
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn-line pre-treatment, separation, and nanoelectrospray mass spectrometric determinations for pesticide metabolites and peptides based on a modular microfluidic platform†
Yinyin Hao‡
ab,
Yajing Bao‡b,
Xueying Huangb,
Yijun Hua and
Bo Xiong *b
*b
aSchool of Mathematics and Statistics, Wuhan University, Wuhan, China
bKey Laboratory of Pesticides & Chemical Biology, Ministry of Education, Institute of Public Health and Molecular Medicine Analysis, College of Chemistry, Central China Normal University, Wuhan, China. E-mail: bx@mail.ccnu.edu.cn
First published on 28th November 2018
Abstract
In order to address time-consuming sample pre-treatment and separation prior to mass spectrometry (MS) identifications, highly integrated chips were developed, but damage to any functional unit in these chips would result in complete replacement. Herein, we propose a modular microfluidic platform comprising pre-treatment, liquid chromatography (LC) separation and nanoelectrospray ionization (nESI) chips for on-line enrichment, separation and nESI MS detection of pesticide metabolites and peptides. The pre-treatment chip is applicable in enriching pyridalyl and its metabolites, and it achieves optimal desalination efficiency, 98.5%, for polymerase chain reaction products. Additionally, the LC separation chip was fully characterised, and it demonstrated satisfactory separation efficiency, quantification ability and pressure durability. Finally, the modular microfluidic platform was used to identify the peptides in trypsin-digested casein. Four additional peptides were identified, indicating an improvement in detection ability compared with using off-line zip tips coupled with MS investigations. Because the proposed modular platform can significantly reduce manual work, it would be a potential tool to achieve high throughput and automatic MS identifications with low sample consumptions.
Introduction
In the past two decades, the development of metabolomics and proteomics has facilitated the improvement of various mass spectrometry (MS) approaches in sensitivity and throughput.1–3 Although liquid chromatography (LC) coupled with nanoelectrospray ionization (nESI) MS achieves enhanced sensitivity with limited samples, its whole workflow is complicated and time-consuming due to comprehensive pre-treatments for samples.4,5 Microfluidic devices being compatible with nESI MS present significant advantages in high throughput and automatic sample manipulations.6–8 Therefore, microfluidic devices coupled with nESI MS, particularly microfluidic LC-nESI MS, are regarded as an ideal tool to investigate metabolites and peptides.9,10Various enrichment pre-treatments, such as solid phase extraction (SPE),11 are generally indispensable in metabolic identifications due to the low content of metabolites.12–14 Nevertheless, complex manual workflow in these strategies reduces their throughputs if enrichment units are off-line coupled with the MS detections.15 Moreover, satisfactory recoveries are generally required because of the low sample content.16–18 In this sense, developing an on-line pre-treatment, separation and MS identification analytical platform should be of great practical significance.19
Additionally, peptide samples in proteomics are generally prepared by enzyme-assisted digestion20 or 2D gel electrophoresis,21 both of which introduce salts into samples.6,22 On-line coupling of microfluidic devices with matrix-assisted laser desorption ionization MS (MALDI MS) is complex, results in relatively poor throughput,23 although the salt tolerance of MALDI MS is excellent.8 In contrast, despite an inevitable sample loss,24 the desalination prior to nESI MS identifications is indispensable, otherwise the ionization suppression would be considerable due to the salts in the samples. Despite several microfluidic desalination strategies,25–28 desalination using zip tips is still one of the most useful approaches in proteomics. It is interesting to note that the stationary phase in both the SPE column and the zip tip is packed silica particles,29 and this provides the feasibility of developing a universal micro-device that not only enriches analytes in metabolomics but also desalinates peptides in proteomics.
Moreover, monolithic nESI nozzles30 attributed to their improved dead volume31–33 have revealed an enhanced reproducibility compared with that of integrating commercial nESI tips in microfluidic devices.34,35 The monolithic nESI emitters have been used in real-time mass calibration,36 investigating perfluoroalkyl acids37 and other pesticides,38–40 all of which indicated their excellent compatibility with various microfluidic applications. In this sense, a monolithic nESI ionization module would be a useful component in a versatile microfluidic platform for metabolic and proteomic identifications.41–43
Furthermore, modular microfluidic platforms have shown preponderances in simplifying their coupling with other functional components, such as the injection valve and mass spectrometer.44 Finally, their modular configuration would also allow the replacement of the damaged functional units, which conventionally result in abandoning the whole highly integrated microfluidic device.45,46
Herein, we propose a microfluidic platform, composed of pre-treatment, separation, and nESI modules, to be coupled with a time-of-flight mass spectrometer (TOF-MS) for the investigations of pesticide metabolites and peptides. A C18 SPE chip was prepared for enriching pesticide metabolites and desalinating peptide samples. Furthermore, microfluidic LC and monolithic nESI modules were prepared and on-line-coupled with the pre-treatment module and a TOF-MS to inhibit sample loss, thus improving the sensitivity. The desalination efficiency of the proposed microfluidic platform and its applicability in peptide identifications were compared with those of its counterparts by inductively coupled plasma atomic emission spectroscopy (ICP AES) and MALDI TOF-MS. Additionally, the proposed platform was applied to determine pyridalyl and its metabolites in the excrement of Helicoverpa armigera (H. armigera) and identify peptides in trypsin-digested casein. Considering the advantages in its user-friendly aspect, the proposed modular microfluidic platform is an ideal potential option to implement metabolic and proteomic identifications.
