
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScope of sulfur dioxide incorporation into alkyldiallylamine–maleic acid–SO2 tercyclopolymer†
Ibrahim Y.
Yaagoob
,
Shaikh A.
Ali
 *,
Hasan A.
Al-Muallem
* and
Mohammad A. J.
Mazumder
*,
Hasan A.
Al-Muallem
* and
Mohammad A. J.
Mazumder
Chemistry Department, King Fahd University of Petroleum & Minerals, Dhahran 31261, Saudi Arabia. E-mail: shaikh@kfupm.edu.sa; Web: http://faculty.kfupm.edu.sa/CHEM/shaikh/ Fax: +966 13 860 4277; Tel: +966 13 860 3830
First published on 22nd November 2018
Abstract
Alternate copolymerization of diallylamine derivatives [(CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CH2)2NR; R = Me, (CH2)3PO(OEt)2, and CH2PO(OEt)2] (I)–maleic acid (MA) and (I·HCl)–SO2 pairs have been carried out thermally using ammonium persulfate initiator as well as UV radiation at a λ of 365 nm. The reactivity ratios of ≈0 for the monomers in each pair I–MA and I·HCl–SO2 ensured their alternation in each copolymer. However, numerous attempted terpolymerizations of I–MA–SO2 failed to entice MA to participate to any meaningful extent. In contrast to reported literature, only 1–2 mol% of MA was incorporated into the polymer chain mainly consisting of poly(I-alt-SO2). Quaternary diallyldialkylammonium chloride [(CH2CH–CH2)2N+R2Cl−; R = Me, Et] (II) also, did not participate in II–MA–SO2 terpolymerizations. Poly((I, R = Me)-alt-SO2) III is a stimuli-responsive polyampholyte; its transformation under pH-induced changes to cationic, polyampholyte-anionic, and dianionic polyelectrolytes has been examined by viscosity measurements. The pKa of two carboxylic acid groups and NH+ in III has been determined to be 2.62, 5.59, and 10.1. PA III, evaluated as a potential antiscalant in reverse osmosis plants, at the concentrations of 5 and 20 ppm, imparted ≈100% efficiency for CaSO4 scale inhibition from its supersaturated solution for over 50 and 500 min, respectively, at 40 °C. The synthesis of PA III in excellent yields from cheap starting materials and its very impressive performance may grant PA III a prestigious place as an environment-friendly phosphate-free antiscalant.
CH–CH2)2NR; R = Me, (CH2)3PO(OEt)2, and CH2PO(OEt)2] (I)–maleic acid (MA) and (I·HCl)–SO2 pairs have been carried out thermally using ammonium persulfate initiator as well as UV radiation at a λ of 365 nm. The reactivity ratios of ≈0 for the monomers in each pair I–MA and I·HCl–SO2 ensured their alternation in each copolymer. However, numerous attempted terpolymerizations of I–MA–SO2 failed to entice MA to participate to any meaningful extent. In contrast to reported literature, only 1–2 mol% of MA was incorporated into the polymer chain mainly consisting of poly(I-alt-SO2). Quaternary diallyldialkylammonium chloride [(CH2CH–CH2)2N+R2Cl−; R = Me, Et] (II) also, did not participate in II–MA–SO2 terpolymerizations. Poly((I, R = Me)-alt-SO2) III is a stimuli-responsive polyampholyte; its transformation under pH-induced changes to cationic, polyampholyte-anionic, and dianionic polyelectrolytes has been examined by viscosity measurements. The pKa of two carboxylic acid groups and NH+ in III has been determined to be 2.62, 5.59, and 10.1. PA III, evaluated as a potential antiscalant in reverse osmosis plants, at the concentrations of 5 and 20 ppm, imparted ≈100% efficiency for CaSO4 scale inhibition from its supersaturated solution for over 50 and 500 min, respectively, at 40 °C. The synthesis of PA III in excellent yields from cheap starting materials and its very impressive performance may grant PA III a prestigious place as an environment-friendly phosphate-free antiscalant.
1. Introduction
Butler's cyclopolymerization protocol1 has etched a place of distinction in the synthesis of polymers of tremendous technological importance.2–4 Free radical-initiated polymerization of diallylammonium salts A led to B having pyrrolidine rings embedded into the macromolecules (Scheme 1). The macromolecule B represents the eighth most important architecture among synthetic high polymers.5,6 Polydiallyldimethylammonium chloride B (R1R2Me), alone accounting for over 1000 patents and publications, has been produced to the tune of millions of pounds for a variety of industrial applications including water treatment.1 Sulfur dioxide has been extensively used to synthesize alternate cyclocopolymers C having numerous applications.7–9 The reactivity ratios of amine (salt) (rA) and rSO2 being zero led to the alternation in C. Likewise, the almost zero reactivity ratios of rA and maleic acid (MA) (rMA) led to the exclusive formation of alternate copolymer D from alkyldiallylamine/MA ion pair A.10,11 Interestingly, diallyldimethylammonium chloride (A)–MA–SO2 is reported in several patents12–14 to give terpolymers in respective ratios of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25:25 (ref. 12) and 70:25:5.14 However, the patents do not describe any micro-structural analysis. To our knowledge, no journal article has appeared to date to describe such terpolymerization involving A–maleic acid–SO2 to give E (Scheme 1).
:25:25 (ref. 12) and 70:25:5.14 However, the patents do not describe any micro-structural analysis. To our knowledge, no journal article has appeared to date to describe such terpolymerization involving A–maleic acid–SO2 to give E (Scheme 1).
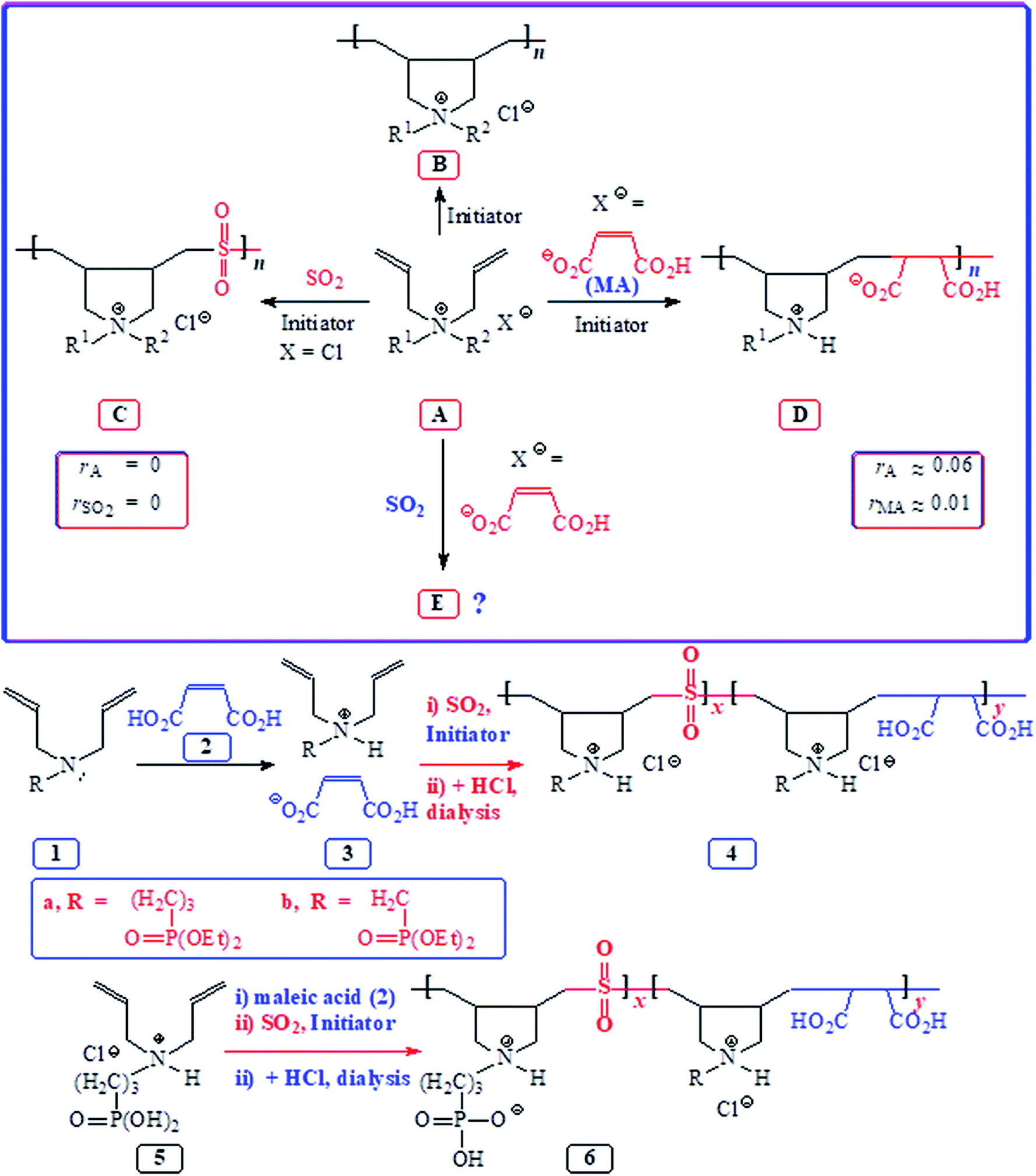 | ||
| Scheme 1 Synthesis of homocyclopolymer and alternate copolymers from monomeric pairs A/maleic acid and A/SO2 and terpolymerization of 1 or 5/MA/SO2. | ||
Amino-carboxylate and -phosphonate are important antiscalant and metal chelation motifs; as such, the decoration of repeating units with a high density of these potent functional groups is expected to impart, for instance, an antiscalant behavior. Looking at the zero reactivity ratios for both the monomer pairs: rA ≈ rSO2 ≈ 0 and rA ≈ rMA ≈ 0 (Scheme 1), we do fancy the success of the terpolymerization to pave the way to a great many interesting tripolymers E. Herein, we report a systematic study involving the cyclopolymerization of A, A–SO2, A–MA, and A–MA–SO2 to investigate the course of action of an amine terminal radical –A˙ whether it adds exclusively or preferentially onto SO2 or MA or both.
2. Experimental section
2.1 Materials
Paraformaldehyde was purchased from Fluka Chemie AG. Diallylamine from Merck Schuchardt OHG, ammonium persulfate (APS), azobisisobutyronitrile (AIBN) and maleic acid from Sigma-Aldrich were used as received. Diallyldimethylammonium chloride (15a) from Sigma-Aldrich, obtained as 65 wt% solution, was freeze-dried and crystallized from methanol/acetone. Phopsphonate ester 1a,151b,16 and 5 (ref. 17) were prepared as described.15–17 For dialysis, Spectra/Por membrane (MWCO: 6000–8000) from Spectrum Laboratories Inc. was utilized.2.2 Physical methods
Elemental analysis was carried out in a Perkin Elmer Elemental Analyzer Series 11 Model 2400 (Waltham, Massachusetts, USA). IR spectra were recorded on a Thermo Scientific FTIR spectrometer (Nicolet 6700, Thermo Electron Corporation, Madison, WI, USA). 1H and 13C NMR spectra of the polymers were measured in D2O on a JEOL LA 500 MHz spectrometer. The residual HOD signal of D2O at δ 4.65 ppm and dioxane 13C resonance at δ 67.4 were used as internal and external references, respectively. Viscosities were measured using an Ubbelohde viscometer (viscometer constant = 0.005718 cSt s−1). An SDT analyzer (Q600: TA instruments, USA) was utilized for Thermogravimetric analysis (TGA) under N2 flowing at a rate of 50 cm3 min−1. For photoinitiated polymerization, the samples were irradiated at 365 nm using UV Lamp 365 nm/254 nm SPECTROLINE (Model: ENF-240C) from Spectronic Corp., Westbury, NY, USA, with LONGLIFE Filter to ensure highest UV intensity and Fluorescence Analysis Cabinet SPECTROLINE CM-10A.2.3 Synthesis of methyldiallylamine (7) and its hydrochloride salt 8
The procedure for the methylation of amines is adapted from the literature.18 Formic acid (35 g, 85%, 0.646 mol) was added slowly (30 min) onto an ice-cold diallylamine (1) (49 g, 0.506 mol) in a 1 L RB flask under stirring. Paraformaldehyde (18.3 g, 0.613 mol) and water (33 mL) were added to the flask. The mixture in the flask, fitted with a long condenser, was stirred at 75–77 °C during which CO2 evolution happened (30 min). Thereafter, the reaction mixture was heated to 90 °C; after the gas evolution stopped (15 min), the reaction mixture was cooled and saturated with the addition of a solution of NaOH (30 g, 0.75 mol) in water (30 mL). The separated organic layer of amine 7 was dried over NaOH and distilled at 107 °C (lit. 19 bp 109 °C) to obtain 7 as a colorless liquid (49 g, 87%).A dry HCl gas was passed to a solution of methyldiallylamine (7) (25 g, 0.225 mol) in ether (150 mL) at 0 °C. Immediate precipitation of white salt 8 occurred. The passage of HCl was continued until the supernatant liquid becomes clear instead of cloudy. After filtration, the white salt was dried under vacuum to obtain 8 (30 g, 90%). δH (D2O) 2.70 (3H, s), 3.65 (4H, d, J 7.3 Hz), 5.50 (4H, m), 5.82 (2H, m); δC (D2O) 39.57 (1C, Me), 58.30 (2C, NCH2), 126.60 (2C, CH), 127.33 (2C, CH2). The 13C spectral assignments was supported by DEPT 135 NMR analysis.
2.4 Polymer syntheses
| Entry | Amine 1 (mmol) | MA 2 (mmol) | SO2 mmol | Initiatorc (mg) | Temp. | Yieldb | [η]c (dL g−1) |
|---|---|---|---|---|---|---|---|
| a Copolymerization reactions were carried out in DMSO (2.6 g) in the presence of ammonium persulfate (APS) or azoisobutyronitrile (AIBN) or tert-butylhydroperoxide for 48 h. b Polymer obtained is written in bold followed by isolated yields; the percent conversion determined by 1H NMR analyses of the crude reaction mixture are written in parentheses. c Viscosity of 1–0.25% polymer solution in 0.1 M NaCl at 30 °C was measured with Ubbelohde viscometer (K = 0.005317 mm2 s−2). d At −10 °C for 2 days, then at 0 and 25 °C for 2 days each. | |||||||
| 1 | 1a: 5 | 6 | 11 | APS (260) | 21 | 4a: 39 (67) | 0.0721 |
| 2 | 1a: 5 | 6 | 11 | APS (70) | 45 | 4a: 34 (68) | 0.0562 |
| 3 | 1a: 5 | 6 | 11 | APS (260) | 45 | 4a: 48 (86) | 0.0571 |
| 4 | 1a: 5 | 10 | 16 | APS (260) | 45 | 4a: 62 (90) | 0.0462 |
| 5 | 1a: 5 | 6 | 11 | APS (96) | 65 | 4a: 11 (72) | 0.0401 |
| 6 | 1a: 5 | 6 | 11 | APS (210) | 65 | 4a: 53 (60) | 0.0454 |
| 7 | 1a: 5 | 6 | 11 | APS (250) | 65 | 4a: 48 (81) | 0.0228 |
| 8 | 1a: 5 | 6 | 11 | AIBN (46) | 65 | 4a: trace (28) | — |
| 9 | 1a: 5 | 6 | 11 | AIBN (100) | 65 | 4a: 27 (60) | 0.0211 |
| 10 | 1b: 5 | 6 | 11 | APS (140) | 65 | 4b: 49 (72) | 0.0355 |
| 11 | 5: 5 | 6 | 11 | APS (200) | 65 | 6: 61 (67) | 0.532 |
| 12 | 5: 5 | 6 | 11 | AIBN (50) | 65 | 6: 67 (71) | 0.519 |
| 13 | — | 6 | 5 | APS (140) | 45 | No polymer | — |
| 14 | — | 6 | 5 | TBHP (100) | Temp.d | No polymer | — |
For the polymerization involving monomer 5,17 the crude reaction mixture was dialyzed against 2 M HCl followed by deionized water. Polymer 6 (y ≈ 0), precipitated out during dialysis, was dried under vacuum. The spectral analysis revealed the identical nature of the material 6 (y = 0) described elsewhere.20
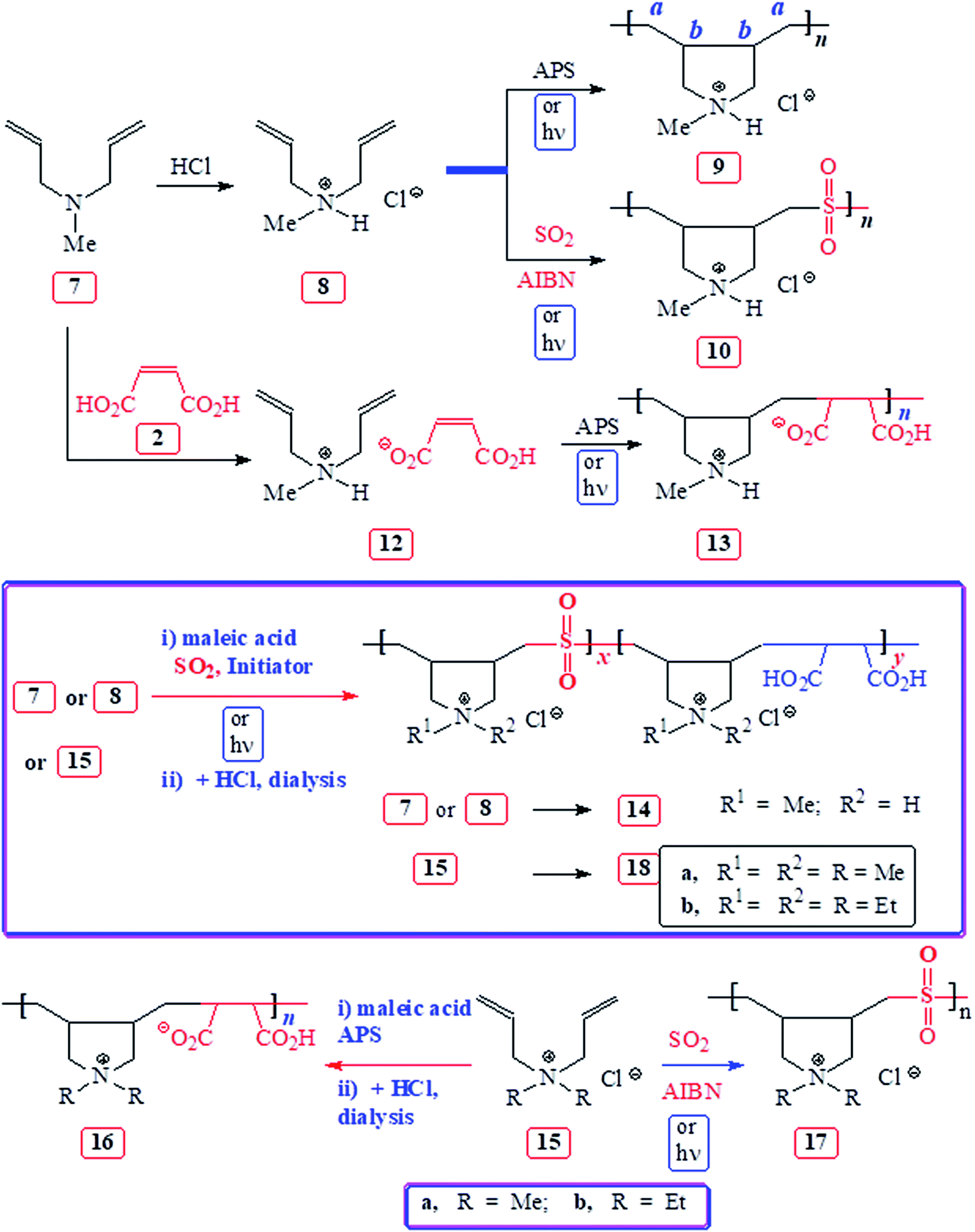 | ||
| Scheme 2 Synthesis of homopolymer from 8, alternate copolymers from 7/MA, 15/MA, 8/SO2, 15/SO2, and terpolymers from 7 or 8/MA/SO2.and 15/MA/SO2. | ||
CH), 127.19 (2C, CH2), 135.09 (CHCH), 171.14 (CO). The 13C spectral assignments were supported by DEPT 135 NMR.
| Entry | 7 or 8 (mmol) | MA 2 (mmol) | SO2 (mmol) | DMSO (g) | Initiator (mg) | Temp (°C) | Time (h) | Yielda | [η]b (dL g−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Isolated yields is followed by the percent conversion written in parentheses. As determined by 1H NMR analyses of the crude reaction mixture. b Viscosity of 1–0.25% polymer solution in 0.1 M NaCl at 30 °C was measured with Ubbelohde viscometer (K = 0.005317 mm2 s−2). c Cloudy reaction mixture after 4 h. d MA remained partially soluble. e Solidified. | |||||||||
| 1c | 7: (5) | 6 | 11 | 2.3 | APS: 85 | 45 | 72 | 14: 21 (63) | 0.141 |
| 2c | 7: (5) | 6 | 11 | 2.3 | APS: 150 | 45 | 72 | 14: 31 (57) | 0.125 |
| 3 | 7: (5) | 11 | 16 | 2.3 | APS: 85 | 45 | 72 | 14: 23 (63) | 0.121 |
| 4 | 7: (5) | 11 | 16 | 2.3 | APS: 175 | 45 | 48 | 14: 47 (60) | 0.182 |
| 5 | 7: (5) | 11 | 16 | 2.3 | APS: 260 | 65 | 24 | 14: 32 (83) | 0.129 |
| 6 | 7: (5) | 22 | 31 | 2.8 | APS: 260 | 65 | 24 | 14: 62 (85) | 0.172 |
| 7c | 7: (5) | 6 | 11 | 2.3 | AIBN: 75 | 65 | 36 | 14: 0 (10) | — |
| 8d | 8: (1) | 10 | 25 | 4.6 | APS: 360 | 45 | 48 | 14: 51 (67) | — |
| 9d | 8: (5) | 5 | 5 | 0 | Dark | 23 | 24 | 14: 0 (0) | — |
| 10d | 7: (5) | 5 | 6 | 0 | UV | 23 | 168 | 14: 26 (43) | 0.278 |
| 11e | 8: (5) | 5 | 10 | 0 | UV | 23 | 24 | 14: 45 (62) | 0.329 |
| 12e | 8: (5) | 5 | 10 | 0 | UV | −15 | 4 | 14: 43 (67) | 0.273 |
| 13e | 8: (5) | 5 | 10 | 1.0 | UV | 23 | 24 | 14: 43 (83) | 0.471 |
| 14 | 8: (5) | 5 | 10 | MeOH: 1.0 | UV | −10 | 6 | 14: 15 (35) | 0.192 |
| 15 | 8: (5) | 0 | 4.5 | 0 | UV | 23 | 24 | 10: 54 (72) | 0.836 |
| 16 | 8: (5) | 0 | 0 | H2O: 0.3 | UV | 23 | 144 | 9: — (7.6) | — |
| Entry | 15 (mmol) | MA 2 (mmol) | SO2 (mmol) | DMSO (g) | Initiator (mg) | Temp (°C) | Time (h) | Yield (%) | [η]a (dL g−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Viscosity of 1–0.0625% polymer solution in 0.1 N NaCl at 30 °C was measured with Ubbelohde viscometer (K = 0.005317 mm2 s−2). b 98% conversion as revealed by 1H NMR of crude mixture. c White solid mixture in the beginning and no phase change throughout. d Not been able to add more SO2. e Slightly turbid in the beginning but hardened towards the end. | |||||||||
| 1 | 15a: 5 | 6 | — | H2O (1.0) | APS: 250 | 100 | 0.25 | 16a: 27b | 0.102 |
| 2c | 15a: 5 | — | 6 | 0 | UV | 23 | 48 | 17a: 80 | 1.86 |
| 3 | 15a: 5 | 6 | 11 | 3.0 | APS: 215 | 55 | 24 | 18a: 87 | 0.317 |
| 4 | 15a: 5 | 6 | 6d,e | 0 | UV | 23 | 48 | 18a: 71 | 1.67 |
| 5 | 15b: 5 | — | 6 | 2.6 | APS: 250 | 55 | 2 | 17b: 96 | 0.552 |
| 6 | 15b: 5 | — | 6 | 2.6 | AIBN: 29 | 55 | 72 | 17b: 87 | 0.253 |
| 7 | 15b: 5 | 6 | 10 | 2.8 | APS: 125 | 55 | 48 | 18b: 88 | 0.401 |
2.5 Determination of basicity constants by titrations
The basicity constant (K) of the three basic centers in PA 13 were determined using reported procedures11,22 and conditions described in Table S1.† logKis were calculated using the Henderson–Hasselbalch equation (eqn (2); Scheme 3).
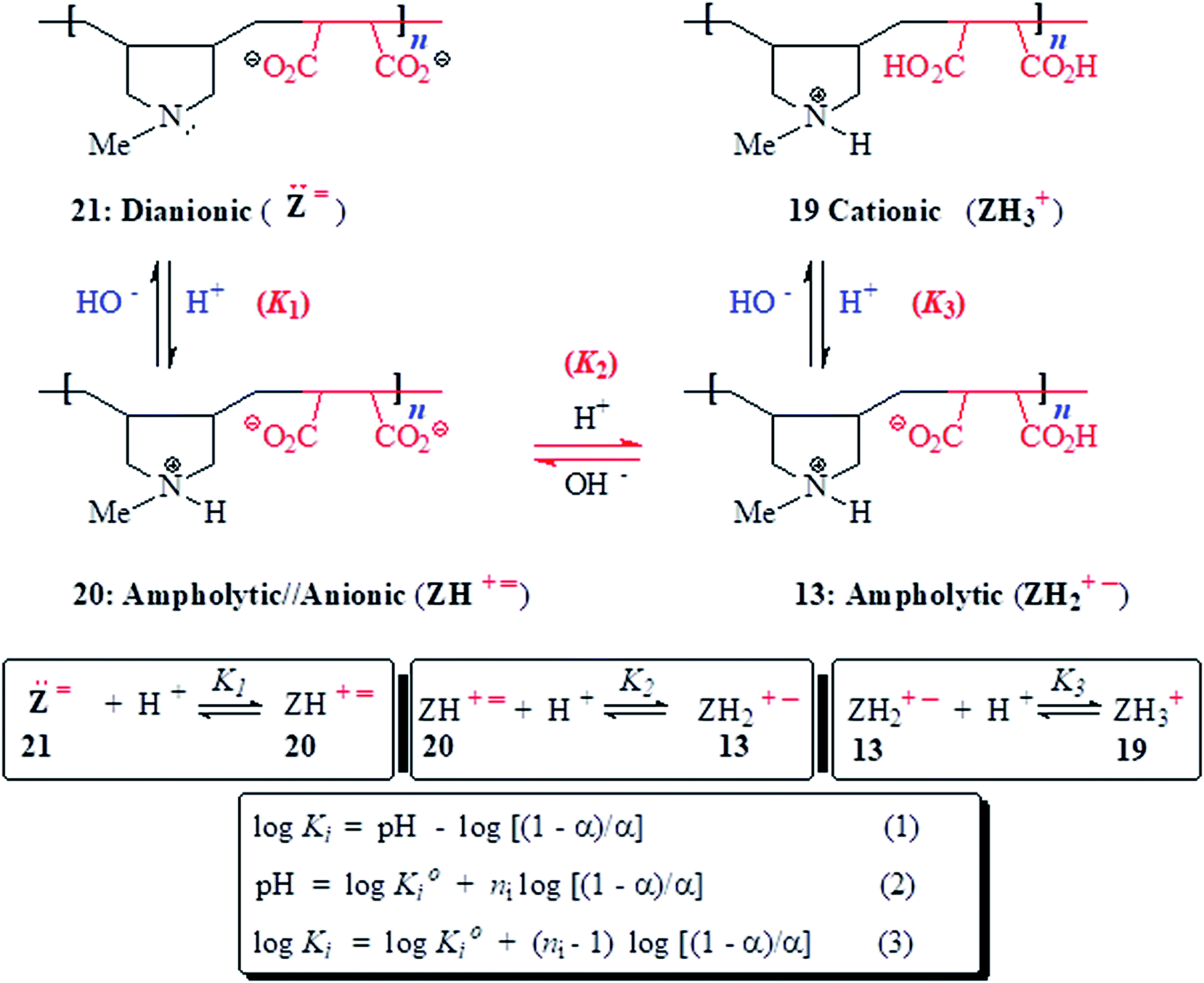 | ||
| Scheme 3 pH Induced changes in the charge types in the backbone of 13, 19–21. | ||
2.6 Use of synthesized PA 13 as an antiscalant
The inhibition of CaSO4 scale formation from its supersaturated solution was examined in the presence of various concentrations of PA 13 in an aqueous solution containing Ca2+ (2600 mg L−1) and SO42− (6300 mg L−1) at 40 ± 1 °C as described.22 The conductivity decreased suddenly after an induction time, owing to the formation of scale as confirmed by visual inspections.3. Results and discussion
3.1 Polymer synthesis
The results of APS-initiated terpolymerization of monomers 1a or 1b or 5 with SO2 and MA 2 are given in Table 1 (Scheme 1). The attempted copolymerization of MA–SO2 was unsuccessful (entries 13 and 14). Terpolymerization of 1a–MA–SO2 was conducted under various conditions using APS or AIBN initiator. The polymer 4a was isolated after extensive dialysis against aqueous HCl to remove unreacted MA and replace the maleate anion with Cl− ions. The spectral (NMR and IR) analysis revealed that terpolymer 4a resembled the 1a–SO2 copolymer 4a (y = 0),20 except a minor 13C CO peak at ≈δ 180 ppm (vide infra) and a minor vibration at ≈1728 cm−1 attributed to CO of CO2H in the IR spectrum. The absence of alkene carbons at δ 135 ppm excluded the presence of MA either as a free acid or the maleate ions; however, the presence of minor CO signals suggests the incorporation of maleic acid into the polymer backbone. The MA does undergo propagation, but the addition is reversible as indicated by the presence of fumaric acid (FA) (trans-HO2CCCHCO2H) in the unreacted reaction mixture, in addition to its original cis-counterpart MA in a ≈ 1:4 ratio. The radical having MA terminal: R–CH(CO2H)–CH(CO2H)· may undergo quick reversion to give R· and MA as well as FA.
Likewise, 1b–MA–SO2 (entry 10) and 5–MA–SO2 (Table 1, entries 11 and 12) terpolymerizations afforded 4b and 6, respectively (Scheme 1), whose spectra are identical to those of 4b (y = 0)21 and 6 (y = 0),20 except that a very minor peak due to CO in the 13C NMR and IR spectra indicate the very minor incorporation of MA.
Monomer 8 was homo- and co-polymerized with SO2 to obtain cationic polyelectrolytes 9 and 10 in excellent yields of 87 and 93%, respectively (see Experimental) (Scheme 2). Homopolymerization of 8 in an aqueous solution under UV light (λ = 365 nm) was very slow; after 6 days, it gave CPE 9 in a very low yield of 7.6% (entry 16, Table 2). The bulk photocopolymerization 8–SO2 (entry 15, Table 2) in the absence of solvent gave CPE 10 having much higher intrinsic viscosity [η] of 0.836 dL g−1 than that of the thermally initiated copolymer with a [η] of 0.276 dL g−1 (Experimental section 2.4.4).
Terpolymerization of 7 or 8–MA–SO2 was extensively studied using free radical initiators APS, AIBN or UV radiation at λ 365 nm (Scheme 2). The results are included in Table 2. Note that the isolated yields after dialysis are low as compared to the NMR conversion since the low molecular weight polymer fractions leave the dialysis bag. The 1H and 13C NMR spectra of the samples from entries 1–14 dealing with the 7 or 8–MA–SO2 terpolymerizations afforded 14, which is almost identical to the 8–SO2 copolymer 10 except the IR has very minor CO vibrational peak at ≈1728 cm−1 indicating the presence of MA repeating units in 14. A series of IR spectra of copolymer 10 containing 0–5 mol% succinic acid (CH2)2(CO2H)2 helped us to estimate the proportion of MA unit in 14 as ≈1–3 mol%. The strong non-overlapping IR vibrations of SO2 unit in 10 at 1304 and 1128 cm−1 and the CO vibration of CO2H at ≈1728 cm−1 were used to estimate the MA mol%.
Next, we focussed our attention to the polymerization of quaternary ammonium salts 15 as outlined in Scheme 2; the polymerization conditions and the results are included in Table 3. The 15a–MA copolymerization afforded 16a in 27% isolated yields, while the crude mixture before dialysis indicated 98% conversion as calculated from 1H NMR spectrum (entry 1). The spectral data matched with that reported for the polymer.23 Bulk copolymerization of 15a–SO2 under UV light24 afforded copolymer 17a in excellent yield (entry 2). The very high [η] value of 1.86 dL g−1 indicated the high molar mass of the polymer. The thermal 15b–SO2 (ref. 19) copolymerization afforded copolymer 17b also in excellent yields (entries 5 and 6). The IR and NMR spectra of 17a, b matched with the reported data.25,26 The terpolymerizations: 15a–MA–SO2 (entries 3 and 4) and 15b–MA–SO2 (entry 7) gave polymers 18a and 18b, respectively; their NMR and IR spectra matched with those of the respective copolymers 17a and 17b except the minor CO vibration at ≈1728 cm−1 indicating the incorporation of ≈1–3 mol% MA.
All the reactivity ratios in the pairs: amine salt–MA and amine salt–SO2 are almost zero (vide supra) thereby implying that under the reaction conditions, amine salt or MA or SO2 cannot undergo homopolymerization, instead they must give alternate copolymers as experimentally observed. MA–SO2 reaction has been shown to give neither homo- nor co-polymer (entries 13 and 14, Table 1). In the numerous amine salt–MA–SO2 terpolymerizations, the extent of MA incorporation is only 1–2 mol%. In every instance, amine salt–SO2 copolymer was obtained and no trace of amine salt–MA copolymer could be identified. The claim in the patented literatures12–14 that the formation of terpolymer: diallyldimethylammonium chloride–maleic acid–SO2 having repeating unit composition of 50:25:25 and 70:25:5 are doubtful. A composition ratio of 50:25:25 may indeed be a result of a mixture of two alternate copolymers of diallyldimethylammonium chloride–maleic acid and diallyldimethylammonium chloride–SO2. The current work confirms that the amine salt–MA–SO2 cannot be terpolymerized to any meaningful extent.
3.2 Solubility behavior
Cationic polymers 4a, b (y ≈ 0), 9, 10 and 14 (y ≈ 0) are water-soluble, while the PZ 6 (y ≈ 0) is insoluble in water but soluble in 0.8 M HCl.20 The water-solubility of 13, unlike most electroneutral polymers,27–29 could be attributed to the less effective participation of the crowded CO2− to undergo electrostatic associations with NH+ for manifestation of ampholytic interactions.2 Moreover, the hydrophilicity and solubility of the ampholytic motifs increase as a result of the increased dipole moment (μ) because of the large separation between NH+ and CO2−.30,313.3 Spectroscopic characterization
The strong IR bands at 1304 and 1128 cm−1 were assigned to the vibrations of SO2 unit in 10. The absorption band at ≈1714 and 1574 cm−1 is attributed to the CO stretch of CO2H and CO2− in (+˙–) 13.32
The NMR spectra of several polymers are displayed in Fig. 1 and 2. For the monomer pair 12, alkene protons of the diallyl amine part and maleate motif appeared at δ 5.5–6 ppm and 6.08 ppm, respectively (Fig. 1a), while the corresponding carbons are displayed at δ 126.4 (CH), 127.2 (2C, CH2), and 135.1 (CHCH) (Fig. 2a). The low molar masses (as suggested by low viscosity values) and the absence of spectral signal for residual alkene protons or carbons of the polymers 9, 10 and 13 are indicative of degradative chain transfer33 involving allylic hydrogens or a termination reaction by coupling.34 The compact coil10 conformation of MA-copolymer (+˙–) 13 is reflected in the broadened 1H as well as 13C NMR spectra (Fig. 1d and 2d), whereas the spectral signals are found to be sharp for homopolymer 9 or SO2-copolymer 10 (Scheme 2) (Fig. 1b or 1c). The incorporation of MA in (+˙–) 13 is confirmed by the presence of a broad signal around δ 180 ppm (Fig. 2d). Elemental analysis and the spectral data confirmed the formation of the alternate copolymer because both the reactivity ratios ramine and rMA, are expected to be close to zero.10,11 Integration of the relevant carbon signals35,36 revealed a 3:1 cis/trans ratio of the ring substituents at Cb,b (Scheme 2).
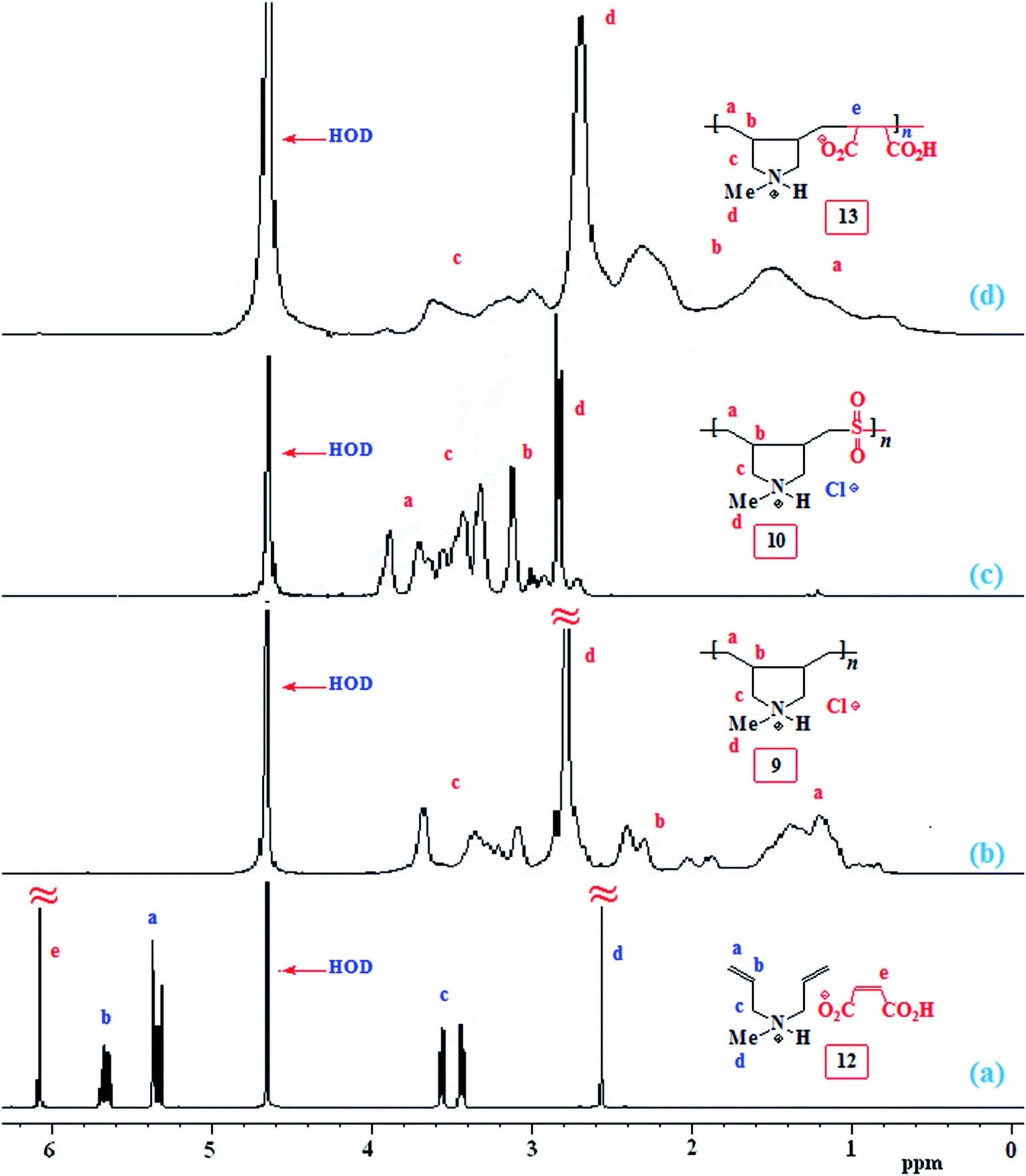 | ||
| Fig. 1 1H NMR spectrum of (a) 12, (b) 9, (c) 10, and (d) 13 in D2O. | ||
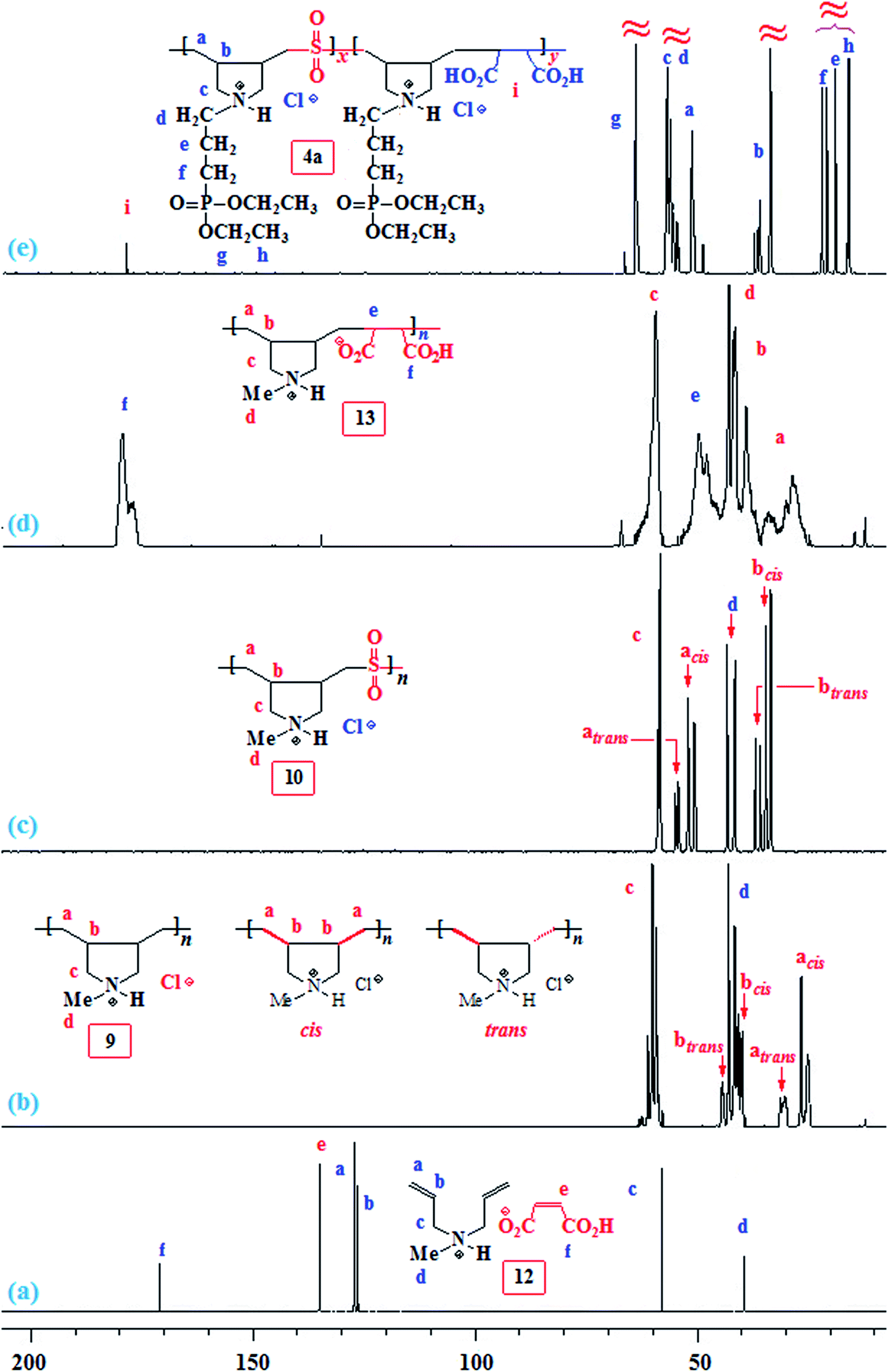 | ||
| Fig. 2 13C NMR spectrum of (a) 12, (b) 9, (c) 10, (d) 13, and (e) 4a in D2O. | ||
3.4 TGA and viscosity behaviors PA (+˙–) 13
TGA plot for a PA 13 (dried under vacuum at 55 °C for 6 h) revealed the weight losses at various temperature ranges (Fig. 3a). The presence of adjacent CO2H groups would lead to the formation of anhydride motifs with the loss of water as reflected by an 8.3% weight loss up to 200 °C. In fact, a loss of 1 molecule of water per repeating unit is calculated to be 7.9%. In the 200–380 °C range, maleic anhydride is itself released from the polymer backbone thereby accounting for the loss of ≈41.2% as compared to a calculated value of 46.9%.10,37 A 48% loss in the 380–800 °C range is attributed to the removal of the amine units.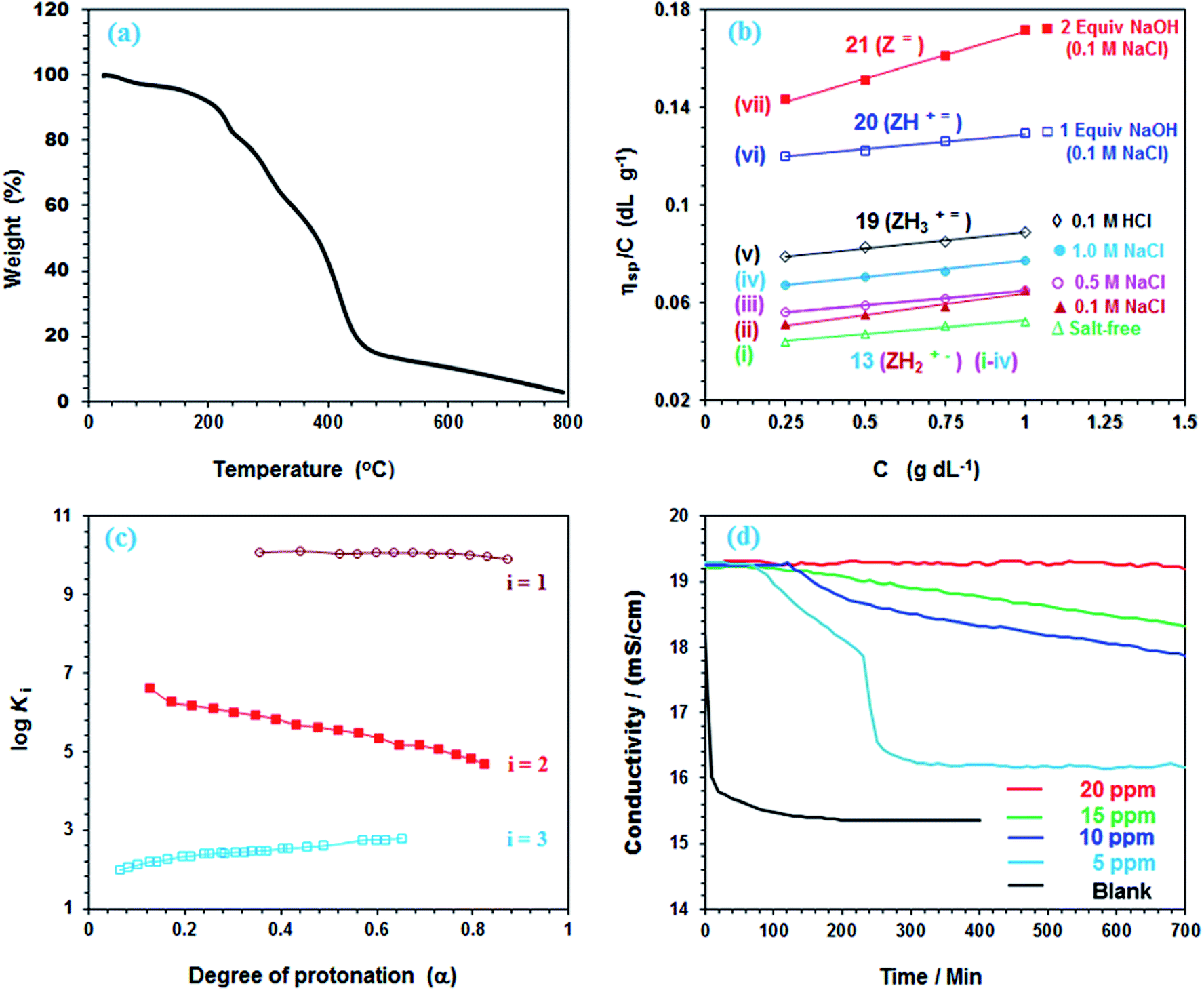 | ||
Fig. 3 (a) TGA curves of 13; (b) the viscosity behavior at 30 °C of polyampholyte 13 (i)  in salt-free water, (ii) in salt-free water, (ii)  in 0.1 M NaCl, (iii) in 0.1 M NaCl, (iii)  in 0.5 M NaCl, (iv) in 0.5 M NaCl, (iv)  in 1.0 M NaCl, (v) in 1.0 M NaCl, (v)  in 0.1 M HCl, and (vi) in 0.1 M HCl, and (vi)  in the presence of 1 equiv. NaOH in 0.1 M NaCl, and (vii) in the presence of 1 equiv. NaOH in 0.1 M NaCl, and (vii)  in the presence of 2 equiv. NaOH in 0.1 M NaCl; (c) plot for the apparent logKiversus degree of protonation (α) in salt-free water in the presence of 2 equiv. NaOH in 0.1 M NaCl; (c) plot for the apparent logKiversus degree of protonation (α) in salt-free water  (logK3, run 2, Table S1†), (logK3, run 2, Table S1†),  (logK2, run 2, Table S1†) and (logK2, run 2, Table S1†) and  (log K3, run 2, Table S1†); (d) conductivity of a supersaturated solution of CaSO4 in the presence (5, 10, 15, 20 ppm) of 13 and in the absence (blank) of antiscalant. (log K3, run 2, Table S1†); (d) conductivity of a supersaturated solution of CaSO4 in the presence (5, 10, 15, 20 ppm) of 13 and in the absence (blank) of antiscalant. | ||
The viscosities of PA (+˙–) 13 are presented in Fig. 3b. The increase in viscosity with the increasing salt concentrations demonstrates the antipolyelectrolytic38 nature of the polyampholyte [cf.Fig. 3b(i–iv)]. The ampholytic dipole is not perfectly electroneutral, rather has a residual negative charge on CO2− since Na+ cannot effectively neutralize it as compared to the greater shielding of N+ by Cl−.39–41 The magnitude of the excess negative charge, hence the viscosity, increases with salt concentrations.
PA (+˙–) 13 becomes a cationic polyelectrolyte 19 by protonation of CO2− and as such its viscosity becomes higher in 0.1 M HCl because of repulsion among positive charges (Fig. 3b-v) (Scheme 3). The neutralization of 13 with one or two equivalents of NaOH leads to polyampholyte/anion (+˙) 20 and polydianion () 21, the greater charge imbalance in the latter makes it more viscous (Fig. 3b-vi and vii).
Strong interactions of materials of GPC columns with functional groups like amine and CO2− prevented the determination of molar masses of the polymers like 13. Similar observation was reported earlier.10 Purification by extensive dialysis with membrane of MWCO of 6000–8000 daltons indicates the products as true polymers not oligomers. The use of higher initiator dose along with degradative chain transfer termination process33 led to PA 13, having low molar mass as indicated by its lower [η] value.
The intrinsic viscosities of the synthesized polymers 4a, 4b, 6, 9, 10, 13, 14, 16–18 are reported in the Tables 1–3. Since the MA incorporation is very minimal, 4a, 4b and 6 with the value of y ≈ 0 resembled the copolymers 4a, 4b and 6 (y = 0) whose molar mass and [η] are amply described.20,21 Similarly, polymers 17a, b which are similar to 18a, b (y ≈ 0)23,25,26 and 16a32 have been extensively characterized in terms of viscosity and molar mass.
3.5 Basicity constant
As outlined in Scheme 3, the protonation constant K of three basic centers in () 21 are calculated using eqn (3) where logK° = pH at α (degree of protonation) = 0.5. The variation from the true basicity constant depends on the value of n which equals 1 for small molecules. The values of ‘n’ and logK° were extracted from the slope and intercept of the pH vs. log[(1 − α)/α] straight-line plot. The respective 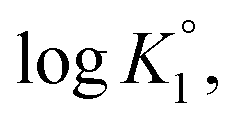
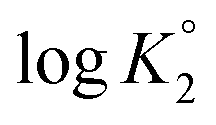 and
and 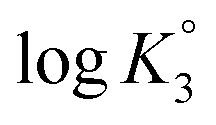 were determined to be 10.1, 5.59 and 2.62, with the corresponding n1, n2 and n3 values of 1.15, 2.19 and 0.48 (Table S1†).
were determined to be 10.1, 5.59 and 2.62, with the corresponding n1, n2 and n3 values of 1.15, 2.19 and 0.48 (Table S1†).
All the n values, especially n2 and n3, are reflective of the “apparent”42,43 nature of the Kis which changes with the α as shown in Fig. 3c. The n value reflects a measure of the polyelectrolyte index. There are significant decrease and increase of logK2 and logK3, respectively, with increasing α, while the logK1 remains almost constant since the associated n value of 1.15 is not far from 1.
With increasing α, the equilibrium: [(ZH+˙) 20 + H+ ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) (ZH2+˙–) 13] is shifted to the right thereby decreasing
(ZH2+˙–) 13] is shifted to the right thereby decreasing 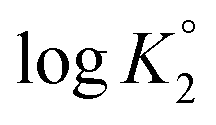 as a consequence of decreasing negative charge density that encourages protonation. This is in contrast to the increase of
as a consequence of decreasing negative charge density that encourages protonation. This is in contrast to the increase of 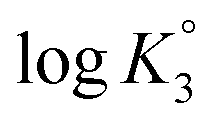 [involving (ZH2+˙–) 13 + H+ (ZH3+˙) 19], whereby protonation of a repeating unit also decreases the overall negative charges of the polymer chain. The n values > or < 1 are caused by entropy effects.42,44 With each protonation, a repeating unit (RU) in ampholytic/anionic (ZH+˙) 20 collapses into an ampholytic RU of (ZH2+˙–) 13. Ampholytic/anionic (ZH+˙) 20 having excess negative charges is more hydrated than PA (+˙–) 13 as supported by the higher viscosity value of the former (cf. Fig. 3b-vivs.3b-ii). The protonation of a RU leads to an entropically favourable release of water of hydration. For (ZH+˙) 20, as the α increases, the average number of water molecules per RU in a polymer chain decreases which leads to a decrease in the magnitude of the positive entropy changes. Since the exothermic ΔH° is independent of α, the ΔG°, hence K, decreases with the increasing α.44
[involving (ZH2+˙–) 13 + H+ (ZH3+˙) 19], whereby protonation of a repeating unit also decreases the overall negative charges of the polymer chain. The n values > or < 1 are caused by entropy effects.42,44 With each protonation, a repeating unit (RU) in ampholytic/anionic (ZH+˙) 20 collapses into an ampholytic RU of (ZH2+˙–) 13. Ampholytic/anionic (ZH+˙) 20 having excess negative charges is more hydrated than PA (+˙–) 13 as supported by the higher viscosity value of the former (cf. Fig. 3b-vivs.3b-ii). The protonation of a RU leads to an entropically favourable release of water of hydration. For (ZH+˙) 20, as the α increases, the average number of water molecules per RU in a polymer chain decreases which leads to a decrease in the magnitude of the positive entropy changes. Since the exothermic ΔH° is independent of α, the ΔG°, hence K, decreases with the increasing α.44
For the protonation of (ZH2+˙–) 13 to (ZH3+˙) 19 (Scheme 3), the latter is more hydrated thus having expanded polymer chain as evinced by its greater viscosity value (cf. Fig. 3b-vvs.3b-ii). The more exposed RU makes the neutralization process easier with the increasing α (Fig. 3c, i = 3).45,46 With increasing α, increasing imbalance in favour of positive charge also increases the degree of hydration which leads to greater entropy changes because of release of a greater number of molecules during progressive protonation.
3.6 Scale inhibition study
In a Reverse Osmosis (RO) process, feed water is converted to product water and rejects brine, in which supersaturation of dissolved salts, like CaSO4 and CaCO3, may lead to precipitation which reduces the efficacy of the membrane separation process.The percent scaling inhibition (PSI) is calculated using eqn (4):
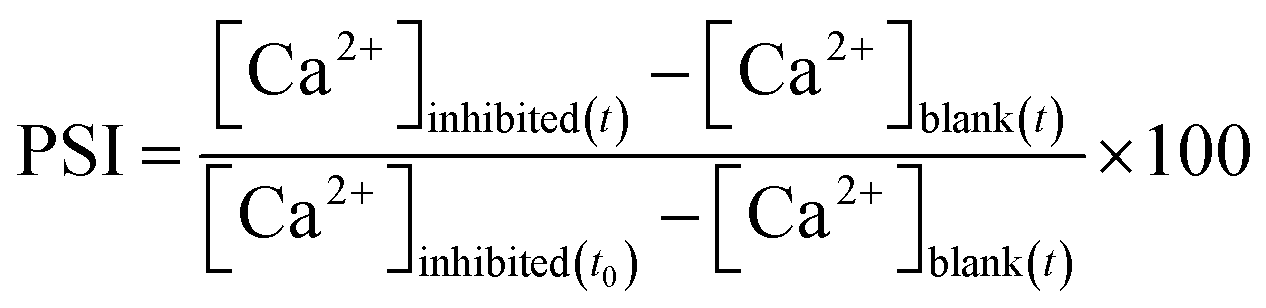 | (4) |
The concentration of Ca2+ and SO42−, present in the reject brine of a RO plant,47 was described as 1-CB (concentrated brine). The scaling behavior in the presence of PA 13 was carried out in a supersaturated solution of CaSO4 comprising 3-CB concentration of Ca2+ (2600 ppm) and SO42− (6300 ppm) by conductivity measurements. A sudden drop in the conductivity indicates the onset of CaSO4 scaling (Fig. 3d).
It is indeed satisfying that in the presence of 5 and 20 ppm 13, a 100% scale inhibition was registered for about 50 and 500 min, respectively (Table 4). Since the reject brine usually has a residence time of ≈30 min in the osmosis chamber, the current antiscalant may thus be very effective in inhibiting CaSO4 scaling. As shown in Fig. 3d, the onset of precipitation occurs after an induction period; an accelerated growth of CaSO4 crystals is indicated by a sudden drop in conductivity. For a duration of 700 min, 20 ppm PA 13 did not show any induction time. The crystal growth starts after nucleation process, which is interfered with the complexation of the metal ions by the chelating ligands of the antiscalant.48,49 The gypsum scale, i.e., CaSO4 in mineral form, occurs during several processes involving production of water.50 PA 13 is found to be remarkably efficient in preventing scale formation, and as such it has the ability to mitigate the membrane fouling in RO plants.
| Entry | Sample (ppm) | Percent inhibition at times (min) | ||||||
|---|---|---|---|---|---|---|---|---|
| 50 | 100 | 200 | 300 | 400 | 500 | 700 | ||
| a Three times the concentration of Ca2+ and SO42− found in the concentrated brine of an RO plant. | ||||||||
| 1 | 5 | 100 | 92 | 71 | 24 | 22 | 17 | 17 |
| 2 | 10 | 100 | 100 | 88 | 81 | 76 | 71 | 63 |
| 3 | 15 | 100 | 99 | 96 | 91 | 88 | 83 | 75 |
| 4 | 20 | 100 | 100 | 100 | 100 | 100 | 100 | 98 |
4. Conclusions
Several diallylamine derivatives 1a, b, 5 and 7 as well as quaternary diallyldialkylammonium chlorides 15a, b has been terpolymerized with MA and SO2 using APS initiator or UV light at a λ of 365 nm in an attempt to obtain amine (salt)–MA–SO2 terpolymer. In contrast to reported literature, only 1–2 mol% of MA was incorporated into the polymer chain mainly consisting of amine (salt)–SO2 copolymers. The non-incorporation of MA is attributed to the far greater rate of propagation of amine (salt)–SO2 than either amine (salt)–MA or MA–SO2 under the polymerization conditions. The reactivity ratios of both monomers in the amine (salt)–MA and MA–SO2 pairs are ≈0; however, in a three-monomer system: amine (salt)–MA–SO2, the very slow reactivity of MA does not allow its incorporation into terpolymer chain to any meaningful extent.The 1c–MA copolymer PA 13 has been synthesized in excellent yield with an anticipation that it could be a potential antiscalant. Under pH-induced changes, stimuli-responsive polyampholyte 13 was transformed to cationic 19, polyampholyte-anionic 20, and dianionic polyelectrolyte 21 to examine their viscosity. The viscosity values of 13 in the presence of salt NaCl confirmed its antipolyelectrolyte behaviour. The logK (i.e. pKa) of two carboxylic acid groups and NH+ in 13 has been determined to be 2.62, 5.59, and 10.1, respectively. PA 13 was evaluated as an antiscalant for potential application in reverse osmosis (RO) plants. At concentrations of 5 and 20 ppm, it demonstrated remarkable efficiency of ≈100% for CaSO4 scale inhibition from its supersaturated solution for 50 and 500 min, respectively. Since an antiscalant should be effective for the duration of brine's residence time (≈30 min) in the osmosis chamber, the synthesis of PA 13 in excellent yields from cheap starting materials and its very impressive performance may accord it a prestigious place among many an environment-friendly phosphate-free antiscalant. Note that polyphosphate additives used for controlling scale formation, when discharged in the sea have deleterious influence over the marine biota picture.51
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The facilities provided by KFUPM are gratefully acknowledged.References
- G. B. Butler, Cyclopolymerization and Cyclocopolymerization, Marcel Dekker, New York, 1992 Search PubMed.
- S. Kudaibergenov, W. Jaeger and A. Laschewsky, Adv. Polym. Sci., 2006, 201, 157–224 CrossRef CAS.
- A. Laschewsky, Polymers, 2014, 6, 1544–1601 CrossRef.
- P. K. Singh, V. K. Singh and M. Singh, e-Polym., 2007, 30, 1–34 Search PubMed.
- G. B. Butler, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3451–3461 CrossRef CAS.
- F. C. McGrew, J. Chem. Educ., 1958, 35, 178–186 CrossRef CAS.
- N. Y. Abu-Thabit, I. W. Kazi, H. A. Al-Muallem and S. A. Ali, Eur. Polym. J., 2011, 47, 1113–1123 CrossRef CAS.
- S. A. Ali, H. A. Al-Muallem and M. A. J. Mazumdar, Polymer, 2003, 44, 1671–1679 CrossRef CAS.
- S. A. Ali, I. B. Rachman and T. A. Saleh, Chem. Eng. J., 2017, 330, 663–674 CrossRef CAS.
- F. Rullens, M. Devillers and A. Laschewsky, Macromol. Chem. Phys., 2004, 205, 1155–1166 CrossRef CAS.
- I. Y. Yaagoob, H. A. Al-Muallem and S. A. Ali, RSC Adv., 2017, 7, 31641–31653 RSC.
- E. Kogure, N. Morii, Y. Makino, T. Hawakawa and K. Tsukuda, JP Pat., 2007106920 A 20070426, 2007.
- Y. Yomogida and H. Torigoe, JP Pat., 2012214539 A 20121108, 2012.
- N. Suzuki, H. Togashi, S. Ito and Y. Yamazaki, JP Pat., 2007238921 A 20070920, 2007.
- S. A. Ali, N. Y. Abu-Thabit and H. A. Al-Muallem, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5693–5703 CrossRef CAS.
- O. C. S. Al-Hamouz and S. A. Ali, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3580–3591 CrossRef CAS.
- S. A. Ali, I. W. Kazi and N. Ullah, Ind. Eng. Chem. Res., 2015, 54, 9689–9698 CrossRef CAS.
- S. H. Pine, J. Chem. Educ., 1968, 45, 118 CrossRef CAS.
- Y. Negi, S. Harada and O. Ishizuka, J. Polym. Sci., Part A: Polym. Chem., 1967, 5, 1951–1965 CrossRef CAS.
- S. A. Ali, I. W. Kazi and F. Rahman, Polym. Int., 2014, 63, 616–625 CrossRef CAS.
- S. A. Ali and O. C. S. Al-Hamouz, Polymer, 2012, 53, 3368–3377 CrossRef CAS.
- S. A. Haladu and S. A. Ali, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 5130–5142 CrossRef CAS.
- A. I. Vorobeva, G. R. Sultanova, A. K. Bulgakov, V. I. Zainchkovskii and S. V. Kolesov, Pharm. Chem. J., 2013, 46, 653–655 CrossRef CAS.
- S. Harada and K. Arai, US Pat., 3585118, 1971.
- S. A. Ali, Y. Umar and B. F. Abu-Sharkh, J. Appl. Polym. Sci., 2005, 97, 1298–1306 CrossRef CAS.
- A. I. Vorobeva, E. V. Vasileva, K. A. Gaisina, Y. I. Puzin and G. V. Leplyanin, Polym. Sci., Ser. A, 1996, 38, 1077–1080 Search PubMed.
- J. C. Salamone, W. Volksen, A. P. Olson and S. C. Israel, Polymer, 1978, 19, 1157–1162 CrossRef CAS.
- T. A. Wielema and J. B. F. N. Engberts, Eur. Polym. J., 1987, 23, 947–950 CrossRef CAS.
- V. M. M. Soto and J. C. Galin, Polymer, 1984, 25, 254–262 CrossRef CAS.
- M. Galin, A. Chapoton and J.-C. Galin, J. Chem. Soc., Perkin Trans. 2, 1993, 545–553 RSC.
- A. W. Lloyd, C. J. Olliff and K. L. Rutt, Int. J. Pharm., 1996, 131, 257–262 CrossRef CAS.
- Z. E. Ibraeva, M. Hahn, W. Jaeger, L. A. Bimendina and S. E. Kudaibergenov, Macromol. Chem. Phys., 2004, 205, 2464–2472 CrossRef CAS.
- G. B. Butler and R. J. Angelo, J. Am. Chem. Soc., 1957, 79, 3128–3131 CrossRef CAS.
- R. M. Pike and R. A. Cohen, J. Polym. Sci., 1960, 44, 531–538 CrossRef CAS.
- S. A. Ali, A. Rasheed and M. I. M. Wazeer, Polymer, 1999, 40, 2439–2446 CrossRef CAS.
- V. D. Vynck and E. J. Goethals, Macromol. Rapid Commun., 1997, 18, 149–156 CrossRef.
- M. Hahn, W. Jaeger, R. Schmolke and J. Behnisch, Acta Polym., 1990, 41, 107–112 CrossRef CAS.
- K. Nishida, K. Kaji, T. Kanaya and N. Fanjat, Polymer, 2002, 43, 1295–1300 CrossRef CAS.
- M. Gao, K. Gawel and B. T. Stokke, Eur. Polym. J., 2014, 53, 65–74 CrossRef CAS.
- Z. Cao and G. Zhang, Phys. Chem. Chem. Phys., 2015, 17, 27045–27051 RSC.
- S. Morozova, G. Hu, T. Emrick and M. Muthukumar, ACS Macro Lett., 2016, 5, 118–122 CrossRef CAS.
- R. Barbucci, M. Casolaro, N. Danzo, V. Barone, P. Ferruti and A. Angeloni, Macromolecules, 1983, 16, 456–462 CrossRef CAS.
- M. S. Bodnarchuk, K. E. B. Doncom, D. B. Wright, D. M. Heyes, D. Dini and R. K. O'Reilly, RSC Adv., 2017, 7, 20007–20014 RSC.
- R. Barbucci, M. Casolaro, P. Ferruti and M. Nocentini, Macromolecules, 1986, 19, 1856–1861 CrossRef CAS.
- R. Barbucci, M. Casolaro, M. Nocentini, S. Correzzi, P. Ferruti and V. Barone, Macromolecules, 1986, 19, 37–42 CrossRef CAS.
- A. Fini, M. Casolaro, M. Nocentini, R. Barbucci and M. Laus, Makromol. Chem., 1987, 188, 1959–1971 CrossRef CAS.
- F. H. Butt, F. Rahman and U. Baduruthamal, Desalination, 1995, 103, 189–198 CrossRef CAS.
- H. David, S. Hilla and S. Alexander, Ind. Eng. Chem. Res., 2011, 50, 7601–7607 CrossRef.
- R. J. Davey, The Role of Additives in Precipitation Processes, Industrial Crystallization, Proc. Symp., ed. S. J. Jancic and E. J. de Jong, North-Holland Publishing Co., 8th edn, 1982, pp. 123–135 Search PubMed.
- G. Mazziotti di Celso and M. Prisciandaro, Desalin. Water Treat., 2013, 51, 1615–1622 CrossRef CAS.
- A. M. Shams El Din, Sh. Aziz and B. Makkawi, Desalination, 1994, 97, 373–388 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08723g |
| This journal is © The Royal Society of Chemistry 2018 |