
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControl of the stereochemistry of C14 hydroxyl during the total synthesis of withanolide E and physachenolide C†
M. Anees a,
S. Nayakb,
K. Afarinkia*a and
V. Vinader*a
a,
S. Nayakb,
K. Afarinkia*a and
V. Vinader*a
aInstitute of Cancer Therapeutics, University of Bradford, Bradford BD7 1DP, UK. E-mail: m.vinader@bradford.ac.uk
bSchool of Chemistry & Biosciences, University of Bradford, Bradford BD7 1DP, UK
First published on 27th November 2018
Abstract
The stereochemical outcome of the epoxidation of Δ14–15 cholestanes with mCPBA is controlled by the steric bulk of a C17 substituent. When the C17 is in the β configuration, the epoxide is formed in the α face, whereas if the C17 is trigonal (flat) or the substituent is in the α configuration, the epoxide is formed in the β face. The presence of a hydroxyl substituent at C20 does not influence the stereochemical outcome of the epoxidation.
Introduction
Recently, two ergostane natural products, physachenolide C,1,2 1 and withanolide E,3–8 2 (Fig. 1) have attracted considerable attention because of their interesting and unusual anti-tumour properties. Key structural features of both molecules include the C14α-hydroxylation (which is relatively less common compared to C14β-hydroxylation in steroids) as well as C17β and pro-R hydroxylation at C20. Neither compound has been totally synthesised previously. So far, through semi-synthesis and derivatisation of authentic samples, a limited amount of information about their structure activity relationships has been gathered. None of these studies however have established the role of the three tertiary hydroxyl groups at C14α, C17β and C20 in the potency of the molecules. Clearly a total synthesis approach to physachenolide C, 1 and its deoxy analogues, which include withanolide E, 2, will allow us to explore the contribution that these various hydroxyl groups make to the biological activity of these molecules.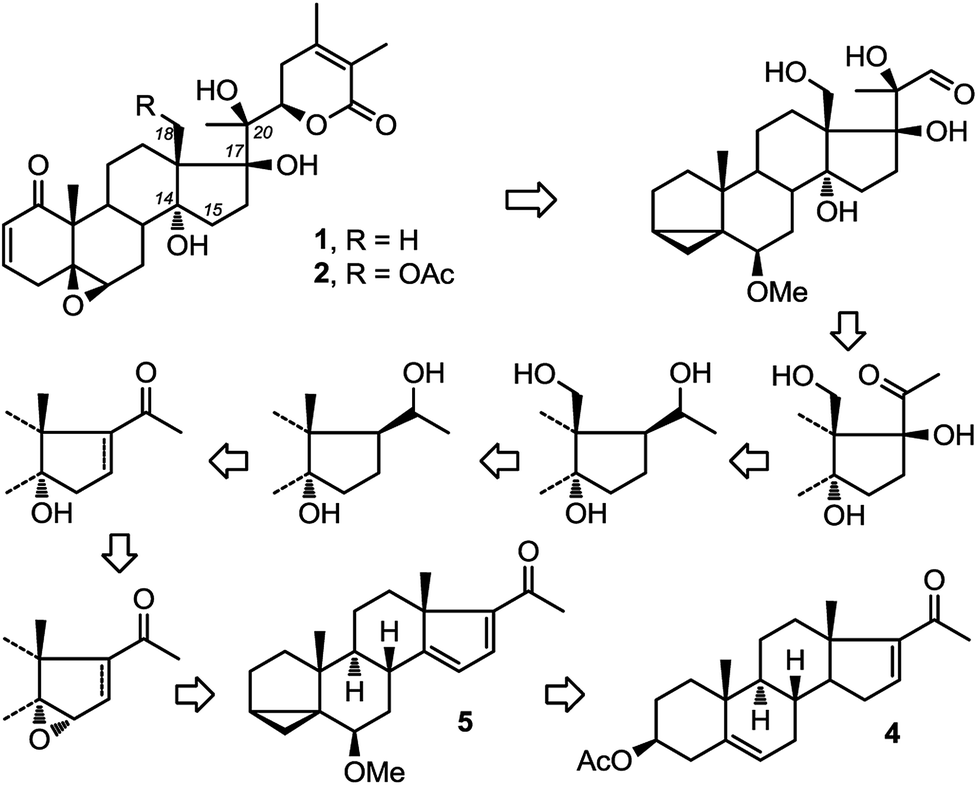 | ||
| Fig. 1 A route to withanolide E, 1 and physachenolide C, 2. | ||
Therefore, we have recently set out on a total synthesis of physachenolide C, 1 and withanolide E, 2. As part of this total synthesis, we envisaged a route consisting of sequential introduction of C-14α hydroxyl, followed by functionalisation of angular C18 methyl group, mediated by the C-20 hydroxyl;9–11 which will lead to a total synthesis of physachenolide C, 1 (Fig. 1). If this step is omitted, the follow on steps, introduction of C-17β hydroxyl and chain extension at C-20, will lead to withanolide E, 2. Here we describe our approach to the stereoselective introduction of the C14α hydroxyl group.
A number of strategies for the stereoselective introduction of a hydroxyl group at the C14 position of steroids have been described; however they mainly lead to the β-OH configuration. These include hydrobromination of Δ14–15 steroid followed by removal of bromine from C-15;12–15 Norish type II reaction of C-12 ketosteroids,16 metal catalysed hydration of Δ14–15 steroid,17 metal18,19 or enzyme catalysed direct conversion of C14–H to C14–OH,20–22 and through tandem construction of steroid's ring D.23
In our synthetic path, we opted for selective introduction of C-14α hydroxylation via epoxidation of Δ14–15, followed by regioselective reduction at C15 to generate the corresponding alcohol (Fig. 1). A survey of the literature reveals only three related examples, one of them being compound 3, where the stereochemical outcome of the epoxidation was assigned as α (Fig. 2, eqn (1)).24–27 However, a rationale for the stereoselectivity was not provided in any of these examples. In contrast, direct epoxidation from the β face is reported only in two cases, where an adjacent (homo-allylic) hydroxyl group can direct the stereoselective introduction of oxygen, presumably through hydrogen bonding to the reagent, mCPBA (Fig. 2, eqn (2)).28,29
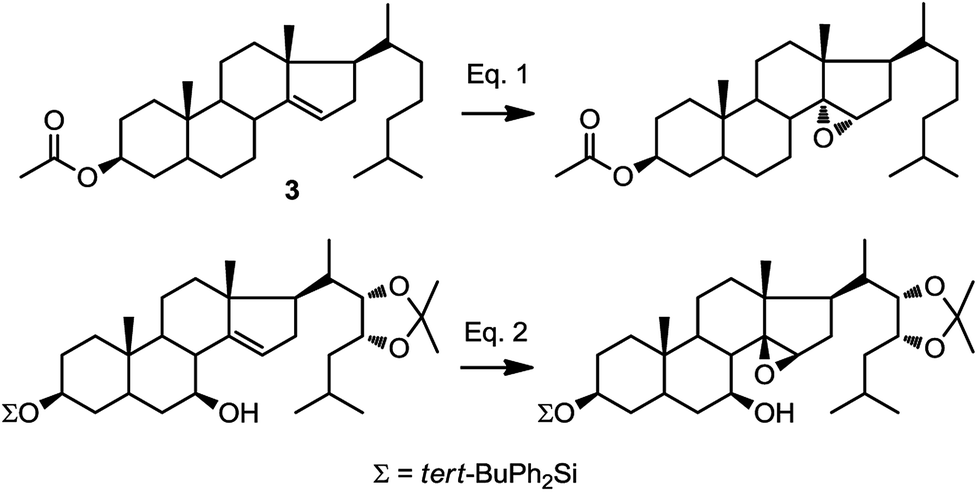 | ||
| Fig. 2 Diastereofacial selectivity in the epoxidation of C14–C15.25,29 | ||
Based on this information, we were confident that installation of the C-14 hydroxyl can be achieved from the α face, however, we discovered steric factors affecting the stereoselectivity of that epoxidation which we describe below.
Results and discussion
Starting from commercially available 4, we first set out to prepare 5 (Fig. 3). Molecule 5 contains two alkene groups, and our expectation was that only the C14–C15 double bond (and not the C16–C17 double bond) will undergo epoxidation, as it is further away from the electron withdrawing carbonyl group. Molecule 5 also contains a 6-methoxy-3,5-cyclochole-stane as a masked 3β-hydroxycholest-5,6-ene (see below).30,31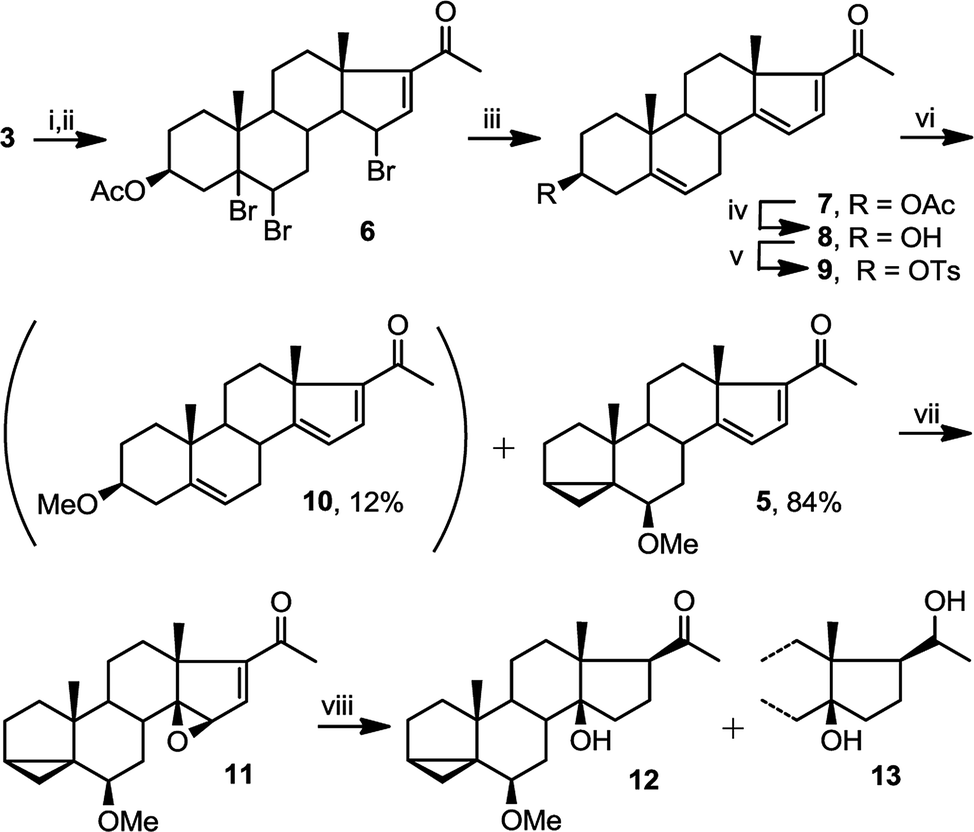 | ||
| Fig. 3 Preparation of epoxide 11 and its reduction to 12. (i) KOAc, Br2 (acetic acid), ether, 0 °C; (ii) NBS, AIBN, EtOAc, reflux; (iii) Nal, reflux (39% over 3 steps); (iv) KOH, t-butanol, 30 °C, 10 h (100%); (v) TsCl, pyridine, 28 h (83%); (vi) MeOH, pyridine (3 eq), reflux (84%); (vii) mCPBA (81%); (viii) LiAlH4, THF, reflux 2 h (12, 42%; 13, 23%). | ||
Treatment of 3 with bromine, in order to temporarily mask the C5–C6 alkene, followed by the bromination of the only remaining allylic position next to the C16–C17 double bond afforded 6, which upon debromination yielded 7. Hydrolysis of the 3β-OAc to alcohol 8 and subsequent conversion of the alcohol to a tosylate afforded 9. Treatment of tosylate 9 with methanol in the presence of mild base (pyridine) mainly results in homo-allylic displacement of the tosylate to afford 5. The reaction also produced 10 (11%) as a by-product, resulting from double substitution with methanol acting as nucleophile and attacking 5 at C3. This sequence of reactions effectively masks and protects the C5–C6 double bond. There is sufficient precedence in the literature to suggest that unmasking of the group can be accompanied by introduction of C2–C3 alkene and derivatisation of the ring A which is required in the total synthesis of physachenolide C.8,32–34
Treatment of 5 with mCPBA afforded a single epoxide, 11, and treatment of 11 with limiting amount of LiAlH4 yielded alcohol, 12 as well as fully reduced product, compound 13. Analysis of the configuration of C14 hydroxyl in 12, using a previously established method based on NMR chemical shifts was inconclusive at that time (see below). However, X-ray crystallography confirmed the configuration of 11 to be the undesired β configuration.35 We concluded therefore, that the C14 hydroxyl in 12 also had a β configuration (Fig. 4 and 5).
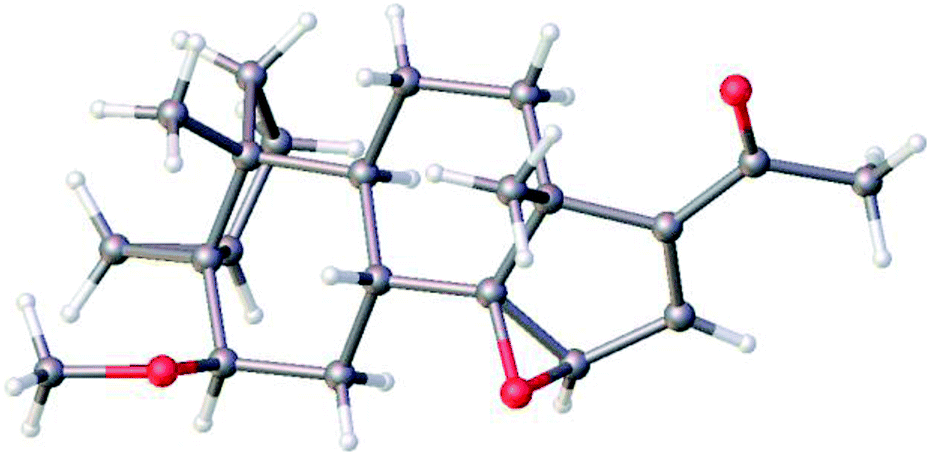 | ||
| Fig. 4 Molecular structure of 11 as determined by single crystal X-ray diffraction showing the configuration of epoxide. Colour code for the atoms: C (grey), H (white), and O (red). | ||
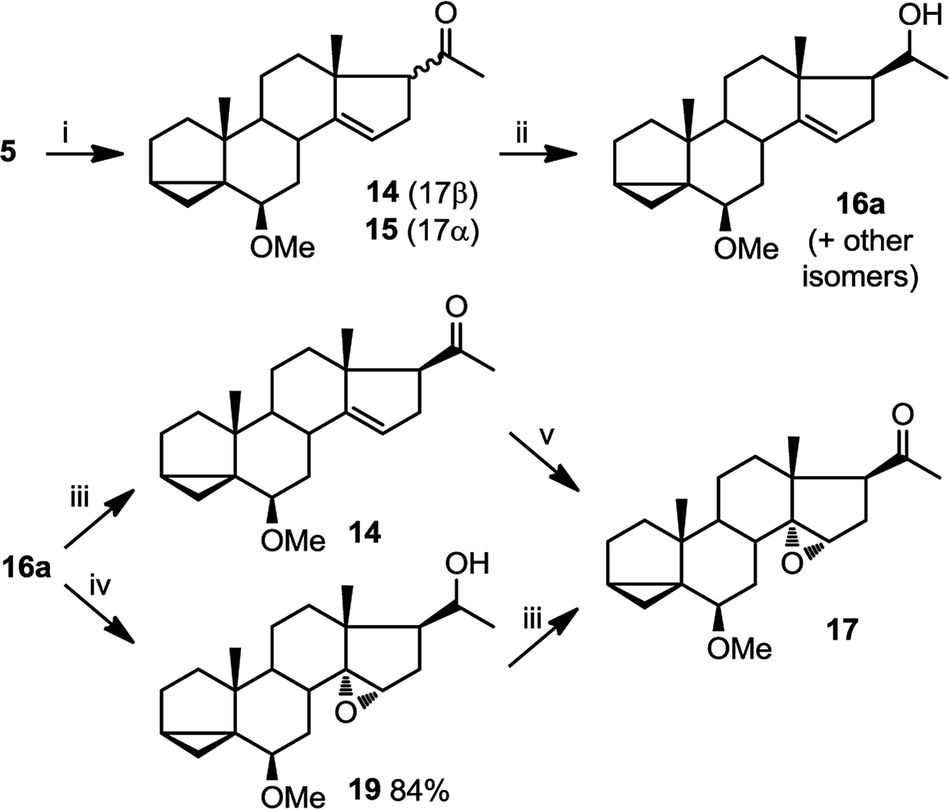 | ||
| Fig. 5 Preparation of epoxide 17. (i) Ph3SnH, AIBN, toluene, reflux, 10 h (39%); (ii) NaBH4, MeOH/CH2Cl2, 40 min (58% for 16aplus 32% combined yield of other isomers); (iii) PDC, CH2Cl2, 48 h (78%); (iv) mCPBA, CHCl3, 3 h (84%); (v) mCPBA, CHCl3, 3 h (93%). | ||
This epoxidation from the β face in 5 is in contrast to the available literature precedence, showing epoxidation occurs from the α face in 4 (Fig. 2).25 Of course, the difference between 5 and 4 is that in the latter, the C17 substituent is at the β configuration. Therefore, we presumed that C17 substituent may provide steric hindrance in the β face, driving the introduction of epoxides from the α face. To confirm our hypothesis, we set out to prepare 14 and to determine whether the configuration of its epoxidation would differ that of 5. Regioselective reduction of C16–C17 double bond in 5 afforded a mixture of two C-17 epimers, compounds 14 with β configuration at C-17, and 15 with α configuration at C-17. This was in contrast to a previous report that indicated that a similar reduction was stereoselective.36 Unfortunately, the two isomers, 14 and 15, were not separable in our hands. This isomeric mixture was reduced with NaBH4, affording a mixture of the four possible stereoisomers. The major isomer, 16a was isolated by chromatography and then converted back to 14 by treatment with PDC. Treatment of 14 with mCPBA afforded 17, in which the epoxide was later shown to have α configuration (vide infra).
The observed α configuration of the epoxide in the oxidation of 14 is in contrast to the observed β configuration of the epoxide in the oxidation of 5. This is presumably due to the differing configuration between the substituents at C17. In compound 14, C17 substituent is in β (up) configuration whereas in 5, C17 substituent is flat. We next set out to show that commensurate to this observation, epoxidation of 15, where the C17 substituent is in α (down) configuration affords an epoxide with β (up) configuration.
We were unable to obtain a sample of 15 which was completely free from 14. However, when epoxidising a mixture of 15 and 14, we obtained only two products: one of which is 17 as expected (from epoxidation of 14). The other, 18, was assigned with a β epoxide configuration (Fig. 6) (vide infra).
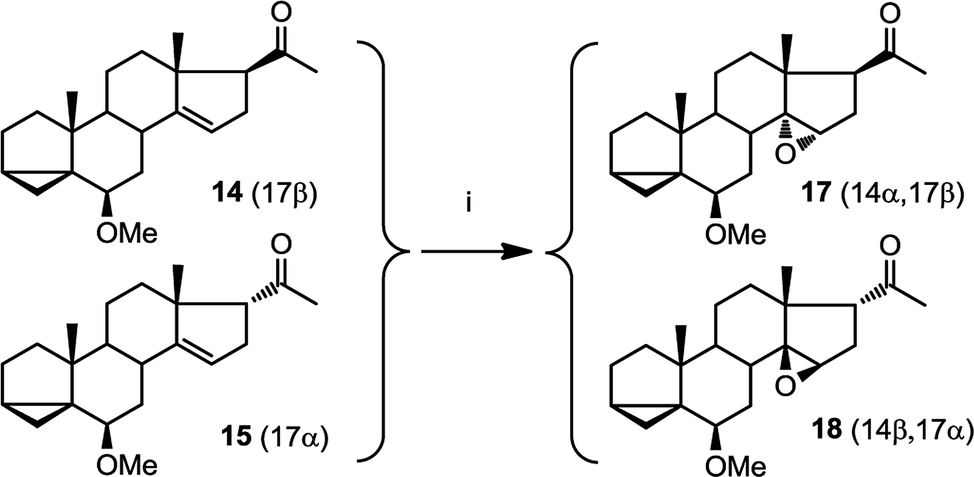 | ||
| Fig. 6 Epoxidation of a mixture of 14 and 15 to 17 and 18. (i) mCPBA, CHCl3, 3 h (93%). | ||
In oxidations with mCPBA, steric encumbrance can sometimes be overcomed by directing the delivery of the oxygen atom through hydrogen bonding of the reagent to an adjacent hydroxyl group. This has been shown in the literature as we have previously indicated (Fig. 2, eqn (2)).29 To investigate this, we carried out an epoxidation of 16a, which contains a hydroxyl group at C20, with mCPBA. The reaction afforded an epoxide 19 which after treatment with PDC, produced compound 17. This means that surprisingly, a hydroxyl group at C20, regardless of its configuration, is unable to direct the mCPBA epoxidation away from the α face.
The assignment of C14β hydroxyl configuration in compound 12 was deduced from compound 11, where X-ray crystallography had confirmed presence of a β epoxide. To unequivocally assign the configuration of epoxides 17–19 and their corresponding alcohols (see above) we used NMR chemical shifts.
A key feature in the NMR spectra that supports the assignment of the epoxide configuration is the value of the chemical shift for C18 protons. In both 17 and 19 (where the configuration of the C14–C15 epoxide is α) and in 14 (where there is no epoxide at C14–C15), the chemical shift for protons on C18 are similar and at 0.81, 1.01 and 0.89 ppm respectively. However, in compound 11 and 18 (where the configuration of epoxide is β) the C18 protons are subject to magnetic anisotropy effect by the oxygen atom in the epoxide and therefore, their chemical shift is increased to 1.28 and 1.32 ppm respectively. These variations in the chemical shifts of C18 protons are consistent with those previously reported in similar structures.37
Another way in which NMR can be used to confirm the configuration of epoxides is by assessing the γ gauche effect in the molecules. It has been previously established that in steroids, a 14α hydroxyl group shields C12 and C17 through γ gauche effect more than a 14β hydroxyl group does.38–40 C17 is easily identifiable in NMR due to its distinctive chemical shift at around 60 ppm. As an example from the literature, in compound 20, containing C14α hydroxylation, the chemical shift of C-17 is 2.8 ppm lower than that for compound 21, containing no C-14 hydroxylation; whereas the difference between the values for 22 containing C14β hydroxylation, and 21 is only 1.6 ppm (Table 1).40 The only limitation to this method is that we require a deoxy analogue to measure the differences in chemical shifts.
| Position | Chemical shifts for compounds | |||||
|---|---|---|---|---|---|---|
| 20 (α) | 21 | 23 (β) | 24 (α) | 25 | 12 | |
| δC-17 (ppm) | 61.1(−2.8) | 63.9 | 62.3(−1.6) | 60.0(−4.9) | 64.9 | 61.4(−3.5) |
Therefore, we set out to prepare the corresponding deoxy and C14α analogues for comparison of their C17 chemical shifts. Compound 19 was first converted into 23 by treatment with LiAlH4 and then to 24 by treatment with PDC (Fig. 7). Compound 25 was obtained as a byproduct from hydrogenation of 11, along with compounds 12 and 26.
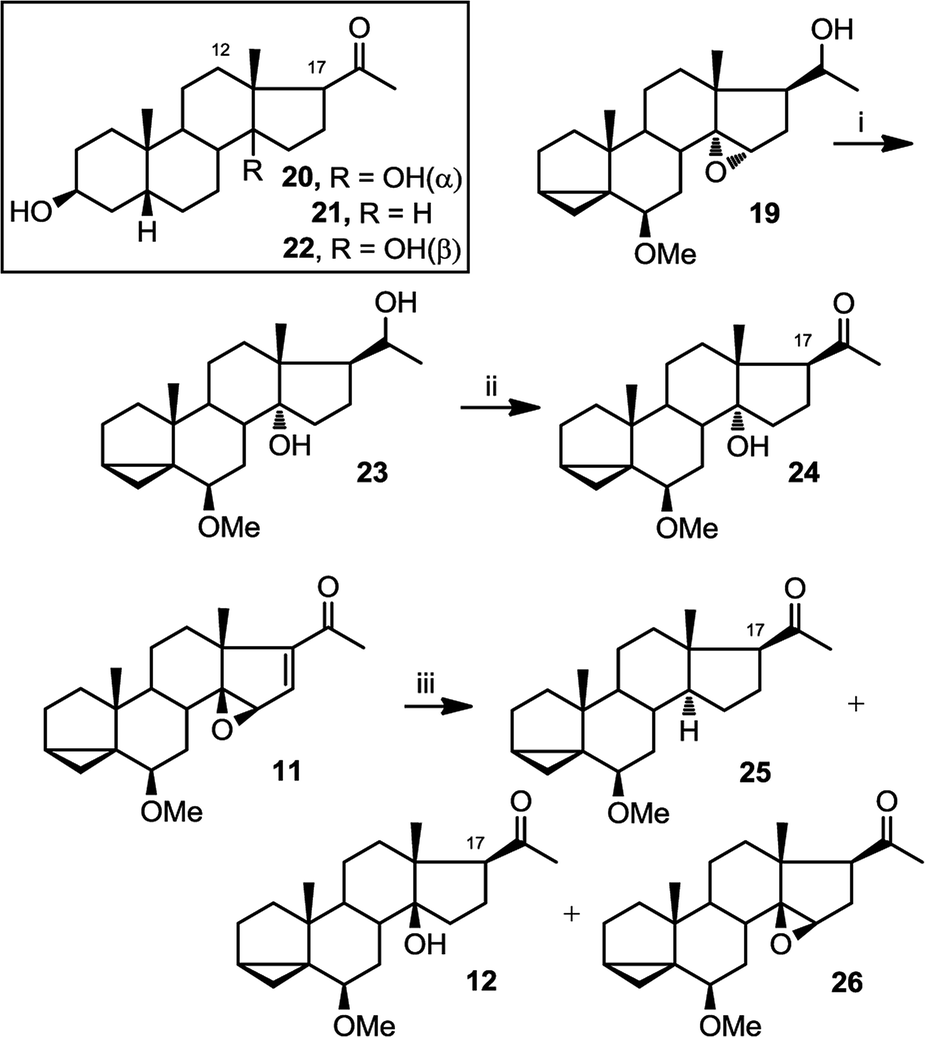 | ||
| Fig. 7 Preparation of 23–26. (i) LiAlH4, THF, reflux, overnight (61%); (ii) PDC, CH2Cl2, overnight (77%); (iii) H2 (g), 10% Pd/C, methanol, r.t.p., 15 h (25, 17%; 12, 37%; 26, 34%). | ||
The comparison between the chemical shift of C17 between compounds 20–22 and 24, 25 and 12 is shown below (Table 1). The larger deshielding of C17 in compound 25 compared to that in compound 24 (4.9 ppm) than 12 (3.5 ppm) supports the assignment of C14α hydroxyl configuration in compound 24.
Conclusions
In summary, the configuration of the epoxide in the mCPBA oxidation of C14–C15 alkenes function during the total synthesis of physachenolide C and withanolide E is determined by the configuration of the C17 substituent. When the C17 is in the β configuration, the epoxide is formed in the α face, whereas if the C17 is trigonal (flat) or the substituent is in the α configuration, the epoxide is formed in the β face.Experimental
Synthetic procedures and full characterisation data are provided in the ESI†.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank University of Bradford for a bursary (MA).Notes and references
- H. Zhang, C. M. Cao, R. J. Gallagher, V. W. Day, K. Kindscher and B. N. Timmermann, Phytochemistry, 2015, 109, 147–153 CrossRef CAS PubMed.
- Y. M. Xu, M. X. Liu, N. Grunow, E. M. Wijeratne, G. Paine-Murrieta, S. Felder, R. M. Kris and A. A. Gunatilaka, J. Med. Chem., 2015, 58, 6984–6993 CrossRef CAS PubMed.
- C. J. Henrich, A. D. Brooks, K. L. Erickson, C. L. Thomas, H. R. Bokesch, P. Tewary, C. R. Thompson, R. J. Pompei, K. R. Gustafson, J. B. McMahon and T. Sayers, Cell Death Dis., 2015, 6, e1666 CrossRef CAS PubMed.
- M. C. Gu, Y. K. Yu, G. M. K. B. Gunaherath, A. A. L. Gunatilaka, D. P. Li and D. X. Sun, Invest. New Drugs, 2014, 32, 68–74 CrossRef CAS PubMed.
- I. Prassas, G. S. Karagiannis, I. Batruch, A. Dimitromanolakis, A. Datti and E. P. Diamandis, Mol. Cancer Ther., 2011, 10, 2083–2093 CrossRef CAS PubMed.
- C. Y. Yen, C. C. Chiu, F. R. Chang, J. Y. F. Chen, C. C. Hwang, Y. C. Hseu, H. L. Yang, A. Y. L. Lee, M. T. Tsai, Z. L. Guo, Y. S. Cheng, Y. C. Liu, Y. H. Lan, Y. C. Chang, Y. C. Ko, H. W. Chang and Y. C. Wu, BMC Cancer, 2010, 10, 46 CrossRef PubMed.
- T. Dasdia and A. Dimarco, Eur. J. Cell Biol., 1980, 22, 548 Search PubMed.
- A. Perez-Medrano and P. A. Grieco, J. Am. Chem. Soc., 1991, 113, 1057–1059 CrossRef CAS.
- P. Tang and B. Yu, Eur. J. Org. Chem., 2009, 259–269 CrossRef CAS.
- J. M. Sonego, A. M. Cirigliano, G. M. Cabrera, G. Burton and A. S. Veleiro, Steroids, 2013, 78, 644–650 CrossRef CAS PubMed.
- H. Suginome, Y. Nakayama and H. Senboku, J. Chem Soc. Perkin Trans. 1, 1992, 1837–1842 RSC.
- P. Kočovský and I. Stieborová, Tetrahedron Lett., 1989, 30, 4295–4298 CrossRef.
- U. Stache, W. Fritsch, W. Haede, K. Radscheit and K. Fachinger, Justus Liebigs Ann. Chem., 1969, 726, 136–144 CrossRef CAS.
- S. Lociuro, T. Y. R. Tsai and K. Wiesner, Tetrahedron, 1988, 44, 35–40 CrossRef CAS.
- U. K. Pati and K. Wiesner, Heterocycles, 1989, 29, 275–282 CrossRef.
- P. Geoffroy, B. Ressault, E. Marchioni and M. Miesch, Steroids, 2011, 76, 1166–1175 CrossRef CAS PubMed.
- H. Renata, Q. Zhou and P. S. Baran, Science, 2013, 339, 59–63 CrossRef CAS PubMed.
- Z. Fang and R. Breslow, Org. Lett., 2006, 8, 252–254 CrossRef PubMed.
- M. D. Kaufman, P. A. Grieco and D. W. Bougie, J. Am. Chem. Soc., 1993, 115, 11648–11649 CrossRef CAS.
- C. S. Bensasson, J. R. Hanson and Y. Le Huerou, Phytochemistry, 1999, 52, 1279–1282 CrossRef CAS PubMed.
- J. Boynton, J. R. Hanson and A. C. Hunter, Phytochemistry, 1997, 45, 951–956 CrossRef CAS.
- J. R. Hanson, P. B. Hitchcock and A. C. Hunter, Phytochemistry, 1998, 49, 1287–1292 CrossRef CAS.
- N. R. Cichowicz, W. Kaplan, Y. Khomutnyk, B. Bhattarai, Z. Sun and P. Nagorny, J. Am. Chem. Soc., 2015, 137, 14341–14348 CrossRef CAS PubMed.
- S. Araki, S. Eguchi and M. Morisaki, Chem. Pharm. Bull., 1990, 38, 1796–1797 CrossRef CAS.
- M. E. Jung and T. W. Johnson, Tetrahedron, 2001, 57, 1449–1481 CrossRef CAS.
- M. S. Bjelakovic, N. M. Krstic, N. Todorovic, A. Krunic, B. Tinant, M. M. Dabovic and V. D. Pavlovic, Tetrahedron, 2009, 65, 9557–9568 CrossRef CAS.
- A. Lardon and T. Reichstein, Helv. Chim. Acta, 1962, 45, 943–962 CrossRef CAS.
- K. Michalak, M. Morawiak and J. Wicha, Org. Lett., 2016, 18(23), 6148–6151 CrossRef CAS PubMed.
- M. Li, P. Zhou and A. Wu, Tetrahedron Lett., 2006, 47, 3409–3412 CrossRef CAS.
- T. Gebreyesus and C. Djerassi, J. Org. Chem., 1985, 50, 154–156 CrossRef CAS.
- H. Suginome and Y. Nakayama, J. Chem. Soc. Perkin Trans. 1, 1992, 1843–1848 RSC.
- T. Kametani, T. Katoh, M. Tsubuki and T. Honda, J. Am. Chem. Soc., 1986, 108, 7055–7060 CrossRef CAS.
- W. S. Zhou, B. Jiang and X. F. Pan, J. Chem. Soc. Chem. Commun., 1988, 791–793 RSC.
- V. Richmond, G. A. G. Santos, A. P. Murray and M. S. Maier, Steroids, 2011, 76, 1160–1165 CrossRef CAS PubMed.
- The crystal structure and data for compound 11 can be retrieved from Cambridge Crystallographic Data Centre (deposition number 1861117).
- E. Yoshii, T. Oribe, K. Tumura and T. Koizumi, J. Org. Chem., 1978, 43, 3946–3950 CrossRef CAS.
- E. Glotter, S. Kumar, M. Sahai, A. Goldman, I. Kirson and M. Mendelovici, J. Chem. Soc. Perkin Trans. 1, 1991, 739–745 RSC.
- E. Glotter, M. Sahai and I. Kirson, J. Chem. Soc. Perkin Trans. 1, 1985, 2241–2245 RSC.
- H. Zhang and B. N. Timmermann, J. Nat. Prod., 2016, 79, 732–742 CrossRef CAS PubMed.
- S. C. Bensasson, R. J. Hanson and L. Y. Huerou, Phytochemistry, 1999, 52, 1279–1282 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: For detailed experimental procedure and characterisation of all compounds. CCDC 1861117. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra08540d |
| This journal is © The Royal Society of Chemistry 2018 |