Materials and methods
Fabrication of three modules
Photomasks (ESI, Fig. S1A and B†) were designed with FreeHand (Adobe, San Jose, USA) and produced by Caifeng Graphic (Wuhan, China). Micro-channels (40 μm in depth) in the pre-treatment module (ESI, Fig. S1A†) comprised two equilateral triangles (30° apex angle, 2.5 mm wide, and 5.4 mm high) and one rectangle region (2 mm × 2.5 mm). The lengths of the rectangle regions in pre-treatment and LC separation modules were 2.5 mm and 20 mm, respectively (ESI, Fig. S1A and B†). Micro-pillar (20 μm diameter) arrays were integrated in both modules to avoid the collapse of channels.Molds of three microfluidic modules were prepared with Su-8 photoresist (MicroChem, Westborough, USA) according to a standard photolithography approach.36,43 Briefly, silicon wafers with photoresist were spun at 1700 rpm for 30 s, and sequentially baked at 65 °C and 95 °C for 2 min and 8 min, respectively. After exposing to 180 mJ cm−2 UV light (350 nm) for 5 min, the wafers were then immersed in the developer solution for 2 min after a post bake process (65 °C and 95 °C for 1 min and 8 min), and finally cleaned with isopropyl alcohol.
Pre-treatment and LC separation chips (Fig. 1A and B) were both composed of a patterned polydimethylsiloxane (PDMS) slab and a glass slide, both of which bonded to each other after treating in oxygen plasma. A holder, consisting of two aluminium plates with drilled holes, a pair of screw nut and mount, a rubber O-ring (1.8 mm i.d., 4.0 mm o.d.) and a porous plate filter were used for maintaining silica particles (the stationary phase) and introducing various solutions from a tubing. Silica particles (5 μm diameter) were dispersed in methanol aqueous solution (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, v/v), which was immediately infused into the enrichment and separation chips. Pre-treatment and LC separation modules (ESI, Fig. S1C and D†) were then obtained after repeatedly infusing the stationary phase. In the as-prepared nESI chip (Fig. 1C), which was fabricated according to a reported method,36,41 a sharp and symmetrical spray nozzle (Fig. 1D) was observed, and it would facilitate a stable electrospray ionization.
:50, v/v), which was immediately infused into the enrichment and separation chips. Pre-treatment and LC separation modules (ESI, Fig. S1C and D†) were then obtained after repeatedly infusing the stationary phase. In the as-prepared nESI chip (Fig. 1C), which was fabricated according to a reported method,36,41 a sharp and symmetrical spray nozzle (Fig. 1D) was observed, and it would facilitate a stable electrospray ionization.
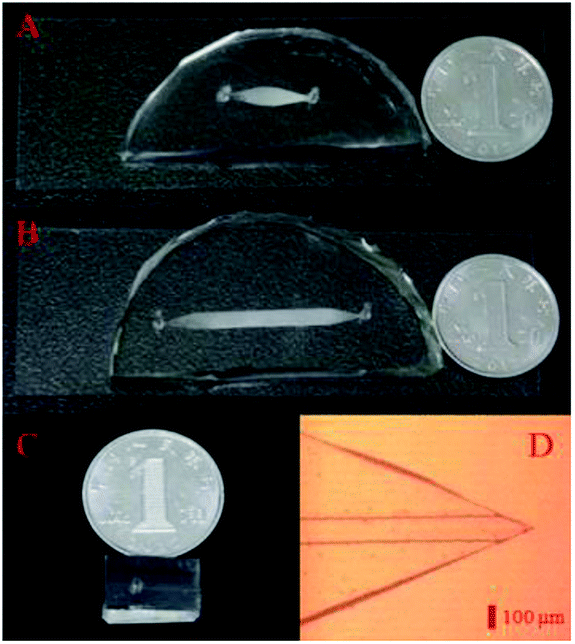 | ||
| Fig. 1 Pre-treatment (A), LC separation (B) and nESI (C) chips in the modular microfluidic platform. (D) Monolithic nESI nozzle in the nESI chip under the microscope. | ||
Instrumentations
Pre-treatment, separation, and nESI chips were coupled with a manual injection valve, a high pressure pump, and a 6224 TOF-MS (Agilent, Santa Clara, USA), as shown in the configuration of the modular microfluidic platform (Fig. 2) and its instrumental setup (ESI, Fig. S2†). In the microfluidic platform, the pre-treatment chip was used to extract or desalinate samples (Fig. 2A), and the sample solution in the sample loop was then infused into the separation chip (Fig. 2B), which was coupled with the TOF-MS by the on-line nESI chip.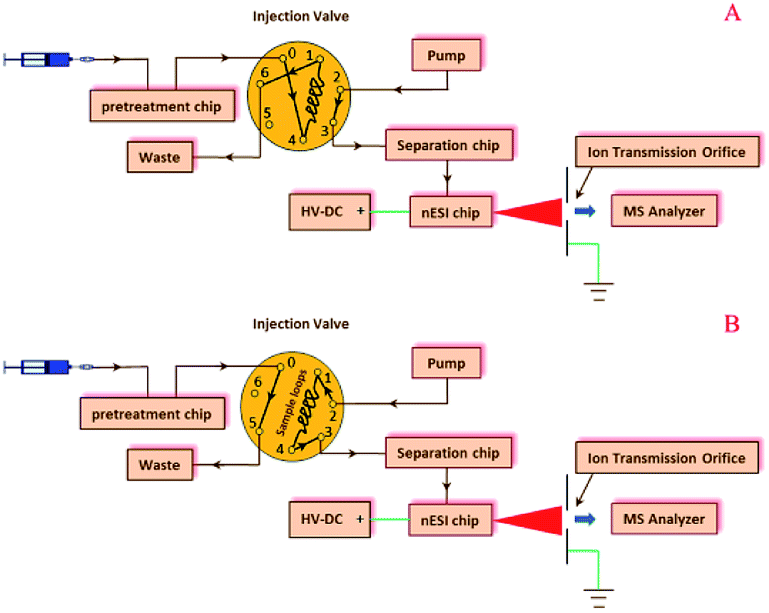 | ||
| Fig. 2 Schematic configurations of the modular microfluidic platform consisting of pretreatments, LC separation and nESI chips, an injection valve, and a TOF-MS when loading (A) and investigating (B) samples. | ||
Evaluations of three modules
The pre-treatment module was evaluated by extracting the excrements of H. armigera fed with pyridalyl. Excrements (1 mg) were ground in liquid nitrogen and dispersed in three-fold volume of acetone. The mixture was treated in an ultrasound generator (Shumei instrument, Kunshan, China) at 26 °C for 5 min, and then centrifuged at 7000 rpm for 10 min. After extracting the residue for another two times, all supernatants were combined and concentrated to 50 μL in a nitrogen concentrator (Do-Chrom, Tianjin, China). Then, the pre-treatment module was used to extract the concentrated solution according to the following procedure. First, the pre-treatment chip was sequentially cleaned by infusing methanol (100 μL), acetonitrile aqueous solution (40 μL, 50:50, v/v), water (20 μL), acetonitrile aqueous solution (40 μL, 50:50, v/v), and methanol (20 μL). The concentrated solution was then loaded in the pre-treatment chip, which was sequentially eluted by methanol (30 μL) and acetone (30 μL). All elution fractions were collected and analyzed by a 1260 HPLC coupled with a 6224 TOF-MS (Agilent, Santa Clara, USA).
Additionally, a polymerase chain reaction (PCR) product (ESI, Table S1†) was desalinated with the pre-treatment module to evaluate its desalination efficiency. The module was first cleaned by infusing a mixture of methanol (50 μL), water (49 μL) and acetic acid (1 μL). Then, two methods were compared to desalinate the PCR product. In the first method, the chip was sequentially rinsed with water (100 μL), ammonia aqueous solution (100 μL), water (100 μL) and ammonium acetate (100 μL). After loading the PCR product (40 μL), the pre-treatment chip was then eluted by 40 mM ammonium hydrogen carbonate (40 μL) and 20% methanol aqueous solution (40 μL). In the second method, the pre-treatment module was rinsed by 0.1% TFA aqueous solution (30 μL) and 0.1% TFA in acetonitrile (30 μL) after loading the PCR product (40 μL). All desalinated elutions were collected and digested in two-fold volume of the mixture composed of the nitric acid, sulfuric acid and water (2:2:1, v/v/v). Digested elutions were dried in a mild nitrogen flow, re-dissolved in water, and probed by ICP-AES (iCAP6000, Thermo Scientific, Waltham, USA) to investigate the achieved desalination efficiencies with the preceding two methods.
The separation module was evaluated by monitoring RhB (5 ng μL−1), reserpine (5 ng μL−1) and juvenile hormone III (5 ng μL−1). Its pressure durability was tested by injecting methanol aqueous solutions (90:10, v/v) at different infusion rates.
Identifications of trypsin-digested peptides
First, the enrichment chip was sequentially rinsed by A (50% acetonitrile aqueous solution with 0.1% formic acid, 100 μL) and B (0.1% formic acid aqueous solution, 100 μL) with a 20 μL min−1 infusion rate. Prior to investigations, peptide samples were conditioned to pH 2.0–3.0 by adding formic acid. After loading the digested casein (40 μL), peptides were desalinated by B (30 μL) and eluted by A (20 μL) with a 15 μL min−1 infusion rate. All eluted peptides were then on-line-introduced to the separation module and identified by the nESI module and the TOF-MS. At each peak in the total ion chromatogram, a deconvolution process was imposed to all the collected m/z of species, thus obtaining identified peptides. As a comparison, the digested casein was desalinated by a zip tip (Waters, Milford, USA) and off-line-determined by a TOF-MS.Moreover, the modular microfluidic platform was used to probe pyridalyl and metabolites in the concentrated solution from the excrements of H. armigera. Finally, pyridalyl solutions at different concentrations were detected to evaluate the quantification with the modular microfluidic platform.
Trypsin-digested α-casein
α-Casein (5 mg) was dissolved in ammonium bicarbonate buffer (1 mL, 50 mM, pH 8.2) and digested by trypsin (125 μg) for 16 h at 37 °C. The solution was then diluted to 10 ng μL−1 and investigated by a MALDI TOF-MS (G2 HDMS, Waters, Milford, USA) for comparison.Chemicals
Poly(dimethylsiloxane) (PDMS), α-casein, silica particles, juvenile hormone III, rhodamine B (RhB) and reserpine were purchased from Sigma-Aldrich Inc. (St. Louis, USA). All organic solvents were obtained from Merck Ltd. (Darmstadt, Germany). Double-distilled water used in this study was acquired from a water purified system (Millipore, Bedford, USA).Results and discussion
Evaluations of the pre-treatment chip
Extracted ion chromatograms (EICs) of pyridalyl and its two potential metabolites, MP2 and MP3, were investigated (Fig. 3), and all peaks corresponding to target substances were observed. Moreover, their retention times were similar to those of their counterparts in a reported work with an SPE column.47 These results demonstrated the applicability of the pre-treatment module as an alternative of SPE to extract pyridalyl and its metabolites in the excrements of H. armigera. MP2 was only observed in the acetone fraction, while pyridalyl and MP3 were detected in both methanol and acetone elutions. This result was probably because MP2 was more polar than pyridalyl and MP3, resulting in its enhanced recovery in acetone.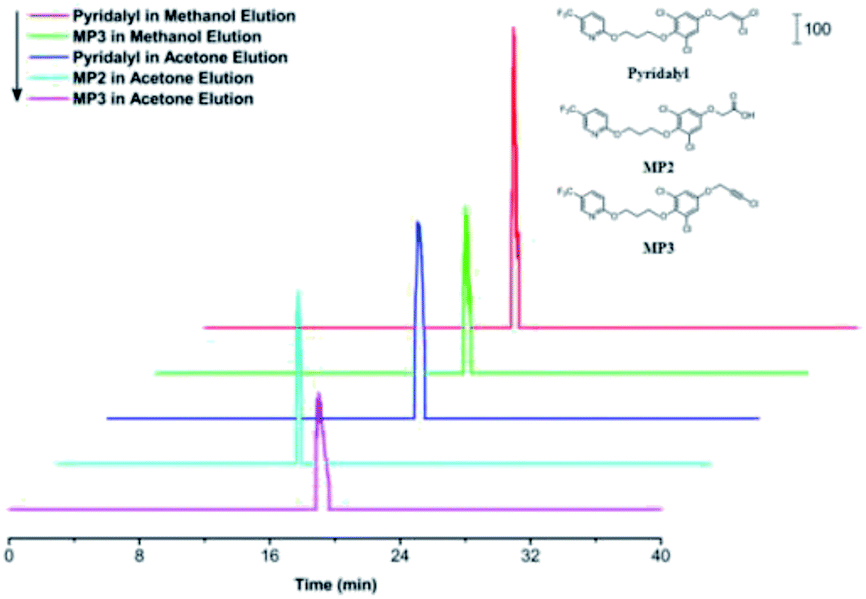 | ||
| Fig. 3 EICs of pyridalyl and two metabolites (MP2 and MP3) in the excrements of H. armigera based on the pre-treatment chip and HPLC-MS. These figures were extracted from total ion chromatograms with m/z 489.975, 440.027 and 453.999, all of which were the mass-to-charge ratio of molecular ions ([M + H]+) for pyridalyl, MP2 and MP3. | ||
The desalination performance of the pre-treatment chip was also evaluated by treating PCR products. The residual K+ contents in the PCR products (Table 1) were compared between two desalination methods. The residual amount obtained by the first method was less than one tenth of that obtained from its counterpart by the second method, which achieved a 21.9% residue relative to its counterpart in the blank control. These results demonstrate that the first method with 98.5% desalination efficiency was superior to the second method (83.1%), and both methods showed decent desalination efficiencies. A zip tip was also used for desalination and achieved a 94.6% clearance rate, indicating the effect of the pre-treatment chip.
Evaluations of the LC separation chip
The LC separation chip was evaluated by detecting RhB under different injection volumes and flow rates (Table 2), and the optimal experimental conditions were studied. Mean retention times and peak widths decreased with an increase i flow rate from 10 μL min−1 to 60 μL min−1 under different injection volumes. This occurred because the greater flow rate inhibited peak broadening within 10–60 μL min−1. Moreover, the best plate numbers under different injection volumes were always achieved with an optimal 30 μL min−1 flow rate, suggesting an appropriate balance between the retention of sample with the stationary phase and its peak broadening under this condition. Furthermore, the mean plate number with a 2 μL injection volume enhanced compared with that of its counterpart with a 5 μL injection volume. This occurred because the limited bonding sites of the stationary phase in the LC separation chip may be relatively saturated when injecting a greater volume of the sample. Thus, the peak broadening deteriorated, resulting in a comparably smaller theoretical plate number.| Flow rate (μL min−1) | Injection volumea (μL) | Mean retention time (min) | RSD (%, n = 3) | Mean half peak width (min) | RSD (%, n = 3) | Mean theoretical plate number | RSD (%, n = 3) |
|---|---|---|---|---|---|---|---|
| a The RhB solution (5 ng μL−1) was injected in all determinations. | |||||||
| 10 | 2 | 4.34 | 2.3 | 0.662 | 4.6 | 238 | 2.2 |
| 20 | 2 | 3.42 | 3.5 | 0.458 | 3.2 | 310 | 7.3 |
| 30 | 2 | 2.70 | 4.7 | 0.338 | 6.2 | 356 | 3.5 |
| 40 | 2 | 2.06 | 3.8 | 0.265 | 3.8 | 333 | 8.1 |
| 50 | 2 | 1.64 | 2.9 | 0.253 | 4.6 | 232 | 2.4 |
| 60 | 2 | 1.38 | 5.1 | 0.215 | 2.7 | 230 | 2.6 |
| 10 | 5 | 4.38 | 3.1 | 0.782 | 2.9 | 174 | 2.5 |
| 20 | 5 | 3.51 | 3.6 | 0.514 | 3.4 | 257 | 4.3 |
| 30 | 5 | 2.81 | 6.7 | 0.376 | 1.7 | 309 | 6.3 |
| 40 | 5 | 2.08 | 4.5 | 0.308 | 7.8 | 252 | 2.9 |
| 50 | 5 | 1.69 | 5.2 | 0.291 | 2.6 | 187 | 3.1 |
| 60 | 5 | 1.44 | 6.3 | 0.268 | 2.1 | 158 | 3.7 |
Additionally, the LC separation module was used to separate a mixture of reserpine ([M + H]+, m/z 609.28) and juvenile hormone III ([M + H]+, m/z 267.19). Their peaks in EICs (Fig. 4) were observed with different retention times. Furthermore, two sample peaks were observed, which revealed that the LC module yielded acceptable separation efficiency and peak broadening, indicating a uniform filling of the silica particles. Theoretical plate numbers for reserpine and juvenile hormone III with the LC separation chip were 16.0 and 187, respectively, and those for their counterparts were 131 and 854, respectively, with a traditional LC, revealing an improved separation performance compared with those for the proposed LC separation chip. This occurred because the LC separation chip was more compatible with low-volume samples (in nL and pL), and a sample injection in microliters would result in peak width broadening, thus deteriorating the separation performance of the LC separation chip.
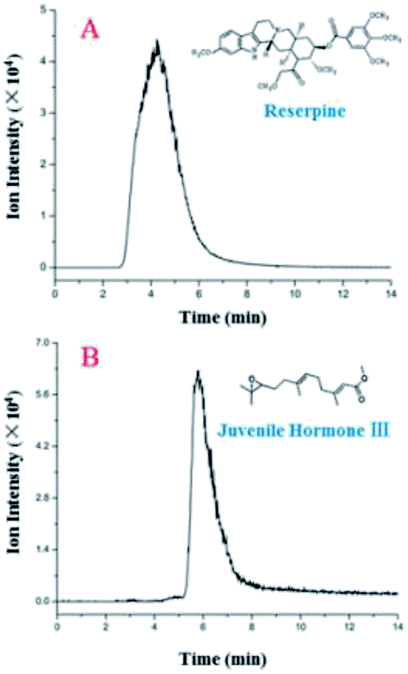 | ||
| Fig. 4 EICs of reserpine (A) and juvenile hormone III (B) with a 2 μL sample (5 ng μL−1) injection and a 30 μL min−1 flow rate. | ||
Pyridalyl solutions at different concentrations were probed to evaluate the quantification with the LC separation module. All peak areas and their errors are presented in a linear calibration curve (Fig. 5). The linear range was 0.26–3.78 ng μL−1, and the limit of detection (s/n = 3) was 0.14 ng μL−1, which was relative to those of its counterpart from a commercial LC column, which achieved a linear range and limit of detection as 0.26–5.67 and 0.11 ng μL−1, respectively. All of these results suggest the applicability of the LC separation module.
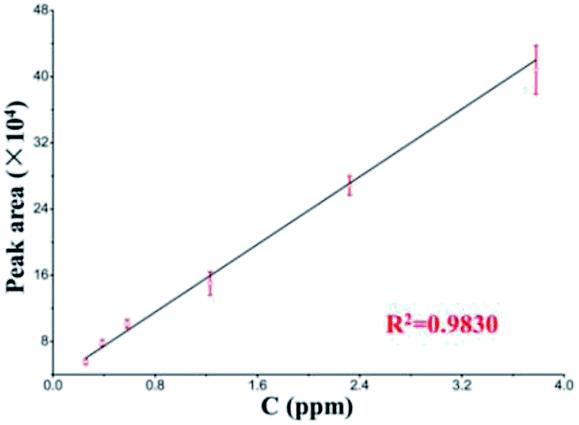 | ||
| Fig. 5 Linear calibration curve of pyridalyl (n = 5) based on the LC separation chip coupled with commercial TOF-MS. | ||
The pressure durability of LC separation module was tested by infusing methanol aqueous solution (90:10, v/v) under different flow rates. The mean pressure within five minutes was 1.07 bar at the optimal flow rate (30 μL min−1), and the pressure increased to 1.42 bar, 1.66 bar and 1.85 bar at 50 μL min−1, 60 μL min−1, and 70 μL min−1, respectively. When applying an 80 μL min−1 flow rate, the pressure was stable within 1.92–2.18 bar for 29 hours. These results indicate a decent pressure durability of the LC separation chip.
Identifications of peptides with the modular microfluidic platform
First, the modular microfluidic platform was evaluated by investigating reserpine. The separation result (ESI, Fig. S3†) and linear range (0.20–4.18 ng μL−1) was decent. These results indicate an appropriate on-line coupling of the nESI chip with the TOF-MS and other modules in the microfluidic platform.Both the pre-treatment module and a zip tip were off-line coupled with TOF-MS to compare their performances in desalinating trypsin-digested casein (Fig. 6). Two peptides (m/z 1266.7 and 1383.7) were identified after the deconvolutions in both strategies, and their intensities (m/z 1266.7, 1383.7) with the pre-treatment module (Fig. 6A) were similar compared with those of their counterparts using the zip tip. Considering both peptides were also identified by the standard method (MALDI TOF-MS), these results supported the applicability of the pre-treatment module in pre-treating peptides.
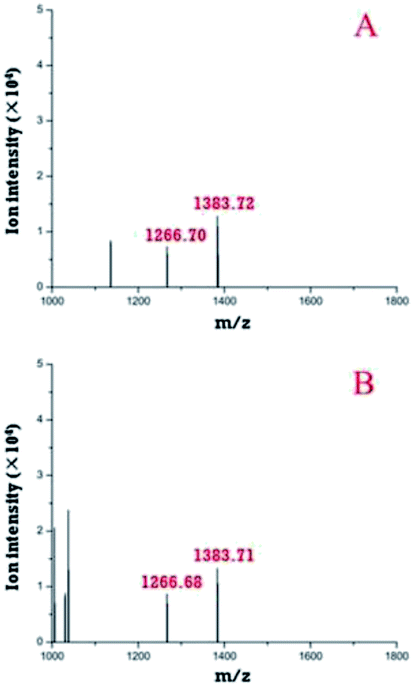 | ||
| Fig. 6 Deconvoluted peptides identified by off-line coupling the pre-treatment chip (A) and a zip tip (B) with the TOF-MS. | ||
Additionally, another peptide (m/z 1760.02) was identified (Fig. 7A) when the pre-treatment module was on-line-coupled with the nESI chip. This result may be attributed to the comparably smaller dead volume and less sample loss relative to their counterparts when off-line-coupling the pre-treatment chip and the TOF-MS. When all three modular chips were on-line-coupled (Fig. 7B), the intensities of three peptides (m/z 1266.8, 1383.79, and 1760.03) increased compared with those of their counterparts in the off-line and partly on-line configurations (Fig. 6A and 7A). Furthermore, additional three peptides (m/z 1192.85, 1208.83 and 1232.81) were also identified from the acetone elution (Fig. 7D) with the proposed modular platform. Moreover, three peptides (m/z 1192.85, 1232.81 and 1266.76) in Fig. 7D were not observed in the acetone elution without the separation chip (Fig. 7C). The total ion chromatogram of the digested casein with the proposed microfluidic platform was similar to that obtained from conventional LC-MS, in which total ion intensities were recorded as a function of time. Preceding results reveal that the existence of the separation chip in the proposed modular platform improved its performance in identifying peptides.
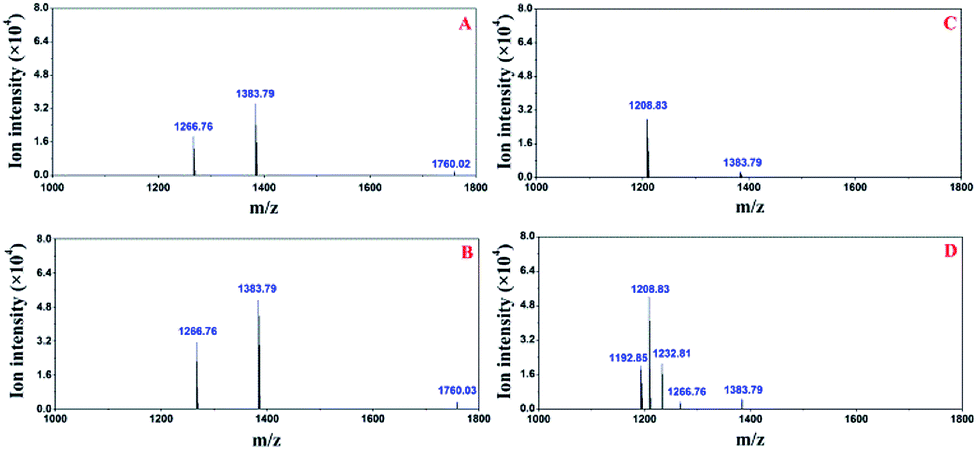 | ||
| Fig. 7 Deconvoluted peptides identified from methanol and acetone elutions with the modular microfluidic platform. (A and B) Identified peptides in the methanol elution without and with the separation chip, (C and D) identified peptides in the acetone elution without and with the separation chip in the proposed microfluidic platform. | ||
Finally, the reproducibility of the modular platform was evaluated by on-line-probing the trypsin-digested casein continuously for twenty times. All peptides were observed in each determination, and the relative standard deviation of their ion intensities was between 7.2–27.3% (n = 20), which was comparable with that of their counterparts with the LC column (5.8–23.6%, n = 20). Moreover, the average duration for each on-line pre-treatment and identification of peptides in various elution fractions was approximately 60 minutes, which was comparable relative to that required for traditional methods, consisting of manual pre-treatment with zip tips and off-line MS identifications. Because the proposed method avoids a great deal of manual operation, the modular microfluidic platform as a conceptual prototype reveals the possibility to pursue completely automatic identification for low volume samples in proteomics and metabolomics.
Conclusions
A modular microfluidic platform composed of independent pre-treatment, LC separation, and nESI chips, was developed for the analysis of pesticide metabolites and peptides. The pre-treatment module showed satisfactory performances in extracting pyridalyl and its metabolites in the excrement of H. armigera and desalinating PCR products. Moreover, the LC separation chip demonstrated its ability in determining RhB, reserpine, juvenile hormone III, and pyridalyl. It also showed decent durability and stability in the pressure test. Finally, improved identification of peptides was achieved with the on-line desalination, separation, and nESI MS detection for trypsin-digested casein by the modular microfluidic platform. These results indicate the potential of this proposed method in elevating the identification throughput and alleviating the laborious work in metabolomics and proteomics.PDMS is an excellent material to facilitate easy fabrications of microfluidic chips, albeit with reported absorbance, which could be inhibited by immersion in solvents42 or surface modifications.36 Additionally, the separation efficiency of the separation chip in the proposed platform was worse compared with that of a commercial LC column due to the comparably poor pressure durability of the chip made of PDMS. If a glass chip with the same configuration could be prepared to replace the PDMS LC chip, the achieved separation efficiency would be effectively improved. If an automated switching valve, which is capable of regulating various solution directions, could be integrated in the proposed platform, it would clearly improve the robustness and the detection throughput of the proposed platform.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
Authors appreciate Prof. Hongying Zhong, Dr Xuemei Tang and Dr Pei Liang for MALDI TOF-MS and ICP-AES determinations. This research was financially supported by the program of introducing talents of discipline to Universities of China (111 Program, B17019), self-determined research funds of CCNU from the colleges' basic research and operation of MOE (CCNU18KYZHSY7), and NSFC of China (21205044).Notes and references
- J. Xie, Y. N. Miao, J. Shih, Y. C. Tai and T. D. Lee, Anal. Chem., 2005, 77, 6947–6953 CrossRef CAS PubMed.
- W. C. Yang and A. T. Woolley, Journal of the Association for Laboratory Automation, 2010, 15, 198–209 CrossRef CAS PubMed.
- A. J. Chetwynd and A. David, Talanta, 2018, 182, 380–390 CrossRef CAS PubMed.
- C. Krisp, H. Yang, R. van Soest and M. P. Molloy, Mol. Cell. Proteomics, 2015, 14, 1708–1719 CrossRef CAS PubMed.
- D. Gao, H. Liu, Y. Jiang and J.-M. Lin, Lab Chip, 2013, 13, 3309–3322 RSC.
- N. Nordman, T. Sikanen, S. Aura, S. Tuomikoski, K. Vuorensola, T. Kotiaho, S. Franssila and R. Kostiainen, Electrophoresis, 2010, 31, 3745–3753 CrossRef CAS PubMed.
- W. A. Black, B. B. Stocks, J. S. Mellors, J. R. Engen and J. M. Ramsey, Anal. Chem., 2015, 87, 6280–6287 CrossRef CAS PubMed.
- X. J. Feng, B. F. Liu, J. J. Li and X. Liu, Mass Spectrom. Rev., 2015, 34, 535–557 CrossRef CAS PubMed.
- M. A. Bynum, H. F. Yin, K. Felts, Y. M. Lee, C. R. Monell and K. Killeen, Anal. Chem., 2009, 81, 8818–8825 CrossRef CAS PubMed.
- J. Gao, J. Xu, L. E. Locascio and C. S. Lee, Anal. Chem., 2001, 73, 2648–2655 CrossRef CAS PubMed.
- J. R. Enders, C. C. Marasco, J. P. Wikswo and J. A. McLean, Anal. Chem., 2012, 84, 8467–8474 CrossRef CAS PubMed.
- S. Mao, D. Gao, W. Liu, H. Wei and J.-M. Lin, Lab Chip, 2012, 12, 219–226 RSC.
- N. Nuchtavorn, W. Suntornsuk, S. M. Lunte and L. Suntornsuk, J. Pharm. Biomed. Anal., 2015, 113, 72–96 CrossRef CAS PubMed.
- D. Gao, H. X. Liu, J. M. Lin, Y. N. Wang and Y. Y. Jiang, Lab Chip, 2013, 13, 978–985 RSC.
- R. G. Wu and Y. S. Fung, Bioanalysis, 2015, 7, 907–922 CrossRef CAS PubMed.
- M. D. Brennan, M. L. Rexius-Hall, L. J. Elgass and D. T. Eddington, Lab Chip, 2014, 14, 4305–4318 RSC.
- Q. Ramadan and M. A. M. Gijs, Lab Chip, 2015, 15, 614–636 RSC.
- P. M. van Midwoud, E. Verpoorte and G. M. M. Groothuis, Integr. Biol., 2011, 3, 509–521 RSC.
- P. Dittrich and A. J. Ibanez, Electrophoresis, 2015, 36, 2196–2206 CrossRef CAS PubMed.
- Z.-Y. Chen, H. N. Abdelhamid and H.-F. Wu, Rapid Commun. Mass Spectrom., 2016, 30, 1403–1412 CrossRef CAS PubMed.
- T. C. Chao and N. Hansmeier, Proteomics, 2013, 13, 467–479 CrossRef CAS PubMed.
- A. A. Dawoud, H. A. Sarvalyal and I. M. Lazar, Electrophoresis, 2007, 28, 4645–4660 CrossRef CAS PubMed.
- T. T. Thuy, M. Inganas, G. Ekstrand and G. Thorsen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 2803–2810 CrossRef CAS PubMed.
- H. Yin, K. Killeen, R. Brennen, D. Sobek, M. Werlich and T. van de Goor, Anal. Chem., 2005, 77, 527–533 CrossRef CAS PubMed.
- S. H. Roelofs, M. van Soestbergen, M. Odijk, J. C. T. Eijkel and A. van den Berg, Ionics, 2014, 20, 1315–1322 CrossRef CAS.
- S. H. Roelofs, B. Kim, J. C. T. Eijkel, J. Han, A. van den Berg and M. Odijk, Lab Chip, 2015, 15, 1458–1464 RSC.
- S. H. Roelofs, A. van den Berg and M. Odijk, Lab Chip, 2015, 15, 3428–3438 RSC.
- V. V. Swaminathan, P. Dak, B. Reddy, E. Salm, C. Duarte-Guevara, Y. Zhong, A. Fischer, Y. S. Liu, M. A. Alam and R. Bashir, Appl. Phys. Lett., 2015, 106, 053105 CrossRef PubMed.
- M. Ghitun, E. Bonneil, M. H. Fortier, H. F. Yin, K. Killeen and P. Thibault, J. Sep. Sci., 2006, 29, 1539–1549 CrossRef CAS PubMed.
- L. Jian, R. Kyung-Won, R. Nayak and D. R. Knapp, Int. J. Mass Spectrom., 2007, 259, 65–72 CrossRef.
- E. Mery, F. Ricoul, N. Sarrut, O. Constantin, G. Delapierre, J. Garin and F. Vinet, Sens. Actuators, B, 2008, 134, 438–446 CrossRef CAS.
- P. Hoffmann, M. Eschner, S. Fritzsche and D. Belder, Anal. Chem., 2009, 81, 7256–7261 CrossRef CAS PubMed.
- W. Kim, M. Guo, P. Yang and D. Wang, Anal. Chem., 2007, 79, 3703–3707 CrossRef CAS PubMed.
- K. Kleparnik, Electrophoresis, 2015, 36, 159–178 CrossRef CAS PubMed.
- X. Wang, L. Yi, N. Mukhitov, A. M. Schrell, R. Dhumpa and M. G. Roper, J. Chromatogr. A, 2015, 1382, 98–116 CrossRef CAS PubMed.
- L. Wang, Y. Wang, S. Jiang, M. Ye, P. Su and B. Xiong, J. Chromatogr. A, 2016, 1470, 1–8 CrossRef CAS PubMed.
- H. Zhang, J. Ou, Y. Wei, H. Wang, Z. Liu and H. Zou, J. Chromatogr. A, 2016, 1440, 66–73 CrossRef CAS PubMed.
- Y. Hua, A. B. Jemere, J. Dragoljic and D. J. Harrison, Lab Chip, 2013, 13, 2651–2659 RSC.
- X. Li, D. Xiao, X.-M. Ou, C. McCullm and Y.-M. Liu, J. Chromatogr. A, 2013, 1318, 251–256 CrossRef CAS PubMed.
- F. Schwarzkopf, T. Scholl, S. Ohla and D. Belder, Electrophoresis, 2014, 35, 1880–1886 CrossRef CAS PubMed.
- X. Sun, R. T. Kelly, K. Tang and R. D. Smith, Analyst, 2010, 135, 2296–2302 RSC.
- X. F. Sun, R. T. Kelly, K. Q. Tang and R. D. Smith, Anal. Chem., 2011, 83, 5797–5803 CrossRef CAS PubMed.
- B. Xiong, L. Wang, Y. Wang, Y. Bao, S. Jiang and M. Ye, Analyst, 2016, 141, 177–182 RSC.
- L. J. Millet, J. D. Lucheon, R. F. Standaert, S. T. Retterer and M. J. Doktycz, Lab Chip, 2015, 15, 1799–1811 RSC.
- K. Churski, M. Nowacki, P. M. Korczyk and P. Garstecki, Lab Chip, 2013, 13, 3689–3697 RSC.
- A. Weltin, K. Slotwinski, J. Kieninger, I. Moser, G. Jobst, M. Wego, R. Ehret and G. A. Urban, Lab Chip, 2014, 14, 138–146 RSC.
- L. Wang, Y. Nie, Y. Wang, Z. Wang and B. Xiong, RSC Adv., 2015, 5, 103474–103479 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: One PDF file provides supporting information for pre-treatment and separation modules configurations, instrumental setup of the modular platform, PCR experimental details, and identification of reserpine with the proposed platform. See DOI: 10.1039/c8ra08276f |
| ‡ Dr Yinyin Hao and Ms Yajing Bao contributes equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |