DOI:
10.1039/C8RA08622B
(Paper)
RSC Adv., 2018,
8, 40243-40251
One-pot synthesis of 3,4-dihydropyrimidin-2(1H)-ones catalyzed by SO3H@imineZCMNPs as a novel, efficient and reusable acidic nanocatalyst under solvent-free conditions
Received
17th October 2018
, Accepted 18th November 2018
First published on 3rd December 2018
Abstract
The synthesis of 3,4-dihydropyrimidin-2(1H)-one derivatives was accomplished efficiently via a three-component reaction between ethyl acetoacetate, various types of aldehydes, and urea in the presence of 10 mg SO3H@imineZCMNPs as a novel, environment friendly, and reusable heterogeneous magnetic nanocatalyst under solvent-free conditions at 90 °C. The desired products were obtained with high quantitative yields. The catalyst was separated by simple isolation from the reaction mixture using a permanent magnet and reused several times without any significant loss of catalytic activity. The synthesized catalyst was fully characterized through various techniques including thermogravimetric analysis, Fourier transform infrared spectroscopy, scanning electron microscopy, transmission electron microscopy, X-ray diffraction, and the Hammett acidity test. This methodology tolerates most substrates and has the salient features of green reaction conditions, lower catalyst loading, high quantitative yields, low cost, the absence of solvents, and easy isolation and reusability of the catalyst.
Introduction
Energy and stability are challenging and vital issues from universal economic and social viewpoints. These issues are connected to the concept of green chemistry and make the science of catalysts more creative. The philosophy behind green chemistry deals with encouraging chemical and engineering researchers to design products and processes with minimum usage and production of harmful materials.1–6 Therefore, green catalysts are considered as an important subset of green chemistry. In recent years, green and environment-friendly syntheses have played a central role in increasing efficiency, preventing the use of volatile solvents and toxic reactants, and reducing reaction times.7–9 In the dissimilation process, the catalysts are trapped by the surface or pores of a solid bed such as silica or alumina.10 The connection of the catalyst to the intended surface can occur by simple absorption of the active molecules of the catalyst and/or covalent bonds as the covalent bond of the catalyst and the surface is of utmost importance. The formation of such bonds results in the connection of the catalyst to the surface. This connection will be tight enough, and washing the catalyst from the surface reaches its minimum in harsh reaction conditions. However, a limited access to the active centers of heterogeneous catalysts leads to the reduction of their activity compared to homogeneous catalysts.11–13 To resolve this issue, all the centers of the porous solid bed should be activated, as in most cases, only the active centers on the external surface of the porous solid bed are available for reaction. A homogeneous system and a heterogeneous one are similar in terms of higher number of active centers and a facile recovery in the catalyst system. Achieving this dual purpose is possible only by using nanocatalysts. The benefit of nanocatalysts is that it allows for the favorable properties of both catalysts by filling the gap between homogeneous and heterogeneous catalysts.14–17 Heterogeneous nanocatalysts, which are prepared by specific methods in nanoscale, have many applications in different industries because of the multiplicity of active surfaces and the selectivity, high stability, and extensive use of the heterogeneous catalyst.18–21 Although nanocatalysts have many benefits compared to typical catalyst systems, the separation and recovery of small nanocatalysts from the reaction mixture does not appear to be an easy task. Moreover, typical methods of filtration do not show good efficacy because of the small size of the nanocatalyst particles.22,23 To solve this problem, a suitable solution is to employ magnetic nanoparticles. The paramagnetic and insoluble natures of nanoparticles make their recovery possible by an external magnet; consequently, the catalyst could be used in another cycle.24–32
The compounds containing dihydropyrimidinone (DHPM) moiety have wide pharmaceutical and biological activities such as anti-bacterial, anti-oxidant, anti-HIV, and anti-cancer activities.33,34 In this regard, 3,4-dihydropyrimidin-2(1H)-ones are a common type of these heterocyclic compounds that can be prepared by one-pot condensation of ethyl acetoacetate, urea and an aldehyde under strongly acidic conditions.35 Previously, different derivatives of 3,4-dihydropyrimidine-2-(1H)-one have exhibited calcium channel modulators for the treatment of cardiovascular diseases.36 Some of the reported catalysts for the preparation of these types of heterocyclic molecules are: Ln(OTf)3,37 p-dodecylbenzenesulfonic acid,38 chiral phosphoric acid,39 sulfated tungstate,40 FeCl3·6H2O,41 Mn(OAc)3·2H2O,42 Ce(LS)3,43 and SnCl2-nano SiO2.44 Although numerous strategies have been reported for the production of 3,4-dihydropyrimidin-2(1H)-ones, some of these protocols suffer from disadvantages such as the need for excess amounts of the catalyst, the need for microwave or ultrasound irradiation, the use of high temperatures, the use of a toxic solvent, require multiple steps, and long reaction times. Therefore, it is essential to develop an improved procedure for the synthesis of 3,4-dihydropyrimidin-2(1H)-ones under mild reaction conditions.
Herein, we decided to investigate the synthesis of 3,4-dihydropyrimidin-2(1H)-ones with various substituents from the reaction of ethyl acetoacetate, aldehyde and urea under solvent-free conditions using SO3H@imineZCMNPs as a novel, eco-friendly, reusable and promising nanocatalyst.
Experimental
General
All reagents and solvents were obtained from Fluka or Merck chemical companies. The purity determination of the substrates and reaction monitoring were carried out with TLC on silica gel 60 F254 (0.25 mm thick). FTIR spectra were recorded using KBr pellets in the range of 400–4000 cm−1 with a PerkinElmer PXI spectrometer. The powder XRD patterns of samples were carried out in the 2θ range of 10–80° at room temperature on a Philips X-pert diffractometer (Holland), using Ni-filtered Co-Kα radiation (λ = 1.54 Å). Scanning electron microphotography (SEM) was performed on an LEO 1430VP instrument. TEM images were obtained with a Zeiss-EM10C transmission electron microscope with an acceleration voltage of 80 kV (Oberkochen, Germany). Thermogravimetric analyses (TGA) were performed from room temperature to 700 °C at a ramp of 10 °C min−1 under N2 atmosphere using a Linseis STA PT 1000 instrument.
Catalyst synthesis
Preparation of the magnetic Fe3O4 nanoparticles (MNPs). The magnetite nanoparticles were synthesized according to the method reported in ref. 45. Briefly, 7.0 g of FeCl3·6H2O and 2.4 g of FeCl2·4H2O were dissolved in 40 mL of distilled water in a three-necked round-bottom flask (250 mL) under stirring at 90 °C and a continuous flow of Ar atmosphere. Thereafter, under intense mechanical stirring, 10 mL of ammonia (25%) was added to the reaction mixture. The resulting nanoparticles were magnetically separated and subsequently washed three times with deionized water and then dried under vacuum at 60 °C for 12 h.
Preparation of zirconia coated magnetic nanoparticles (ZCMNPs). The Fe3O4@ZrO2 core–shell nanoparticles were prepared according to the method proposed by Wu with slight modifications.46 Briefly, 1 g of ZrOCl·8H2O was dissolved in 20 mL of a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (v/v) mixture of ethanol and water and heated at 70 °C for 2 h. Then, under continuous mechanical stirring, 5.0 g of Fe3O4 nanoparticles was added into the zirconia sol. The suspension was homogenized under ultrasonication for 1 h. After 12 h, the coated particles were finally separated from the liquid by magnetic decantation and dried under vacuum at 300 °C for 2 h.
:3 (v/v) mixture of ethanol and water and heated at 70 °C for 2 h. Then, under continuous mechanical stirring, 5.0 g of Fe3O4 nanoparticles was added into the zirconia sol. The suspension was homogenized under ultrasonication for 1 h. After 12 h, the coated particles were finally separated from the liquid by magnetic decantation and dried under vacuum at 300 °C for 2 h.
Functionalization of Fe3O4@ZrO2 with 3-aminopropyltriethoxysilane (AmpZCMNPs). Core–shell Fe3O4@ZrO2 nanoparticles (1 g) were dispersed in dry toluene (20 mL) for 20 min using ultrasound. Then, 3-aminopropyltriethoxysilane (Amp, 2 mL) was added to the resulting mixture. The mixture was refluxed for 24 h under argon atmosphere. After 24 h, the functionalized magnetite nanoparticles (AmpZCMNPs) were separated from the reaction mixture using a permanent magnet, washed several times with ethanol and distilled water, and dried in a vacuum oven.
Immobilization of anthranilic acid on the surface of zirconia-coated magnetite nanoparticles (anthZCMNPs). For the preparation of anthZCMNPs, AmpZCMNPs (2 g) was dispersed in ethanol (100 mL) for 30 min using ultrasonication. About 4 mmol of excess glutaraldehyde was added to the mixture and refluxed for 24 h. After this period of time, the resultant solid (named as gluZCMNPs) was separated by an external magnet and washed with ethanol several times; the unreacted glutaraldehyde residue was removed, and the solid was then dried in a vacuum oven. Afterward, the prepared gluZCMNPs (2 g) were suspended in 100 mL of ethanol. Anthranilic acid (4 mmol) was added to this solution under dry nitrogen atmosphere, and the mixture was refluxed for 24 h. After 24 h, the resultant solid, named as anthZCMNPs, was filtered, and 500 mL ethanol was added to it in order to remove the unreacted reagents. The resultant material was then dried in a vacuum oven.
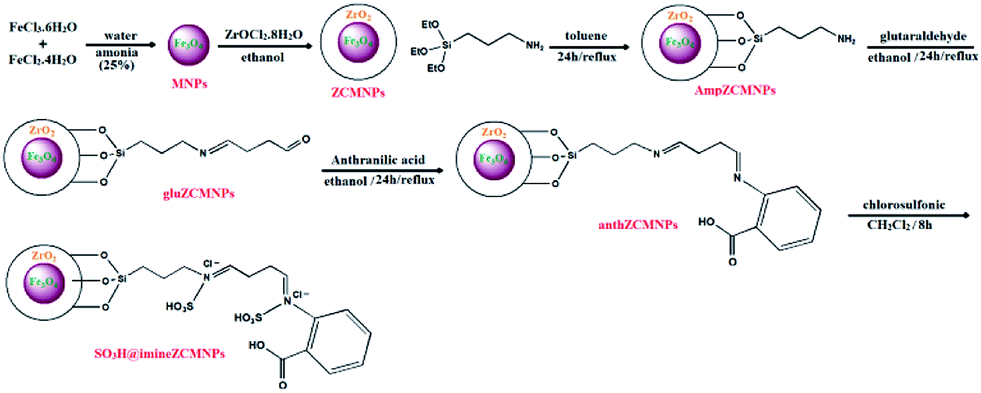
Immobilization of SO3H on the surface of magnetite nanoparticles (SO3H@imineZCMNPs). For the preparation of SO3H@imineZCMNPs, 2 g of anthZCMNPs was suspended in 50 mL dry dichloromethane; 10 mmol chlorosulfonic acid was added dropwise to the vessel, and the reaction mixture was stirred for 8 h. After separation with an external magnet, the product was washed with dry dichloromethane to remove the unreacted chlorosulfonic acid. The resultant SO3H@imineZCMNPs material was then dried under vacuum at 80 °C. All stages of the SO3H@imineZCMNPs synthesis are shown in Scheme 1.
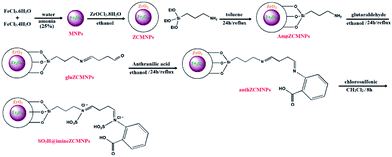 |
| | Scheme 1 All stages of the SO3H@imineZCMNPs synthesis. | |
General procedure for the synthesis of 3,4-dihydropyrimidine-2-(1H)-one. A mixture of ethyl acetoacetate (1 mmol), aldehyde (1 mmol), urea (1.2 mmol), and SO3H@imineZCMNPs (10 mg) was stirred under solvent-free conditions at 90 °C for the required period of time (Table 3). After completion of the reaction (monitored by TLC), it was observed that most of the nanoparticles were adsorbed onto the magnetic stirring bar when the magnetic stirring was turned off. After cooling to room temperature, the catalyst was separated using a permanent magnet and reused for the next experiment. The pure product was obtained via recrystallization from ethanol.
Results and discussion
Catalyst characterization
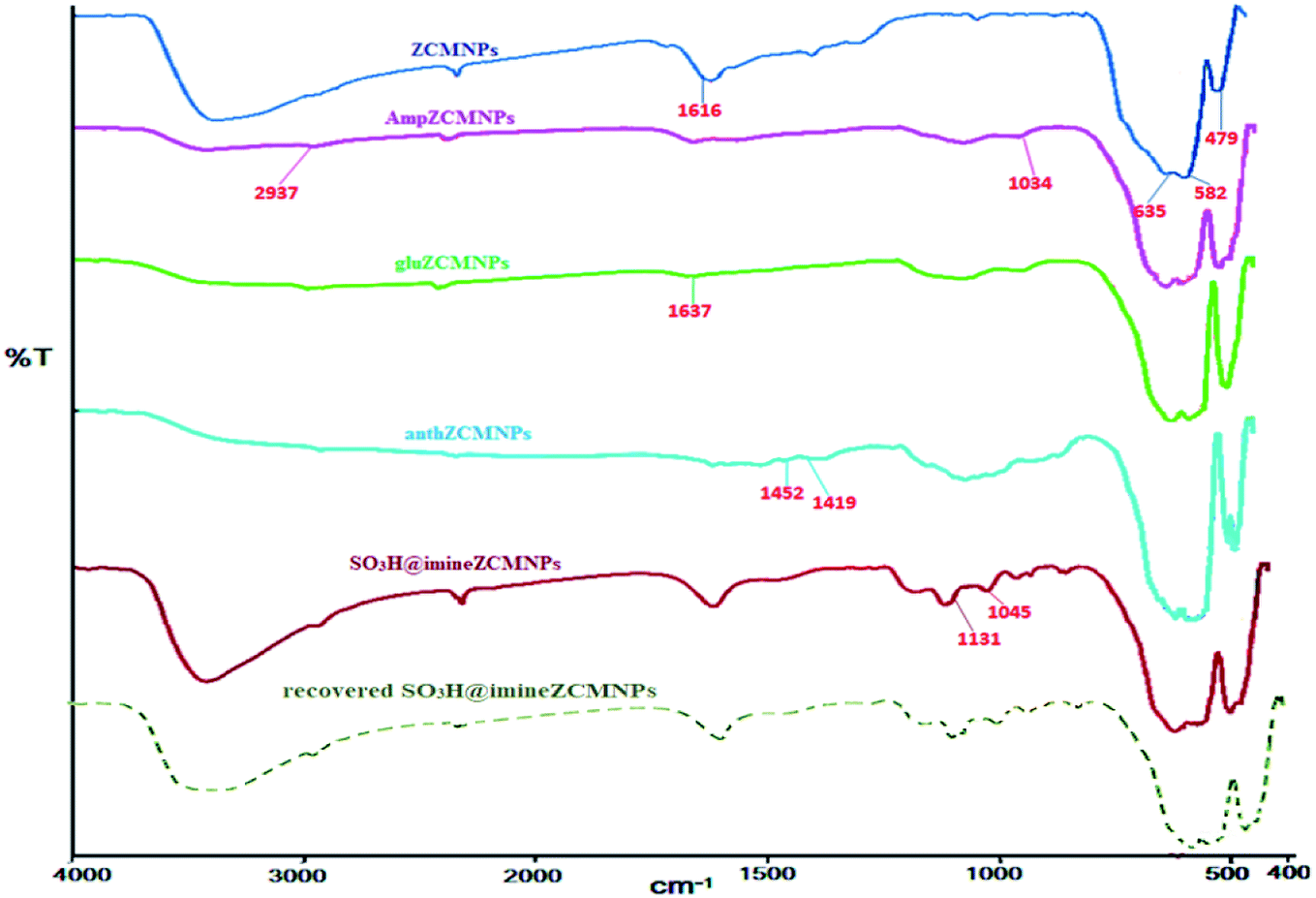
IR analysis of SO3H@imineZCMNPs. The infrared spectra of the ZCMNPs, AmpZCMNPs, gluZCMNPs, anthZCMNPs, and SO3H@imineZCMNPs samples in the region of 400–4000 cm−1 are presented in Fig. 1. In the ZCMNPs spectrum, the absorption peaks at 479 cm−1 and 582 cm−1 can be indexed to the Fe–O bonds in the crystalline lattice of Fe3O4. The peak observed at 635 cm−1 is ascribed to the Zr–O groups. The peak observed at 1616 cm−1 is related to the H–OH bending vibrations of the physically adsorbed water, while the peaks appearing in the 3100–3500 cm−1 region can be attributed to the stretching vibrations of the OH bonds in water molecules. In the case of the AmpZCMNPs, the bands at 1034 cm−1 can be attributed to Si–O stretching vibration mode. The absorption bands appearing at 2937 cm−1 (aliphatic C–H group) demonstrate the successful modification of the magnetite nanoparticles. In the gluZCMNPs spectrum, the stretching vibration of the imine C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group appears at 1637 cm−1, which confirms the successful modification of the surface of the AmpZCMNPs. In the case of the anthZCMNPs, the absorption peaks at 1419 and 1452 cm−1 can be assigned to the stretching vibration of the CC bond. The presence of the SO3H groups was confirmed by the stretching vibrations appearing at 1045 and 1131 cm−1 in the FTIR spectrum of the compound named SO3H@imineZCMNPs. Also, the increase in the intensities of the peaks at 2700–3600 cm−1 suggests that there were more OH functional groups under the magnetic nanoparticle surface after sulfonation. It should be noted that the spectrum of recovered SO3H@imineZCMNPs after the first recovery and reuse did not show any change.
N group appears at 1637 cm−1, which confirms the successful modification of the surface of the AmpZCMNPs. In the case of the anthZCMNPs, the absorption peaks at 1419 and 1452 cm−1 can be assigned to the stretching vibration of the CC bond. The presence of the SO3H groups was confirmed by the stretching vibrations appearing at 1045 and 1131 cm−1 in the FTIR spectrum of the compound named SO3H@imineZCMNPs. Also, the increase in the intensities of the peaks at 2700–3600 cm−1 suggests that there were more OH functional groups under the magnetic nanoparticle surface after sulfonation. It should be noted that the spectrum of recovered SO3H@imineZCMNPs after the first recovery and reuse did not show any change.
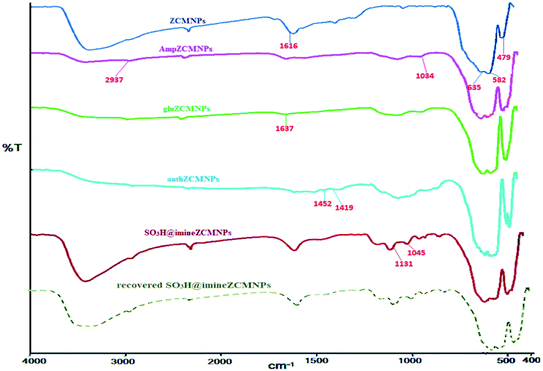 |
| | Fig. 1 FT-IR spectra of SO3H@imineZCMNPs. | |
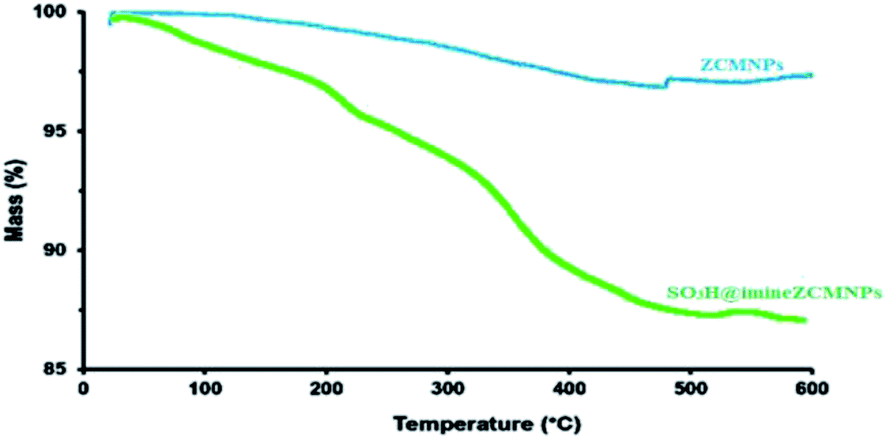
Thermal studies of SO3H@imineZCMNPs. TGA analyses were carried out to characterize the ZCMNPs and SO3H@imineZCMNPs samples toward each other. The results of these analyses are presented in Fig. 2. In the TGA curve of ZCMNPs, the weight loss observed at temperatures below 320 °C is related to water desorption from the surface of the zirconia layer while weight loss appearing above 600 °C can be attributed to the release of the water structure. The TGA of SO3H@imineZCMNPs illustrates an approximately 9% weight loss in the temperature range from 200 to 600 °C apart from two water loss events, resulting from the decomposition of the organic groups grafted to the zirconia surface and the degradation of the sulfonic acid parts.
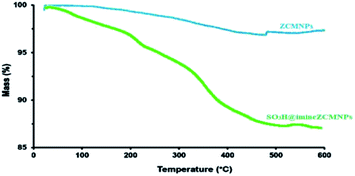 |
| | Fig. 2 TGA curves of ZCMNPS and SO3H@imineZCMNPs. | |
SEM analysis of SO3H@imineZCMNPs. The surface morphology, particle shape and size distribution of the SO3H@imineZCMNPs were examined by SEM (scanning electron microscopy) as shown in Fig. 3. The SEM image of the SO3H@imineZCMNPs clearly demonstrates variations in the surfaces of these nanoparticles. The SEM images illustrate that these nanoparticles are of nearly spherical morphology with a mean diameter of about 45 nm.
 |
| | Fig. 3 SEM images of ZCMNPs (a), anthZCMNPs (b), and SO3H@imineZCMNPs (c). | |
TEM analysis of SO3H@imineZCMNPs. The transmission electron micrograph of the prepared SO3H@imineZCMNPs determined that they appear to have an almost spherical shape with a narrow size distribution (the average particle size is 35 nm, Fig. 4), thereby retaining a nanocrystalline appearance. Therefore, the major active sites of the SO3H@imineZCMNPs may allow for high activity in organic transformations, even with a low catalyst loading.
 |
| | Fig. 4 TEM images of SO3H@imineZCMNPs. | |
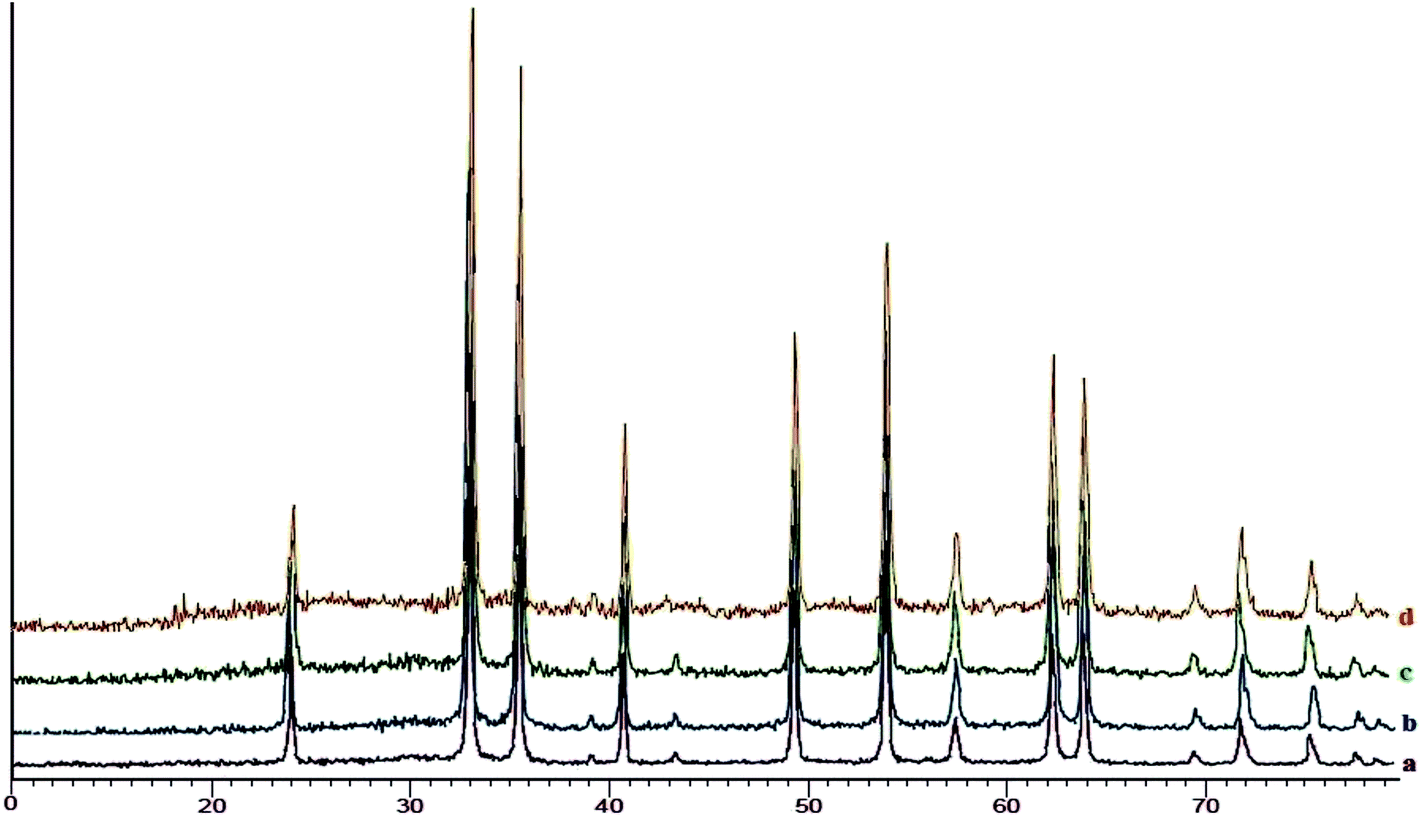
XRD analysis of SO3H@imineZCMNPs. The X-ray diffraction patterns (XRD) for ZCMNPs, anthZCMNPs, and SO3H@imineZCMNPs are given in Fig. 5. As can be seen, the XRD patterns of anthZCMNPs and SO3H@imineZCMNPs are similar to those of ZCMNPs, suggesting that the modification does not have any considerable effect on the phase of ZCMNPs. Also, the XRD patterns of recovered SO3H@imineZCMNPs after the first recovery and reuse do not show any change.
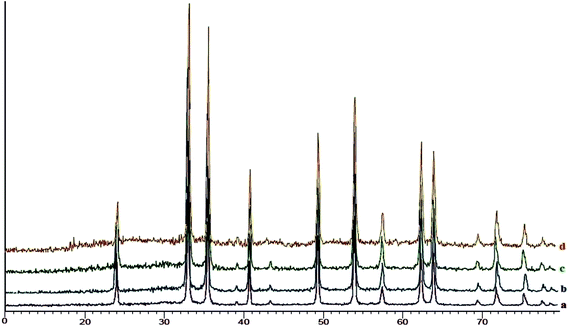 |
| | Fig. 5 XRD patterns of ZCMNPs (a), anthZCMNPs (b), SO3H@imineZCMNPs (c) and recovered SO3H@imineZCMNPs (d). | |
The acidity strength of our catalyst can be expressed by the Hammett acidity function (H0). It can be obtained from the following equation:
| H0 = pK(I)aq + log([I]s/[IH+]s) |
where ‘I’ is the indicator base and [I]
s and [IH
+]
s are the unprotonated and protonated indicator molar concentrations, respectively. The p
K(I)
aq value of 4-nitroaniline as a basic indicator to abstract proton is 0.99. Also, the value of [I]
s/[IH
+]
s according to the Lambert–Beer's law could be determined using the UV-visible spectrum. In our experiment, an unprotonated form of the 4-nitroaniline in CCl
4 showed an absorbance maximum at 330 nm.
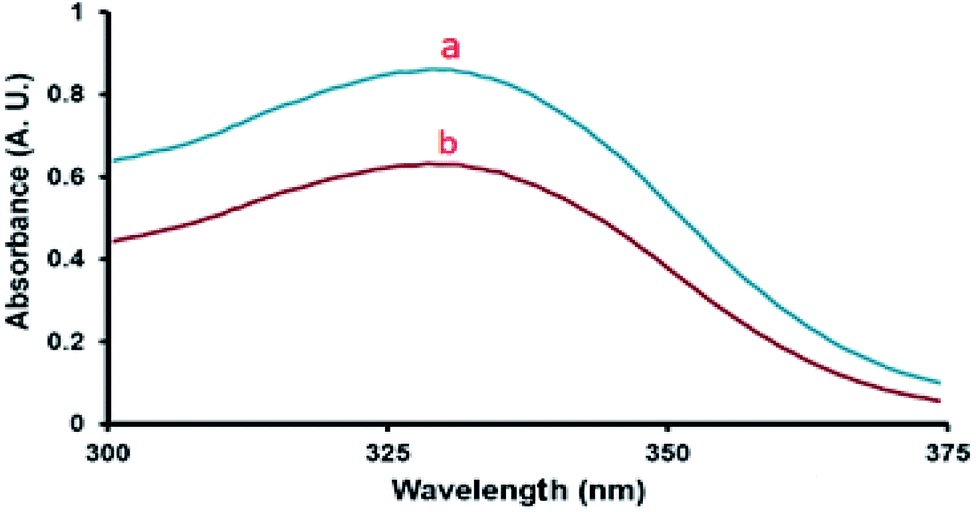
As can be seen from Fig. 6, the absorbance of the unprotonated form of the indicator in SO3H@imineZCMNPs was weak compared with the indicator sample in CCl4, suggesting that the indicator was partially in the [IH+] form. The obtained results of the acidity strength of SO3H@imineZCMNPs are listed in Table 1.
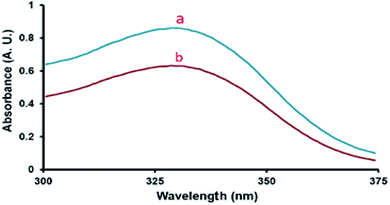 |
| | Fig. 6 Absorption spectra of (a) 4-nitroaniline (indicator) and (b) SO3H@imineZCMNPs (catalyst) in CCl4. | |
Table 1 Calculation of the Hammett acidity function (H0) of SO3H@imineZCMNPsa
| Entry |
Catalyst |
[I]s (%) |
[IH+]s (%) |
H0 |
| Conditions for UV-visible spectrum measurement: solvent, CCl4; indicator, p-nitroaniline (pK(I)aq = 0.99), 1.44 × 10−4 mol L−1; catalyst (10 mg) 25 °C. |
| 1 |
— |
100.0 |
0.0 |
— |
| 2 |
SO3H@imineZCMNPs |
75.0 |
25.0 |
1.47 |
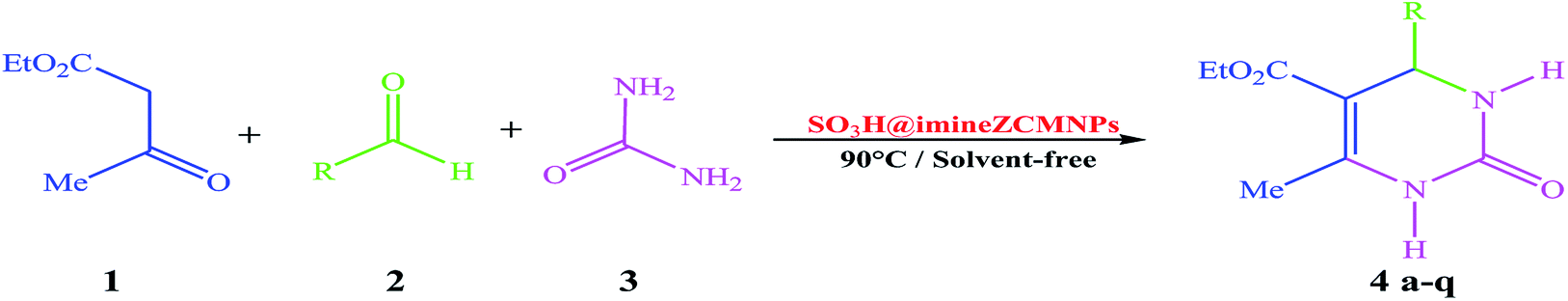
In continuation of our efforts on the synthesis of novel catalysts in performing organic transformations,47–49 we decided to investigate the synthesis of 3,4-dihydropyrimidin-2(1H)-one derivatives from the reaction of ethyl acetoacetate, various aldehyde, and urea under solvent-free conditions using SO3H@imineZCMNPs as a new, environment friendly, and reusable heterogeneous magnetic nanocatalyst (Scheme 2).
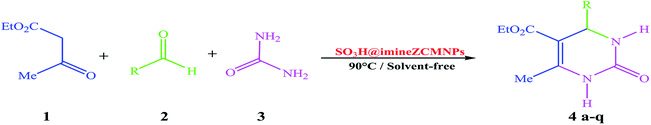 |
| | Scheme 2 One-pot three-component reaction of ethyl acetoacetate (1), arylaldehyde (2) and urea (3) catalyzed by SO3H@imineZCMNPs under solvent-free conditions. | |
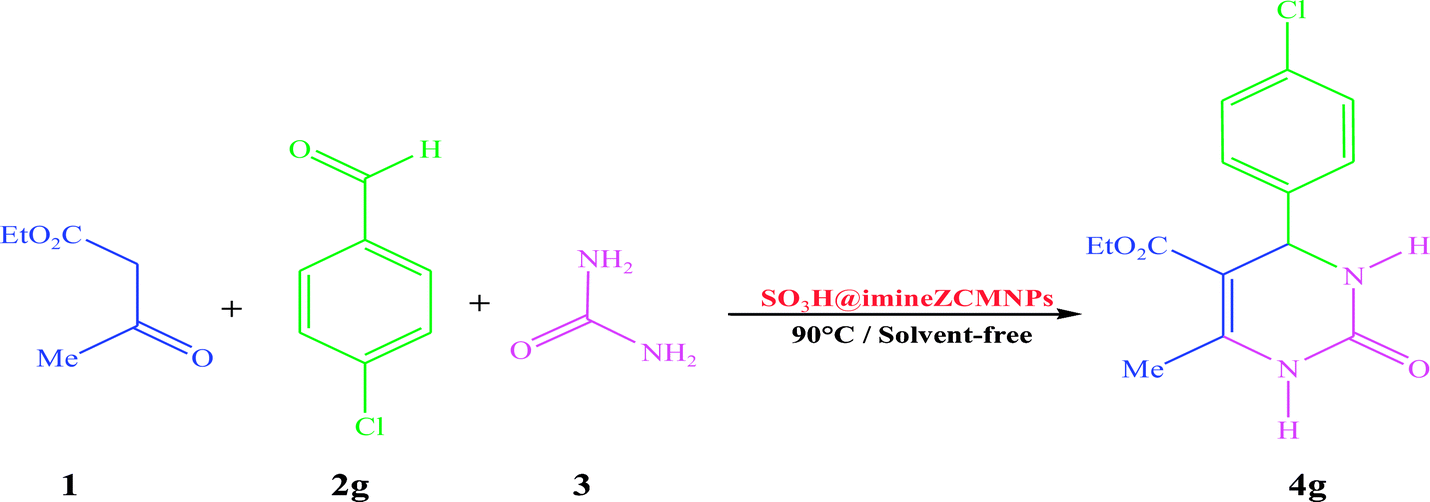
Solvents are widely applied in a myriad of applications as reactants, reaction mediums or carriers in chemical transformations.2,50 In order to screen the reaction conditions for the synthesis of 3,4-dihydro-5-etoxycarbonyl-4-(4-chlorophenyl)-6-methylpyrimidine-2(1H)-one (4g), the reaction of ethyl acetoacetate (1), 4-chlorobenzaldehyde (2g) and urea (3) (molar ratio 1.0:1.0:1.2) toward the corresponding product was considered as a model reaction. Next, different variables affecting the reaction yield, including the amount of the catalyst, solvent, and temperature were investigated. The results are summarized in Table 2. To obtain the optimal reaction media, different solvents such as H2O, CH2Cl2, CHCl3, DMSO, DMF, EtOH, toluene, and solvent-free conditions in the presence of a catalytic amount (10 mg) of SO3H@imineZCMNPs were used. Surprisingly, a short reaction time and the highest yield of the product (98%) was achieved when the same reaction was carried out while utilizing the same amount of catalysts at 90 °C under solvent-free conditions (entry 8). The initial optimization experiments revealed that low yields of the desired products were obtained in the presence of DMSO and DMF as coordinating solvents even after 1 h under reflux conditions (entries 3 and 4). Moderate to good yields were obtained when the same reaction was performed while utilizing H2O and EtOH, respectively, instead of coordinating solvents (entries 1 and 2). It is also remarkable that with EtOH, a lower reaction time was required compared to H2O. These solvents play a negative role by retarding the multi-component pathway. This might be due to the solvent adsorption on the acidic catalyst surface or the solvent–reactant interactions. In contrast, using CH2Cl2 and CHCl3 gave the desired product in lower yields (entries 5 and 6). A closer examination indicated that when toluene was replaced with the above-mentioned chlorinated solvents, the yield was markedly improved (entry 7). In the next phase of the survey, the effect of different catalyst amounts on the completion of the reaction was evaluated (entries 8–10). As noted above, the best result was obtained for 10 mg loading (entry 8). Decreasing the amount of catalyst required for the reaction produced a lower yield of the product, while higher catalyst loading did not considerably affect the duration of the reaction or the product yield (entries 9 and 10). To explore the effect of temperature with the same amount of catalyst (10 mg) under solvent-free conditions, different temperatures (25, 80, 90, and 100 °C) were used to compare the reaction efficiency (entries 8 and 11–13). It was observed that room temperature was not suitable for the formation of the desired product 4g under solvent-free conditions (entry 11). It is noteworthy that the use of heat with the same conditions leads to a faster reaction and a higher yield (entries 8, 12 and 13). Increasing the temperature beyond this point (90 °C) led to no substantial improvement in the yield (entry 13).
Table 2 Optimization of the three-component reaction of ethyl acetoacetate (1), 4-chlorobenzaldehyde (2g) and urea (3) under various conditionsa
Using the optimized reaction conditions (Table 2, entry 8) in order to generalize the effectiveness and acceptability of the strategy, a variety of aryl aldehydes containing electron-withdrawing as well as electron-donating groups in the ortho-, meta-, and para- positions of the benzene ring (2a–q) were selected for the reaction with ethyl acetoacetate (1) and urea (3), furnishing the desired products with high to excellent isolated yields (Table 3, entries 1–17). It is observed that with electron-withdrawing substituents, a relatively higher reaction yield of the products was achieved compared to electron-donating substituents.
Table 3 The SO3H@imineZCMNPs catalyzed three-component Biginelli couplinga
| Entry |
Product |
R |
Yieldb (%) |
Time (min) |
Mpc (°C) found |
Mp (°C) (Lit.) |
| Reaction conditions: ethyl acetoacetate (1), aldehyde (2), urea (3) (molar ratio 1:1:1.2) and SO3H@imineZCMNPs (10 mg), 90 °C. Isolated yield. Melting points are uncorrected. |
| 1 |
4a |
C6H5– |
93 |
8 |
202–204 |
202–203 (57) |
| 2 |
4b |
2-(NO2)–C6H4– |
95 |
8 |
218–220 |
220–222 (42) |
| 3 |
4c |
3-(NO2)–C6H4– |
95 |
8 |
226–228 |
226–228 (57) |
| 4 |
4d |
4-(NO2)–C6H4– |
98 |
8 |
211–213 |
208–211 (63) |
| 5 |
4e |
2-(Cl)–C6H4– |
97 |
8 |
217–219 |
215–218 (44) |
| 6 |
4f |
3-(Cl)–C6H4– |
96 |
8 |
192–195 |
192–193 (56) |
| 7 |
4g |
4-(Cl)–C6H4– |
98 |
8 |
211–214 |
212–214 (51) |
| 8 |
4h |
2,4-(Cl)2–C6H3– |
98 |
8 |
252–255 |
251–252 (60) |
| 9 |
4i |
3,4-(Cl)2–C6H3– |
97 |
8 |
222–225 |
222–223 (62) |
| 10 |
4j |
4-(F)–C6H4– |
95 |
8 |
178–180 |
175–177 (35) |
| 11 |
4k |
4-(CF3)–C6H4– |
98 |
8 |
177–179 |
173–175 (58) |
| 12 |
4l |
3-(Br)–C6H4– |
94 |
8 |
186–189 |
185–186 (61) |
| 13 |
4m |
2-(OH)–C6H4– |
94 |
8 |
198–201 |
199–201 (59) |
| 14 |
4n |
4-(OH)–C6H4– |
95 |
8 |
229–231 |
231–233 (57) |
| 15 |
4o |
2-(OCH3)–C6H4– |
93 |
8 |
258–260 |
259–260 (53) |
| 16 |
4p |
3-(OCH3)–C6H4– |
91 |
8 |
208–210 |
207–208 (52) |
| 17 |
4q |
4-(OCH3)–C6H4– |
95 |
8 |
203–205 |
203–204 (57) |
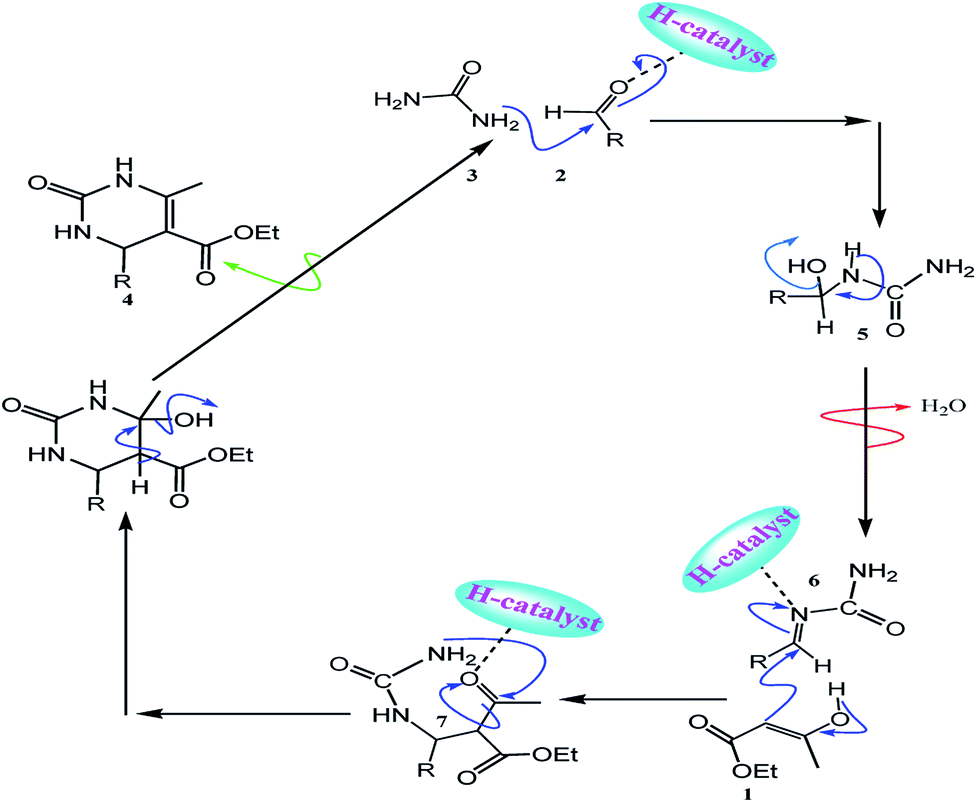
A reasonable pathway for the synthesis of 3,4-dihydropyrimidin-2(1H)-one derivatives is presented in Scheme 3. Initially, aldehyde 2 and urea 3 as the reactant components react with each other by nucleophilic addition. After forming the acylimine intermediate 5, it creates an open-chain ureide 6 after a dehydration step. The intermediate 6 may further undergo nucleophilic addition with ethyl acetoacetate 1 to give the intermediate 7. The final product 4 was then formed by the intramolecular cyclization and dehydration of the intermediate 7.
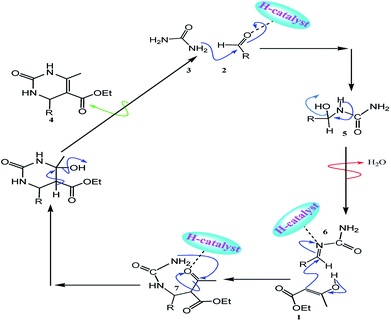 |
| | Scheme 3 A plausible mechanism for the one-pot three-component reaction of ethyl acetoacetate (1), aldehyde (2), urea (3) catalyzed by the SO3H@imineZCMNPs under solvent-free conditions. | |
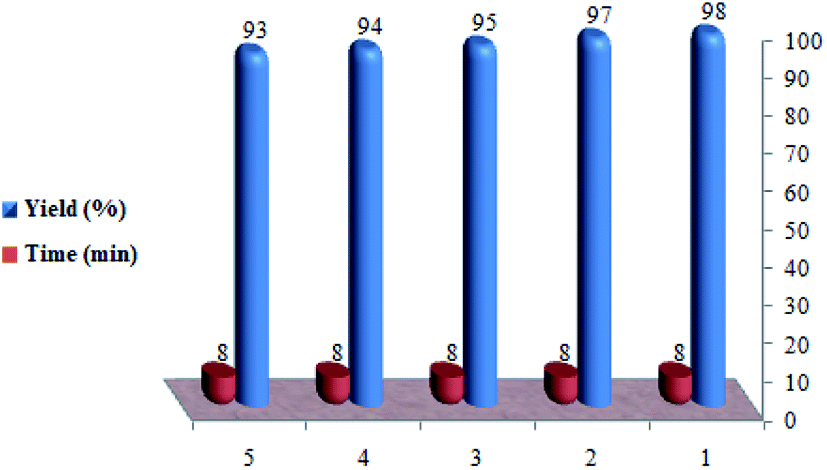
In the next phase of the study, the feasibility of the recovery and reuse cycle of the catalyst was checked by performing the reaction of ethyl acetoacetate, 4-chlorobenzaldehyde, and urea in the presence of SO3H@imineZCMNPs under solvent-free conditions. After reaction completion, as monitored by TLC, the catalyst was separated from the reaction mixture using a permanent magnet, and the mixture was washed with ethanol (2 × 15 mL). As shown in Fig. 7, SO3H@imineZCMNPs can be recycled five times with a negligible change in terms of the reaction time and with a yield range of 98 to 93%.
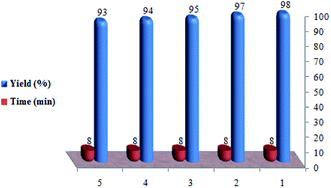 |
| | Fig. 7 Reusability of the catalyst. | |
Furthermore, to investigate and compare the presented method for the synthesis of 3,4-dihydro-5-etoxycarbonyl-4-(4-phenyl)-6-methylpyrimidine-2(1H)-one (4a), a comparison was made with some of the other earlier reported homogeneous and heterogeneous catalysts. The obtained results are shown in Table 4. The collected data demonstrate our reported protocol in terms of using solvent-free conditions, product yield, and reaction time.
Table 4 Comparison of the results of the synthesis of compound 4a using different catalysts
| Entry |
Catalyst and conditions |
Amount of catalyst |
Reaction time |
Yield (%) |
Ref. |
| 1 |
BF3·OEt2/CuCl/THF/reflux |
10 mol% |
18 h |
94 |
52 |
| 2 |
PPA-SiO2/CH3CN/reflux |
0.3 g |
30 min |
85 |
55 |
| 3 |
Zeolite/toluene/reflux |
15 mol% |
12 h |
80 |
65 |
| 4 |
CD-SO3H/solvent-free/100 °C |
0.04 g |
2 h |
83 |
54 |
| 5 |
FeCl3·6H2O/solvent-free/reflux |
0.16 g |
4 h |
94 |
56 |
| 6 |
Silica sulfuric acid/EtOH/heat |
0.23 g |
6 h |
91 |
53 |
| 7 |
SnCl2-nano SiO2/EtOH/reflux |
0.45 mol% |
40 min |
92 |
64 |
| 8 |
Sulfated tungstate/solvent-free/80 °C |
10 mol% |
1 h |
92 |
40 |
| 9 |
SO3H@imineZCMNPs/solvent-free/90 °C |
10 mg |
8 min |
93 |
This work |
Conclusion
In conclusion, we described a simple, efficient, and green methodology for the one-pot preparation of a wide range of biologically and pharmacologically important 3,4-dihydropyrimidin-2(1H)-one derivatives in the presence of an eco-friendly and reusable nanocatalyst (SO3H@imineZCMNPs). The most important advantages of this procedure are green reaction conditions, low catalyst loading, high quantitative yields, low cost, the absence of solvents, and facile isolation and reusability of the catalyst.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge the financial support of this study by the Research Council of the University of Guilan.
References
- V. Poshettiwar and R. S. Varma, Chem. Soc. Rev., 2008, 37, 1546 RSC.
- P. T. Anastas and J. C. Warner, Green Chemistry Theory and Practice, 1998 Search PubMed.
- A. S. Matlack, Introduction to Green Chemistry, 2001 Search PubMed.
- J. H. Clark and D. J. Macquarrie, Handbook of Green Chemistry and Technology, 2002 Search PubMed.
- M. Lancaster, Green Chemistry: An Introductory Text, RSC Editions, 2002 Search PubMed.
- M. Poliakoff, J. M. Fitzpatrick, T. R. Farren and P. T. Anastas, Science, 2002, 297, 807 CrossRef CAS PubMed.
- M. Doble and A. K. Kruthiventi, Green Chemistry and Engineering, Elsevier Science & Technology Books, 2007 Search PubMed.
- J. A. Loch and R. H. Crabtree, Pure Appl. Chem., 2001, 73, 119 CAS.
- R. A. Sheldon, I. Arends and U. Hanefeld, Green Chemistry and Catalysis, 2007 Search PubMed.
- R. T. Baker and W. Tumas, Science, 1999, 284, 1477 CrossRef CAS.
- F. Hoffmann, M. Cornelious, J. Moreli and M. Froba, Angew. Chem., 2006, 3216 CrossRef CAS PubMed.
- M. Jaroniec, T. W. Kim, M. Kruk and R. Ryoo, Chem. Mater., 2003, 15, 2815 CrossRef.
- H. Yang and D. Zhao, J. Mater. Chem., 2005, 15, 1217 CAS.
- H. U. Blaser, A. Indolese and A. Schnyder, Curr. Sci., 2000, 78, 1336 CAS.
- Y. Chauvin and J. C. Vedrine, Actual. Chim., 1996, 58, 7 Search PubMed.
- J. P. Collman, L. S. Hegedus, M. P. Cooke, J. R. Norton, G. Doleeti and D. N. Marquardt, J. Am. Chem. Soc., 1972, 94, 1789 CrossRef CAS.
- W. Dumont, J. C. Poulin, T. P. Daud and H. B. Kagan, J. Am. Chem. Soc., 1973, 95, 8295 CrossRef CAS.
- L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133 CrossRef CAS PubMed.
- H. U. Blaser, Tetrahedron: Asymmetry, 1991, 9, 843 CrossRef.
- P. McMorn and G. J. Hutchings, Chem. Soc. Rev., 2004, 33, 108 RSC.
- M. Heitbaum, F. Glorius and I. Escher, Angew. Chem., 2006, 45, 4732 CrossRef CAS PubMed.
- D. Astruc, Nanoparticles and Catalysis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- G. A. Somorjia, H. Frei and J. Y. Park, J. Am. Chem. Soc., 2009, 131, 16589 CrossRef PubMed.
- S. Shylesh, L. Wang and W. R. Thiel, Adv. Synth. Catal., 2010, 352, 425 CrossRef CAS.
- A. J. Amali and R. K. Rana, Green Chem., 2011, 11, 1781 RSC.
- J. Hu, Y. Wang, M. Han, Y. Zhou, X. Jiang and P. Sun, Catal. Sci. Technol., 2011, 2, 2332 RSC.
- Q. Zhang, H. Su, J. Luo and Y. Wei, Catal. Sci. Technol., 2013, 3, 235 RSC.
- A. H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222 CrossRef CAS PubMed.
- S. Shylesh, V. Schünemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428 CrossRef CAS PubMed.
- Y. Zhu, L. P. Stubbs, F. Ho, R. Liu, C. P. Ship, J. A. Maguire and N.
S. Hosmane, ChemCatChem, 2010, 2, 365 CrossRef CAS.
- V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J. M. Basset, Chem. Rev., 2011, 111, 3036 CrossRef CAS PubMed.
- R. B. N. Baig and R. S. Varma, Chem. Commun., 2013, 49, 752 RSC.
- S. S. Kim, B. S. Choi, J. H. Lee, K. K. Lee, T. H. Lee, Y. H. Kim and S. Hyunik, Synlett, 2009, 599 CAS.
- C. O. Kappe, Eur. J. Med. Chem., 2000, 35, 1043 CrossRef CAS PubMed.
- P. G. Biginelli, Gazz. Chim. Ital., 1893, 23, 360 Search PubMed.
- K. S. Atwal, B. N. S. Wanson, S. E. Unger, D. M. Floyd, S. Mereland, A. Hedberg and B. C. J. O'Reilly, J. Med. Chem., 1991, 34, 806 CrossRef CAS PubMed.
- Y. Huang, F. Yang and C. Zhu, J. Am. Chem. Soc., 2005, 127, 16386 CrossRef CAS PubMed.
- M. A. Bigdeli, G. Gholami and E. Sheikhhosseini, Chin. Chem. Lett., 2011, 22, 903 CrossRef CAS.
- X. H. Chen, X. Y. Xu, H. Liu, L. F. Cun and L. Z. Gong, J. Am. Chem. Soc., 2006, 128, 14802 CrossRef CAS PubMed.
- S. D. Salim and K. G. Akamanchi, Catal. Commun., 2011, 12, 1153 CrossRef CAS.
- S. Rostamnia and A. Morsali, RSC Adv., 2014, 4, 10514 RSC.
- K. A. Kumar, M. Kasthuraiah, C. S. Reddy and C. D. Reddy, Tetrahedron Lett., 2001, 42, 7873 CrossRef.
- Y. Qiu, H. Sun, Z. Ma and W. Xia, J. Mol. Catal. A: Chem., 2014, 392, 76 CrossRef CAS.
- C. O. Kappe, D. Kumar and R. S. Varma, Synthesis, 1999, 10, 1799 CrossRef.
- M. Yamaura, R. L. Camilo, L. C. Sampaio, M. A. Macedo, M. Nakamura and H. E. Toma, J. Magn. Magn. Mater., 2004, 279, 210 CrossRef CAS.
- Y. W. Wu, J. Zhang, J. F. Liu, L. Chen, Z. L. Deng, M. X. Han, X. S. Wei, A. M. Yu and H. L. Zhang, Appl. Surf. Sci., 2012, 258, 6772 CrossRef CAS.
- E. Abbaspour-Gilandeh, M. Aghaei-Hashjin, A. Yahyazadeh and H. Salemi, RSC Adv., 2016, 6, 55444 RSC.
- A. Yahyazadeh, E. Abbaspour-Gilandeh and M. Aghaei-Hashjin, Catal. Lett., 2018, 148, 1254 CrossRef CAS.
- M. Sheykhan, A. Yahyazadeh and Z. Rahemizadeh, RSC Adv., 2016, 6, 34553 RSC.
- J. Zhang and N. Yan, ChemCatChem, 2017, 9, 2790 CrossRef CAS.
- U. B. More, Asian J. Chem., 2012, 24, 1906 CAS.
- E. H. Hu, D. R. Sidler and U. H. Dolling, J. Org. Chem., 1998, 63, 3454 CrossRef CAS.
- P. Salehi, M. Dabiri, M. A. Zolfigol and M. A. B. Fard, Tetrahedron Lett., 2004, 44, 2889 CrossRef.
- S. Asghari, M. Tajbakhsh, B. Jafarzadeh-Kenari and S. Khaksar, Chin. Chem. Lett., 2011, 22, 127 CrossRef CAS.
- M. Zeinali-Dastmalbaf, A. Davoodnia, M. M. Heravi, N. Tavakoli-Hoseini, A. Khojastehnezhad and H. A. Zamani, Bull. Korean Chem. Soc., 2011, 32, 656 CrossRef CAS.
- J. Lu and H. Ma, Synlett, 2000, 63 CAS.
- R. Ghosh, S. Maiti and A. Chakraborty, J. Mol. Catal. A: Chem., 2004, 217, 47 CrossRef CAS.
- Y. Ma, C. Qian, L. Wang and M. Yang, J. Org. Chem., 2000, 65, 3864 CrossRef CAS PubMed.
- D. Russowsky, F. A. Lopes, V. S. S. Silva, K. F. S. Canto, M. G. Montes and M. N. Godoi, J. Braz. Chem. Soc., 2004, 165 CrossRef CAS.
- A. D. Patil, N. V. Kumar, W. C. Kokke, M. F. Bean, A. J. Freyer, C. D. Brossi, S. Mai, A. Truneh, D. J. Faulkner, B. Carte, A. L. Breen, R. P. Hertzberg, R. K. Johnson, J. W. Westly and B. C. M. Potts, J. Org. Chem., 1995, 60, 1182 CrossRef CAS.
- N. S. Nandurkar, M. J. Bhanushali, M. D. Bhor and B. M. Bhanage, J. Mol. Catal. A: Chem., 2007, 271, 14 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy, R. Srinivas, C. V. enugopal and T. Ramalingam, Synthesis, 2001, 1341 CrossRef CAS.
- K. K. Pasunooti, H. Chai, C. N. Jensen, B. K. Gorityala, S. Wang and X. W. Liu, Tetrahedron Lett., 2011, 52, 80 CrossRef CAS.
- J. Safaei-Ghomi, R. Teymuri and A. Ziarati, Monatsh. Chem., 2013, 144, 1865 CrossRef CAS.
- V. Radha Rani, N. Srinivas, M. Radha Kishan, S. J. Kulkarni and K. V. Raghavan, Green Chem., 2001, 3, 305 RSC.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Asieh Yahyazadeh*a and
Mehraneh Aghaei-Hashjinb
*a,
Asieh Yahyazadeh*a and
Mehraneh Aghaei-Hashjinb