min (LSD1, LSD2 and MAO-A) or 0 to 30 min (MAO-B) were used for fitting analysis with the equation of competitive inhibition via steady-state kinetic analysis to obtain Ki with GraphPad Prism 6 software (version 6.0e).40Crystallization and dataset collection of LSD1·CoREST complex soaked with polyamines
Human LSD1 (residues 172–833) and CoREST (residues 308–440) were expressed, purified and crystallized as previously described.30 Crystals of LSD1·CoREST were obtained by the hanging drop method with 100 mM N-(carbamoylmethyl)iminodiacetic acid (pH 5.5) and 1.18–1.28 M potassium sodium tartrate tetrahydrate. The crystals were soaked in a solution composed of the reservoir solution supplemented with 10% glycerol and 2 mM polyamine 2, 8 or H3(1–20)K4me2 peptide, and flash-cooled in a stream of N2 gas (100 K) for data collection. In the case of the crystals soaked with polyamine 2 or H3(1–20)K4me2 peptide, the datasets were collected at beamline BL26B2 of the synchrotron radiation facility SPring-8 (Harima, Japan). XAFS experiments with LSD1·CoREST·polyamine 8 were performed at the synchrotron beamline BL26B2 with wavelengths from 0.9154 to 0.9254 Å, and the X-ray diffraction data set was collected at 0.9190 Å.Molecular modeling study
The docking program BIOVIA Discovery Studio software package was used for the docking study. The crystal structure of LSD1·CoREST complex soaked with polyamine 2 was used as a docking template, which was prepared by adding hydrogen atoms to the structure, removing the electron density of polyamine and water molecules, while keeping FAD as an integrated component of the protein. The binding conformations of polyamine 2 were estimated using CHARMm-based MD docking algorithm (CDOCKER).41 The binding conformations obtained were ranked based on CDOCKER energy parameter.Cell proliferation inhibition assay
Human promyelocytic leukemia HL-60 cells (RCB0041) were obtained from RIKEN BioResource Research Center (Tsukuba, Ibaraki, Japan) and seeded in 96-well plates at 4000 cells per well. After culture for 4 h, serial dilutions of test compound were added. The plates were incubated for 96 h in an atmosphere of 5% CO2 in air at 37 °C. The half-maximal inhibitory concentration (IC50) was determined by alamarBlue cell viability assay (Bio-Rad). AlamarBlue reagent corresponding to 10% of the sample volume was added to each well. After incubation for 2 h in an atmosphere of 5% CO2 in air at 37 °C, the fluorescence was measured with the EnVision multimode plate reader (PerkinElmer Japan) in triplicate.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank Dr Hideaki Niwa (RIKEN BDR) for a critical reading of the manuscript. This work was supported in part by JSPS KAKENHI Grant Numbers 15K08027 (N. U.) and 16H05089 (T. U.).Notes and references
- A. E. Pegg, IUBMB Life, 2009, 61, 880–894 CrossRef CAS PubMed.
- A. E. Pegg, J. Biol. Chem., 2016, 291, 14904–14912 CrossRef CAS PubMed.
- V. Battaglia, C. DeStefano Shields, T. Murray-Stewart and R. A. Casero, Amino Acids, 2014, 46, 511–519 CrossRef CAS PubMed.
- A. E. Pegg, Am. J. Physiol.: Endocrinol. Metab., 2008, 294, E995–E1010 CrossRef CAS PubMed.
- F. Lan, A. C. Nottke and Y. Shi, Curr. Opin. Cell Biol., 2008, 20, 316–325 CrossRef CAS PubMed.
- Y. Shi, F. Lan, C. Matson, P. Mulligan, J. R. Whetstine, P. A. Cole, R. A. Casero and Y. Shi, Cell, 2004, 119, 941–953 CrossRef CAS PubMed.
- E. Metzger, M. Wissmann, N. Yin, J. M. Muller, R. Schneider, A. H. Peters, T. Gunther, R. Buettner and R. Schule, Nature, 2005, 437, 436–439 CrossRef CAS PubMed.
- B. Laurent, L. Ruitu, J. Murn, K. Hempel, R. Ferrao, Y. Xiang, S. Liu, B. A. Garcia, H. Wu, F. Wu, H. Steen and Y. Shi, Mol. Cell, 2015, 57, 957–970 CrossRef CAS PubMed.
- T. Wada, D. Koyama, J. Kikuchi, H. Honda and Y. Furukawa, Blood, 2015, 125, 3731–3746 CrossRef CAS PubMed.
- Y. J. Shi, C. Matson, F. Lan, S. Iwase, T. Baba and Y. Shi, Mol. Cell, 2005, 19, 857–864 CrossRef CAS PubMed.
- A. Karytinos, F. Forneris, A. Profumo, G. Ciossani, E. Battaglioli, C. Binda and A. Mattevi, J. Biol. Chem., 2009, 284, 17775–17782 CrossRef CAS PubMed.
- C. Binda, A. Mattevi and D. E. Edmondson, J. Biol. Chem., 2002, 277, 23973–23976 CrossRef CAS PubMed.
- M. G. Lee, C. Wynder, N. Cooch and R. Shiekhattar, Nature, 2005, 437, 432–435 CrossRef CAS PubMed.
- D. M. Z. Schmidt and D. G. McCafferty, Biochemistry, 2007, 46, 4408–4416 CrossRef CAS PubMed.
- M. Yang, J. C. Culhane, L. M. Szewczuk, P. Jalili, H. L. Ball, M. Machius, P. A. Cole and H. Yu, Biochemistry, 2007, 46, 8058–8065 CrossRef CAS PubMed.
- S. Mimasu, T. Sengoku, S. Fukuzawa, T. Umehara and S. Yokoyama, Biochem. Biophys. Res. Commun., 2008, 366, 15–22 CrossRef CAS PubMed.
- S. Mimasu, N. Umezawa, S. Sato, T. Higuchi, T. Umehara and S. Yokoyama, Biochemistry, 2010, 49, 6494–6503 CrossRef CAS PubMed.
- T. E. McAllister, K. S. England, R. J. Hopkinson, P. E. Brennan, A. Kawamura and C. J. Schofield, J. Med. Chem., 2016, 59, 1308–1329 CrossRef CAS PubMed.
- H. Niwa and T. Umehara, Epigenetics, 2017, 12, 340–352 CrossRef PubMed.
- Y. Wang, W. Devereux, P. M. Woster, T. M. Stewart, A. Hacker and R. A. Casero, Cancer Res., 2001, 61, 5370–5373 CAS.
- M. Bianchi, F. Polticelli, P. Ascenzi, M. Botta, R. Federico, P. Mariottini and A. Cona, FEBS J., 2006, 273, 1115–1123 CrossRef CAS PubMed.
- S. K. Sharma, Y. Wu, N. Steinbergs, M. L. Crowley, A. S. Hanson, R. A. Casero and P. M. Woster, J. Med. Chem., 2010, 53, 5197–5212 CrossRef CAS PubMed.
- Y. Huang, E. Greene, T. Murray Stewart, A. C. Goodwin, S. B. Baylin, P. M. Woster and R. A. Casero, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8023–8028 CrossRef CAS PubMed.
- S. L. Nowotarski, B. Pachaiyappan, S. L. Holshouser, C. J. Kutz, Y. Li, Y. Huang, S. K. Sharma, R. A. Casero and P. M. Woster, Bioorg. Med. Chem., 2015, 23, 1601–1612 CrossRef CAS PubMed.
- Y. Huang, T. M. Stewart, Y. Wu, S. B. Baylin, L. J. Marton, B. Perkins, R. J. Jones, P. M. Woster and R. A. Casero, Clin. Cancer Res., 2009, 15, 7217 CrossRef CAS PubMed.
- Y. Yoshikawa, N. Umezawa, Y. Imamura, T. Kanbe, N. Kato, K. Yoshikawa, T. Imanaka and T. Higuchi, Angew. Chem., Int. Ed., 2013, 52, 3712–3716 CrossRef CAS PubMed.
- N. Umezawa, Y. Horai, Y. Imamura, M. Kawakubo, M. Nakahira, N. Kato, A. Muramatsu, Y. Yoshikawa, K. Yoshikawa and T. Higuchi, ChemBioChem, 2015, 16, 1811–1819 CrossRef CAS PubMed.
