
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, and bioactivity evaluation of antitumor sorafenib analogues
Shiyang Zhou ab and
Guangying Chen*ab
ab and
Guangying Chen*ab
aCollege of Chemistry and Chemical Engineering, Hainan Normal University, Haikou 571158, China. E-mail: chgying123@163.com
bKey Laboratory of Tropical Medicinal Plant Chemistry of Ministry of Education, Hainan Normal University, Haikou 571158, China
First published on 8th November 2018
Abstract
Malignant tumors are a serious threat to human health and are generally treated with chemical therapy. This chemical therapy uses agents that act on signal transduction pathway mechanism of tumor with good selectivity and low toxicity. Sorafenib is a multikinase target inhibitor with good tumor inhibitory activity and a protein kinase inhibitor. In this research, a novel series of sorafenib analogues and derivatives were designed, synthesized, and evaluated as tumor inhibitors. These compounds used sorafenib as the lead compound and achieved modifications using bioisosteres and the alkyl principle. The in vitro the results showed that compounds 3c, 3d, 3h, 3n, 3r, and 3z had good inhibitory effects on human cervical cancer cells (Hela), while compounds 3t and 3v had good inhibitory effects on human lung cancer cells (H1975 and A549). Among these, compound 3d had an inhibitory activity (IC50) of 0.56 ± 0.04 μmol L−1 against Hela cells (human cervical cancer), the compound 3t had an IC50 of 2.34 ± 0.07 μmol L−1 against H1975 cells (human lung cancer), and compound 3v had an IC50 of 1.35 ± 0.03 μmol L−1 against A549 cells (human lung cancer). The in vivo results showed that these compounds had good antitumor effects and low acute toxicity.
1. Introduction
A malignant tumor is a common disease that seriously threatens human health. The number of deaths caused by malignant tumors is second only to cerebrovascular disease among all diseases.1–3 General treatment methods for tumors include surgical, radiation, chemical (drug therapy), and biological treatments. However, chemotherapy and surgical treatment remain the most common treatment methods.4–7 Antitumor drugs have advanced considerably since the discovery of mechlorethamine in the 1940s, which was used to treat malignant tumors.8–13 In the last twenty years, the development of molecular biology and cell biology have further improved understanding of tumor biological mechanisms, and research into antitumor drugs has provided new targets.14–18 The batch production of new chemical structures or drugs with unique mechanisms of action for clinical tumor treatment has provided highly efficient and low-toxicity drugs.19–21Antitumor drugs currently used in clinical practice can be divided into four categories according to their mechanism of action: (i) agents that directly act on DNA; (ii) agents that interfere with DNA synthesis; (iii) antimitotic agents; and (iv) agents that affect the signal transduction pathway mechanism of tumors.22–24 Among these antitumor drugs, types (i)–(iii) all influence DNA synthesis or cell mitosis.25 Therefore, these antitumor drugs have strong effects, but lack selectivity and have significant toxic effects. More selective, efficient, and less toxic antitumor drugs that interfere with or directly act on specific biological processes in tumor cells have long been sought.26–28 With the development of life science, biological mechanisms related to the occurrence and development of tumors have gradually been elucidated.29 As a result, research into antitumor drugs has begun to move towards targeted rational drug design, with some new high-selectivity drugs produced.30
The reversible phosphorylation of protein amino acid side chains is an important mechanism for the regulation of enzyme and signal protein activity.31 Protein kinase and protein phosphatase are involved in reversible phosphorylation and play key roles in regulating metabolism, gene expression, cell growth, cell division, and cell differentiation. Protein kinase is a phosphotransferase that catalyzes the transfer of phosphate groups from ATP to protein receptor amino acids.32,33 Tyrosine kinase is the most important protein kinase, while protein tyrosine kinase is a type of protein that shows tyrosine kinase activity, including receptor-type and nonreceptor-type proteins.34 Receptor-type proteins include epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PGFR), insulin receptor (InsR), and fibroblast growth factor receptor (FGFR). Nonreceptor-type proteins include Src, Abl, Jak, Csk, Fak, and Fes. Protein tyrosine kinase dysfunction can cause many diseases.35–37 Some data have shown that over 50% of protooncogenes and oncogene products have protein tyrosine kinase activity, and that their abnormal expression leads to cell proliferation and regulation disorders, which cause tumors. Furthermore, abnormal expression of tyrosine kinase is closely related to tumor invasion and metastasis, tumor angiogenesis, and tumor chemotherapy resistance.38–41 In recent years, protein tyrosine kinase has become the target of drug action. The design of protein kinase inhibitors that can interfere with the cell signal transduction pathway has been used in the search for disease drug treatments.42–45
Agents that act on the signal transduction pathway mechanism in tumors include protein kinase inhibitors and proteasome inhibitors. Protein kinase inhibitors can be divided into Bcr-Abl protein kinase inhibitors, epidermal growth factor receptor tyrosine kinase inhibitors, and multiple kinase target inhibitors.46–50 Currently, protein kinase inhibitors commonly used in clinical practice include imatinib, dasatinib, gefitinib, erlotinib, sorafenib, and sunitinib (Fig. 1). These drugs all contain aniline structures. Herein, using sorafenib as the lead compound, sorafenib analogues and derivatives with core aniline structures were designed (Fig. 2) and synthesized. The results of in vitro and in vivo experiments showed that these compounds had good antitumor activities. In the target molecule design process, bioisosteres and the alkyl principle were used to prepare different compounds. The target compounds were synthesized from 2-aminobenzoic acid (1a) or 6-methyl-2-aminobenzoic acid (1b) by amination and condensation (N-alkylation). The synthetic route involved simple operations and mild reaction conditions, and afforded high total yields (Scheme 1).
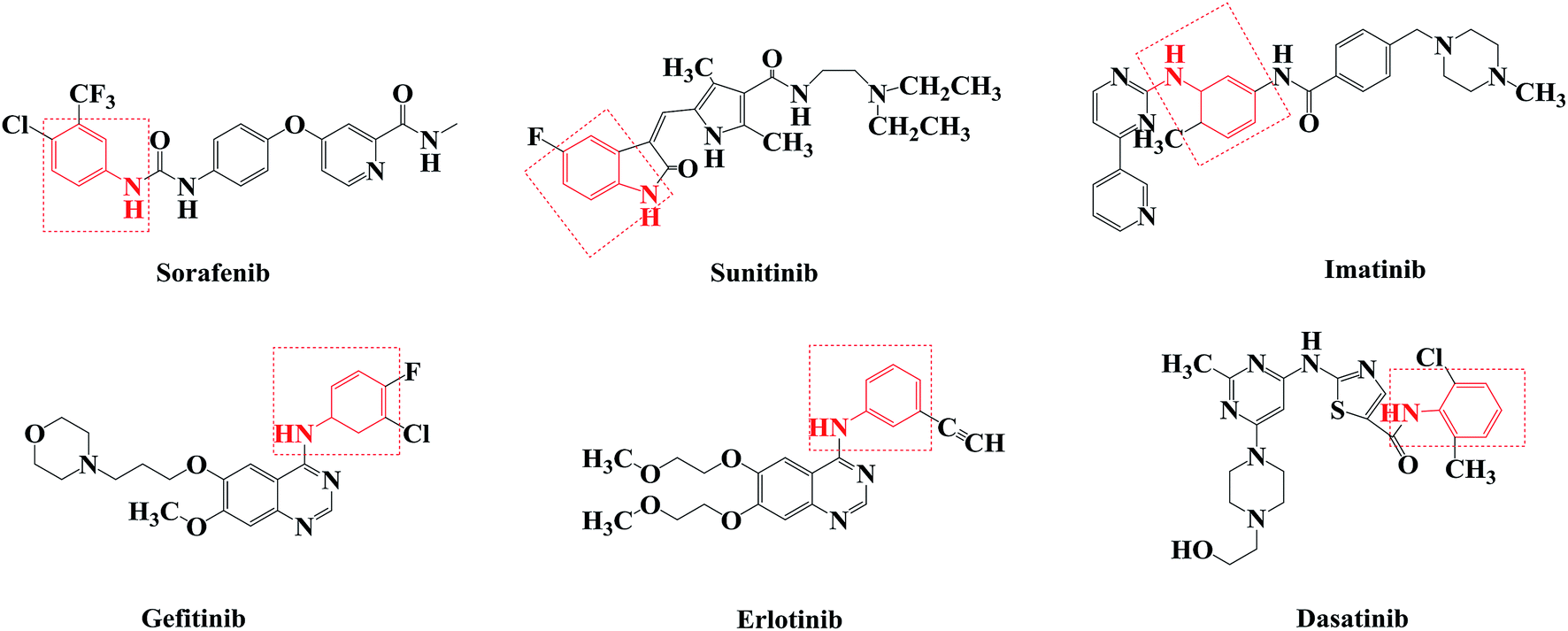 | ||
| Fig. 1 Structures of protein kinase inhibitors. | ||
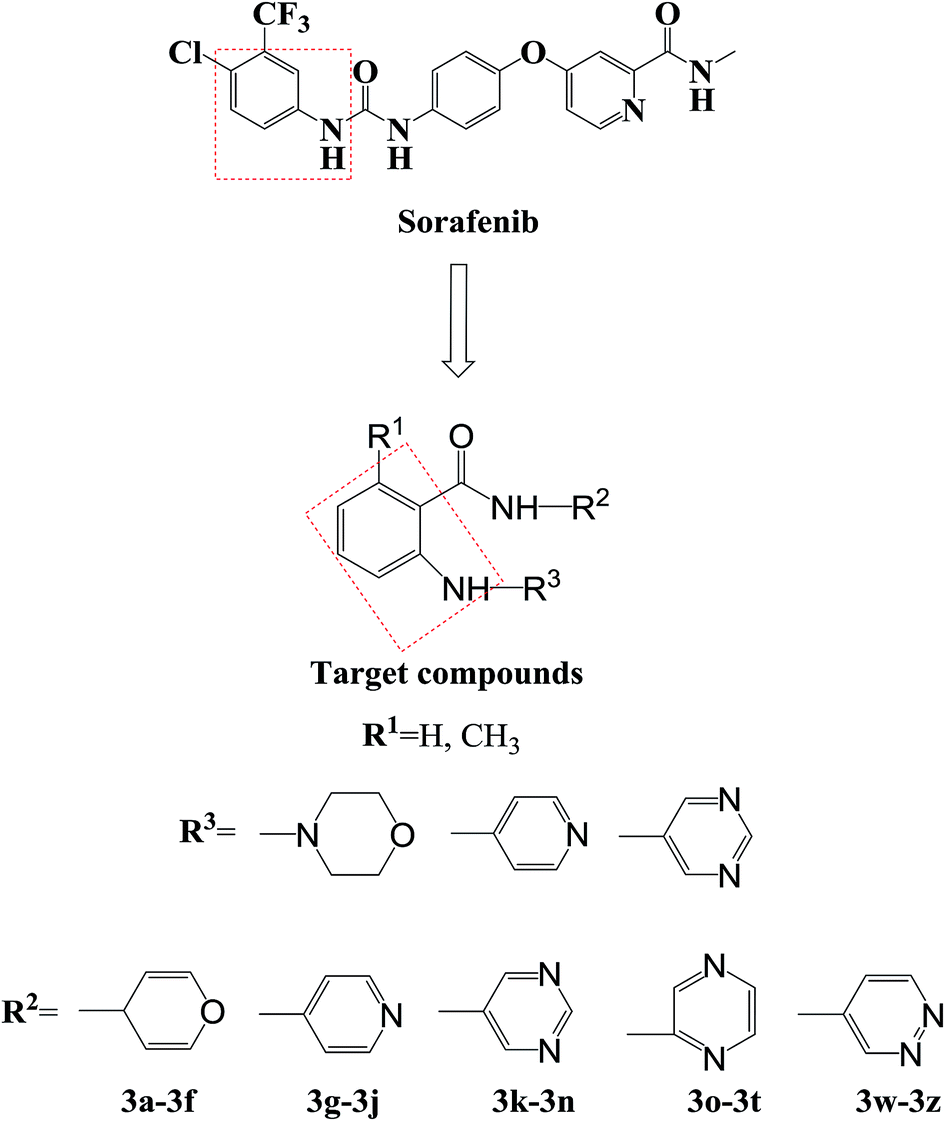 | ||
| Fig. 2 Design of sorafenib analogues and derivatives. | ||
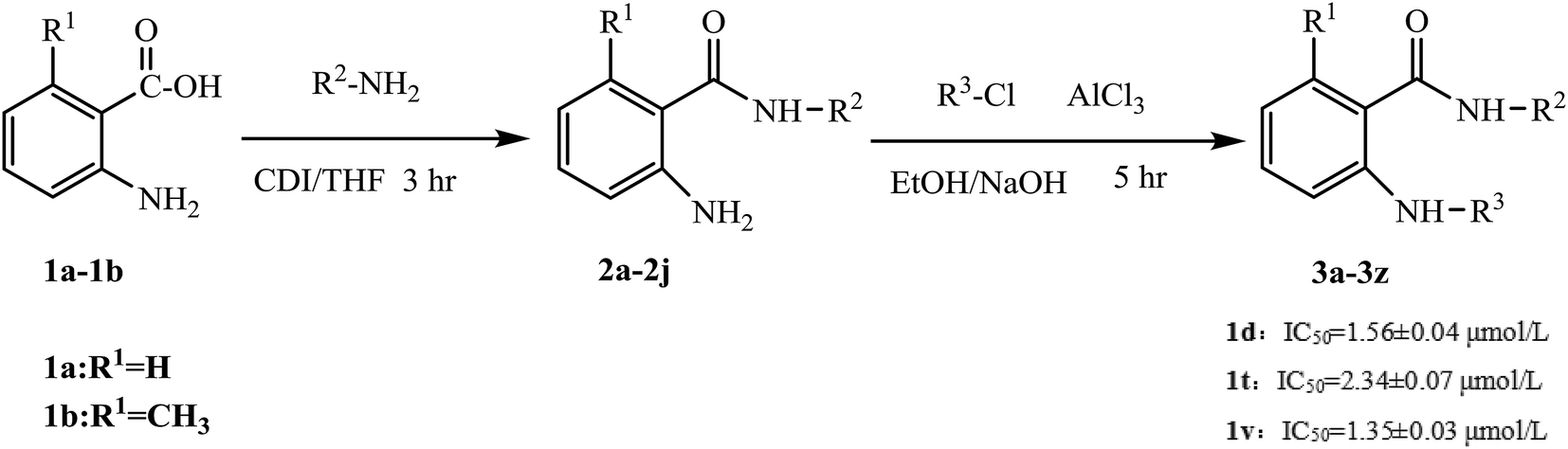 | ||
| Scheme 1 Synthetic route for sorafenib analogues. | ||
2. Results and discussion
2.1. Design and synthesis of sorafenib analogues and derivatives
Sorafenib (protein kinase inhibitor) has a novel structure and is used as an antitumor drug against multiple kinase targets. Herein, the design of protein kinase inhibitors was based on sorafenib as the lead compound. A series of novel sorafenib analogues and derivatives were prepared using bioisosteres and the alkyl principle (Fig. 2). As shown in Fig. 2, the target compounds and sorafenib were found to contain the same aniline structure. According to structure–activity relationship (SAR) studies, this novel series of compounds might have anti-tumor activities. During structural modification of the lead compound (sorafenib), substituents R1, R2, and R3 on the aniline structure were modified. The following substituents were selected and tested: H and CH3 at R1; pyran, pyridine, pyrimidine, pyrazine, and pyridazine at R2; and morpholine, pyridine, and pyrimidine at R3. These R1, R2, and R3 substituents were chosen to alter the physical and chemical properties (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P and pKa) of the target compounds and achieve good biological activity. To prepare the target compounds, we chose a synthesis route that involved simple operations and mild reaction conditions, and afforded high total yields (Scheme 1). As shown in Scheme 1, the target compounds were synthesized by amination and condensation (N-alkylation). Compounds 2a–2j were prepared using a general amination method, with 2-aminobenzoic acid (1a) or 6-methyl-2-aminobenzoic acid (1b) as the starting material, tetrahydrofuran (THF) as solvent, and 1,1′-carbonyldiimidazole (CDI) as catalyst under reflux for 3 h to achieve reaction completion. Compounds 3a–3z were prepared using a general condensation (N-alkylation) method with AlCl3 as a catalyst in EtOH/NaOH under reflux for 5 h. The resulting products were characterized by 1H NMR, 13C NMR, HR-ESI-MS, and elemental analysis.
P and pKa) of the target compounds and achieve good biological activity. To prepare the target compounds, we chose a synthesis route that involved simple operations and mild reaction conditions, and afforded high total yields (Scheme 1). As shown in Scheme 1, the target compounds were synthesized by amination and condensation (N-alkylation). Compounds 2a–2j were prepared using a general amination method, with 2-aminobenzoic acid (1a) or 6-methyl-2-aminobenzoic acid (1b) as the starting material, tetrahydrofuran (THF) as solvent, and 1,1′-carbonyldiimidazole (CDI) as catalyst under reflux for 3 h to achieve reaction completion. Compounds 3a–3z were prepared using a general condensation (N-alkylation) method with AlCl3 as a catalyst in EtOH/NaOH under reflux for 5 h. The resulting products were characterized by 1H NMR, 13C NMR, HR-ESI-MS, and elemental analysis.
2.2. In vitro antitumor activity
Biological activity experiments were based on the MTT assay (a colorimetric assay for assessing cell metabolic activity). For the in vitro experiments, human lung cancer cells (H1975 and A549) and cervical cancer cells (Hela) were used as inhibitory targets. The inhibitory activity was measured as the half maximal inhibitory concentration (IC50). Sorafenib and sunitinib were used as positive drugs and DMSO was used as the blank control. The results of the biological activity experiments are shown in Table 1. The data were analyzed by linear regression using statistical software SPSS (version 13.0), with the results showing a linear fit, as shown in Table 1. Compounds 3c, 3d, 3h, 3n, 3r, and 3z showed better inhibitory effects against human cervical cancer cells (Hela), while compounds 3t and 3v showed better inhibitory effects against human lung cancer cells (H1975 and A549). Among these compounds, compound 3d had an inhibitory activity (IC50) of 1.56 ± 0.04 μmol L−1 against Hela cells (human cervical cancer), compound 3t had an IC50 of 2.34 ± 0.07 μmol L−1 against H1975 (human lung cancer), and compound 3v had an IC50 of 1.35 ± 0.03 μmol L−1 against A549 (human lung cancer). As shown in Table 1, these compounds showed good antitumor activity in vitro. Compounds 3d and 3r showed better biological activity than positive control sunitinib (IC50 = 2.06 ± 0.34 μmol L−1) against Hela cells, compound 3t showed better biological activity than positive control sorafenib (IC50 = 4.20 ± 0.21 μmol L−1) against H1975 cells, and compound 3v showed similar biological activity to positive control sunitinib (IC50 = 1.05 ± 0.04 μmol L−1) against A549 cells.| Compounds | IC50a (μmol L−1) ± SD | ||
|---|---|---|---|
| Hela | A549 | H1975 | |
| a IC50 values are displayed as means ± standard deviations, n ≥ 8. | |||
| 3a | 10.20 ± 0.88 | 12.40 ± 1.20 | 11.13 ± 1.11 |
| 3b | 7.02 ± 0.85 | 10.31 ± 1.00 | 8.90 ± 0.96 |
| 3c | 2.01 ± 0.11 | 25.33 ± 2.05 | 43.12 ± 3.22 |
| 3d | 1.56 ± 0.04 | 20.46 ± 2.20 | 40.39 ± 2.11 |
| 3e | 34.78 ± 1.55 | 12.66 ± 1.03 | 10.18 ± 1.06 |
| 3f | 30.69 ± 1.33 | 10.23 ± 1.09 | 9.45 ± 0.55 |
| 3g | 3.08 ± 0.011 | 10.26 ± 0.65 | 8.02 ± 0.36 |
| 3h | 2.07 ± 0.33 | 7.45 ± 0.45 | 6.96 ± 0.66 |
| 3i | 12.67 ± 0.89 | 56.09 ± 3.00 | 45.99 ± 2.98 |
| 3j | 10.33 ± 1.09 | 54.37 ± 2.16 | 43.56 ± 3.11 |
| 3k | 56.88 ± 3.12 | 4.60 ± 0.45 | 4.33 ± 0.24 |
| 3l | 54.70 ± 4.00 | 3.04 ± 0.73 | 2.66 ± 0.29 |
| 3m | 4.04 ± 0.34 | 24.67 ± 2.23 | 13.56 ± 1.32 |
| 3n | 2.54 ± 0.23 | 20.45 ± 2.10 | 11.34 ± 1.34 |
| 3o | 12.45 ± 1.32 | 15.67 ± 1.58 | 20.49 ± 1.99 |
| 3p | 10.45 ± 0.78 | 13.55 ± 2.10 | 18.48 ± 2.09 |
| 3q | 4.22 ± 0.26 | 23.47 ± 3.12 | 26.68 ± 2.11 |
| 3r | 1.90 ± 0.10 | 19.55 ± 3.10 | 23.40 ± 2.89 |
| 3s | 18.34 ± 2.04 | 4.45 ± 0.21 | 4.36 ± 0.15 |
| 3t | 16.23 ± 1.23 | 3.78 ± 0.21 | 2.34 ± 0.11 |
| 3u | 15.04 ± 1.45 | 2.44 ± 0.14 | 4.22 ± 0.22 |
| 3v | 13.58 ± 1.67 | 1.35 ± 0.02 | 2.67 ± 0.12 |
| 3w | 25.55 ± 2.55 | 13.33 ± 1.89 | 12.29 ± 2.00 |
| 3x | 23.68 ± 3.11 | 10.29 ± 0.90 | 10.04 ± 0.89 |
| 3y | 3.46 ± 0.09 | 46.33 ± 3.11 | 56.48 ± 4.66 |
| 3z | 2.11 ± 0.23 | 44.60 ± 4.21 | 54.69 ± 5.10 |
| Sorafenib | 6.02 ± 0.21 | 2.10 ± 0.10 | 4.20 ± 0.21 |
| Sunitinib | 2.06 ± 0.34 | 1.05 ± 0.04 | 16.02 ± 0.85 |
| DMSO | None | None | None |
2.3. In vivo antitumor activity
In vivo antitumor test results are the most important indicators in evaluating the effectiveness of candidate antitumor compounds. In vivo experiments were conducted to evaluate the inhibitory effect and intensity of the compounds on the growth of xenograft tumors in nude mice with human carcinoma. During the experiment, human lung cancer (A549) and human cervical cancer (Hela) were inoculated under the right armpit of nude mice. After tumor growth to a certain stage, high (100 mg kg−1 d−1), medium (50 mg kg−1 d−1), and low (10 mg kg−1 d−1) doses of compounds 3c, 3d, 3h, 3n, 3r, 3t, 3v, and 3z were administered (Table 2). Data were analyzed by linear regression using statistical software SPSS (version 13.0), with the results of data analysis showing a linear fit. As shown in Table 2, the data showed that these compounds had good antitumor effects in vivo and showed low relative tumor proliferation rates (T/C, %). Most of the compounds were effective at high, medium, and low doses (T/C > 40% was ineffective and T/C ≤ 40% was effective). As shown in Table 2, compounds 3c, 3d, 3h, 3n, 3r, and 3z showed general inhibitory activities at low doses (10 mg kg−1 d−1) against Hela cells. However, at medium doses (50 mg kg−1 d−1) or high doses (100 mg kg−1 d−1), their inhibitory activities were significantly increased above those of positive controls sorafenib and sunitinib. Compounds 3t and 3v showed general inhibitory activity at low doses (10 mg kg−1 d−1) against A549 cells, but better activities than positive controls sorafenib and sunitinib at medium (50 mg kg−1 d−1) and high doses (100 mg kg−1 d−1). Therefore good inhibitory activities were obtained at medium doses and high doses against Hela and A549 cells in vivo. We also studied the acute toxicity of these target compounds in vivo (Table 2), which found that target compounds 3c, 3d, 3h, 3n, 3r, 3t, 3v, and 3z had low acute toxicity.| Compounds | Doses (mg kg−1 d−1) | T/Ca (%) | LD50 ± SDb (mg kg−1) | |
|---|---|---|---|---|
| Hela | A549 | |||
| a T/C values are displayed as means ± standard deviations, n ≥ 8.b LD50 values are displayed as means ± standard deviations, n ≥ 8. | ||||
| 3c | 10 | 4.78 ± 0.13 | 56.78 ± 3.21 | 2781.2 ± 3.2 |
| 50 | 1.92 ± 0.09 | 43.11 ± 3.45 | ||
| 100 | 0.25 ± 0.13 | 17.23 ± 1.39 | ||
| 3d | 10 | 3.56 ± 0.08 | 45.91 ± 3.56 | 2692.1 ± 4.3 |
| 50 | 1.26 ± 0.05 | 37.80 ± 1.34 | ||
| 100 | 0.03 ± 0.01 | 13.22 ± 1.06 | ||
| 3h | 10 | 5.11 ± 0.87 | 38.90 ± 3.10 | 2469.3 ± 2.8 |
| 50 | 2.67 ± 0.06 | 23.53 ± 1.01 | ||
| 100 | 0.30 ± 0.02 | 9.21 ± 1.31 | ||
| 3n | 10 | 5.89 ± 0.23 | 83.26 ± 5.23 | 1823.2 ± 4.1 |
| 50 | 2.90 ± 0.10 | 58.21 ± 3.34 | ||
| 100 | 0.45 ± 0.07 | 38.33 ± 2.11 | ||
| 3r | 10 | 4.11 ± 0.19 | 76.22 ± 4.62 | 2236.7 ± 3.1 |
| 50 | 1.87 ± 0.08 | 52.17 ± 3.12 | ||
| 100 | 0.09 ± 0.01 | 30.32 ± 3.18 | ||
| 3t | 10 | 69.45 ± 4.32 | 5.78 ± 0.56 | 1803.7 ± 2.3 |
| 50 | 40.23 ± 2.39 | 2.26 ± 0.34 | ||
| 100 | 10.12 ± 1.01 | 0.23 ± 0.02 | ||
| 3v | 10 | 42.34 ± 3.43 | 5.38 ± 0.67 | 2013.5 ± 2.2 |
| 50 | 36.45 ± 1.22 | 2.11 ± 0.05 | ||
| 100 | 6.55 ± 0.33 | 0.11 ± 0.03 | ||
| 3z | 10 | 6.35 ± 0.56 | 71.31 ± 3.42 | 1749.1 ± 3.2 |
| 50 | 2.93 ± 0.08 | 58.51 ± 2.32 | ||
| 100 | 0.31 ± 0.01 | 41.12 ± 1.23 | ||
| Sorafenib | 50 | 9.56 ± 1.05 | 3.11 ± 0.23 | 2210.5 ± 2.3 |
| Sunitinib | 50 | 2.89 ± 0.02 | 2.21 ± 0.15 | 2331.2 ± 2.1 |
| DMSO | 50 | 100 | 100 | |
3. Conclusions
We have reported the design, synthesis, and evaluation of a series of novel sorafenib analogues and derivatives as tumor inhibitors. Using sorafenib as the lead compound, various compounds were designed and prepared using bioisosteres and the alkyl principle. The target compounds were synthesized from 2-aminobenzoic acid (1a) or 6-methyl-2-aminobenzoic acid (1b) by amination and condensation (N-alkylation). This synthetic route involved simple operations and mild reaction conditions, and afforded high total yields. In vitro activity results showed that compounds 3c, 3d, 3h, 3n, 3r, and 3z had good inhibitory effects on human cervical cancer cells (Hela), and that compounds 3t and 3v had good inhibitory effects on human lung cancer cells (H1975 and A549). Among these compounds, compound 3d had an inhibitory activity (IC50) of 1.56 ± 0.04 μmol L−1 against Hela cells (human cervical cancer), compound 3t had an IC50 of 2.34 ± 0.07 μmol L−1 against H1975 cells (human lung cancer), and compound 3v had an IC50 of 1.35 ± 0.03 μmol L−1 against A549 cells (human lung cancer). In vivo activity results showed that these compounds had good antitumor effects and low acute toxicity.4. Experimental
4.1. Chemistry
2-(Morpholinoamino)-N-(4H-pyran-4-yl)benzamide (3a). 84.8% yield; mp 145–146 °C; 1H NMR (300 MHz, CDCl3) δ: 9.18 (s, 1H, –NH–), 7.69 (m, 1H, Ph-H), 7.50 (m, 1H, Ph-H), 7.08 (m, 1H, Ph-H), 6.78 (m, 1H, Ph-H), 6.17 (d, J = 8.4 Hz, 2H, C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–O), 4.76 (m, 1H, –CH–), 4.63 (m, 2H, –CHC), 3.65 (m, 4H, C–CH2–O), 3.00 (m, 4H, –CH2-C); 13C NMR (75 MHz, CDCl3) δ: 167.8, 141.0, 140.1, 132.6, 128.0, 119.2, 114.8, 112.1, 104.1, 65.5, 58.4, 51.1; HR-ESI-MS m/z: calcd for C16H19N3O3 ([M + H]+), 301.3503; found, 301.3501. Anal. calcd for C16H19N3O3: C, 63.77; H, 6.36; N, 13.94; O, 15.93; found: C, 63.76; H, 6.37; N, 13.93; O, 15.94%.
CH–O), 4.76 (m, 1H, –CH–), 4.63 (m, 2H, –CHC), 3.65 (m, 4H, C–CH2–O), 3.00 (m, 4H, –CH2-C); 13C NMR (75 MHz, CDCl3) δ: 167.8, 141.0, 140.1, 132.6, 128.0, 119.2, 114.8, 112.1, 104.1, 65.5, 58.4, 51.1; HR-ESI-MS m/z: calcd for C16H19N3O3 ([M + H]+), 301.3503; found, 301.3501. Anal. calcd for C16H19N3O3: C, 63.77; H, 6.36; N, 13.94; O, 15.93; found: C, 63.76; H, 6.37; N, 13.93; O, 15.94%.
2-Methyl-6-(morpholinoamino)-N-(4H-pyran-4-yl)benzamide (3b). 89.2% yield; mp 153–156 °C; 1H NMR (300 MHz, CDCl3) δ: 9.18 (s, 1H, –NH–), 7.36 (m, 1H, Ph-H), 6.98 (m, 1H, Ph-H), 6.69 (m, 1H, Ph-H), 6.17 (d, J = 8.4 Hz, 2H, C
CH–O), 4.76 (m, 1H, –CH–), 4.63 (m, 2H, –CHC), 3.65 (m, 4H, C–CH2–O), 3.00 (m, 4H, –CH2–C), 2.48 (s, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ: 167.8, 140.9, 140.1, 137.7, 132.5, 121.9, 117.7, 104.1, 65.5, 58.4, 51.1; HR-ESI-MS m/z: calcd for C17H21N3O3 ([M + H]+), 315.3701; found, 315.1583l; anal. calcd for C17H21N3O3: C, 64.74; H, 6.71; N, 13.32; O, 15.22; found: C, 64.73; H, 6.72; N, 13.33; O, 15.21%.
N-(4H-Pyran-4-yl)-2-(pyridin-4-ylamino)benzamide (3c). 90.1% yield; mp 173–174 °C; 1H NMR (300 MHz, CDCl3) δ: 11.02 (s, 1H, –NH–N), 9.18 (s, 1H, –NH–), 8.46 (m, 2H, –CH
N), 8.39 (m, 1H, Ph-H), 7.73 (m, 1H, Ph-H), 7.66 (m, 1H, Ph-H), 6.99 (m, 1H, Ph-H), 6.99 (m, 2H, CCH–), 6.17 (d, J = 8.4 Hz, 2H, CCH–O), 4.76 (m, 1H, –CH–), 4.63 (m, 2H,–CHC); 13C NMR (75 MHz, CDCl3) δ: 167.8, 155.3, 151.9, 150.2, 140.1, 132.9, 128.3, 118.8, 117.9, 116.4, 109.0, 104.1, 51.1; HR-ESI-MS m/z: calcd for C17H15N3O2 ([M + H]+), 293.3301; found, 293.1164; anal. calcd for C17H15N3O2: C, 69.61; H, 5.15; N, 14.33; O, 10.91; found: C, 69.60; H, 5.17; N, 14.32; O, 10.91%.
2-Methyl-N-(4H-pyran-4-yl)-6-(pyridin-4-ylamino)benzamide (3d). 92.5% yield; mp 182–185 °C; 1H NMR (300 MHz, CDCl3) δ: 11.02 (s, 1H, –NH–N), 9.18 (s, 1H, –NH–), 8.46 (m, 2H, –CH
N), 8.29 (m, 1H, Ph-H), 7.40 (m, 1H, Ph-H), 6.99 (m, 2H, CCH–), 6.90 (m, 1H, Ph-H), 6.17 (d, J = 8.4 Hz, 2H, CCH–O), 4.76 (m, 1H, –CH–), 4.63 (m, 2H, –CHC), 2.48 (s, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ: 167.8, 155.3, 150.2, 143.8, 143.1, 140.1, 138.0, 121.5, 120.8, 113.4, 109.0, 104.1, 51.1, 18.1; HR-ESI-MS m/z: calcd for C18H17N3O2 ([M + H]+), 307.3505; found, 307.1320; anal. calcd for C18H17N3O2: C, 70.34; H, 5.58; N, 13.67; O, 10.41; found: C, 70.35; H, 5.59; N, 13.66; O, 10.40%.
N-(4H-Pyran-4-yl)-2-(pyrimidin-5-ylamino)benzamide (3e). 93.4% yield; mp 201–203 °C; 1H NMR (300 MHz, CDCl3) δ: 9.23 (s, 1H, –NH–C), 9.18 (s, 1H, –NH–C), 8.90 (s, 1H, N
CH–N), 8.50 (s, 1H, –CHN), 8.39 (m, 1H, Ph-H), 7.73 (m, 1H, Ph-H), 7.66 (m, 1H, Ph-H), 6.99 (m, 1H, Ph-H), 6.17 (d, J = 8.4 Hz, 2H, CCH–O), 4.76 (m, 1H, –CH–), 4.63 (m, 2H, –CHC); 13C NMR (75 MHz, CDCl3) δ: 167.8, 151.9, 147.4, 143.7, 142.9, 140.1, 132.9, 128.3, 118.8, 117.9, 116.4, 104.1, 51.1; HR-ESI-MS m/z: calcd for C16H14N4O2 ([M + H]+), 294.3101; found, 294.1117; anal. calcd for C16H14N4O2: C, 65.30; H, 4.79; N, 19.04; O, 10.87; found: C, 65.31; H, 4.79; N, 19.03; O, 10.87%.
2-Methyl-N-(4H-pyran-4-yl)-6-(pyrimidin-5-ylamino)benzamide (3f). 94.0% yield, mp 222–223 °C, 1H NMR (300 MHz, CDCl3) δ: 9.23 (s, 1H, –NH–C), 9.18 (s, 1H, –NH–C), 8.90 (s, 1H, N
CH–N), 8.50 (s, 1H, –CHN), 8.29 (m, 1H, Ph-H), 7.40 (m, 1H, Ph-H), 6.90 (m, 1H, Ph-H), 6.17 (d, J = 8.4 Hz, 2H, CCH–O), 4.76 (m, 1H, –CH–), 4.63 (m, 2H,–CHC), 2.48 (s, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ: 167.8, 147.4, 143.8, 143.7, 143.1, 140.1, 138.0, 121.5, 120.8, 113.4, 104.1, 51.1, 18.1; HR-ESI-MS m/z: calcd for C17H16N4O2 ([M + H]+), 308.3411; found, 308.1273; anal. calcd for C17H16N4O2: C, 66.22; H, 5.23; N, 18.17; O, 10.38; found: C, 66.23; H, 5.22; N, 18.18; O, 10.37%.
