
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic study of the ligand controlled regioselectivity in iridium catalyzed C–H borylation of aromatic imines†
Yuhua Liu‡
a,
Jipei Chen‡a,
Kangsheng Zhana,
Yiqiang Shena,
Hui Gao *bc and
Lingmin Yao*a
*bc and
Lingmin Yao*a
aSchool of Physics and Electronic Engineering, Guangzhou University, Guangzhou, 510006, China. E-mail: emmiyao@163.com
bKey Laboratory of Molecular Target & Clinical Pharmacology, School of Pharmaceutical Sciences & the Fifth Affiliated Hospital, Guangzhou Medical University, Guangzhou, 511436, China. E-mail: gaoh9@gzhmu.edu.cn
cGuangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Guangzhou 510640, PR China
First published on 16th October 2018
Abstract
As a major challenge in C–H borylation, how to control the selectivity has attracted lots of attention, however, the related mechanistic information still needs to be uncovered. Herein, density functional theory (DFT) has been used to study the mechanism for the ligand controlled regioselectivity in the iridium-catalyzed C–H borylation of aromatic imines, which is inspired by experimental observations (R. Bisht, B. Chattopadhyay, J. Am. Chem. Soc., 2016, 138, 84–87). The proposed Ir(I)–Ir(III) catalytic cycle includes (i) the oxidative addition of the C–H bond to iridium(I); (ii) the reductive elimination of a C–B bond; (iii) the oxidative addition of B2pin2 to an iridium(I) hydride complex; and (iv) the reductive elimination of a B–H bond. The oxidative addition of a C–H bond to the iridium center is the determining step. For the ligand AQ, ortho-selectivity is proposed to be attributed to the decreased steric hindrance and increased electron donating effect of AQ (8-aminoquinoline) which promotes proton-transfer in the ortho-transition state of C–H activation. While, for the TMP ligand, the steric repulsion between the TMP (4,5,7,8-tetramethyl-1, 10-phenanthroline) ligand and the ortho-substituted imine hinders the ortho C–H activation and favors meta borylation. Our calculations provide insights into further ligand design to achieve different regioselective borylation of aromatics. Guided by the results, the regioselectivity in the borylation of aromatics may be achieved by accordingly modifying the electronic and steric substituents of the ligand.
Introduction
Transition metal catalyzed C–H borylation of aryl rings is one of the most important methods used to activate C–H bonds and functionalize organic molecules.1 In the past years, the C–H borylation reaction catalyzed by different transition metal complexes such as Ir, Rh, and Co has been developed well.2–6 In particular, the number of iridium catalyzed aromatic C–H borylation reactions have grown considerably,4–7 in which an important issue to be solved is how to control the regioselectivity. One of the most employed strategies is the combination of iridium complexes with directing groups to allow selective ortho-borylation.8–10 Smith reported an example of sterically directed ortho-borylation.11 On the other hand, meta-selective C–H bond borylation of aryl rings is more difficult. Only a few methods for the sterically controlled meta selective borylations have been reported.12,13Understanding the experimentally observed ligand controlled regioselectivity is essential for clarification of the detailed influence of ligands and guiding further catalysis design. Previous studies have proposed several hypothesises on the regioselective C–H borylation,14–19 though debate is still existed. Maseras and co-workers suggested that in the borylation of methyl benzoate catalyzed by iridium complexes supported by PPh3 (triphenyl phosphine) or dtbpy (4,4-di-tert-butyl bipyridine), the possible mechanism is iridium(III)/iridium(V) catalytic cycle17(Fig. 1, green). Alternatively, Lan and co-workers suggested that in the borylation of aromatic C–H catalyzed by [Ir(cod)OH]2/Xyl-MeO-BIPHEP, iridium(I)/iridium(III)-based catalytic cycle exists18 (Fig. 1, aubergine).
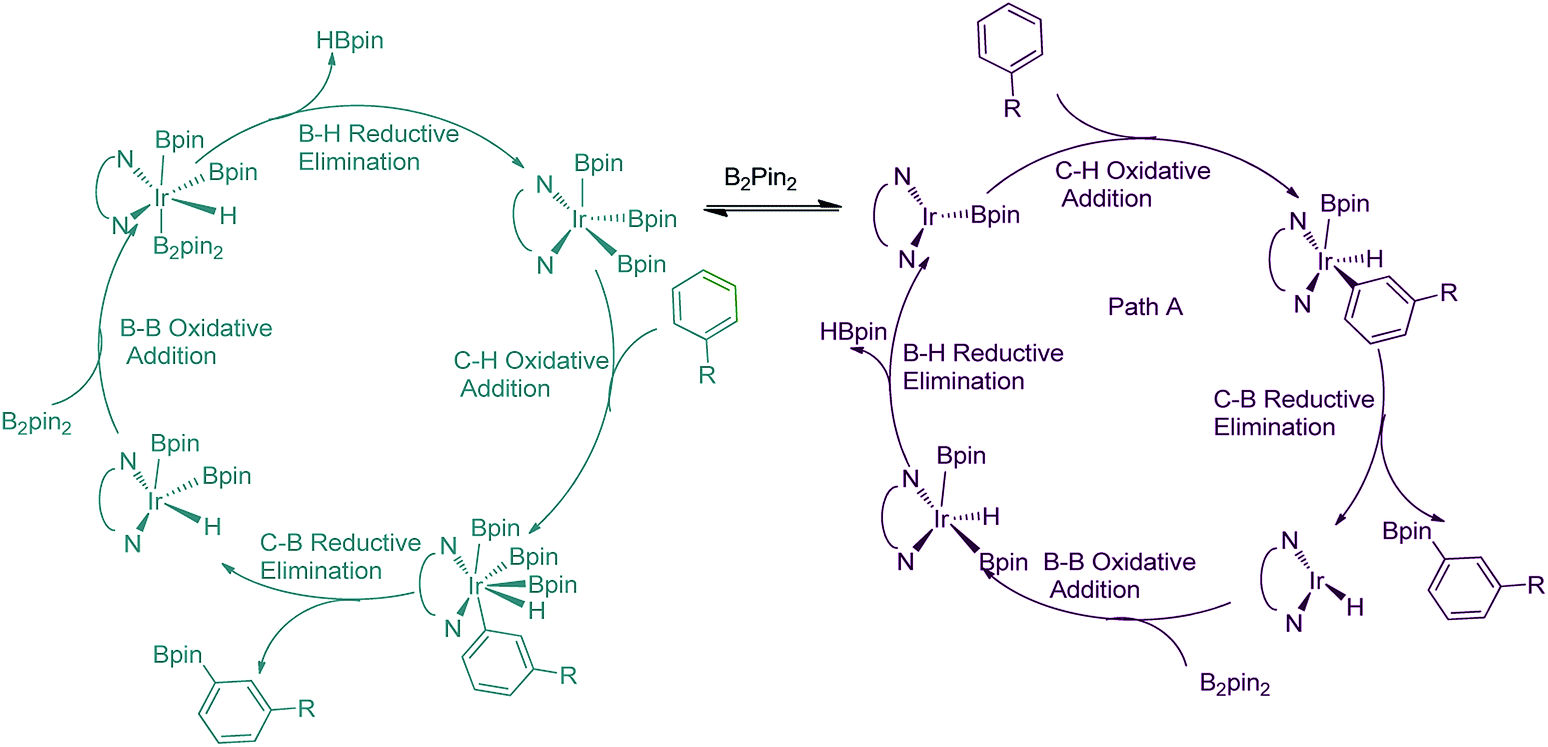 | ||
| Fig. 1 Potential mechanisms of iridium(III)/iridium(V) and iridium(I)/iridium(III) catalytic cycles for aromatic C–H borylation. | ||
Recently, Chattopadhyay reported a series of iridium catalyzed borylation of aromatics with diboron reagents (Scheme 1).20 The original report described that the Ir catalysts supported by AQ showed the capability of borylation on ortho position of aromatic imine while the Ir/TMP system preferred meta-selectivity, however, no explanation on the regioselectivity was given. There are some issues to be answered: (1) what are the details of the catalytic cycle? (2) What is the regioselective determine step in the cycle? (3) How does the structure of ligand influence the reactivity and regioselectivity? With the aim of answering these questions, we conducted computational study by using the prototypical reaction between phenyl imine and pinacolborane as the model reaction (Scheme 1).