- A. Muramatsu, Y. Shimizu, Y. Yoshikawa, W. Fukuda, N. Umezawa, Y. Horai, T. Higuchi, S. Fujiwara, T. Imanaka and K. Yoshikawa, J. Chem. Phys., 2016, 145, 235103 CrossRef PubMed.
- M. Niitsu, H. Sano and K. Samejima, Chem. Pharm. Bull., 1992, 40, 2958–2961 CrossRef CAS.
- Y. Amano, M. Kikuchi, S. Sato, S. Yokoyama, T. Umehara, N. Umezawa and T. Higuchi, Bioorg. Med. Chem., 2017, 25, 2617–2624 CrossRef CAS PubMed.
- Y. Amano, N. Umezawa, S. Sato, H. Watanabe, T. Umehara and T. Higuchi, Bioorg. Med. Chem., 2017, 25, 1227–1234 CrossRef CAS PubMed.
- C. Binda, S. Valente, M. Romanenghi, S. Pilotto, R. Cirilli, A. Karytinos, G. Ciossani, O. A. Botrugno, F. Forneris, M. Tardugno, D. E. Edmondson, S. Minucci, A. Mattevi and A. Mai, J. Am. Chem. Soc., 2010, 132, 6827–6833 CrossRef CAS PubMed.
- P. Vianello, O. A. Botrugno, A. Cappa, R. Dal Zuffo, P. Dessanti, A. Mai, B. Marrocco, A. Mattevi, G. Meroni, S. Minucci, G. Stazi, F. Thaler, P. Trifiró, S. Valente, M. Villa, M. Varasi and C. Mercurio, J. Med. Chem., 2016, 59, 1501–1517 CrossRef CAS PubMed.
- M. Tortorici, M. T. Borrello, M. Tardugno, L. R. Chiarelli, S. Pilotto, G. Ciossani, N. A. Vellore, S. G. Bailey, J. Cowan, M. O'Connell, S. J. Crabb, G. Packham, A. Mai, R. Baron, A. Ganesan and A. Mattevi, ACS Chem. Biol., 2013, 8, 1677–1682 CrossRef CAS PubMed.
- M. G. Lee, C. Wynder, D. M. Schmidt, D. G. McCafferty and R. Shiekhattar, Chem. Biol., 2006, 13, 563–567 CrossRef CAS PubMed.
- D. M. Gooden, D. M. Z. Schmidt, J. A. Pollock, A. M. Kabadi and D. G. McCafferty, Bioorg. Med. Chem. Lett., 2008, 18, 3047–3051 CrossRef CAS PubMed.
- C. Binda, A. Mattevi and D. E. Edmondson, Int. Rev. Neurobiol., 2011, 100, 1–11 CrossRef CAS PubMed.
- T. Schenk, W. C. Chen, S. Göllner, L. Howell, L. Jin, K. Hebestreit, H.-U. Klein, A. C. Popescu, A. Burnett, K. Mills, R. A. Casero Jr, L. Marton, P. Woster, M. D. Minden, M. Dugas, J. C. Y. Wang, J. E. Dick, C. Müller-Tidow, K. Petrie and A. Zelent, Nat. Med., 2012, 18, 605–612 CrossRef CAS PubMed.
- D. Yang, M.-K. Wong and Z. Yan, J. Org. Chem., 2000, 65, 4179–4184 CrossRef CAS PubMed.
- J. M. Burg, J. J. Gonzalez, K. R. Maksimchuk and D. G. McCafferty, Biochemistry, 2016, 55, 1652–1662 CrossRef CAS PubMed.
- G. Wu, D. H. Robertson, C. L. Brooks and M. Vieth, J. Comput. Chem., 2003, 24, 1549–1562 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra and HPLC chromatogram of novel polyamines, molecular docking simulation of 2 against LSD1 crystal structure, kinetic analysis of LSD1, LSD2, MAO-A, and MAO-B activities by polyamines. See DOI: 10.1039/c8ra07879c |
| This journal is © The Royal Society of Chemistry 2018 |