2-(Morpholinoamino)-N-(pyridin-4-yl)benzamide (3g). 88.4% yield; mp 168–170 °C; 1H NMR (300 MHz, CDCl3) δ: 10.35 (s, 1H, –NH–N), 10.22 (s, 1H, –NH–), 8.46 (d, J = 7.4 Hz, 2H, –CH
N), 7.69 (m, 1H, Ph-H), 7.50 (m, 1H, Ph-H), 7.36 (d, J = 7.2 Hz, 2H, –CHC), 7.08 (m, 1H, Ph-H), 3.65 (m, 4H, C–CH2–O), 3.00 (m, 4H, –CH2–C); 13C NMR (75 MHz, CDCl3) δ: 167.5, 155.3, 150.2, 141.0, 132.6, 128.0, 119.2, 114.8, 112.1, 109.0, 65.5, 58.4; HR-ESI-MS m/z: calcd for C16H14N4O2 ([M + H]+), 294.3111; found, 294.1116; anal. calcd for C16H14N4O2: C, 65.30; H, 4.79; N, 19.04; O, 10.87; found: C, 65.31; H, 4.78; N, 19.04; O, 10.87%
2-Methyl-6-(morpholinoamino)-N-(pyridin-4-yl)benzamide (3h). 90.2% yield; mp 177–178 °C; 1H NMR (300 MHz, CDCl3) δ: 10.35 (s, 1H, –NH–N), 10.22 (s, 1H, –NH–), 8.46 (d, J = 7.4 Hz, 2H, –CH
N), 7.36 (d, J = 7.2 Hz, 2H, –CHC), 7.36 (m, 1H, Ph-H), 6.98 (m, 1H, Ph-H), 6.69 (m, 1H, Ph-H), 3.65 (m, 4H, C–CH2–O), 3.00 (m, 4H, –CH2–C), 2.48 (s, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ: 164.7, 155.3, 150.2, 140.9, 137.7, 132.5, 121.9, 117.7, 109.1, 109.0, 65.5, 58.4, 18.1; HR-ESI-MS m/z: calcd for C17H16N4O2 ([M + H]+), 308.3400; found, 308.1273; anal. calcd for C17H16N4O2: C, 66.22; H, 5.23; N, 18.17; O, 10.38; found: C, 66.23; H, 5.24; N, 18.15; O, 10.38%.
N-(Pyridin-4-yl)-2-(pyrimidin-5-ylamino)benzamide (3i). 92.1% yield; mp 188–190 °C; 1H NMR (300 MHz, CDCl3) δ: 10.22 (s, 1H, –NH–N), 9.23 (s, 1H, –NH–), 8.90 (s, 1H, –N–CH–), 8.50 (s, 2H, –CH
N), 8.46 (d, J = 7.4 Hz, 2H, –CHN), 8.39 (m, 1H, Ph-H), 7.73 (m, 1H, Ph-H), 7.66 (m, 1H, Ph-H), 7.36 (d, J = 7.2 Hz, 2H, –CHC), 6.99 (m, 1H, Ph-H); 13C NMR (75 MHz, CDCl3) δ: 167.5, 155.3, 151.9, 150.2, 147.4, 143.7, 142.9, 132.9, 128.3, 118.8, 117.9, 116.4, 109.0; HR-ESI-MS m/z: calcd for C16H13N5O ([M + H]+), 291.3100; found, 291.1120; anal. calcd for C16H13N5O: C, 65.97; H, 4.50; N, 24.04; O, 5.49; found: C, 65.97; H, 4.51; N, 24.04; O, 5.48%.
2-Methyl-N-(pyridin-4-yl)-6-(pyrimidin-5-ylamino)benzamide (3j). 92.9% yield; mp 201–204 °C; 1H NMR (300 MHz, CDCl3) δ: 10.22 (s, 1H, –NH–N), 9.23 (s, 1H, –NH–), 8.90 (s, 1H, –N–CH–), 8.50 (s, 2H, –CH
N), 8.46 (d, J = 7.4 Hz, 2H, –CHN), 8.29 (m, 1H, Ph-H), 7.40 (d, J = 7.2 Hz, 2H, –CHC), 7.36 (d, J = 7.2 Hz, 2H, –CHC), 6.90 (m, 1H, Ph-H); 2.48 (s, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ: 164.7, 155.3, 150.2, 147.4, 143.8, 143.7, 143.1, 142.9, 138.0, 121.5, 120.8, 113.4, 109.0, 18.1; HR-ESI-MS m/z: calcd for C17H15N5O ([M + H]+), 305.3401; found, 305.1277; anal. calcd for C17H15N5O: C, 66.87; H, 4.95; N, 22.94; O, 5.24; found: C, 66.88; H, 4.95; N, 22.94; O, 5.23%.
2-(Morpholinoamino)-N-(pyrimidin-5-yl)benzamide (3k). 87.1% yield; mp 178–180 °C; 1H NMR (300 MHz, CDCl3) δ: 10.35 (s, 1H, –NH–N), 10.22 (s, 1H, –NH–), 9.21 (s, 2H, –CH
N), 9.10 (s, 1H, –NCH–), 7.69 (m, 1H, Ph-H), 7.50 (m, 1H, Ph-H), 7.08 (m, 1H, Ph-H), 3.65 (m, 4H, C–CH2–O), 3.00 (m, 4H, –CH2–C); 13C NMR (75 MHz, CDCl3) δ: 167.5, 147.4, 143.7, 141.1, 141.0, 132.6, 128.0, 119.2, 114.8, 112.1, 65.5, 58.4; HR-ESI-MS m/z: calcd for C15H17N5O2 ([M + H]+), 299.3301; found, 299.1382; anal. calcd for C15H17N5O2: C, 60.19; H, 5.72; N, 23.40; O, 10.69; found: C, 60.19; H, 5.73; N, 23.40; O, 10.68%.
2-Methyl-6-(morpholinoamino)-N-(pyrimidin-5-yl)benzamide (3l). 89.0% yield; mp 192–194 °C; 1H NMR (300 MHz, CDCl3) δ: 10.35 (s, 1H, –NH–N), 10.22 (s, 1H, –NH–), 9.21 (s, 2H, –CH
N), 9.10 (s, 1H, –NCH–), 7.36 (m, 1H, Ph-H), 6.98 (m, 1H, Ph-H), 6.69 (m, 1H, Ph-H), 3.65 (m, 4H, C–CH2–O), 3.00 (m, 4H, –CH2–C), 2.48 (s, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ: 164.7, 147.4, 143.7, 141.1, 140.9, 137.7, 132.5, 121.9, 117.7, 109.1, 65.5, 58.4, 18.1; HR-ESI-MS m/z: calcd for C16H19N5O2 ([M + H]+), 313.3602; found, 313.1539; anal. calcd for C16H19N5O2: C, 61.33; H, 6.11; N, 22.35; O, 10.21; found: C, 61.35; H, 6.10; N, 22.35; O, 10.20%.
2-(Pyridin-4-ylamino)-N-(pyrimidin-5-yl)benzamide (3m). 90.2% yield; mp 192–193 °C; 1H NMR (300 MHz, CDCl3) δ: 11.02 (s, 1H, –NH–N), 10.21 (s, 1H, –NH–), 9.21 (s, 2H, –CH
N), 9.10 (s, 1H, –NCH–), 8.46 (m, 2H, –CH–N), 8.39 (m, 1H, Ph-H), 7.73 (m, 1H, Ph-H), 7.66 (m, 1H, Ph-H), 6.99 (m, 1H, Ph-H), 6.99 (m, 2H, –C–CH–); 13C NMR (75 MHz, CDCl3) δ: 167.5, 155.3, 151.9, 150.2, 147.4, 143.7, 141.1, 132.9, 128.3, 118.8, 117.9, 116.4, 109.0; HR-ESI-MS m/z: calcd for C16H13N5O ([M + H]+), 291.3112; found, 291.1120; anal. calcd for C16H13N5O: C, 65.97; H, 4.50; N, 24.04; O, 5.49; found: C, 65.98; H, 4.51; N, 24.03; O, 5.48%.
2-Methyl-6-(pyridin-4-ylamino)-N-(pyrimidin-5-yl)benzamide (3n). 93.1% yield; mp 201–202 °C; 1H NMR (300 MHz, CDCl3) δ: 11.02 (s, 1H, –NH–N), 10.21 (s, 1H, –NH–), 9.21 (s, 2H, –CH
N), 9.10 (s, 1H, –NCH–), 8.46 (m, 2H, –CH–N), 8.29 (m, 1H, Ph-H), 7.40 (m, 1H, Ph-H), 6.99 (m, 2H, –C–CH–), 6.90 (m, 1H, Ph-H), 2.48 (s, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ: 164.7, 155.3, 150.2, 147.4, 143.8, 143.7, 143.1, 141.1, 138.0, 121.5, 120.8, 113.4, 109.0, 18.1; HR-ESI-MS m/z: calcd for C17H15N5O ([M + H]+), 305.3411; found, 305.1276; anal. calcd for C17H15N5O: C, 66.87; H, 4.95; N, 22.94; O, 5.24; found: C, 66.88; H, 4.94; N, 22.93; O, 5.25%.
2-(Morpholinoamino)-N-(pyrazin-2-yl)benzamide (3o). 86.3% yield; mp 181–183 °C; 1H NMR (300 MHz, CDCl3) δ: 12.32 (s, 1H, –NH–N), 11.17 (s, 1H, –NH–), 8.59 (s, 1H, –CH–N), 8.40 (s, 1H, –N
CH–), 8.35 (s, 1H, –NCH–), 7.69 (m, 1H, Ph-H), 7.50 (m, 1H, Ph-H), 7.08 (m, 1H, Ph-H), 6.78 (m, 1H, Ph-H), 3.65 (m, 4H, C–CH2–O), 3.00 (m, 4H, –CH2–C); 13C NMR (75 MHz, CDCl3) δ: 164.7, 149.7, 141.8, 139.5, 137.4, 136.1, 132.6, 128.0, 119.2, 114.8, 112.1, 65.5, 58.4; HR-ESI-MS m/z: calcd for C15H17N5O2 ([M + H]+), 299.3312; found, 299.1382; anal. calcd for C15H17N5O2: C, 60.19; H, 5.72; N, 23.40; O, 10.69; found: C, 60.19; H, 5.73; N, 23.41; O, 10.67%.
2-Methyl-6-(morpholinoamino)-N-(pyrazin-2-yl)benzamide (3p). 89.1% yield; mp 194–196 °C; 1H NMR (300 MHz, CDCl3) δ: 12.32 (s, 1H, –NH–N), 11.17 (s, 1H, –NH–), 8.59 (s, 1H, –CH–N), 8.40 (s, 1H, –N
CH–), 8.35 (s, 1H, –NCH–), 7.36 (m, 1H, Ph-H), 6.98 (m, 1H, Ph-H), 6.69 (m, 1H, Ph-H), 3.65 (m, 4H, C–CH2–O), 3.00 (m, 4H, –CH2–C), 2.48 (s, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ: 164.7, 149.7, 140.9, 139.5, 137.4, 137.7, 137.4, 136.1, 132.5, 121.9, 117.7, 109.1, 65.5, 58.4; HR-ESI-MS m/z: calcd for C16H19N5O2 ([M + H]+), 313.3611; found, 313.1538; anal. calcd for C16H19N5O2: C, 61.33; H, 6.11; N, 22.35; O, 10.21; found: C, 61.34; H, 6.10; N, 22.35; O, 10.21%.
N-(Pyrazin-2-yl)-2-(pyridin-4-ylamino)benzamide (3q). 86.3% yield; mp 196–198 °C; 1H NMR (300 MHz, CDCl3) δ: 11.17 (s, 1H, –NH–N), 11.02 (s, 1H,–NH–), 8.59 (s, 1H, –CH–N), 8.46 (m, 2H, –CH–N), 8.40 (s, 1H, –N
CH–), 8.39 (m, 1H, Ph-H), 8.35 (s, 1H, –NCH–), 7.73 (m, 1H, Ph-H), 7.66 (m, 1H, Ph-H), 6.99 (m, 1H, Ph-H), 6.99 (m, 2H, –C–CH–); 13C NMR (75 MHz, CDCl3) δ: 164.7, 155.3, 151.9, 150.2, 149.7, 139.5, 137.4, 136.1, 132.9, 128.3, 118.8, 117.9, 116.4, 109.0; HR-ESI-MS m/z: calcd for C16H13N5O ([M + H]+), 291.3122; found, 291.1120; anal. calcd for C16H13N5O: C, 65.97; H, 4.50; N, 24.04; O, 5.49; found: C, 65.98; H, 4.50; N, 24.05; O, 5.47%.
2-Methyl-N-(pyrazin-2-yl)-6-(pyridin-4-ylamino)benzamide (3r). 88.4% yield; mp 210–212 °C; 1H NMR (300 MHz, CDCl3) δ: 11.17 (s, 1H, –NH–N), 11.02 (s, 1H, –NH–), 8.59 (s, 1H, –CH–N), 8.46 (m, 2H, –CH–N), 8.40 (s, 1H, –N
CH–), 8.29 (m, 1H, Ph-H), 8.35 (s, 1H, –NCH–), 7.40 (m, 1H, Ph-H), 6.99 (m, 2H, –C–CH–), 6.90 (m, 1H, Ph-H), 2.48 (s, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ: 164.7, 155.3, 150.2, 149.7, 143.8, 143.1, 139.5, 138.0, 137.4, 136.1, 121.5, 120.8, 113.4, 109.0, 18.1; HR-ESI-MS m/z: calcd for C17H15N5O ([M + H]+), 305.3402; found, 305.1277; anal. calcd for C17H15N5O: C, 66.87; H, 4.95; N, 22.94; O, 5.24; found: C, 66.88; H, 4.95; N, 22.93; O, 5.24%.
N-(Pyrazin-2-yl)-2-(pyrimidin-5-ylamino)benzamide (3s). 90.2% yield, mp 212–214 °C; 1H NMR (300 MHz, CDCl3) δ: 11.17 (s, 1H, –NH–N), 9.20 (s, 1H, –NH–), 8.90 (s, 1H, -N–CH–), 8.59 (s, 1H, –CH–N), 8.50 (s, 2H, –CH
N), 8.40 (s, 1H, –NCH–), 8.39 (m, 1H, Ph-H), 8.35 (s, 1H, –NCH–), 7.73 (m, 1H, Ph-H), 7.66 (m, 1H, Ph-H), 6.99 (m, 1H, Ph-H); 13C NMR (75 MHz, CDCl3) δ: 164.7, 151.9, 149.7, 147.4, 143.7, 142.9, 139.5, 137.4, 136.1, 132.9, 128.3, 118.8, 117.9, 116.4; HR-ESI-MS m/z: calcd for C15H12N6O ([M + H]+), 292.3002; found, 292.1072; anal. calcd for C15H12N6O: C, 61.64; H, 4.14; N, 28.75; O, 5.47; found: C, 61.65; H, 4.15; N, 28.74; O, 5.46%.
2-Methyl-N-(pyrazin-2-yl)-6-(pyrimidin-5-ylamino)benzamide (3t). 91.4% yield; mp 222–224 °C; 1H NMR (300 MHz, CDCl3) δ: 11.17 (s, 1H, –NH–N), 9.20 (s, 1H, –NH–), 8.90 (s, 1H, –N–CH–), 8.59 (s, 1H, –CH–N), 8.50 (s, 2H, –CH
N), 8.40 (s, 1H, –NCH–), 8.35 (s, 1H, –NCH–), 8.29 (m, 1H, Ph-H), 7.40 (m, 1H, Ph-H), 6.90 (m, 1H, Ph-H), 2.48 (s, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ: 164.7, 149.7, 147.4, 143.8, 143.7, 143.1, 142.9, 139.5, 138.0, 137.4, 136.1, 121.5, 120.8, 113.4, 18.1; HR-ESI-MS m/z: calcd for C16H14N6O ([M + H]+), 306.3302; found, 306.1229; anal. calcd for C16H14N6O: C, 62.74; H, 4.61; N, 27.43; O, 5.22; found: C, 62.75; H, 4.61; N, 26.43; O, 5.22%.
2-(Morpholinoamino)-N-(pyridazin-4-yl)benzamide (3u). 87.3% yield; mp 167–168 °C; 1H NMR (300 MHz, CDCl3) δ: 11.20 (s, 1H, –NH–N), 10.37 (s, 1H, –NH–), 9.61 (s, 1H, –CH–N), 9.30 (m, 1H, –CH–N), 8.05 (m, 1H, –C–CH–), 7.69 (m, 1H, Ph-H), 7.50 (m, 1H, Ph-H), 7.08 (m, 1H, Ph-H), 6.78 (m, 1H, Ph-H), 3.65 (m, 4H, C–CH2–O), 3.00 (m, 4H, –CH2–C); 13C NMR (75 MHz, CDCl3) δ: 167.5, 150.4, 144.2, 141.0, 139.4, 132.6, 128.0, 119.2, 114.8, 114.2, 112.1, 65.5, 58.4; HR-ESI-MS m/z: calcd for C15H17N5O2 ([M + H]+), 299.3302; found, 299.1382; anal. calcd for C15H17N5O2: C, 60.19; H, 5.72; N, 23.40; O, 10.69; found: C, 60.20; H, 5.71; N, 23.39; O, 10.70%.
2-Methyl-6-(morpholinoamino)-N-(pyridazin-4-yl)benzamide (3v). 88.5% yield; mp 182–184 °C; 1H NMR (300 MHz, CDCl3) δ: 11.20 (s, 1H, –NH–N), 10.37 (s, 1H, –NH–), 9.61 (s, 1H, –CH–N), 9.30 (m, 1H, –CH–N), 8.05 (m, 1H, –C–CH–), 7.36 (m, 1H, Ph-H), 6.98 (m, 1H, Ph-H), 6.69 (m, 1H, Ph-H), 3.65 (m, 4H, C–CH2–O), 3.00 (m, 4H, –CH2–C), 2.48 (s, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ: 164.7, 150.4, 144.2, 140.9, 139.4, 137.7, 132.5, 121.9, 117.7, 114.2, 109.1, 65.5, 58.4, 18.1; HR-ESI-MS m/z: calcd for C16H19N5O2 ([M + H]+), 313.3611; found, 313.1538; anal. calcd for C16H19N5O2: C, 61.33; H, 6.11; N, 22.35; O, 10.21; found: C, 61.34; H, 6.12; N, 22.34; O, 10.20%.
N-(Pyridazin-4-yl)-2-(pyridin-4-ylamino)benzamide (3w). 88.5% yield; mp 182–184 °C; 1H NMR (300 MHz, CDCl3) δ: 11.02 (s, 1H, –NH–N), 10.37 (s, 1H, –NH–), 9.61 (s, 1H, –CH–N), 9.30 (m, 1H, –CH–N), 8.46 (m, 2H, –CH–N), 8.39 (m, 1H, Ph-H), 8.05 (m, 1H, –C–CH–), 7.73 (m, 1H, Ph-H), 7.66 (m, 1H, Ph-H), 6.99 (m, 1H, Ph-H), 6.99 (m, 2H, –C–CH–); 13C NMR (75 MHz, CDCl3) δ: 167.5, 155.3, 151.9, 150.4, 150.2, 144.2, 139.4, 132.9, 128.3, 118.8, 117.9, 116.4, 114.2, 109.0; HR-ESI-MS m/z: calcd for C16H13N5O ([M + H]+), 291.3102; found, 291.1120; anal. calcd for C16H13N5O: C, 65.97; H, 4.50; N, 24.04; O, 5.49; found: C, 65.98; H, 4.51; N, 24.03; O, 5.48%.
2-Methyl-N-(pyridazin-4-yl)-6-(pyridin-4-ylamino)benzamide (3x). 90.2% yield; mp 193–195 °C; 1H NMR (300 MHz, CDCl3) δ: 11.02 (s, 1H, –NH–N), 10.37 (s, 1H, –NH–), 9.61 (s, 1H, –CH–N), 9.30 (m, 1H, –CH–N), 8.46 (m, 2H, –CH–N), 8.29 (m, 1H, Ph-H), 8.05 (m, 1H, –C–CH–), 7.40 (m, 1H, Ph-H), 6.99 (m, 1H, Ph-H), 6.99 (m, 2H, –C–CH–), 2.48 (s, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ: 164.7, 155.3, 150.4, 150.2, 144.2, 143.8, 143.1, 139.4, 138.0, 121.5, 120.8, 114.2, 113.4, 109.0, 18.1; HR-ESI-MS m/z: calcd for C17H15N5O ([M + H]+), 305.3402; found, 305.1276; anal. calcd for C17H15N5O: C, 66.87; H, 4.95; N, 22.94; O, 5.24; found: C, 66.88; H, 4.95; N, 22.93; O, 5.24%.
N-(Pyridazin-4-yl)-2-(pyrimidin-5-ylamino)benzamide (3y). 91.1% yield; mp 198–200 °C; 1H NMR (300 MHz, CDCl3) δ: 11.02 (s, 1H, –NH–N), 10.37 (s, 1H, –NH–), 9.61 (s, 1H, –CH–N), 9.30 (m, 1H, –CH–N), 8.90 (s, 1H, –N–CH–), 8.50 (s, 2H, –CH
N), 8.39 (m, 1H, Ph-H), 7.73 (m, 1H, Ph-H), 7.66 (m, 1H, Ph-H), 6.99 (m, 1H, Ph-H); 13C NMR (75 MHz, CDCl3) δ: 167.5, 151.9, 150.4, 147.4, 144.2, 143.7, 142.9, 139.4, 132.9, 128.3, 118.8, 117.9, 116.4, 114.2; HR-ESI-MS m/z: calcd for C15H12N6O ([M + H]+), 292.3012; found, 292.1072; anal. calcd for C15H12N6O: C, 61.64; H, 4.14; N, 28.75; O, 5.47; found: C, 61.65; H, 4.13; N, 28.76; O, 5.46%.
2-Methyl-N-(pyridazin-4-yl)-6-(pyrimidin-5-ylamino)benzamide (3z). 92.6% yield; mp 223–235 °C; 1H NMR (300 MHz, CDCl3) δ: 10.37 (s, 1H, –NH–N), 9.61 (s, 1H, –CH–N), 9.30 (m, 1H, –CH–N), 9.23 (s, 1H, –NH–), 8.90 (s, 1H, –N–CH–), 8.50 (s, 2H, –CH
N), 8.29 (m, 1H, Ph-H), 7.40 (m, 1H, Ph-H), 6.90 (m, 1H, Ph-H), 2.48 (s, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ: 164.7, 150.4, 147.4, 144.2, 143.8, 143.7, 143.1, 142.9, 139.4, 138.0, 121.5, 120.8, 114.2, 113.4, 18.1; HR-ESI-MS m/z: calcd for C16H14N6O ([M + H]+), 306.3302; found, 306.1229; anal. calcd for C16H14N6O: C, 62.74; H, 4.61; N, 27.43; O, 5.22; found: C, 62.75; H, 4.60; N, 27.42; O, 5.23%.
4.2. Biological activity
Live subject statement
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of “Chongqing Institute of Chinese Materia Medica” and experiments were approved by the Animal Ethics Committee of “Chongqing Institute of Chinese Materia Medica”.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was sponsored by the Innovation Team Project of Universities in Chongqing (No. CXTDX201601018), the Chongqing Scientific and Technological Innovation Special Project of Social Undertakings and People’s Livelihood Guarantee (No. cstc2015shmszx80060), the Chongqing University Students' Training Project of Innovation and Undertaking (201510637085), the Doctoral Program of Chongqing Normal University (No. 12XLB006), and the Outstanding Achievements Transformation Project of Chongqing Normal University (No. 15XZH08), and the National Natural Science Foundation (21662012, 41866005), China.References
- A. Birbrair, T. Zhang and Z. Wang, et al., Type-2 pericytes participate in normal and tumoral angiogenesis, Am. J. Physiol.: Cell Physiol., 2014, 307, C25–C38 CrossRef CAS PubMed.
- E. Lee, J. Locker and M. Nalesnik, et al., The association of Epstein-Barr virus with smooth-muscle tumors occurring after organ transplantation, N. Engl. J. Med., 1995, 332, 19–25 CrossRef CAS PubMed.
- D. Ambrosi and F. Mollica, On the mechanics of a growing tumor, Int. J. Eng. Sci., 2002, 40, 1297–1316 CrossRef.
- K. Volokh, Stresses in growing soft tissues, Acta Biomater., 2006, 2, 493–504 CrossRef CAS PubMed.
- C. Bernstein, A. Prasad, V. Nfonsam, et al., DNA Damage, DNA Repair and Cancer, New Res Dir DNA Repair, 2013, Vol. 5, pp. 413–465 Search PubMed.
- M. Kastan, DNA damage responses: mechanisms and roles in human disease: 2007 G.H.A. Clowes Memorial Award Lecture, Mol. Cancer Res., 2008, 6, 517–524 CrossRef CAS PubMed.
- F. Cunningham, S. Fiebelkorn and M. Johnson, et al., A novel application of the Margin of Exposure approach: segregation of tobacco smoke toxicants, Food Chem. Toxicol., 2011, 49, 2921–2933 CrossRef CAS PubMed.
- H. Kanavy and M. Gerstenblith, Ultraviolet radiation and melanoma, Semin. Cutaneous Med. Surg., 2011, 30, 222–228 CrossRef CAS PubMed.
- O. Handa, Y. Naito and T. Yoshikawa, Redox biology and gastric carcinogenesis: the role of Helicobacter pylori, Redox Rep., 2011, 16, 1–7 CrossRef CAS PubMed.
- C. Bernstein, H. Holubec and A. Bhattacharyya, et al., Carcinogenicity of deoxycholate, a secondary bile acid, Arch. Toxicol., 2011, 85, 863–871 CrossRef CAS PubMed.
- M. Katsurano, T. Niwa and Y. Yasui, et al., Early-stage formation of an epigenetic field defect in a mouse colitis model, and non-essential roles of T-cells and B-cells in DNA methylation induction, Oncogene, 2012, 31, 342–351 CrossRef CAS PubMed.
- D. Malkin, Li-fraumeni syndrome, Genes Cancer, 2011, 2, 475–484 CrossRef CAS PubMed.
- P. Lichtenstein, N. Holm and P. Verkasalo, et al., Environmental and heritable factors in the causation of cancer-analyses of cohorts of twins from Sweden, Denmark, and Finland, N. Engl. J. Med., 2000, 343, 78–85 CrossRef CAS PubMed.
- L. Shen, Y. Kondo and G. Rosner, et al., MGMT promoter methylation and field defect in sporadic colorectal cancer, J. Natl. Cancer Inst., 2005, 97, 1330–1338 CrossRef CAS PubMed.
- K. Truninger, M. Menigatti and J. Luz, et al., Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer, Gastroenterology, 2005, 128, 1160–1171 CrossRef CAS.
- N. Valeri, P. Gasparini and M. Fabbri, et al., Modulation of mismatch repair and genomic stability by miR-155, Proc. Natl. Acad. Sci. U. S. A., 2005, 107, 6982–6987 CrossRef PubMed.
- A. Facista, H. Nguyen and C. Lewis, et al., Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer, Genome Integr., 2012, 3, 3 CrossRef CAS PubMed.
- C. Bernstein, H. Bernstein and C. Payne, et al., Field defects in progression to gastrointestinal tract cancers, Cancer Lett., 2008, 260, 1–10 CrossRef CAS PubMed.
- H. Rubin, Fields and field cancerization: the preneoplastic origins of cancer: asymptomatic hyperplastic fields are precursors of neoplasia, and their progression to tumors can be tracked by saturation density in culture, BioEssays, 2011, 33, 224–231 CrossRef PubMed.
- B. Vogelstein, N. Papadopoulos and V. Velculescu, et al., Cancer genome landscapes, Science, 2013, 339, 1546–1558 CrossRef CAS PubMed.
- P. Lochhead, A. Chan and R. Nishihara, et al., Etiologic field effect: reappraisal of the field effect concept in cancer predisposition and progression, Mod. Pathol., 2014, 28, 14–29 CrossRef PubMed.
- G. Manning and D. Whyte, The protein kinase complement of the human genome, Science, 2002, 298, 1912–1934 CrossRef CAS PubMed.
- V. Reiterer, P. Eyers and H. Farhan, Day of the dead: pseudokinases and pseudophosphatases in physiology and disease, Trends Cell Biol., 2014, 24, 489–505 CrossRef CAS PubMed.
- J. Murphy, A robust methodology to subclassify pseudokinases based on their nucleotide-binding properties, Biochem. J., 2014, 457, 323–334 CrossRef CAS PubMed.
- P. Eyers and J. Murphy, The evolving world of pseudoenzymes: proteins, prejudice and zombies, BMC Biol., 2016, 14, 98 CrossRef PubMed.
- N. Dhanasekaran and R. Premkumar, Signaling by dual specificity kinases, Oncogene, 1998, 17, 1447–1455 CrossRef CAS PubMed.
- P. Besant, E. Tan and P. Attwood, Mammalian protein histidine kinases, Int. J. Biochem. Cell Biol., 2003, 35, 297–309 CrossRef CAS PubMed.
- T. Stout, P. Foster and D. Matthews, High-throughput structural biology in drug discovery: protein kinases, Curr. Pharm. Des., 2004, 10, 1069–1082 CrossRef CAS PubMed.
- O. Linden, A. Kooistra and R. Leurs, et al., KLIFS: A knowledge-based structural database to navigate kinase-ligand interaction space, J. Med. Chem., 2013, 5 7, 249–277 Search PubMed.
- S. Hanks, Genomic analysis of the eukaryotic protein kinase superfamily: a perspective, Genome Biol., 2003, 4, 111 CrossRef PubMed.
- D. Knighton, J. Zheng and E. Ten, et al., Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase, Science, 1991, 253, 407–414 CrossRef CAS PubMed.
- S. Vlahopoulos and V. Zoumpourlis, JNK: a key modulator of intracellular signaling, Biochemistry, 2004, 69, 844–854 CAS.
- Q. Xu, K. Malecka and L. Fink, et al., Identifying three-dimensional structures of autophosphorylation complexes in crystals of protein kinases, Sci. Signaling, 2015, 8, rs13 CrossRef PubMed.
- P. Wolanin, P. Thomason and J. Stock, Histidine protein kinases: key signal transducers outside the animal kingdom, Genome Biol., 2002, 3, 1–8 CrossRef.
- F. Yakes, J. Chen and J. Tan, et al., Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth, Mol. Cancer Ther., 2011, 10, 2298–2308 CrossRef CAS PubMed.
- G. Roth, A. Heckel and F. Colbatzky, et al., Design, synthesis, and evaluation of indolinones as triple angiokinase inhibitors and the discovery of a highly specific 6-methoxycarbonyl-substituted indolinone(BIBF 1120), J. Med. Chem., 2009, 52, 4466–4480 CrossRef CAS PubMed.
- Y. Oguro, N. Miyamoto and K. Okada, et al., Design, synthesis, and evaluation of 5-methyl-4-phenoxy-5H-pyrrolo [3, 2-d] pyrimidine derivatives: novel VEGFR2 kinase inhibitors binding to inactive kinase conformation, Bioorg. Med. Chem., 2010, 18, 7260–7273 CrossRef CAS PubMed.
- K. Sanphanya, S. Wattanapitayakul and S. Phowichit, et al., Novel VEGFR2 kinase inhibitors identified by the back-to-front approach, Bioorg. Med. Chem. Lett., 2013, 23, 2962–2967 CrossRef CAS PubMed.
- W. Lin, J. Hsu and S. Hsieh, et al., Discovery of 3-phenyl-1H-5-pyrazolylamine derivatives containing a urea pharmacophore as potent and efficacious inhibitors of FMS-like tyrosine kinase-3 (FLT3), Bioorg. Med. Chem., 2013, 21, 2856–2867 CrossRef CAS PubMed.
- T. Cho, S. Dong and F. Jun, et al., Novel potent orally active multitargeted receptor tyrosine kinase inhibitors: synthesis, structure-activity relationships, and anti-tumor activities of 2-indolinone derivatives, J. Med. Chem., 2010, 53, 8140–8149 CrossRef PubMed.
- R. Hudkins, A. Zulli and T. Underiner, et al., 8-THP-DHI analogs as potent type I dual TIE-2/VEGFR2 receptor tyrosine kinase inhibitors, Bioorg. Med. Chem. Lett., 2010, 20, 3356–3360 CrossRef CAS PubMed.
- B. Yu, L. Tang and Y. Li, et al., Design, synthesis and anti-tumor activity of 4-aminoquinazoline derivatives targeting VEGFR2 tyrosine kinase, Bioorg. Med. Chem. Lett., 2012, 22, 110–114 CrossRef CAS PubMed.
- P. Plé, F. Jung and S. Ashton, et al., Discovery of AZD2932, a new quinazoline ether inhibitor with high affinity for VEGFR2 and PDGFR tyrosine kinases, Bioorg. Med. Chem. Lett., 2012, 22, 262–266 CrossRef PubMed.
- S. Rizvi, H. Siddiqui and M. Nisar, et al., Discovery and molecular docking of quinolyl-thienyl chalcones as antiangiogenic agents targeting VEGFR2 tyrosine kinase, Bioorg. Med. Chem. Lett., 2012, 22, 942–944 CrossRef CAS PubMed.
- A. Kiselyov, M. Semenova and V. Semenov, 3, 4-Disubstituted isothiazoles: novel potent inhibitors of VEGF receptors 1 and 2, Bioorg. Med. Chem. Lett., 2009, 19, 1195–1198 CrossRef CAS PubMed.
- Y. Oguro, D. Cary and N. Miyamoto, et al., Design, synthesis, and evaluation of novel VEGFR2 kinase inhibitors: discovery of [1, 2, 4] triazolo [1, 5-a] pyridine derivatives with slow dissociation with slow dissociation kinetics, Bioorg. Med. Chem., 2013, 21, 4714–4729 CrossRef CAS PubMed.
- A. Gangjee, N. Zaware and S. Raghavan, et al., Synthesis and biological activity of 5-chloro-N-4- substituted phenyl-9H-pyrimido [4, 5-b] indole-2, 4-diamines as vascular endothelial growth factor receptor-2 inhibitors and antiangiogenic agents, Bioorg. Med. Chem., 2013, 21, 1857–1864 CrossRef CAS PubMed.
- Q. Liu, Y. Sabnis and Z. Zhao, et al., Developing irreversible inhibitors of the protein kinase cysteinome, Chem. Biol., 2013, 20, 146–159 CrossRef CAS PubMed.
- A. Wissner, H. Fraser and C. Ingalls, et al., Dual irreversible kinase inhibitors: quinazoline-based inhibitors incorporating two independent reactive centers with each targeting different cysteine residues in the kinase domains of EGFR and VEGFR2, Bioorg. Med. Chem., 2007, 15, 3635–3648 CrossRef CAS PubMed.
- S. Kumar and M. Tiwari, Topomer-CoMFA-based predictive modelling on 2,3-diaryl substituted-1,3-thiazolidin-4-ones as non-nucleoside reverse transcriptase inhibitors, Med. Chem. Res., 2015, 24, 245–257 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |