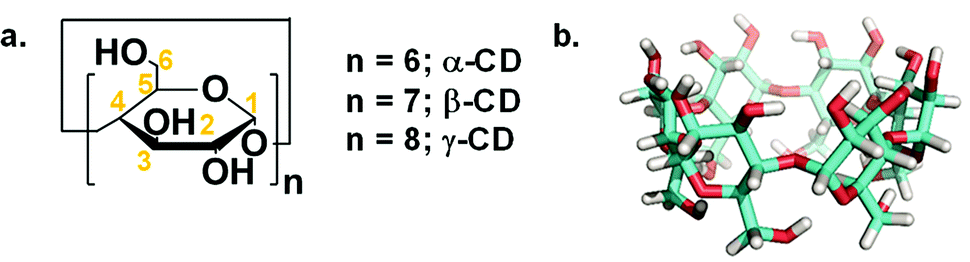DOI:
10.1039/C8RA08040B
(Paper)
RSC Adv., 2018,
8, 37757-37764
A green sorbent for CO2 capture: α-cyclodextrin-based carbonate in DMSO solution†
Received
27th September 2018
, Accepted 26th October 2018
First published on 9th November 2018
Abstract
Cyclodextrin (α-CD)/KOH pellet dissolved in DMSO was utilized to capture CO2. KOH has a dual function of enhancing the nucleophilicity of the hydroxyl groups on the α-CD rims and acting as a desiccant. 13C NMR spectroscopy provided evidence for the chemisorption of CO2 through the formation of organic carbonate (RO-CO2−·K+). This was supported by the spectral changes obtained using ex situ ATR-FTIR spectroscopy upon bubbling CO2. Activation of α-CD with NaH or bubbling with 13CO2 verified that chemisorption occurred solely via RO-CO2−·K+ rather than inorganic bicarbonate. Volumetric gas uptake demonstrated a sorption capacity of 21.3 wt% (4.84 mmol g−1). To the best of our knowledge, this is the highest chemisorption value reported to date for CD-based sorbents. DFT calculations of the Gibbs free energies indicated that the formation of RO-CO2−·K+ was more favoured at the primary carbinol rather than its secondary counterpart.
1. Introduction
Since its early discovery by Bottoms in 1930, monoethanolamine (MEA) has matured into a material that is heavily used as a wet scrubbing agent in industry.1 However, amines tend to be corrosive and require high regeneration temperatures in the range of 100 to 150 °C, which encouraged scientists to search for more benign strategies with lower regeneration temperatures, higher sorption capacity, and long-lived sorbents.2 A new finding in the literature was devoted to a simple acid–base reaction, which yielded organic ionic alkyl carbonate in a reversible fashion, i.e., switchable solvents. This concept has inspired new systems for capturing CO2 as wet scrubbing agents.3 However, the most recently utilized superbases still have corrosion limitations. They exploit volatile organic compounds, which are considered as non-eco-friendly materials and limit their utilization from an environmental point of view.4 Paul Anastas5 defined bio-renewables as one of the twelve green chemistry guidelines to provide alternatives to conventional industrial processes or academic perspectives. Implementation of green design of sorbents was reported via solid state6 and simply activation in non-aqueous solvent to overcome the high heat capacity of water.7–10 This was further enhanced with an-easy-to-manipulate, non-toxic, less concentrated, less corrosive wet sorbent such as chitosan oligosaccharide.11 Therefore, our approach extends this premise through the use of oligomers such as cyclodextrin (CD, Scheme 1a).
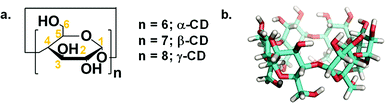 |
| | Scheme 1 (a) Chemical structure of CDs. The numbers across the glucose repeating unit designate the carbon atoms. (b) 3-D model of α-CD (white, red and cyan colours correspond to hydrogen, oxygen and carbon atoms, respectively). | |
Herein, we report on the exploitation of a bio-renewable material, namely, α-CD (Scheme 1b), dissolved in DMSO7–11 in the presence of KOH to activate the α-CD hydroxyl groups for the ultimate production of ionic organic carbonates (RO-CO2−). The affinity of the explored substrate towards CO2 was investigated using nuclear magnetic resonance (NMR) and ex situ attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy. The sorption capacity was measured volumetrically and gravimetrically using an in situ ATR-FTIR autoclave and thermogravimetric analysis (TGA), respectively, and the performance characteristics of the investigated material were measured and compared with related systems in the literature. Furthermore, the thermodynamics of the carbonation reaction was investigated using density functional theory (DFT) calculations to probe the mechanism and the reaction energetics of CO2 sorption. To the best of our knowledge, the studied sorbent exhibits the highest CO2 uptake among CD-based sorbents in terms of chemisorption.
2. Results and discussion
Following our previous work on CO2 capture using non-aqueous solvents,7–11 the utilization of DMSO as a polar aprotic solvent is anticipated to decrease the energy required for regeneration and increase the non-volatile character of the sorbent system due to its lower heat capacity and higher boiling point than water, respectively. Moreover, the absence of water eliminates the formation of inorganic bicarbonate, which is an energy-expensive species in terms of regeneration. In this context, our group reported the role of DMSO in facilitating the supramolecular chemisorption of CO28 and indicated it has no impact on biodegradability, which together with its low cytotoxicity demonstrated its potential as a green solvent.11 We hope that these efforts highlight the importance of the titled material in the field of carbon capture and sequestration. α-CD was chosen over other commercially available CDs consisting of 7 and 8 glucopyranoside units (i.e. β-and γ-CD) due to the poor solubility of the former and the high cost of the latter.
2.1 1H/13C NMR spectroscopy
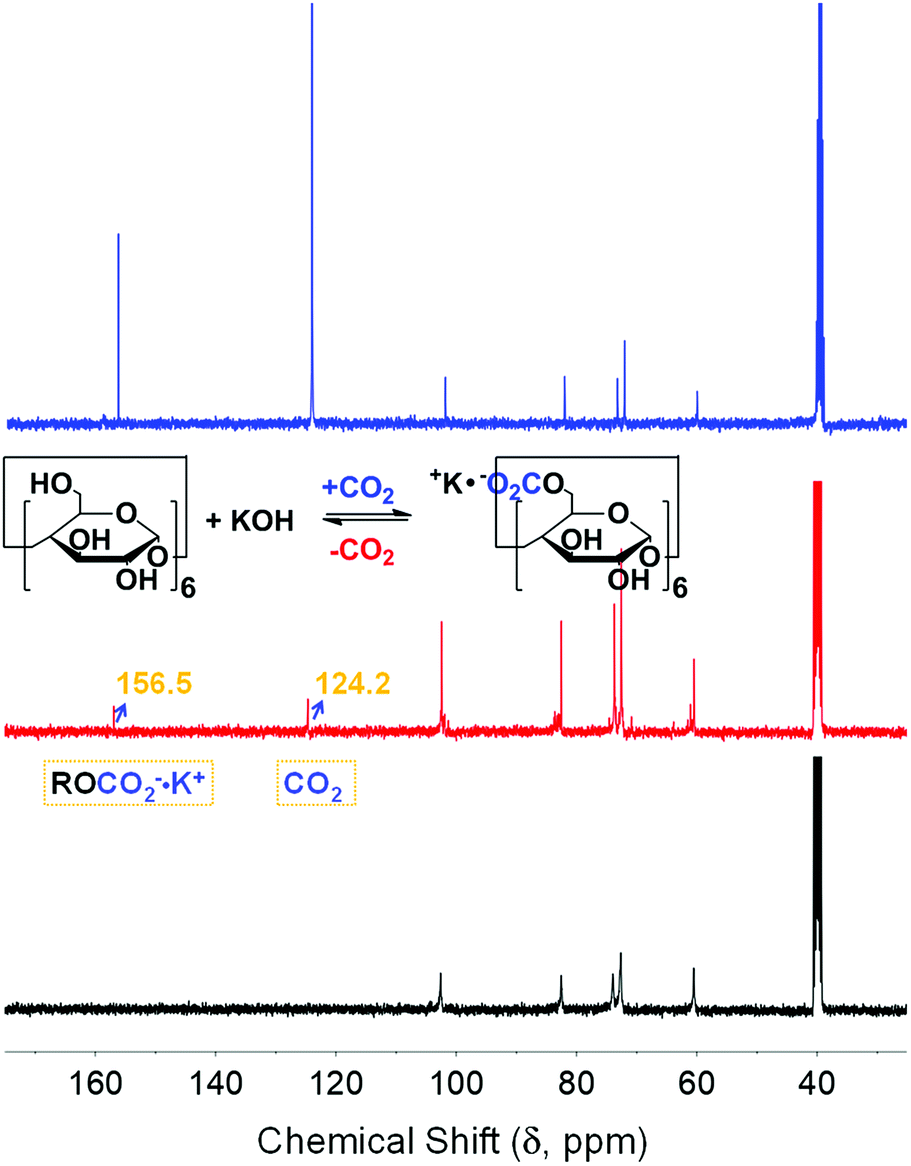
Previous studies have reported on the solvation of CDs by DMSO through hydrogen bonding between the hydrogen atom of the OH moieties in CDs and the oxygen atom of DMSO,12 which is a prerequisite for the supramolecular chemisorption of CO2.7 However, preliminary NMR experiments on the chemistry of α-CD dissolved in DMSO-d6 and CO2 showed no reaction, which indicated the necessity of triggering the nucleophilicity of the α-CD hydroxyl groups using a strong base. Consistent with previous reports on the role of counterions within the CD-based metal–organic framework (CD-MOF) to bind with CO2,13 α-CD solution was bubbled with CO2 after sonication either with KF or KOH. The use of a weakly basic anion (F−) did not result in any difference in the NMR spectrum before and after bubbling CO2. A clear indication of CO2 fixation was observed in the presence of KOH. The 1H NMR spectrum of α-CD solution showed significant changes in the presence of KOH, in which peak broadening was observed together with the disappearance of the OH protons positioned between 5.4–5.6 ppm the spectra of both α-CD and α-CD/KOH in DMSO-d6 are shown in the ESI, Fig. S1.† These results indicated the reaction of the hydroxyl groups with KOH, which led to the formation of alkoxide. Upon bubbling the solution with CO2, the chemical shift of the α-CD proton became resolved with the appearance of a new set of peaks. Furthermore, the broad peak centred at 4.35 ppm before the introduction of CO2 experienced a significant downfield shift, indicating a change in the surrounding environment (Fig. S1, ESI†).
The 13C NMR spectrum of α-CD/KOH/DMSO-d6 bubbled with CO2 (red trace, Fig. 1) showed the emergence of two new peaks at 124.2 and 156.5 ppm, which correspond to the physisorbed CO2 and the formation of organic carbonate (RO-CO2−), respectively. The carbonation of α-CD was also confirmed by the splitting pattern of the α-CD carbon peaks. To eliminate the possibility of inorganic carbonate formation, KOH/DMSO-d6 was bubbled with CO2, which resulted in the formation of a precipitate and no signal for the mother liquor was obtained by 13C NMR spectroscopy. To refute the formation of the bicarbonate species, sodium hydride was used as a base to abstract the activate CD-based OH moieties. The 13C NMR spectrum (Fig. S2, ESI†) of the solution after bubbling CO2 showed the same assigned peaks (vide supra) with no signal for bicarbonate. Moreover, a negative control experiment was conducted using permethylated α-CD (all hydroxyls are replaced with methoxy groups). The 1H and 13C NMR spectra exhibited no changes upon the addition of KOH and no response toward CO2 (Fig. S3, ESI†). Other inorganic and organic bases, namely, NaOH and DBU, were also investigated. CO2 fixation on α-CD was also observed in the presence of NaOH and DBU, as indicated by 13C NMR experiments (Fig. S4, ESI†).
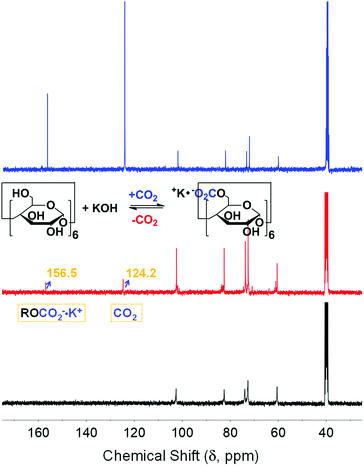 |
| | Fig. 1 13C NMR spectra of α-CD/KOH/DMSO-d6 before (black) and after bubbling CO2 (red) and its labelled counterpart, 13CO2 (blue). | |
The labelled 13CO2 experiment showed evidence for the origin/identity of the chemisorbed species upon increasing the 13C-abundance to eliminate the possibility of the formation of multiple chemisorbed ions. The relative intensities of the newly emerged chemical shifts upon bubbling were observed at ca. 125 and 157 ppm (blue trace, Fig. 1), compared to the previously recorded spectrum, where a substantial fraction of the 13C-labelled gas was involved in both sorption processes (physisorption and chemisorption, respectively). This provided clear evidence for the formation of an exclusive alkyl carbonate adduct.
2.2 Ex situ ATR-FTIR spectroscopy
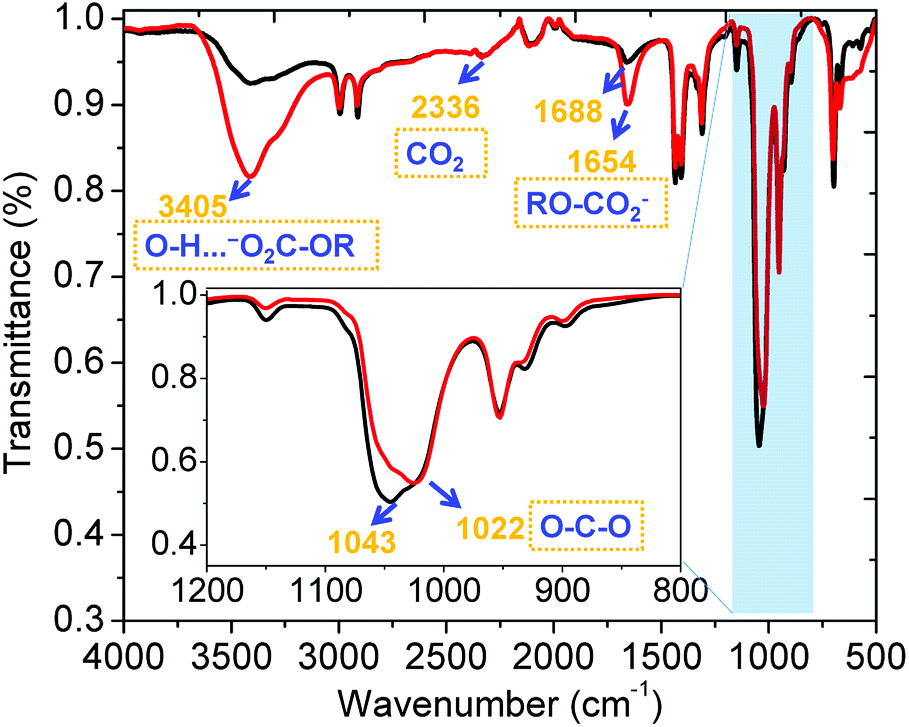
The sorption of CO2 by α-CD/KOH/DMSO was further explored using ex situ ATR-FTIR spectroscopy, as shown in Fig. 2. There are three distinct regions between the spectra before and after bubbling CO2 as follows: band A (3000–3600 cm−1): intensive intramolecular hydrogen bonding within the CD ring (red trace, Fig. 2 and 6, vide infra) was observed upon chemisorption of CO2, which can be seen by the broadening of the band centred at 3400 cm−1, (O–H⋯−O2C-OR). Band B (1500–1700 cm−1): demonstrated a prominent peak centred at 1688 cm−1, which was red-shifted to 1654 cm−1 and can be attributed to one of three unique chemistries: (i) the symmetric stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O within RO-CO2−;14 (ii) the symmetrical bending band of the high-energy, cavity water molecules15 that were released upon organic carbonate formation; and (iii) the frequency of the hydrated bicarbonate anion (HCO3−·(H2O)n),16 which was fortified and not present, as shown via 13C NMR (vide supra). Band C (950–1100 cm−1): attributed to the (C–O–C) resulting from the coordination of CO2. In summary, the peak at 1654 cm−1 is characteristic of the potassium ionic organic carbonate, which is accompanied by a peak at 1022 cm−1 (C–O–C) upon coordination (red trace, Fig. 2) and is characteristic of alkyl carbonates.
O within RO-CO2−;14 (ii) the symmetrical bending band of the high-energy, cavity water molecules15 that were released upon organic carbonate formation; and (iii) the frequency of the hydrated bicarbonate anion (HCO3−·(H2O)n),16 which was fortified and not present, as shown via 13C NMR (vide supra). Band C (950–1100 cm−1): attributed to the (C–O–C) resulting from the coordination of CO2. In summary, the peak at 1654 cm−1 is characteristic of the potassium ionic organic carbonate, which is accompanied by a peak at 1022 cm−1 (C–O–C) upon coordination (red trace, Fig. 2) and is characteristic of alkyl carbonates.
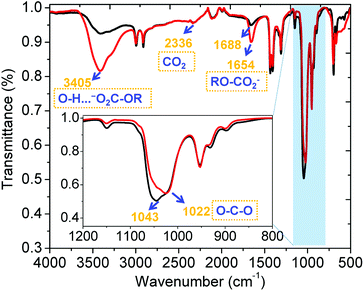 |
| | Fig. 2 Ex situ ATR-FTIR spectra of α-CD/KOH dissolved in DMSO before (black) and after (red) bubbling CO2. | |
2.3 Volumetric CO2 uptake measurements
The sorption capacity of α-CD/KOH dissolved in DMSO solution was measured volumetrically using an in situ ATR-FTIR autoclave. The measurements were carried out under dry conditions at 298 K in a 50.0 mL autoclave. The amount of CO2 adsorbed was calculated using the ideal gas equation of state. The results indicated that neat DMSO9 and DMSO/KOH solution (Table 1, entry 2) adsorbed the same amount of CO2, which designated that physisorption was the dominant CO2 sorption mechanism even in the presence of a base. A similar behavior was observed when low concentrations of α-CD (up to 1.25%, Table 1, entries 3 and 4) were employed. It appears that the extent of KOH consumption at a low molar ratio of substrate to base (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) was insufficient to induce the dissolution of a significant amount of KOH. Therefore, the critical concentration of α-CD was deduced to be 2.5% (Table 1, entry 5), where 0.6 bar CO2 was chemisorbed, which is equivalent to 21.3 wt% (4.84 mmol g−1). Doubling and quadrupling the concentration of α-CD (Table 1, entries 6 and 7) led to a lower sorption pressure, which leveled off at 0.4 bar. This may be attributed to the ion–dipole interaction between the alkoxide-bearing α-CD (ROH + KOH ⇌ ROK + H2O) and other hydroxyl groups of α-CD, which possibly blocked the reactive hotspots (RO−), thus preventing any further reaction with CO2. This hypothesis was verified by exposing a 10% α-CD solution to a two-fold pressure of CO2 (Table 1, entry 8), which resulted in the same sorption capacity. To evaluate the activation efficacy of KOH, NaH (Table 1, entry 9) was used as a base under the same conditions. The obtained volumetric CO2 uptake (3.6 wt%, equivalent to 0.8 mmol g−1) confirmed the effectiveness of the employed base.
:5) was insufficient to induce the dissolution of a significant amount of KOH. Therefore, the critical concentration of α-CD was deduced to be 2.5% (Table 1, entry 5), where 0.6 bar CO2 was chemisorbed, which is equivalent to 21.3 wt% (4.84 mmol g−1). Doubling and quadrupling the concentration of α-CD (Table 1, entries 6 and 7) led to a lower sorption pressure, which leveled off at 0.4 bar. This may be attributed to the ion–dipole interaction between the alkoxide-bearing α-CD (ROH + KOH ⇌ ROK + H2O) and other hydroxyl groups of α-CD, which possibly blocked the reactive hotspots (RO−), thus preventing any further reaction with CO2. This hypothesis was verified by exposing a 10% α-CD solution to a two-fold pressure of CO2 (Table 1, entry 8), which resulted in the same sorption capacity. To evaluate the activation efficacy of KOH, NaH (Table 1, entry 9) was used as a base under the same conditions. The obtained volumetric CO2 uptake (3.6 wt%, equivalent to 0.8 mmol g−1) confirmed the effectiveness of the employed base.
Table 1 Volumetric uptake measurements as obtained via an in situ ATR-FTIR autoclave
| Entry |
Conc. (w/v)% |
Pi (bar) |
Pressure Drop (bar)a |
Chemisorption capacity (mmol CO2 g−1 sorbent) |
| Initial and final pressures of CO2 recorded by a digital manometer coupled with the ATR-FTIR autoclave for different concentrations of α-CD dissolved in DMSO and sonicated in the presence of a KOH pellet. Control experiments were conducted for correction purposes. The pressure drop recorded for the α-CD solutions was corrected with respect to KOH/DMSO solutions at the corresponding pressure. The absorption of CO2 by neat DMSO at 4.6 bar and 298 K was 1.4 bar.9 Control sample (KOH/DMSO), no α-CD was used. NaH was used as the base. |
| 1 |
0b |
5.4 |
1.6 |
n.a. |
| 2 |
0b |
10.4 |
2.6 |
n.a. |
| 3 |
0.625 |
5.8 |
0 |
0 |
| 4 |
1.25 |
5.8 |
0 |
0 |
| 5 |
2.5 |
5.4 |
0.6 |
4.84 |
| 6 |
5 |
5.4 |
0.4 |
1.61 |
| 7 |
10 |
5.4 |
0.4 |
0.80 |
| 8 |
10 |
10.4 |
0.4 |
0.80 |
| 9 |
10c |
10.3 |
0.4 |
0.80 |
The sorption capacities of affiliated systems in the literature were measured using different techniques and sorption conditions, which may hinder a fair comparison among the sorbents investigated by our research group.17,18 However, it is useful to compare systems that have been measured in the same temperature range with a similar mechanism of action (i.e. chemisorption) to ultimately rank the investigated materials in terms of their potential to capture CO2 for real-life applications. As shown in Table 2 (entries 1–4), the sorption capacity of α-CD/KOH (21.3 wt%) is superior over other wet or dry sorbents13 that function via a similar mechanism of action (e.g. n-alcohols/DBU (RO-CO2−·DBU-H+),3 tetra-tethered, hydroxyl-functionalized pentaerythritol/DBU dissolved in DMSO,9 glucosamine-based oligomeric sorbent material11 and the corresponding β-CD/DBU binary system).19 In a similar context, the inclusion complex of β-CD-aniline has a comparable sorption efficiency of 3.0 wt% (entry 6, Table 2) through the formation of an anilinium bicarbonate (C6H5–NH3+ HCO3−) adduct.20 Besides chemical fixation, CD-based porous sorbents have been regarded as promising materials for CO2 storage via physisorption.21–24
Table 2 Different values of sorption capacities reported in the literature
| Entry |
Scrubbing agent |
Mechanism of action |
Sorption capacity (wt%) |
Reference |
| This is an average value for aliphatic n-alcohols ranging from C1 to C6. |
| 1 |
α-CD/KOH |
Chemisorption |
21.3 |
This work |
| 2 |
n-alcohol/DBU |
Chemi/physisorption |
19a |
3 |
| 3 |
Pentaerythritol/DBU |
Chemisorption |
18.5 |
9 |
| 4 |
Chitosan/NaOH |
Chemisorption |
7.04 |
11 |
| 5 |
β-CD/DBU |
Chemisorption |
7.9 |
19 |
| 6 |
CD-based microporous carbon |
Physisorption |
2.8–3.3 |
21 |
| 7 |
β-CD-aniline |
Chemisorption |
3.0 |
20 |
2.4 Gravimetric CO2 uptake measurements
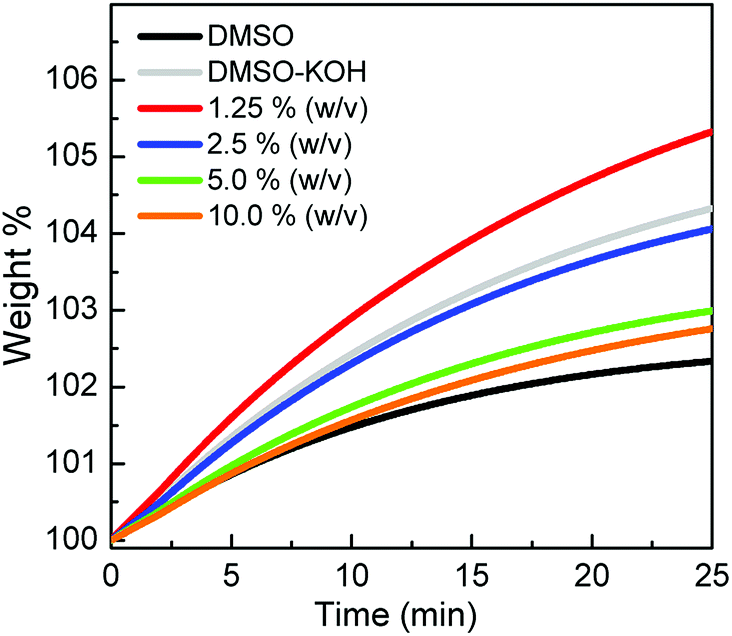
The gravimetric CO2 uptake was measured with different concentrations of α-CD solutions and compared with neat DMSO and DMSO/KOH control samples to eliminate the physisorption contribution/bicarbonate formation, respectively. The CO2 uptake capacities were evaluated relative to the original weight of the sample before bubbling (represented by 100 wt%) between zero and 25 min, as shown in Fig. 3. The CO2 sorption capacity increased upon dissolving KOH in the DMSO (4.33 wt% CO2, gray trace) compared to the neat DMSO (2.33 wt%, black trace) due to the formation of bicarbonate. Since this behaviour was not detected during the volumetric uptake measurements but was viable to a certain degree in the gravimetric CO2 uptake measurement, a rational hypothesis was made. The limited surface area of the platinum crucible exposed to CO2 and its associated mass transport issues made the KOH a strong competitor compared to the alkoxide active sites in α-CD. The use of 1.25% α-CD improved the CO2 uptake (5.33 wt% CO2), but when the α-CD concentration was increased by 2, 4 and 8 folds, the CO2 sorption capacity decreased to 4.06, 2.99 and 2.76 wt%, respectively. The increase in the CO2 sorption when 1.25% solution was used confirmed the chemisorption of CO2, as inferred from the spectroscopic data (vide supra). At a higher concentration of α-CD (>1.25%), it was anticipated that its higher surface coverage coupled with the limited surface area exposed to CO2 hindered the further diffusion of CO2 toward the solution, thus lowering its sorption capacity. The inverse relationship between the α-CD concentration and the uptake measurements well agreed with the volumetric uptake results (Table 1). For comparison, the gravimetric sorption capacity in mmol g−1 of neat DMSO, DMSO/KOH and different solution concentrations is listed in Table 3. Clearly, the volumetric uptake results were larger than the gravimetric equivalent. This disparity can be explained by the inherent characteristics associated with each technique, such as the limited area of the exposed solution within the crucible holding the α-CD/KOH/DMSO solution with no stirring. A similar discrepancy between the two sorption techniques was also reported by Cooper and co-workers.25
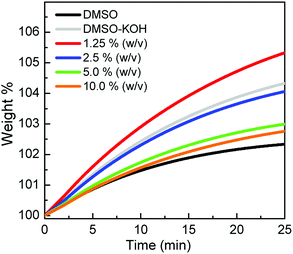 |
| | Fig. 3 Gravimetric sorption profile of CO2 versus time for different α-CD concentrations compared to the neat DMSO and DMSO-KOH solutions. | |
Table 3 Gravimetric sorption capacity of different CD solutions
| Entry |
Composition (w/v)% |
Gravimetric sorption capacity |
| wt% |
(mmol g−1 sorbent) |
| 1 |
Neat DMSO |
2.33 |
0.53 |
| 2 |
DMSO/KOH |
4.33 |
0.98 |
| 3 |
1.25 |
5.33 |
1.21 |
| 4 |
2.5 |
4.06 |
0.92 |
| 5 |
5 |
2.99 |
0.68 |
| 6 |
10 |
2.76 |
0.63 |
2.5 Recyclability and stability
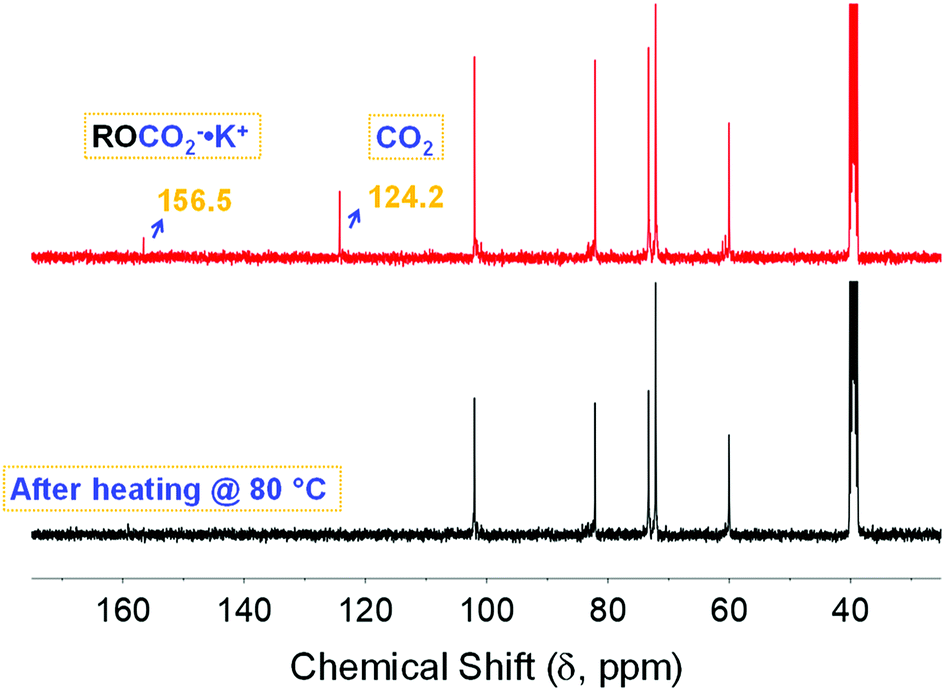
To gain insight into the nature of the sequestered species and identify factors that may enhance the performance of the investigated sorbent, CO2 stripping and the influence of water are key parameters. Additionally, understanding the associated phenomena including both reusability and stability is necessary. CO2 stripping was achieved by heating the solution after bubbling with CO2 at 80 °C for 5 min using a heat gun. The desorption was confirmed via 13C NMR spectroscopy (black trace, Fig. 4) through the disappearance of the chemical shifts associated with the physi- and chemisorbed CO2 at ca. 125 and 157 ppm, respectively. This was in good agreement with the desorption temperature of the pelletized CD-MOF-2 reported by Jeong and co-workers,26 which indicates the solvent has no significant impact on the stripping process and reflects the genuine characteristic of α-CD based carbonate species toward heating. Importantly, the restored chemical shifts after successive bubbling of the stripped solution (red trace, Fig. 4) showed that the chemisorption process was reversible, thus demonstrating the reusability of the investigated system. The heating was done as a proof of concept experiment for the reversible sorption of CO2. We believe that sorption/desorption cycles are important to measure for reusability issues. However, we did not measure several cycles because it would be more beneficial to do such measurements using in situ rather than ex situ techniques.
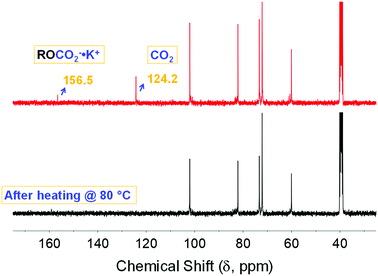 |
| | Fig. 4 13C NMR spectra of the α-CD sequestered species after heating the solution at 80 °C for 5 minutes (black), followed by bubbling with CO2 for another 30 minutes (red). | |
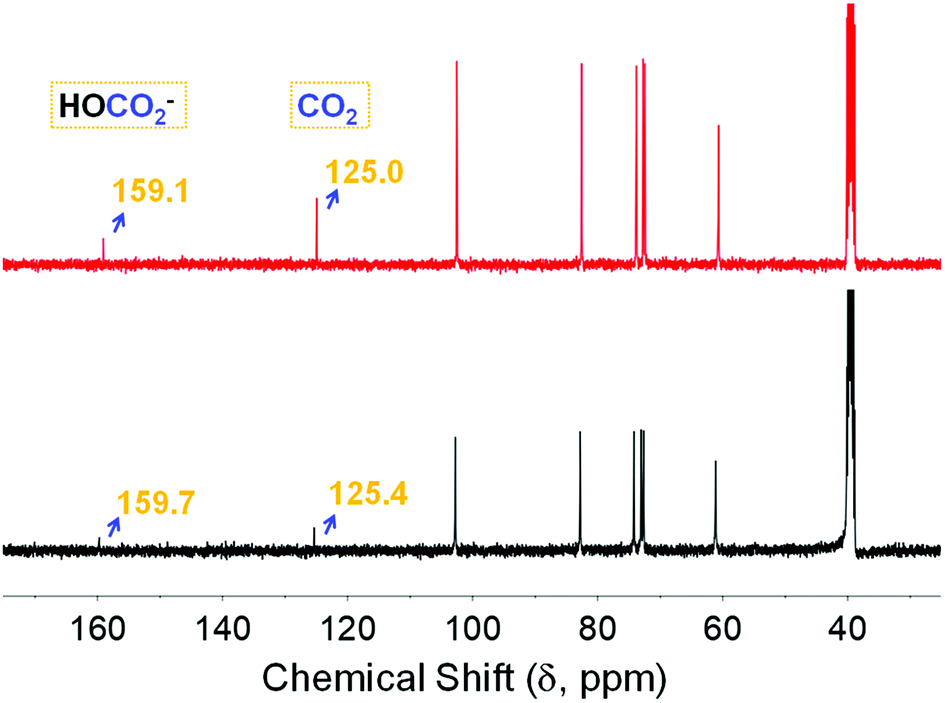
Deuterium oxide (D2O) showed a substantial influence on the stability of the ionic organic carbonate due to the evolution of a new peak at 159.7 ppm assigned to the bicarbonate anion (HCO3−), loss of a peak assigned to ROCO2− at 156.3 ppm and the persistence of the physisorbed CO2 chemical shift (black, Fig. 5). Further bubbling of CO2 did not have any influence on the former spectrum with no recovery of the carbonate adduct despite the presence of CO2 dissolved in DMSO (red trace, Fig. 5). Notably, Forgan et al.27 reported on interstitial water molecules incorporated into CD-MOFs after their synthesis, while all sorption/desorption measurements were carried out after drying the explored CD motifs in vacuo. This may point to presumed competition between water and CO2, which is yet to be explored. It is noteworthy that there are several recent reports on enhancing the stability of CD-MOFs in humid conditions for exploitation in drug delivery applications through the incorporation of fullerene28 and grafting cholesterol.29
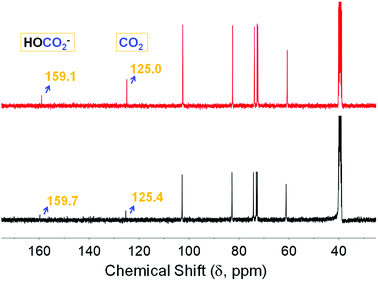 |
| | Fig. 5 13C NMR spectra of the α-CD sequestered species after adding 0.2 mL of D2O to 0.5 mL of α-CD solution (black), followed by bubbling with CO2 for 20 minutes (red). | |
2.6 Density functional theory (DFT) calculations
To gain more insight into the activation mechanism of the hydroxyl-bearing carbons at C-2 and C-6 in the corresponding oligosaccharide, DFT calculations were used to ultimately understand the carbonation reaction of α-CD. As shown in Table S1 (ESI†) and correlated to theory, the pKa value of C-2 is lower than that on C-6 (23.2 vs. 29.2), therefore, it has more acidic character. This is further confirmed via the proton affinity (PA) values of the corresponding alkoxide (i.e. 332.3 vs. 349.6 kcal mol−1). This means the RO− associated with C-2 will have lower basicity and larger nucleophilicity toward carbon dioxide (vide infra).
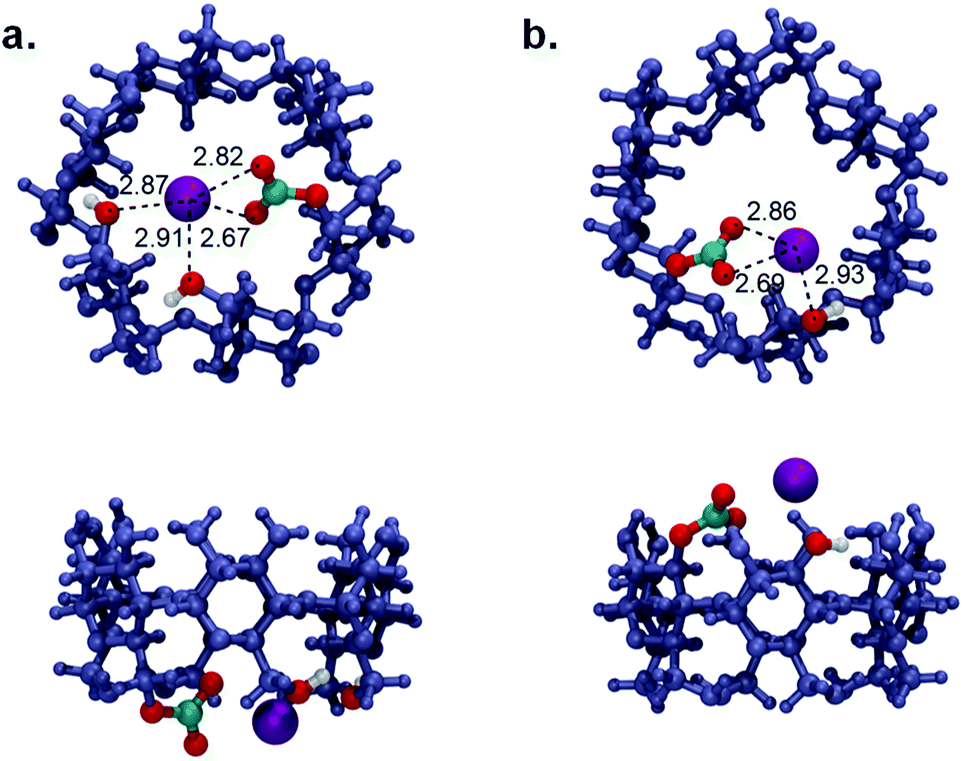
To understand the thermodynamics of this process, the energetics associated with two principal steps for the formation of the carbonate species, Step 1: ROH + KOH ⇌ ROK + H2O and Step 2: ROK + CO2 ⇌ RO-CO2−·K+ were calculated, as shown in Table 4. The results indicated that the enthalpic contribution (ΔH) has a more noticeable impact in comparison with the entropic factor (TΔS) to the overall process. This emphasizes the importance of the electronic factor in directing the CO2 capturing process. Regarding Step 1, the more negative Gibbs free energy (ΔG) values confirmed the more favourable formation of the alkoxide anion in the case of the C-2 carbinol, which is in good agreement with the pKa and PA values. However, the strong enthalpic driving force accompanying Step 2 (1.5 times higher in the case of C-6) strongly supports the formation of RO-CO2−·K+ at C-6 over C-2. This can be understood by taking a closer look at the optimized chemical structures of the ionic alkyl carbonate at the two-potential sites, as shown in Fig. 6. Although the ion pair interaction distances between K+ and RO-CO2− are very close in the two proposed structures, it seems the free movement of the ionic carbonate group at C-6 provides the proximity necessary to establish an ion–dipole interaction between K+ and the adjacent hydroxyl groups along the rim of the CD moiety (Fig. 6a), which is not satisfied when carbonating the C-2 carbinol. It seems this non-bonding interaction is a key factor in stabilizing the CO2 adduct and justifies the favourable energetics of the carbonation reaction at the C-6 position. The possibility of the formation host-guest complexation, that is, the encapsulation of CO2 inside the cavity of α-CD, was also considered. The calculations indicated an unfavorable binding free energy for the entrapped CO2 molecule in the hydrophobic cavity of CD.
Table 4 Thermodynamic data (in kcal mol−1) for the reaction of α-CD with KOH and CO2
| |
C-2 |
C-6 |
| Step 1a |
Step 2b |
Step 1a |
Step 2b |
| ROH + KOH ⇌ ROK + H2O. ROK + CO2 ⇌ RO-CO2−·K+; ΔG = ΔH − TΔS. |
| ΔH |
−29.7 |
−15.6 |
−27.3 |
−23.4 |
| −TΔS |
1.2 |
12.9 |
1.6 |
11.8 |
| ΔG |
−28.5 |
−2.7 |
−25.7 |
−11.6 |
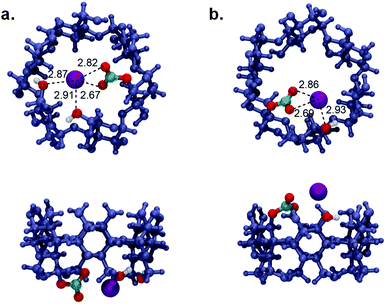 |
| | Fig. 6 Top and side views of the optimized structures of the ionic alkyl carbonate at α-CD: (a) Reaction at the primary C-6 position. (b) Reaction at the secondary C-2 position. | |
3. Conclusions
In summary, we reported a highly efficient, wet scrubbing agent for CO2 capture using α-CD/KOH dissolved in DMSO. Upon activation of the hydroxyl groups on the CD rims with KOH, an alkyl organic carbonate adduct is formed, which was confirmed by a combination of spectroscopic techniques including 13C NMR and ex situ ATR-FTIR spectroscopy. The carbonation reaction showed reasonable reversibility and recyclability upon heating and successive bubbling of CO2. Moreover, 13C NMR measurements indicated the instability of the ionic adduct in the presence of D2O. In addition, the volumetric sorption capacity via chemisorption of the solution system was almost five times greater than that reported for the corresponding CD based MOFs (4.48 vs. 0.91 mmol g−1, respectively), which supported our hypothesis for the loss of active sites upon anchoring hydroxyls to metals within MOFs. Furthermore, the mechanism of action toward CO2 was investigated using DFT calculations, considering all the possible interactions between CO2 and α-CD either through the formation of alkyl organic carbonate at C-2 and C-6 or the entrapment of CO2 inside the hydrophobic cavity of α-CD. The former reaction was found to be more favourable, especially at the primary carbinol, while the formation of a host–guest complex was less favoured, as anticipated from the hydrophobic character of the CD cavity.
4. Experimental
4.1 Chemicals
α-Cyclodextrin (α-CD) was purchased from Cyclolab, Hungary (for spectroscopic measurements) and Sigma Aldrich (for uptake measurements). For experimental manipulations, CD was dried overnight at 40 °C. CO2 (99.95%, Food Grade) for NMR and ex situ IR measurements was purchased from Advanced Technical Gases Co., Jordan, while CO2 (grade 4.6) for in situ IR measurements and volumetric uptake was procured from Westfalen, Germany. Potassium hydroxide (KOH, ACS grade) and potassium fluoride (KF, extra pure) were obtained from BDH Chemicals and Scharlau, respectively. Sodium hydride (NaH, 60% in mineral oil), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), DMSO-d6 (99.5 atom% D), D2O (99 atom% D), and 13CO2 were purchased from Sigma-Aldrich.
4.2 Instruments
Solution 1H and 13C nuclear magnetic resonance (NMR) spectra were collected at ambient temperature using an AVANCE-III 400 MHz FT-NMR NanoBay Spectrometer Bruker, Switzerland (1H: 400.13 MHz and 13C: 100.61 MHz). Ex situ attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectra were recorded on a Bruker Vertex 70-FT-IR spectrometer at room temperature coupled with a Vertex Pt-ATR accessory. In situ ATR-FTIR measurements were carried out using an MMIR45 m RB04-50 (Mettler-Toledo, Switzerland) with an MCT detector with a diamond-window probe connected via a pressure vessel. Sampling was done from 3500 to 650 cm−1 at 16 wavenumber resolution; scan option: 64 and gain: 1×. For the reusability studies, a digital heat gun was used. Thermogravimetric analysis (TGA) was conducted using a TA Q500 with the sample held in a platinum pan under N2 gas.
4.3 Computational method
Calculations were performed using Gaussian 09.30 Geometry optimizations and frequency calculations were carried out using the B3LYP density functional with the 6-31G* basis set. Minima were characterized by the absence of imaginary frequencies. A polarizable continuum model (PCM) was used for implicit solvent calculations. The pKa values and proton affinities were calculated according to previous reports.31
4.4 Solution preparation
A 30.0 mg solution of oven dried α-CD (30.8 μmol) was dissolved in 500 μL DMSO-d6 in a 10 mL glass vial. Upon dissolution, a KOH pellet (ca. 1000 mg, 17.8 mmol) was introduced, then the solution was sonicated for 20 min to ensure alkoxide formation. KOH has limited solubility in DMSO (0.13 g L−1), therefore the rest of the KOH pellet was removed after sonication before conducting the bubbling experiments. For the proof of concept experiment, NaH was used instead of KOH as follows: 60.0 mg of α-CD (61.7 μmol) was dissolved in 1.0 mL of DMSO, then 0.024 g of NaH (1000 μmol, washed with three 20 mL-portions of hexane prior to its addition to the CD solution) was added.
4.5 CO2 bubbling and recycling experiments
A decanted clear solution was bubbled with CO2 for 20 minutes. The NMR spectrum of the entire solution was measured before and after bubbling CO2. Regarding sorption quantification via both volumetric and gravimetric uptake, see Tables 1 and 3, respectively. The recyclability of the α-CD-CO2 solutions was measured by heating the solution after bubbling at 80 °C for 5 min using a digital heat gun. Further bubbling of CO2 was performed to check the restoration of the sequestered species in solution using 13C NMR.
4.6 Stability studies
The stability of the ionic organic carbonate was examined by adding 200 μL D2O to 500 μL of the original solution. For recycling purposes, CO2 was reintroduced into the solution to measure the decomposition of the carbonate-terminated oligosaccharide (vide supra) and followed by 13C NMR.
4.7 ATR-FTIR measurements
DMSO was used for the ATR-FTIR measurements. For the in situ experiments, α-CD solutions were prepared according to the previously described method with different amounts of α-CD (0.625, 1.25, 2.5, 5, and 10% (w/v)). A 10 mL-aliquot of each solution was introduced to the autoclave and after stirring the solution for 3 min, CO2 (5 or 10 bar) was purged for 30 min into the autoclave upon reaching a constant pressure drop.
4.8 Gravimetric CO2 uptake experiments
For the TGA measurements, a saturated solution of DMSO/KOH was prepared by sonicating 149 mg KOH (2.66 mmol) in 15 mL DMSO for 20 min. The clear supernatant was decanted in a clean vial and the obtained DMSO/KOH solution was kept as a stock solution for the preparation of the wet scrubbing agents. Varying masses of α-CD (1.25, 2.5, 5.0 and 10.0 mg) were dissolved separately in DMSO-KOH (10 mL) to obtain 1.25, 2.5, 5.0 and 10.0% (w/v), respectively. The gravimetric CO2 uptake experiments of the liquid samples were performed using a thermal gravimetric analyser. Initially, the setup was conditioned for the measurements by purging with N2 at 15 mL min−1. The liquid sample was loaded into a platinum pan and exposed to CO2 at a flow rate of 15 mL min−1 at 25 °C. The change in the sample weight due to CO2 sorption was monitored as a function of time. After the measurement was completed, the furnace was opened to ensure the removal of any residual CO2.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
AFE acknowledges the Deanship of Scientific Research at the Hashemite University (HU) for the financial support (Grant number: 23/2018). The authors are grateful to Prof. Omar M. Yaghi and Mr Kyle E. Cordova (Berkeley Global Science Institute, UC Berkeley) for their support and fruitful advice. Mrs Hiba S. Alshamaly (HU) is acknowledged for the routine sample preparations and practicing examination for a few spectral data guided by mentorship and pre-assigned laboratory protocols during her probation period in our research group.
References
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- B. Dutcher, M. Fan and A. G. Russell, ACS Appl. Mater. Interfaces, 2015, 7, 2137–2148 CrossRef CAS PubMed.
- D. J. Heldebrant, C. R. Yonker, P. G. Jessop and L. Phan, Energy Environ. Sci., 2008, 1, 487–493 RSC.
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- R. A. Smaldone, R. S. Forgan, H. Furukawa, J. J. Gassensmith, A. M. Z. Slawin, O. M. Yaghi and J. F. Stoddart, Angew. Chem., Int. Ed., 2010, 49, 8630–8634 CrossRef CAS PubMed.
- A. F. Eftaiha, F. Alsoubani, K. I. Assaf, C. Troll, B. Rieger, A. H. Khaled and A. K. Qaroush, Carbohydr. Polym., 2016, 152, 163–169 CrossRef CAS PubMed.
- A. F. Eftaiha, F. Alsoubani, K. I. Assaf, W. M. Nau, C. Troll and A. K. Qaroush, RSC Adv., 2016, 6, 22090–22093 RSC.
- A. K. Qaroush, K. I. Assaf, A. Al-Khateeb, F. Alsoubani, E. Nabih, C. Troll, B. Rieger and A. F. Eftaiha, Energy Fuels, 2017, 31, 8407–8414 CrossRef CAS.
- A. F. Eftaiha, A. K. Qaroush, K. I. Assaf, F. Alsoubani, T. Markus Pehl, C. Troll and M. I. El-Barghouthi, New J. Chem., 2017, 41, 11941–11947 RSC.
- A. K. Qaroush, K. I. Assaf, S. K. Bardaweel, A. Al-Khateeb, F. Alsoubani, E. Al-Ramahi, M. Masri, T. Bruck, C. Troll, B. Rieger and A. F. Eftaiha, Green Chem., 2017, 19, 4305–4314 RSC.
- T. Shikata, R. Takahashi, T. Onji, Y. Satokawa and A. Harada, J. Phys. Chem. B, 2006, 110, 18112–18114 CrossRef CAS PubMed.
- J. J. Gassensmith, H. Furukawa, R. A. Smaldone, R. S. Forgan, Y. Y. Botros, O. M. Yaghi and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 15312–15315 CrossRef CAS PubMed.
- K. J. Hartlieb, A. W. Peters, T. C. Wang, P. Deria, O. K. Farha, J. T. Hupp and J. F. Stoddart, Chem. Commun., 2017, 53, 7561–7564 RSC.
- P. J. Salústio, G. Feio, J. L. Figueirinhas, J. F. Pinto and H. M. Cabral Marques, Eur. J. Pharm. Biopharm., 2009, 71, 377–386 CrossRef PubMed.
- E. Garand, T. Wende, D. J. Goebbert, R. Bergmann, G. Meijer, D. M. Neumark and K. R. Asmis, J. Am. Chem. Soc., 2010, 132, 849–856 CrossRef CAS PubMed.
- A. K. Qaroush, H. S. Alshamaly, S. S. Alazzeh, R. H. Abeskhron, K. I. Assaf and A. F. Eftaiha, Chem. Sci., 2018, 9, 1088–1100 RSC.
- A. K. Qaroush, D. A. Castillo-Molina, C. Troll, M. A. Abu-Daabes, H. M. Alsyouri, A. S. Abu-Surrah and B. Rieger, ChemSusChem, 2015, 8, 1618–1626 CrossRef CAS PubMed.
- G. V. S. M. Carrera, N. Jordao, L. C. Branco and M. Nunes da Ponte, Faraday Discuss., 2015, 183, 429–444 RSC.
- D. L. Sivadas, K. P. Vijayalakshmi, R. Rajeev, K. Prabhakaran and K. N. Ninan, RSC Adv., 2013, 3, 24041–24045 RSC.
- Y.-C. Zhao, L. Zhao, L.-J. Mao and B.-H. Han, J. Mater. Chem. A, 2013, 1, 9456–9461 RSC.
- B. Meng, H. Li, S. Mahurin, H. Liu and S. Dai, RSC Adv., 2016, 6, 110307–110311 RSC.
- T. Guo, A. H. Bedane, Y. Pan, H. Xiao and M. Eić, Mater. Lett., 2017, 189, 114–117 CrossRef CAS.
- T. Guo, A. H. Bedane, Y. Pan, B. Shirani, H. Xiao and M. Eić, Energy Fuels, 2017, 31, 4186–4192 CrossRef CAS.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973 CrossRef CAS PubMed.
- J. J. Gassensmith, J. Y. Kim, J. M. Holcroft, O. K. Farha, J. F. Stoddart, J. T. Hupp and N. C. Jeong, J. Am. Chem. Soc., 2014, 136, 8277–8282 CrossRef CAS PubMed.
- R. S. Forgan, R. A. Smaldone, J. J. Gassensmith, H. Furukawa, D. B. Cordes, Q. Li, C. E. Wilmer, Y. Y. Botros, R. Q. Snurr, A. M. Z. Slawin and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 406–417 CrossRef CAS PubMed.
- H. Li, M. R. Hill, R. Huang, C. Doblin, S. Lim, A. J. Hill, R. Babarao and P. Falcaro, Chem. Commun., 2016, 52, 5973–5976 RSC.
- V. Singh, T. Guo, H. Xu, L. Wu, J. Gu, C. Wu, R. Gref and J. Zhang, Chem. Commun., 2017, 53, 9246–9249 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J.
E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- K. I. Assaf, A. K. Qaroush and A. F. Eftaiha, Phys. Chem. Chem. Phys., 2017, 19, 15403–15411 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08040b |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Abdussalam K. Qaroush
*a,
Abdussalam K. Qaroush