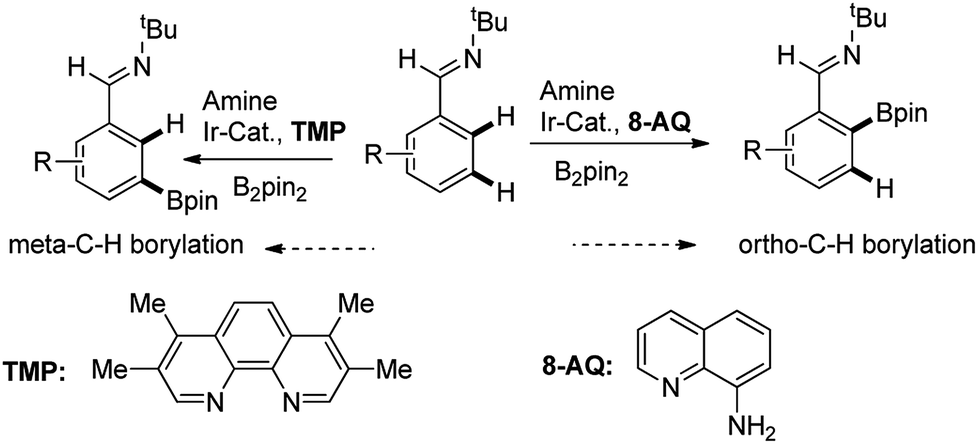 | ||
| Scheme 1 Computational model for aromatic C–H borylation. | ||
For the special reaction system in the model reaction (Scheme 1), three possible mechanistic pathways have been demonstrated and the related reaction profiles have been examined. All the pathways involve C–H activation, C–B reductive elimination and B–B oxidative addition and B–H reductive elimination steps. The only difference lies in that path I proceeds in the sequence of C–H activation, C–B reductive elimination, B–B oxidative addition and B–H reductive elimination (Scheme 2a); while path II occurs in the sequence of C–H activation, B–H reductive elimination, B–B oxidative addition and C–B reductive elimination (Scheme 2b). Both path I and path II proceed through Ir(I)–Ir(III) catalytic cycle. Path III undergoes the analogues reaction sequence with path I but via Ir(III)–Ir(V) catalytic cycle (Scheme 2c). Herein, we report our investigations on the above possible pathways with special emphasis on the ligand effects on regioselectivity.
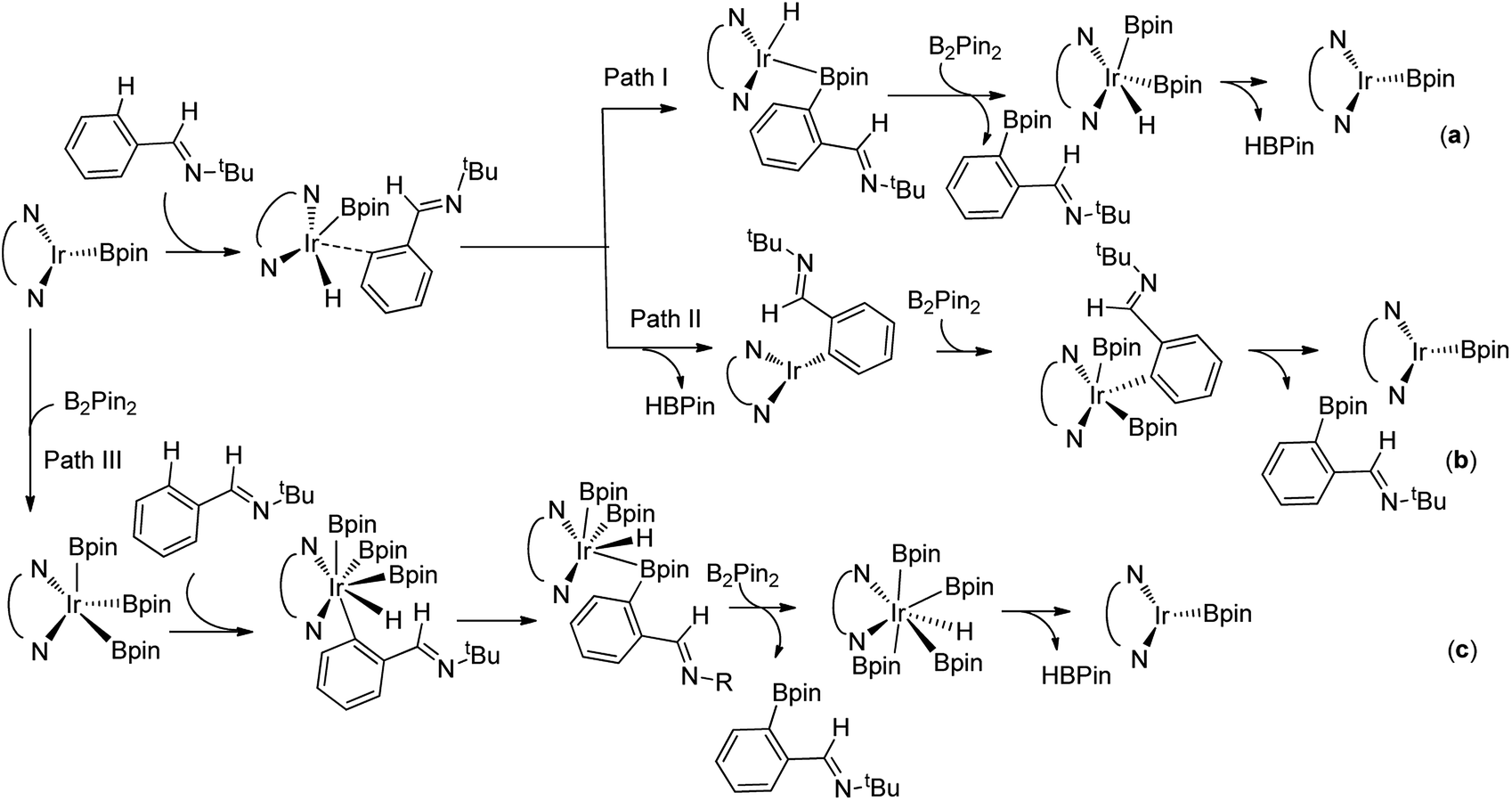 | ||
| Scheme 2 Three types of potential mechanisms for C–H borylation of aromatic imine. | ||
Computational method
The followed DFT calculations have been conducted using Gaussian 09 program.21 Geometries were optimized using the B3LYP functional.22 SDD23 basis set was used for Ir, a standard 6-31G(d)24 basis set was used for other atoms. Frequency analyses were carried out to ensure theses stationary points were at either a minimum or transition state. Solvation free energies were calculated with SMD25 solvation model (solvent = tetrahydrofuran). Additional single point calculations were performed with M11L functional.26 SDD basis set was employed for Ir while standard 6-311++G(d,p)24 basis set level for other atoms. The solvation model for the single point calculation was the same as described above. The energies presented in this paper are the M11L calculated single point energies added with B3LYP optimized thermodynamic corrections.Results and discussion
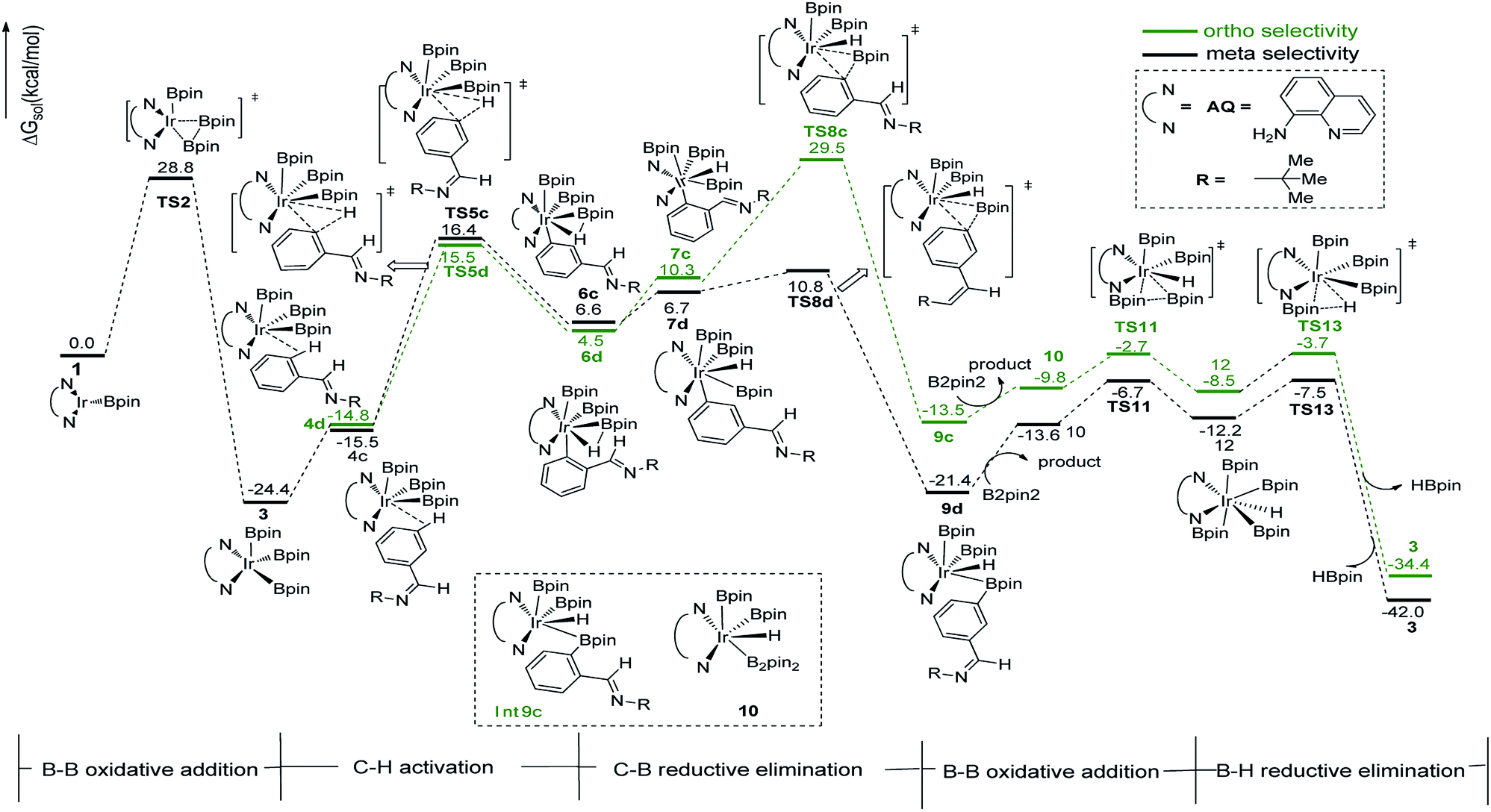
We have first examined the borylation of aromatic imine with B2pin2 catalyzed by the Ir/AQ system (Scheme 1). The catalytic cycle starts with the step of C–H activation of substrate aromatic imine. The C–H bond is splitted upon the addition of H and C to the Ir center, affording intermediate 4a or 4b (Fig. 2). The corresponding barrier of C–H activation is 33.0 kcal mol−1 and 28.0 kcal mol−1 for the meta-C–H activation (TS3b, Fig. 2, black) and ortho-C–H activation (TS3a, Fig. 2, aubergine), respectively. Thereafter, we try to figure out which is more likely the second step in the catalytic cycle, the C–B reductive elimination or B–H reductive elimination?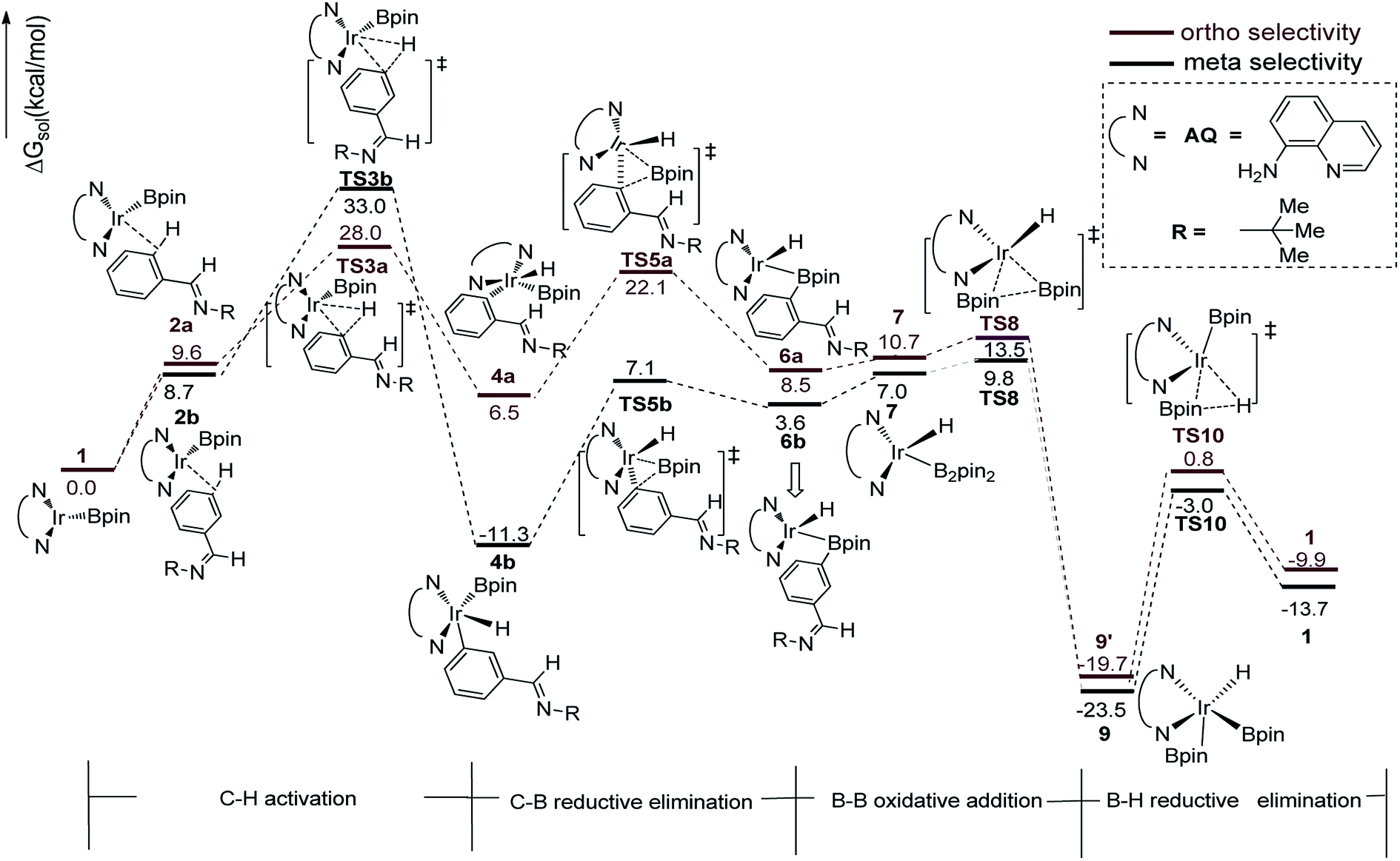 | ||
| Fig. 2 Energy profile of path I in the borylation of aromatic imine catalyzed by AQ/Ir system. | ||
We have calculated the possibility of C–B reductive elimination followed with the initial step of C–H activation. Upon C–H activation, B–C reductively eliminate from the intermediate 4a or 4b via TS5a or TS5b (Fig. 2), the barrier is 18.4 kcal mol−1 and 15.6 kcal mol−1 for meta- and ortho-selectivity, respectively (Fig. 2).
Then B–B oxidative addition to Ir center takes place to generate the iridium(III) complex. Two Ir–B distances of TS8 (Fig. 2) are different (2.40 vs. 2.55 Å). The barrier for TS8 is as low as 2.8 kcal mol−1. The catalytic cycle is closed by B–H reductive elimination to regenerate catalyst 1 (TS10, Fig. 2), which requires 20.5 kcal mol−1. Given that the barrier of transition states in initial C–H activation step is the highest along the route, C–H activation is likely the RDS step (regioselectivity determining step) in path I. Comparing meta-selectivity with ortho-selectivity in path I, our calculations indicate that ortho selectivity is favored over meta selectivity by 5.0 kcal mol−1 (33.0 kcal mol−1 vs. 28.0 kcal mol−1) in the RDS step which is in consistent with experimental observation.20
Thereafter, we investigated an alternative of the second step, the B–H reductive elimination followed with C–H activation step (Scheme 2, path II). The barrier of B–H reductive elimination is 36.7 kcal mol−1 (Scheme 3). Because the barrier is too high to overcome under the experimental condition,20 this pathway is ruled out, and further calculation along path II is abandoned.
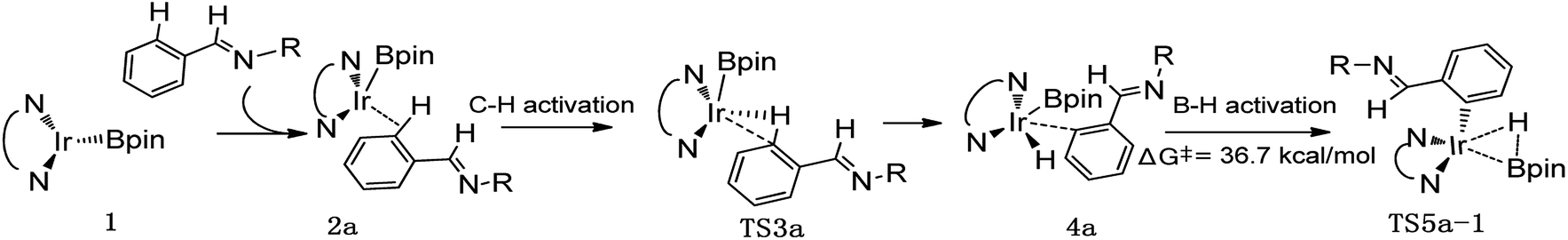 | ||
| Scheme 3 Energy change in the second step of B–H reductive elimination for path II in AQ/Ir(I)-assisted C–H borylation of aromatic imine. | ||
The transition states of C–H activation and the related intermediates in path I are located and detailed in Fig. 3. For C–H activation, C–H of the phenyl ring approaches trans to boron and the C–H bond is splitted upon interaction between H, C and Ir atoms, resembling the previous studies by Sakaki.27 According to the previous reports, boryl nucleophilicity promotes proton transfer in the transition state, thereby accelerates the subsequent C–H borylation.28 During the process of C–H activation, both the length of Ir–C and Ir–H bond become shorter, and the length of C–H bond becomes longer. Comparing the transition states of TS3a and TS3b, which are ortho- and meta-selectivity respectively, the latter is less steric hindrance. Surprisingly, the barrier of the larger hindered TS3a is lower. The lower energy of TS3a may due to the enhanced electron donating ability of AQ which accelerates the proton-transfer from ortho carbon to iridium atom.
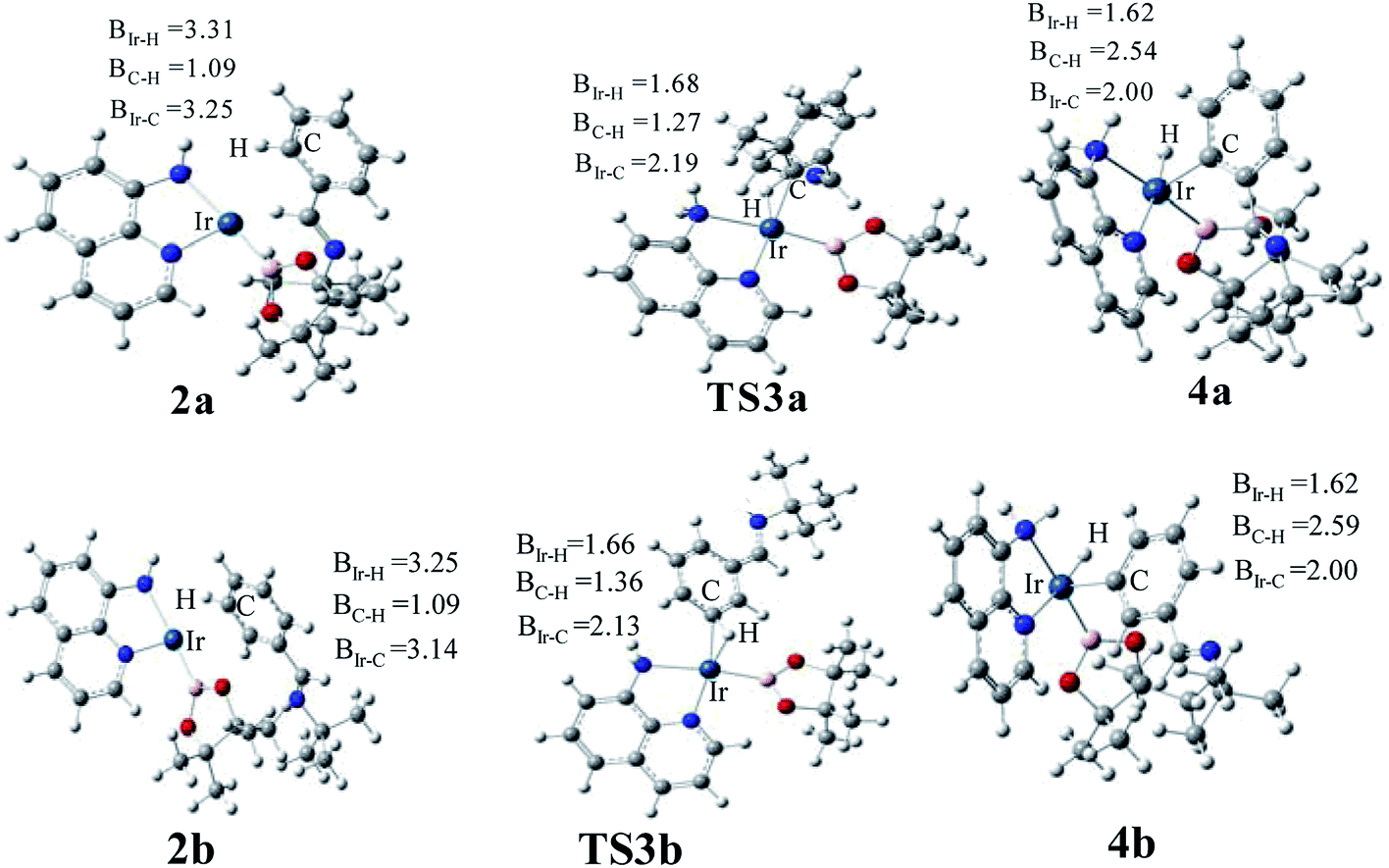 | ||
| Fig. 3 Key transition states and intermediates in Ir(I)/AQ catalyzed C–H activation in path I. | ||
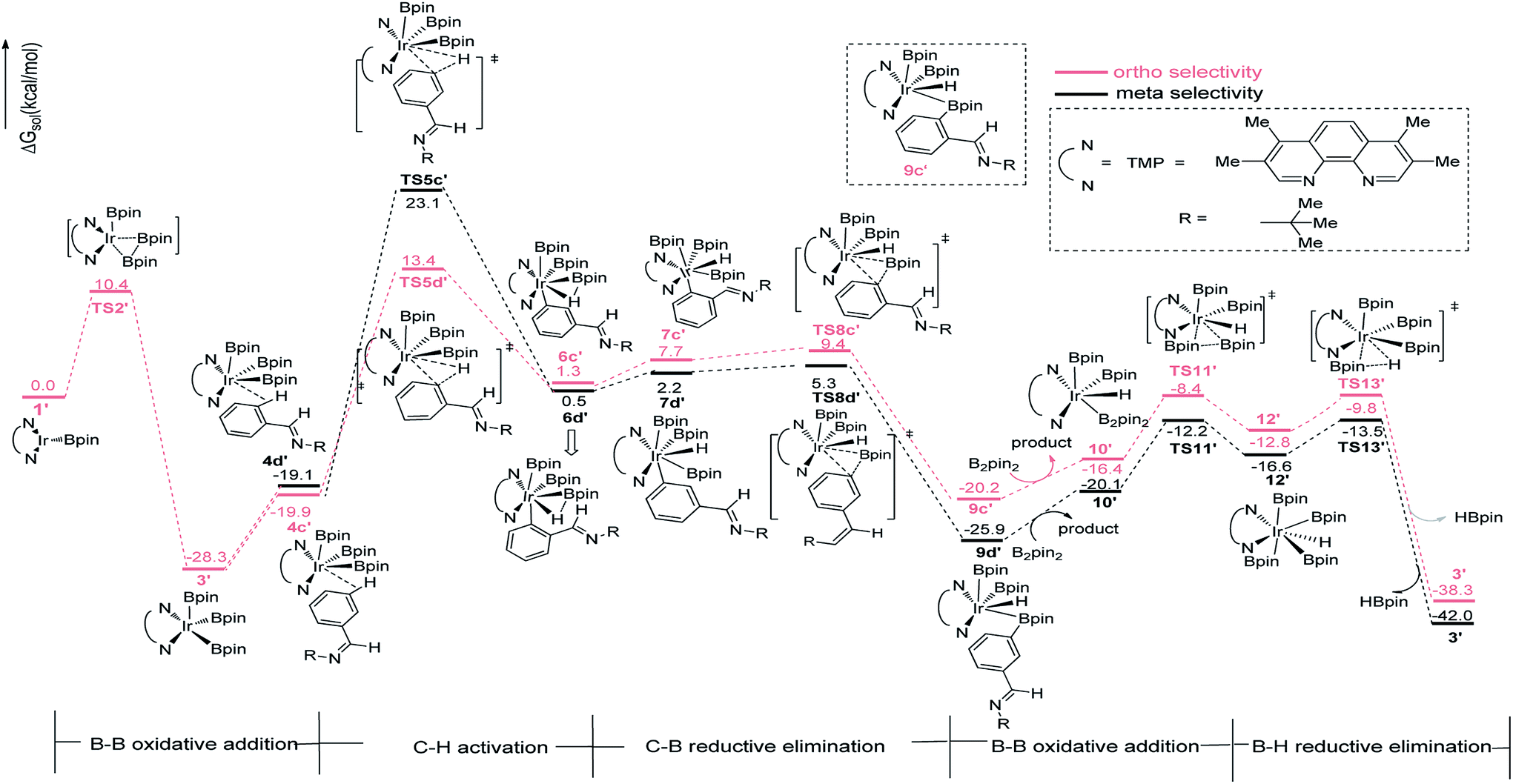
The Ir(I)–Ir(III) catalytic cycle in path I bring us to envisage whether an analogous Ir(III)–Ir(V) pathway exists (Scheme 1c). The iridium center of precatalyst 1 is possible subjected to B–B oxidative addition with B2pin2 (Fig. 4). The barrier of the initial B–B oxidative addition step is slightly higher than that of ortho C–H activation in Ir(I)/Ir(III) cycle (TS2 in Fig. 4 vs. TS3a in Fig. 2, 28.8 kcal mol−1 vs. 28.0 kcal mol−1). Consequently, the B–B oxidative addition to iridium(I) should be a little more difficult to proceed than ortho C–H activation, therefore the B–B oxidative addition may not occur. Furthermore, we have calculated along the whole Ir(III)/Ir(V) route to testify the possibility of path III. The energy required for the initial aromatic C–H activation is higher than 35.0 kcal mol−1 for both meta- and ortho-selectivity (Fig. 4), much higher than the barrier for the analogous C–H activation step in Ir(I)–Ir(III) cycle. Therefore, this route is ruled out. We reach the conclusion that with ligand AQ, path I with ortho-selectivity is favoured over meta-selectivity, in consistent with the experimental observation.20
 | ||
| Fig. 4 Energy profile of path III in the borylation of aromatic imine catalyzed by TMP/Ir(I) system. | ||
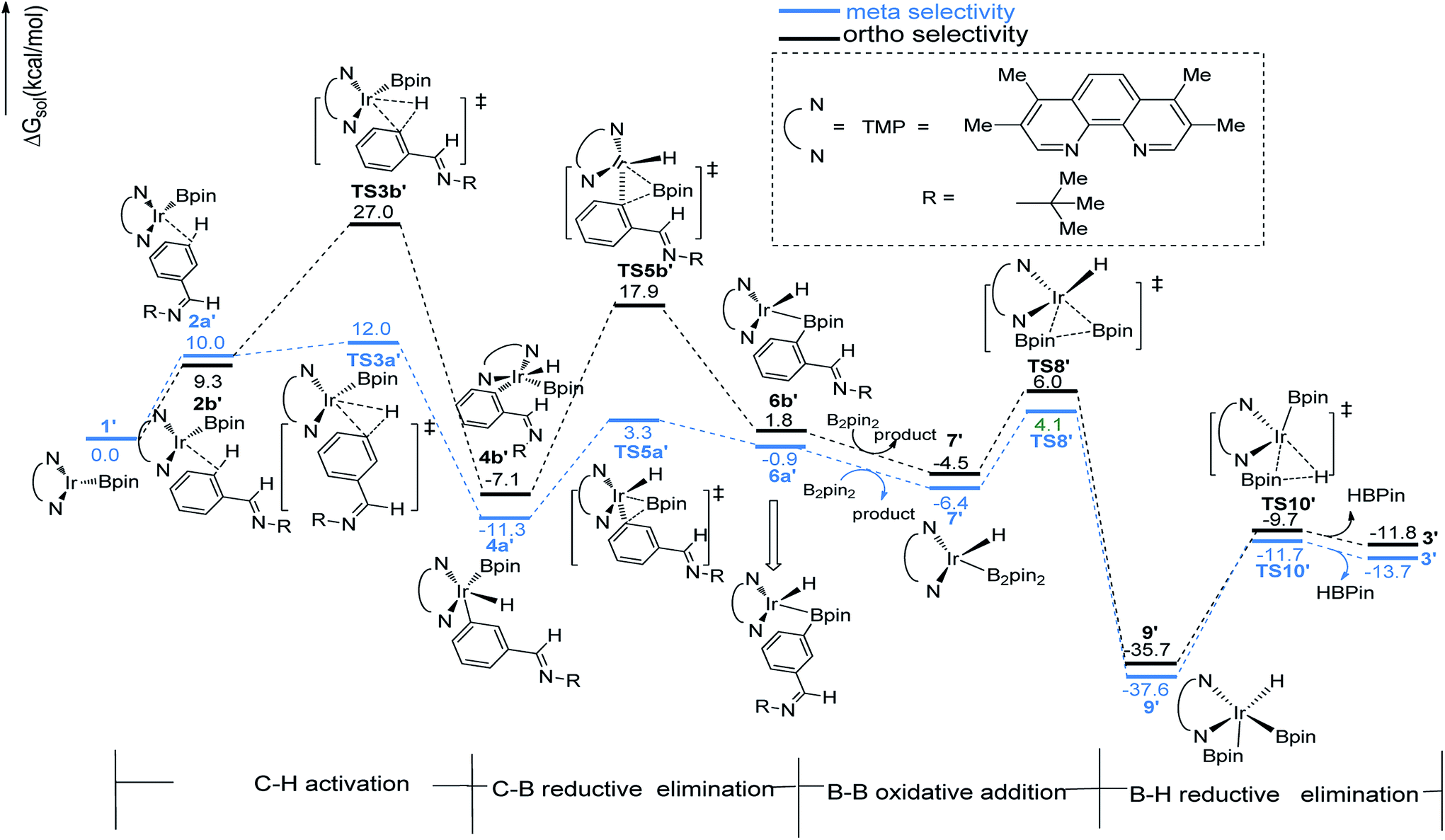
The similar pathways have been computed for the Ir(I)/TMP system. The energy profiles for the ortho-, and meta-borylation of aryl imine are shown in Fig. 5. In path I′, the total energy required for C–H activation is 12.0 kcal mol−1 and 27.0 kcal mol−1 for meta- and ortho borylation respectively (TS3a′ vs. TS3b′, Fig. 5). The followed C–B reductive elimination proceeds with a low barrier of 14.6 kcal mol−1 and 25.0 kcal mol−1 for meta- and ortho-selectivity. The subsequent B–B oxidative and B–H reductive elimination requires energy of 10.5 kcal mol−1 and 25.9 kcal mol−1 respectively, and finally regenerate catalyst 1′. Given that the C–H oxidative addition step is the regioselectivity determining step, the lower energy barrier of meta-C–H activation than ortho-C–H activation indicates that meta-path I′ is preferred (27.0 kcal mol−1 vs. 12.0 kcal mol−1).
 | ||
| Fig. 5 Energy profile of path I′ in the borylation of aromatic imine catalyzed by TMP/Ir(I) system. | ||
We then check another possibility that B–H reductive elimination is followed with the initial meta-C–H activation (Scheme 1, path II). As shown in Scheme 4, for meta-path II, the barrier of B–H reductive elimination is up to 30.2 kcal mol−1, which is not possible to occur under experimental condition.20 Therefore, path II′ is ruled out, and we have not continued to calculate this route.
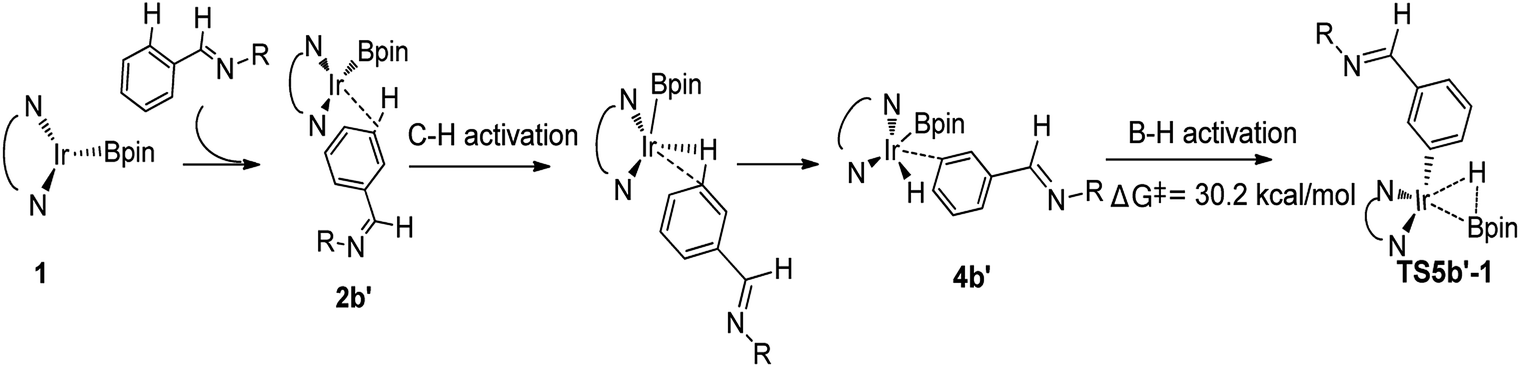 | ||
| Scheme 4 Energy change in the second step of B–H reductive elimination for path II′ in TMP/Ir(I) assisted C–H borylation of aromatic imine. | ||
In path III′, after B–B oxidative addition to Ir(I) which is the same as the first step in path III, the catalytic cycle proceeds with C–H activation, which requires energy of 42.2 kcal mol−1 and 33.3 kcal mol−1 for meta- and ortho-C–H activation. Thereafter, B–C bond reductively eliminates from intermediate 7a′ or 7b′ to produce 9a′ or 9b′ (Fig. 6). The required energy for this process is 3.1 kcal mol−1 and 1.7 kcal mol−1 for meta- and ortho-selectivity respectively. Then, B2pin2 adds to iridium center and the overall barrier for B–B oxidative addition is 8.0 kcal mol−1. B–H reductive elimination via 13TS′ leads to the regeneration of catalyst 1′, with a low barrier of 3.0 kcal mol−1. Overall, both ortho- and meta-path III′ have barriers higher than 30 kcal mol−1 for both ortho- and meta-selectivity, and it is not possible to occur under experimental conditions.20 Therefore, path III′ is also ruled out.
 | ||
| Fig. 6 Energy profile of path III′ in the borylation of aromatic imine catalyzed by TMP/Ir(III) system. | ||
The structure of transition states of C–H activation and related intermediates in path I′ are shown in Fig. 7. Structure comparison between TS3a′ and TS3b′ indicates that the lengths of the Ir–H, C–H, and Ir–C bonds in these two transition states are very similar to each other. The main difference lies in the steric repulsion between the boryl group and the substituted imine group. For TS3b′, the imine group are bent to retain stable structure, while for TS3a′, the imine substitutent on the aryl ring is unfolding, implying less steric hindrance than TS3b′. Therefore, the greater steric repulsion in transition states of orth C–H activation (TS3b′, Fig. 7) leads to higher energy barrier, making orth C–H activation more difficult than meta borylation.
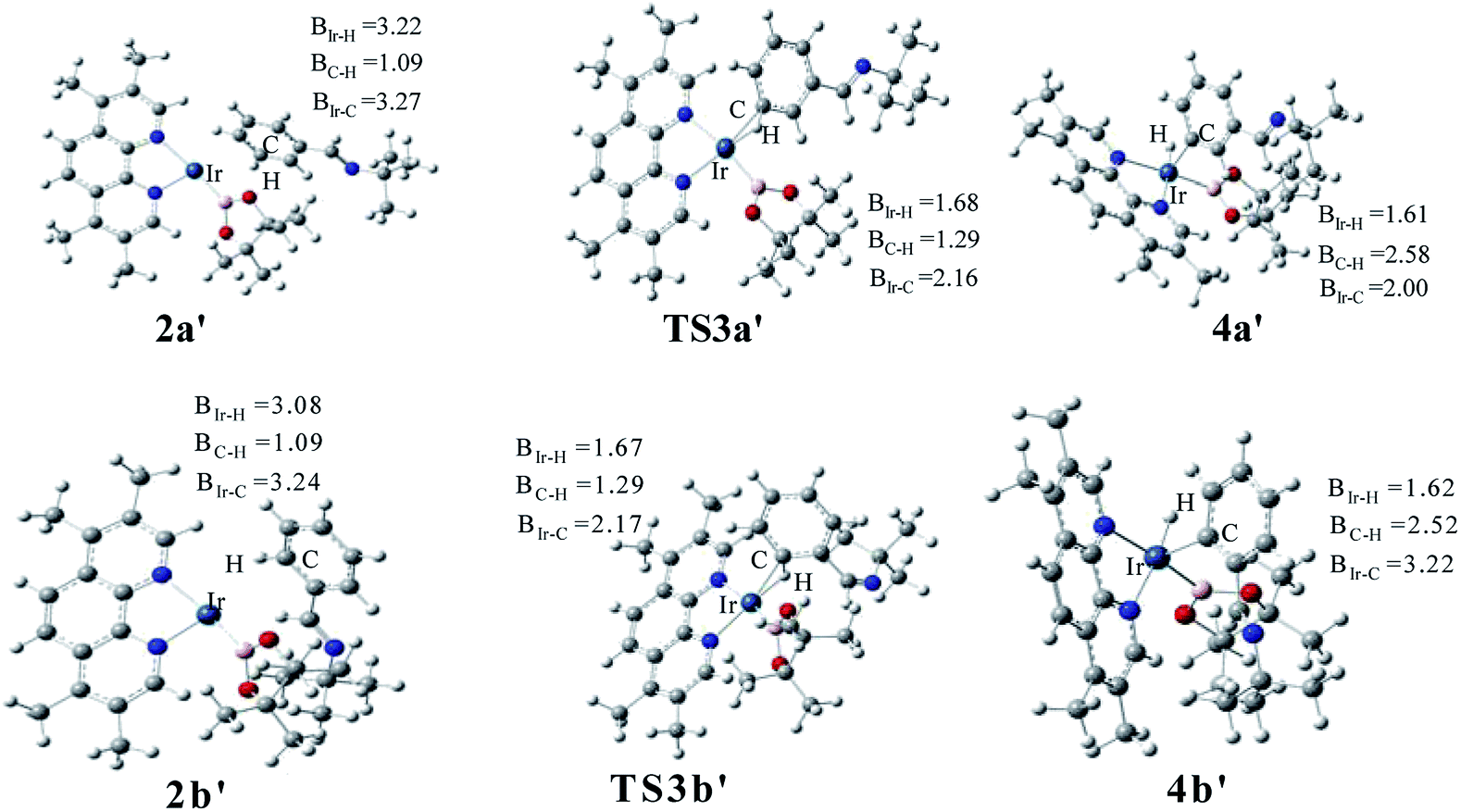 | ||
| Fig. 7 Key transition states and intermediates in Ir(I)/TMP system catalyzed C–H activation in path I′. | ||
The ligand-controlled selectivity can be rationalized by the combination of steric and electronic effects. The key factors that determine RDS step (C–H activation) are those accelerating transfer of hydrogen to Ir center. For TMP ligand, the meta product is obtained due to steric disfavor at ortho position. For AQ ligand, the decreased steric hindrance and strong electron donating ability of AQ promote ortho hydrogen transferring to iridium atom, while the electronic effects could be verified by different mulliken charge on atoms of TS3a and TS3b′ (Table 1, ESI†). Accordingly, the ortho-selectivity attributes to the decreased steric hindrance and increased electron donating ability of AQ ligand while the meta-selectivity is due to the steric of TMP. This is in good consistent with the experimental observation.20
Conclusions
In summary, we have conducted DFT calculations to investigate the origins of the ligand controlled regioselectivity in iridium-catalyzed borylation of aromatic imine. The calculations are in satisfactory agreement with the experimentally observation. The results reveal a Ir(I)–Ir(III) catalytic cycle including four steps: (1) C–H oxidative addition, (2) B–C reductive elimination, (3) B–B oxidative addition and (4) B–H reductive elimination. The C–H oxidative addition is the regioselectivity determining step of the reaction. The structures of transition states in C–H activation step are analyzed to illustrate the different regioselectivity. For AQ ligand, the ortho product is shown to be favored over the meta product because of the decreased steric hindrance and increased electron donating effect of AQ ligand which promote proton transfer in ortho C–H activation. For TMP ligand, the unfavorable steric repulsion between TMP and the ortho substituted imine group results in a higher energy barrier for ortho transition state of C–H activation, therefore meta borylation is preferred.The new mechanistic insights into the iridium catalyzed borylation of aromatic imines might be helpful to the understanding of the origin of ligand controlled regioselectivity in this kind of reaction, also provide guide for the design of new catalyst.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Natural Science Foundation of Guangdong Province (2017A030313020 and 2018A030313274) for financial support on this study. We also thank National Natural Science Foundation of China (11604059), the Priority Academic Program Development of Jiangsu Higher Education Institutions, China, Scientific Research Project of Guangzhou Municipal Colleges and Universities (1201630455) and the Guangzhou University's Training Program for Excellent New-recruited Doctors (YB201715).Notes and references
- (a) A. Ros, R. Fernandez and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229 RSC; (b) J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2 CrossRef CAS PubMed; (c) L. Xu, G. Wang, S. Zhang, H. Wang, L. Wang, L. Liu, J. Jiao and P. Li, Tetrahedron, 2017, 73, 7123 CrossRef CAS; (d) I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed; (e) D. G. Hall, Wiley-VCH: Weinheim, Germany, 2005; (f) G. R. Dick, E. M. Woerly and M. D. Burke, Angew. Chem., Int. Ed., 2012, 51, 2667 CrossRef CAS PubMed.
- C. Cheng and J. F. Hartwig, Science, 2014, 343, 853 CrossRef CAS PubMed.
- J. V. Obligacion, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 4133 CrossRef CAS PubMed.
- C. Xue, Y. Luo, H. L. Teng, Y. H. Ma, M. Nishiura and Z. M. Hou, ACS Catal., 2018, 8, 5017 CrossRef CAS.
- G. Wang, L. Liu, H. Wang, Y. Ding, J. Zhou, S. Mao and P. Li, J. Am. Chem. Soc., 2017, 139, 91 CrossRef CAS PubMed.
- B. Chattopadhyay, J. E. Dannatt, I. L. A. D. Sanctis, K. A. Gore, R. E. Maleczka, D. A. Singleton and M. R Smith, J. Am. Chem. Soc., 2017, 139, 7864 CrossRef CAS PubMed.
- (a) H. L. Li, M. Kanai and Y. Kuninobu, Org. Lett., 2017, 19, 5944 CrossRef CAS PubMed; (b) M. R. Smith, R. Bisht, C. Haldar, G. Pandey, J. E. Dannatt, B. Ghaffari, R. E. Maleczka and B. Chattopadhyay, ACS Catal., 2018, 8, 6216 CrossRef CAS PubMed; (c) C. Haldar, M. E. Hoque, R. Bisht and B. Chattopadhyay, Tetrahedron Lett., 2018, 59, 1269 CrossRef CAS; (d) M. T. Mihai, H. J. Davis, G. R. Genov and R. J. Phipps, ACS Catal., 2018, 8, 3764 CrossRef CAS.
- (a) A. Ros, B. Estepa, R. Lopez-Rodroguez, E. Alvarez, R. Fernandez and J. M. Lassaletta, Angew. Chem., Int. Ed., 2011, 50, 11724 CrossRef CAS PubMed; (b) P. C. Roosen, V. A. Kallepalli, B. Chattopadhyay, D. A. Singleton, R. E. Maleczka and M. R. Smith, J. Am. Chem. Soc., 2012, 134, 11350 CrossRef CAS PubMed; (c) R. Bisht and B. Chattopadhyay, J. Am. Chem. Soc., 2016, 138, 84 CrossRef CAS PubMed.
- (a) S. Kawamorita, H. Ohmiya, K. Hara, A. Fukuoka and M. Sawamura, J. Am. Chem. Soc., 2009, 131, 5058 CrossRef CAS PubMed; (b) T. Ishiyama, H. Isou, T. Kikuchi and N. Miyaura, Chem. Commun., 2010, 46, 159 RSC.
- M. A. Larsen, S. H. Cho and J. Hartwig, J. Am. Chem. Soc., 2016, 138, 762 CrossRef CAS PubMed.
- G. A. Chotana, M. A. Rak and M. R. Smith, J. Am. Chem. Soc., 2005, 127, 10539 CrossRef CAS PubMed.
- R. E. Maleczka, F. Shi, D. Holmes and M. R. Smith, J. Am. Chem. Soc., 2003, 125, 7792 CrossRef CAS PubMed.
- J. M. Murphy, X. Liao and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 15434 CrossRef CAS PubMed.
- H. Tamura, H. Yamazaki, H. Sato and S. Sakaki, J. Am. Chem. Soc., 2003, 125, 16114 CrossRef CAS PubMed.
- T. M. Boller, J. M. Murphy, M. Hapke, T. Ishiyama, N. Miyaura and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 14263 CrossRef CAS PubMed.
- A. G. Green, P. Liu, C. A. Merlic and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 4575 CrossRef CAS PubMed.
- J. Jover and F. Maseras, Organometallics, 2016, 35, 3221 CrossRef CAS.
- L. Zhu, X. T. Qi, Y. Z. Li, M. Duan, L. F. Zou, R. P. Bai and Y. Lan, Organometallics, 2017, 36, 2107 CrossRef CAS.
- T. M. Boller, J. M. Murphy, M. Hapke, T. Ishiyama, N. Miyaura and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 14263 CrossRef CAS PubMed.
- R. Bisht and B. Chattopadhyay, J. Am. Chem. Soc., 2016, 138, 84 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng,J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. N. akai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866 CrossRef CAS.
- (a) P. C. Hariharan and J. A. Pople, Theor. Chem. Acc., 1973, 28, 213 Search PubMed; (b) M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- R. Peverati and D. G. Truhlar, J. Phys. Chem. Lett., 2012, 3, 117 CrossRef CAS.
- H. Tamura, H. Yamazaki, H. Sato and S. Sakaki, J. Am. Chem. Soc., 2003, 125, 16114 CrossRef CAS PubMed.
- B. A. Vanchura, S. M. Preshlock, P. C. Roosen, V. A. Kallepalli, R. J. Staples, R. E. Maleczka and M. R. Smith, Chem. Commun., 2010, 46, 7724 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07886f |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |