
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRationalization of hydrogen production by bulk g-C3N4: an in-depth correlation between physico-chemical parameters and solar light photocatalysis†
Andrea Speltini *a,
Ambra Pisanua,
Antonella Profumoa,
Chiara Milanesea,
Luigi Sangalettib,
Giovanni Drerab,
Maddalena Patrinic,
Marzia Pentimallid and
Lorenzo Malavasi*a
*a,
Ambra Pisanua,
Antonella Profumoa,
Chiara Milanesea,
Luigi Sangalettib,
Giovanni Drerab,
Maddalena Patrinic,
Marzia Pentimallid and
Lorenzo Malavasi*a
aDepartment of Chemistry and INSTM, University of Pavia, via Taramelli 12, 27100 Pavia, Italy. E-mail: andrea.speltini@unipv.it; lorenzo.malavasi@unipv.it
bI-LAMP, Dipartimento di Matematica e Fisica, Università Cattolica del Sacro Cuore, via dei Musei 41, 25121 Brescia, Italy
cDepartment of Physics and CNISM, University of Pavia, via Bassi 6, 27100 Pavia, Italy
dENEA – Italian National Agency for Energy, New Technologies and Sustainable Economic Development, Casaccia Research Centre, via Anguillarese 301, 00123 Roma, Italy
First published on 26th November 2018
Abstract
The aim of this work is the systematic study of the photocatalytic activity of bulk graphitic carbon nitride (g-C3N4) in relation with the physical–chemical, structural and optical properties of the semiconductor. Fourteen g-C3N4 samples have been prepared by thermal condensation starting from three different precursor (melamine, dicyandiamide and urea) and exploring various temperatures (in the range 500–700 °C). The materials obtained have been deeply characterized by high resolution scanning electron microscopy, thermogravimetric analysis, X-ray diffraction, nitrogen adsorption measurements (BET method), X-ray photoelectron spectroscopy and diffuse reflectance spectroscopy. Each semiconductor, coupled with Pt co-catalyst, was tested for hydrogen gas production from aqueous triethanolamine as model sacrificial agent, under simulated solar light. The hydrogen evolution profiles turned out to be strictly dependent on precursor type and synthesis temperature, with the highest evolution rate observed for the samples series produced from urea (up to ca. 4400 μmol g−1 h−1). The results, corroborated by the excellent inter-day precision of irradiation tests (RSD < 5%, n = 3) together with the good batch-to-batch reproducibility (RSD < 11%, n = 3), were critically discussed. Apart from the appealing production values obtained using the as-prepared materials, it was importantly pointed out that, besides crystallinity and visible light absorption, the photocatalytic behavior is definitely correlated to the surface area, which is dependent on the synthesis conditions, that is polymerization temperature and nature of g-C3N4 precursor. Overall, this systematic investigation demonstrated that, contrary to the polymerization degree (sp2/sp3 carbon ratio), surface area is the real determinant parameter for g-C3N4 hydrogen evolution activity.
1. Introduction
Graphitic carbon nitride, known as g-C3N4, is nowadays considered the most appealing visible-light photocatalyst for a variety of applications ranging from hydrogen evolution, overall water splitting, organic pollutants degradation, to CO2 photoreduction. More recently, this material has been proposed also for biomedical applications, and analytical sample preparation and sensing.1–5 All these possibilities are due to the peculiar electronic structure of g-C3N4, the relatively narrow band gap (ca. 2.7 eV) and conduction and valence band positions suitable to run relevant oxidation and reduction processes.6 In addition, g-C3N4 is stable at high temperatures (up to 600 °C in air), it can be easily prepared starting from cheap precursors, it is stable in several organic solvents, and it has a high chemical stability in both acidic and alkaline environments.1–4 Usually, g-C3N4 is prepared by a simple thermal condensation of a nitrogen-rich precursor such as cyanamide,7 dicyandiamide (DCD),8 melamine (MLM),9 thiourea,10 or urea,11 which provide graphitic-like layered structures consisting of 2D sheets composed of tris-s-triazines interconnected via tertiary amines.Most of the studies available in the literature dealing with g-C3N4 are focused on the photocatalytic hydrogen evolution underlining that g-C3N4 is a viable alternative to the known materials for hydrogen production under visible-light irradiation.1–4 The photocatalytic activity of as-prepared g-C3N4 is considered of relatively low-efficiency and several new synthetic strategies have been recently proposed to improve the H2 production. Among these, for pure g-C3N4 we found supramolecular pre-assembly, molten salt, ionic liquid, preparation of 2D nanosheets, and several modifications of carbon nitride by heteroatomic doping, creation of hybrid structures, heteromolecular doping, co-catalysis2 and thermal oxidation.12 In addition, other strategies are focused on the preparation of semiconductor–semiconductor heterojunctions as a way to enhance the photocatalytic performance by promoting the separation of photoinduced carriers. In this context, many semiconductors have been coupled with g-C3N4 to form such junctions.1
Focusing on the photocatalytic hydrogen evolution promoted by g-C3N4, it can be observed that, despite a very intensive investigation carried out in the last years, very conflicting results are reported in the current literature. Even in the case of the same g-C3N4 precursor and reaction temperature, significant differences in the photocatalytic hydrogen production were observed.10,13–16 As a matter of fact, the experimental conditions during the precursor condensation may affect the final characteristics of g-C3N4 in terms of crystal structure, degree of polymerization, protonation etc. These features, in turn, influence the photocatalytic hydrogen production efficiency. For example, a recent investigation on g-C3N4 prepared by urea, DCD or thiourea, put in prominence the relevant role of protonation and degree of polymerization as key parameters in the preparation of highly efficient g-C3N4.17 In particular, the increase in proton concentration was identified as a fundamental parameter,17 even more important than surface area. On the same line, other authors focused on the chemical composition and structure of g-C3N4 prepared by cyanuric acid and MLM and highlighted the key role of chemical composition in terms of s-triazine or tri-s-triazine units in the photocatalytic performance.18 Another relevant parameter affecting the catalytic ability of g-C3N4 has been identified in the defects that can be present in the final product, which may limit the electron–hole separation and transport.19–21 Other key features concern with the morphology of the final g-C3N4 prepared, in particular with reference to the layered structure. It has been pointed out that if layered g-C3N4 is prepared into mono- or few-layer g-C3N4 sheets, the photocatalytic performance is greatly improved.20,22–26 However, most methods to achieve mono- or few-layers g-C3N4 require relatively complex post-synthesis treatments and may result in very low amounts of g-C3N4 nanosheets.20 Finally, very promising results in terms of photoactivity have been recently obtained through the use of molten salts to accelerate the polymerization process and further tailor the properties of carbon nitride polymers.27–29 Materials prepared in this way have a rich variety of structural features that can be modulated through the use of different alkaline halides.27
Overall, notwithstanding the rich literature appeared on g-C3N4 in the last years, there is still a significant spread in the correlation of the reported photocatalytic behavior with synthetic procedure and physico-chemical properties. In order to try to shed some light on this issue, in the present paper we are reporting a systematic and extensive investigation on bulk g-C3N4 prepared by thermal condensation of three different precursors, i.e. urea, DCD, and MLM, at different temperatures (500, 550, 600, 650 and 700 °C). All syntheses were carried out under the same and reproducible conditions in order to be able to compare the characterization results among the different samples series. All samples have been characterized by means of X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), nitrogen adsorption measurements at −196.15 °C (BET method), scanning electron microscopy (SEM), thermogravimetric analysis (TGA), and then they were tested for hydrogen photoproduction under simulated solar light.
We highlight that in this work we intentionally selected a very simple synthetic method and the most common precursors in order to show that high photocatalytic activities can be obtained with no need for complex post-synthesis treatment and co-dopings. This is an essential result to make g-C3N4 a real large-scale photocatalyst for several applicative purposes where the ease of synthesis and the time and cost savings represent key factors. Most importantly, through the extensive physico-chemical characterization of a large number of samples, we were able to highlight which are the most relevant parameters affecting the properties of g-C3N4 and to provide a correlation of hydrogen production with morphology, crystal structure and optical features.
2. Experimental
2.1. Preparation of g-C3N4
The g-C3N4 samples have been synthesized by polymerization of DCD (NH2C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH)NHCN, Aldrich, 99%), urea (CH4N2O, Aldrich > 99.5%), and MLM (C3H6N6, Aldrich 99%) by the following thermal treatment (under N2 flow): heating (10 °C min−1) to the selected temperature (500, 550, 600, 650 or 700 °C), isothermal step for 4 h followed by cooling to room temperature (1 °C min−1). Syntheses have been carried out in a partially closed alumina crucible.
NH)NHCN, Aldrich, 99%), urea (CH4N2O, Aldrich > 99.5%), and MLM (C3H6N6, Aldrich 99%) by the following thermal treatment (under N2 flow): heating (10 °C min−1) to the selected temperature (500, 550, 600, 650 or 700 °C), isothermal step for 4 h followed by cooling to room temperature (1 °C min−1). Syntheses have been carried out in a partially closed alumina crucible.
2.2. Characterization of the g-C3N4 samples
The crystal structure of each sample has been characterized by room temperature Cu-radiation XRD acquired with a Bruker D8 diffractometer in the 5–35° angular range (5 s per step, step size 0.02°). Microstructural characterization was made using a high-resolution scanning electron microscope (SEM, TES-CAN Mira 3) operated at 15 kV.Nitrogen adsorption isotherms at −196.15 °C were acquired for each sample by using a Quantachrome Nova 2200e volumetric device. Preliminarily to the analysis, samples were outgassed at 250 °C for 3 h. The specific surface areas were obtained by applying the BET method to the collected data.
XPS measurements have been collected through a properly calibrated VG-Scienta R3000 electron analyzer, with the Al kα (1486.6 eV) emission line of a dual-anode, non-monochromatized PsP X-ray source.30 The final overall energy resolution was about 0.9 eV. For this analysis, the samples have been pressed into pellets and mounted with carbon tape. Due to mild electrical charge accumulation effects, the energy scale has been defined through the position of the main C 1s peak component, which corresponds in this case to the NC–N bond. Given the relatively large pellets thickness, only the C3N4 photoemission signal has been probed.
Thermogravimetric analyses have been performed by heating 5–8 g of each sample from room temperature up to 1000 °C at 10 °C min−1 under N2 atmosphere in a Pt pan by using a Q5000 equipment by TA Instrument (USA).
The optical diffuse reflectance spectra of the different samples were measured from 0.8 to 4.5 eV (250–1500 nm, in steps of 1 nm) on a Varian Cary 6000i spectrophotometer equipped with an integrating sphere. For this type of measurement, polycrystalline powders were compacted into pellets of about 10 mm in diameter, and reflectance spectra were calibrated using a standard reference disk.
2.3. Photocatalytic hydrogen gas production tests
Experiments were conducted on triethanolamine (TEOA, ≥99% Sigma-Aldrich) aqueous solutions (10%, v/v) irradiated in Pyrex glass containers (28 mL capacity, 21 mL sample31). After addition of the catalyst (1 g L−1), the suspension was deoxygenated by Ar (99.999%, Sapio S.r.l.) bubbling (20 min) to obtain anoxic conditions, and irradiated under magnetic stirring for 6 h. Pt was loaded on the catalyst surface (3 wt%) by in situ photodeposition using H2PtCl6 (∼38% Pt basis, Sigma-Aldrich).12,32 Transmission Electron Microscopy (TEM) inspection of Pt-loaded samples is reported in the ESI,† showing a spherical shape of Pt particles with average grain-size around 40–50 nm. Irradiation was performed under simulated solar light using a Solar Box 1500e (CO.FO.ME.GRA S.r.l., Milan, Italy) set at a power factor 500 W m−2, and equipped with UV outdoor filter of soda lime glass IR-treated. The photon flux, determined experimentally by 2-nitrobenzaldehyde actinometry (300–410 nm),33 was 1.53 × 10−7 moles per photons per s. Triplicate irradiation experiments were always performed. The headspace evolved gas was quantified by gas chromatography coupled with thermal conductivity detection (GC-TCD), as previously described.34 All results throughout the manuscript are expressed in terms of H2 evolution rate (HER), as μmoles of evolved gas per gram of catalyst per hour (μmoles g−1 h−1).3. Results and discussion
The g-C3N4 samples, prepared according to the Experimental section, underwent a full characterization to provide a correlation between physico-chemical properties and photocatalytic activity.3.1. Reaction yields
Samples from DCD and MLM precursors have been obtained at five different temperatures, i.e. at 500, 550, 600, 650 and 700 °C. On the other hand, only four samples (at 500, 550, 600 and 650 °C) have been obtained from the urea precursor after the thermal treatment. This is related to the different reaction yields of the thermal condensation reaction starting from the three different precursors, which are indicated in Fig. 1.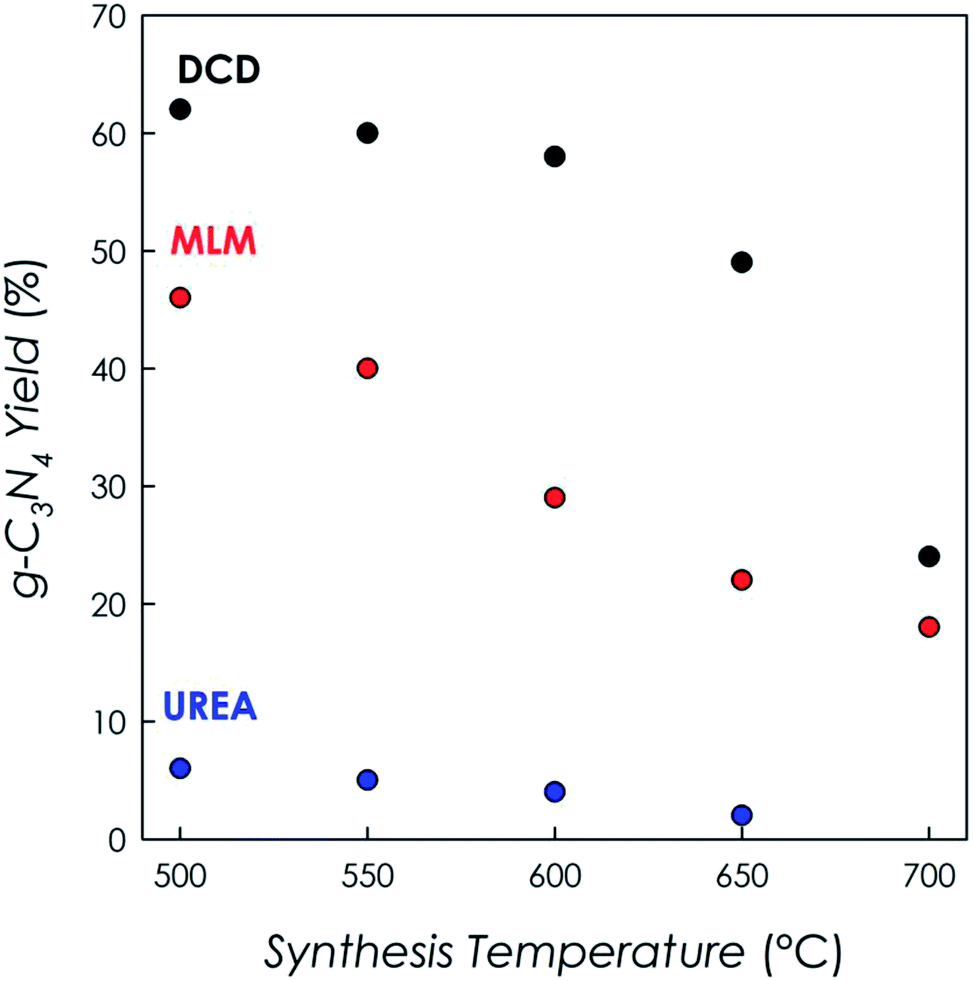 | ||
| Fig. 1 Reaction yields of g-C3N4 for the three different precursors at the different temperatures as determined by TGA measurements. | ||
All results presented in Fig. 1 are relative to the same batch size for all syntheses (about 5 grams of precursor) and show that the highest yields are obtained by using DCD as precursor, followed by MLM and urea. For all precursors the yield reduced by increasing temperature and, in the case of urea, which had the lowest yields at any temperature, it was not possible to obtain any product above 650 °C. As a consequence, in the remaining of the paper, the data at 700 °C for g-C3N4 prepared from this precursor are not available.
3.2. X-ray diffraction
Fig. 2a–c reports the XRD patterns for all g-C3N4 samples investigated in the present work. Temperatures in the three panels represent the reaction temperatures for the materials preparation.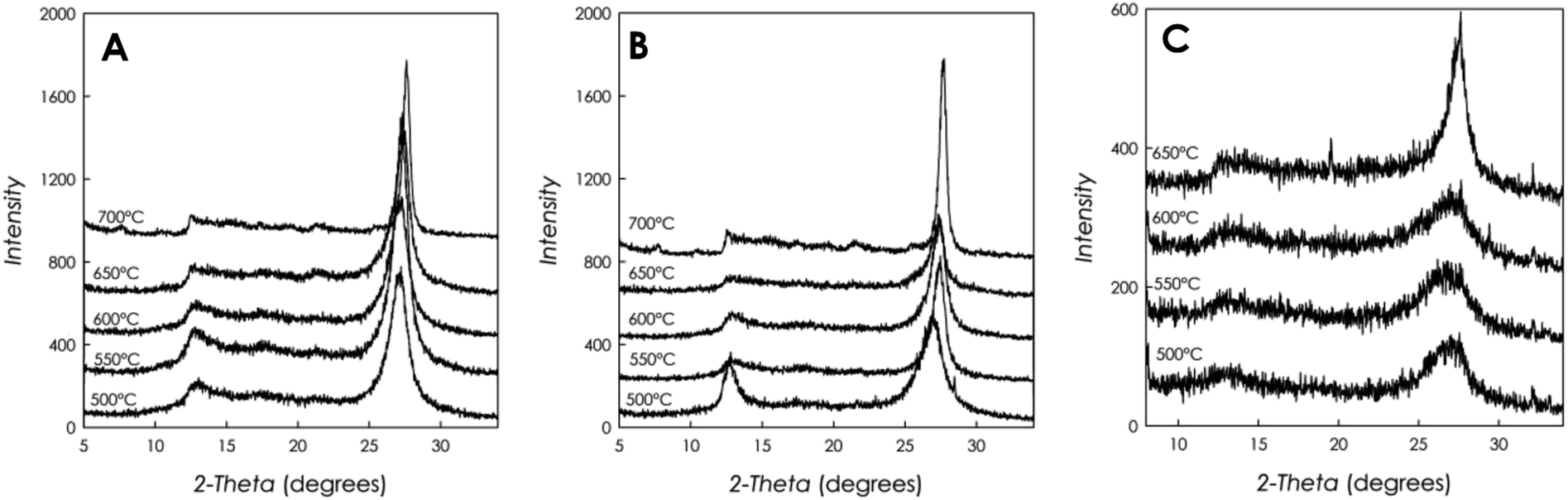 | ||
| Fig. 2 XRD patterns for the g-C3N4 samples prepared starting from different precursors and at different polymerization temperatures: (a) DCD; (b) MLM; and (c) urea. | ||
The patterns reported in Fig. 2a–c show the typical appearance of g-C3N4 diffraction, characterized by a first peak around 12.5°, attributed to the (100) plane, due to the intra-layer d-spacing, and a main peak at 2θ ca. 27°, reported as the (002) plane, ascribed to the distance between the layers of the graphitic material. From the simple visual inspection of Fig. 2, in particular for the samples prepared starting from DCD and MLM, it is possible to observe some trends as a function of increasing reaction temperature, namely, the first peak at about 12.5° becomes progressively less intense and nearly undetectable for the sample prepared at 600 °C while the main peak at about 27° becomes sharper and shifts at higher angles. For instance, the shift of the main peak observed for the DCD-derived sample is from about 26.85° at the synthesis temperature of 500 °C to about 27.60° for the sample prepared at 700 °C.
These observations suggest that the increase of condensation temperature provides a more crystalline solid phase, together with a decrease of the intra-layer spacing caused by a higher polymerization degree, which also evidences a greater disorder in the plane. At the same time, smaller inter-layer d-spacing values are observed at high temperature. The signal-to-noise ratio for the patterns in panels (a) and (b) in Fig. 2 is similar, while for the samples prepared starting from urea (panel (c)), for the same XRD acquisition parameters, such ratio is worse, suggesting a less scattering material, i.e., less crystalline. In addition, the FWHM (Full Width at Half Maximum) for the g-C3N4 samples prepared from DCD and MLM are similar (e.g., about 2° at 500 °C) while for the samples prepared staring from urea precursor the FWHM are higher (e.g., >3° at 500 °C).
3.3. Morphology and surface area
The morphological characterization of the prepared materials was performed by SEM inspection. For the sake of brevity, in Fig. 3 we show the images acquired on the g-C3N4 samples prepared from DCD (a, b), MLM (c, d) and urea (e, f) at the lowest (left) and highest (right) synthesis temperatures. Highest synthesis temperatures are 700 °C for DCD and MLM precursors and 650 °C for urea (see previous discussion on reaction yields). | ||
| Fig. 3 SEM images collected on the g-C3N4 materials prepared from DCD (a, b), MLM (c, d) and urea (e, f) by thermal condensation at 500 °C (a, c, e), 700 °C (b, d) and 650 °C (f). | ||
As it can be appreciated from the representative images selected in Fig. 3, a layered structured is evident in all samples for the powders obtained from DCD and MLM, with the size of the single layers decreasing by increasing the temperature (see the samples prepared at 700 °C). In the case of urea-derived g-C3N4, the powder appears made of less extended layers already at low temperature while at high temperature the microstructure seems made of small grains compared to the above materials (the photocatalyst produced from urea is indeed the most fluffy among all samples). Essentially, by increasing the reaction temperature, a sort of “opening” of the structure occurs leaving more surface available for the catalytic reaction. These observations are in agreement with the surface area values, obtained by the BET method and reported in Table 1 for each sample as function of both synthesis temperature and precursor.
For all the samples the BET area increases with the temperature of the synthesis. Samples obtained from DCD and MLM precursors at 500, 550 and 600 °C exhibit very low specific areas, close to the minimal detectable value of 3 m2 g−1. This is in accordance with the SEM observations (Fig. 3a and c) that reveal a multilayered structure constituted by agglomerated sheets. For higher synthesis temperatures (650, 700 °C) the increase of the BET areas up to 36 m2 g−1 is associated to the observed change in the samples morphology with a much-disordered structure constituted by smaller sheets (Fig. 3b and d). The samples obtained from the urea precursor at temperature of 500–600 °C present BET areas in the range 31–41 m2 g−1 while the double value of 85 m2 g−1 is measured for samples synthetized at 650 °C. These results can be attributed to the morphologies observed by SEM. The urea samples are constituted by poorly agglomerated particles and the particles size decrease for higher synthesis temperature is responsible of the increase in the surface area (Fig. 3e and f).
The surface area (around 85 m2 g−1) observed for the urea sample prepared at the highest temperature can be considered a high value for bulk g-C3N4.35 These findings can be justified considering that, during polymerization, urea (that contains oxygen) generates CO2 and H2O, which prevent full condensation; this hampers long-range in-plain structural packing, thus providing a material with more available surface area.36
3.4. Optical properties
Fig. 4a–c report the diffuse reflectance spectra as a function of wavelength recorded for each material, as function of synthesis temperature and precursor type.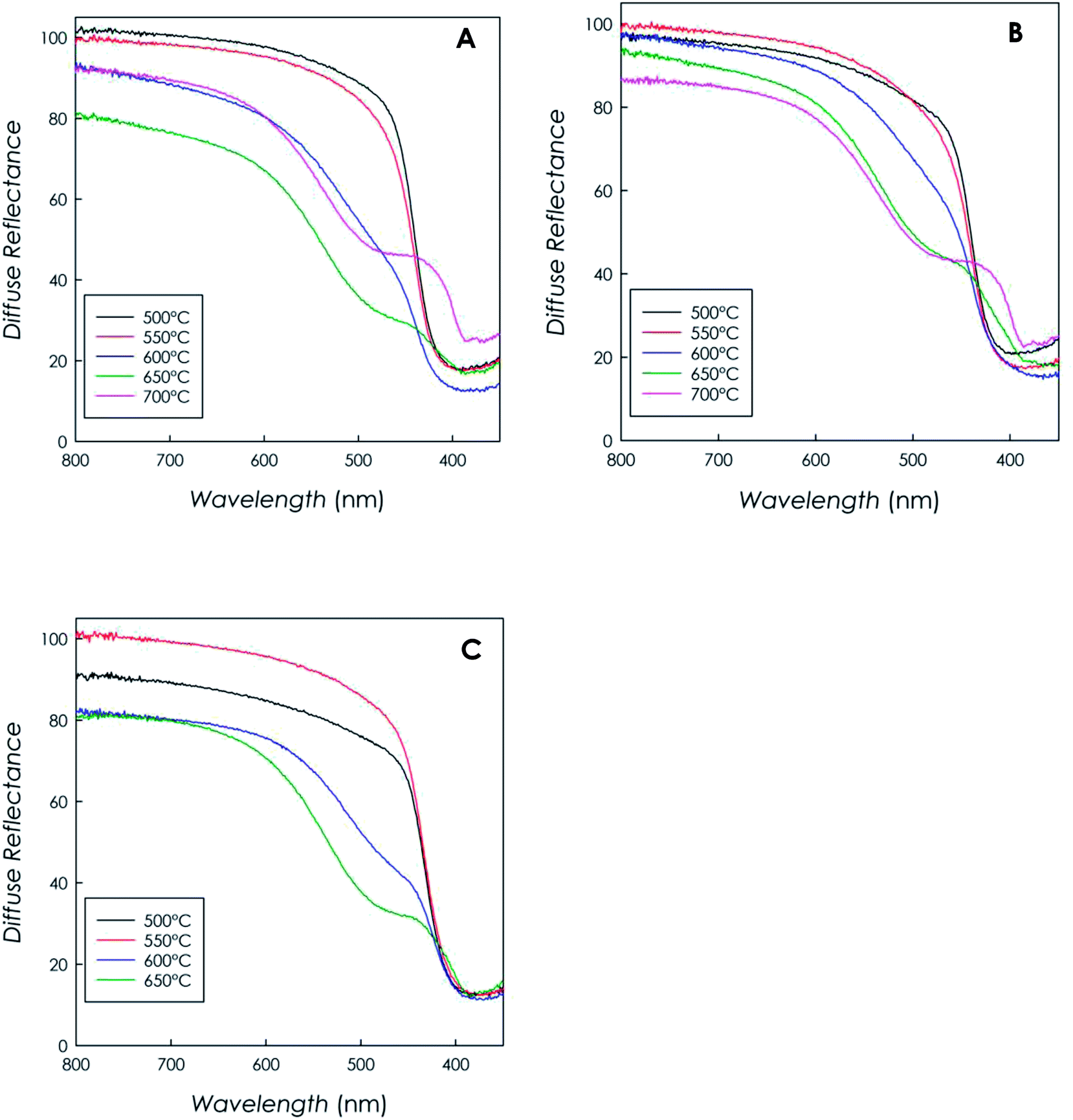 | ||
| Fig. 4 DRS spectra recorded on the g-C3N4 samples obtained from (a) DCD, (b) MLM, (c) urea as function of synthesis temperature. | ||
The curves clearly indicate that all materials are capable of visible-light harvesting, in fact they exhibit absorption edges above 400 nm; for instance, the band gap of the sample synthesized from DCD (at 500 °C) resulted to be 2.70(5) eV, in full agreement with the typical bad gap of g-C3N4 semiconductors (around 2.7 eV), corresponding to an absorption edge of ca. 460 nm.6,35
It is interesting to note that, for synthesis temperatures in the range 600–700 °C, the profiles generally show an enhanced absorption above 400 nm and they show a less defined absorption edge due to the formation of a remarkable band almost centered at 500 nm. This behavior, observed independently of the precursor type, is ascribable to the typical n–π* transition of the asymmetric and nonplanar conjugated heptazine rings,12,37,38 and it suggests a greater absorption capability in the visible region for the high-temperature-polymerization samples.
3.5. XPS
The XPS analysis was carried out on the three samples prepared at lower temperatures (500 °C), as well as on the three samples prepared at the highest common temperature (650 °C). For the sample grown from urea also the sample prepared at 600 °C was considered, to confirm the trend observed in the C–C/C–N ratio with hydrogen production.The N 1s spectra for samples with several precursors and various growth temperatures are shown in Fig. 5.
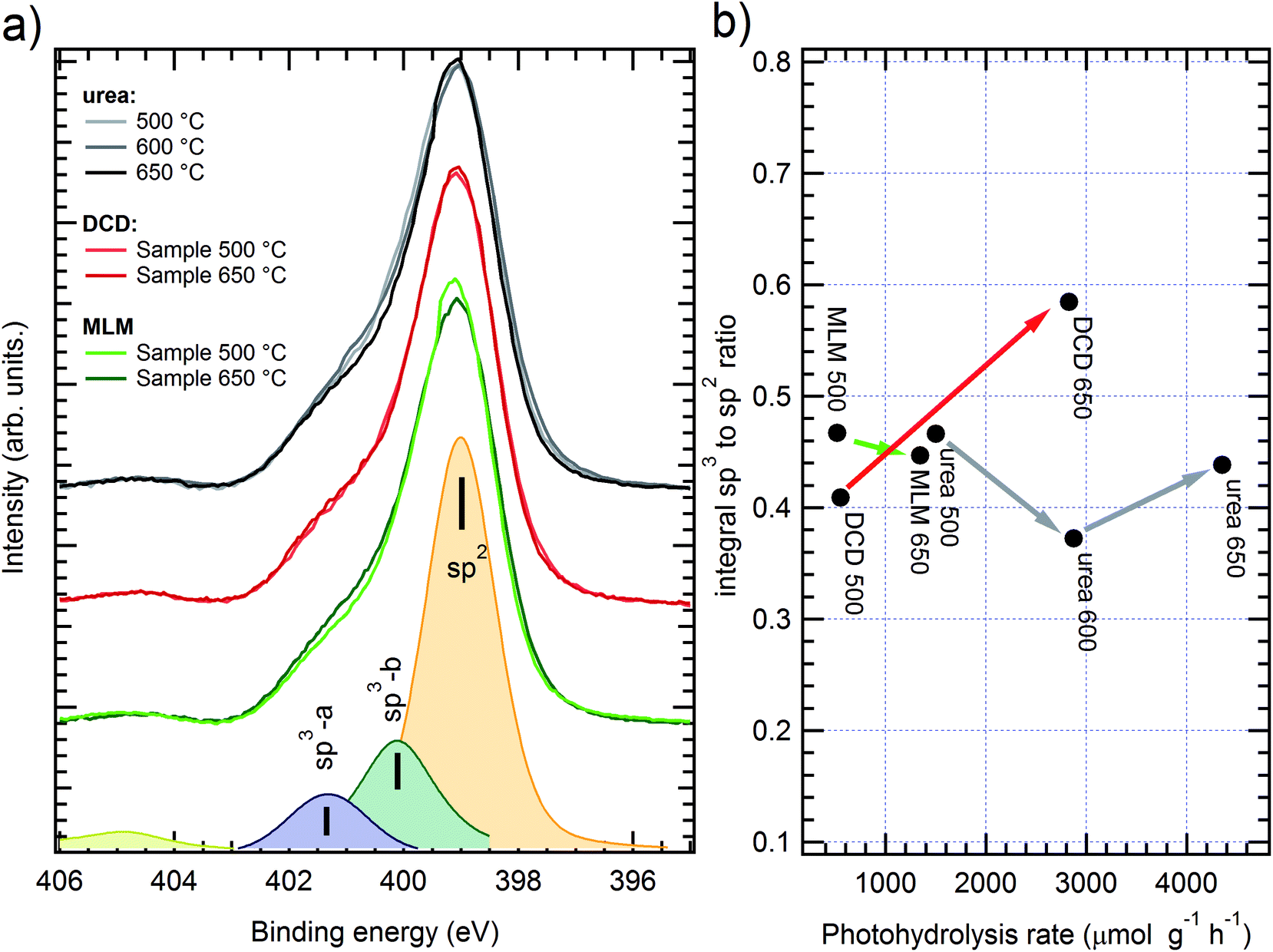 | ||
| Fig. 5 (a) XPS spectra of N 1s peak on g-C3N4 samples obtained from urea (grey scale colors), DCD (red) and melamine (MLM, green); darker color corresponds to higher polymerization temperature. Peak fitting results for MM sample grown at 500 °C are shown on the bottom. (b) sp2/sp3 peaks area ratio dependence on photocatalytic activity; arrows connect samples grown from the same precursor with the raising synthesis temperature. | ||
In each spectrum it is possible to discriminate the nitrogen sp2 and sp3 components, consistently with the literature;17 such ratio has been evaluated through curve fitting with three Voigt peaks (an example is shown at the bottom of Fig. 5a). The main component is ascribed to C–N–C bond in the sp2 configuration (BE = 399 ± 0.2 eV) while the other two peaks are usually ascribed to sp3 configuration, corresponding to N–C3 (labelled as sp3-a in Fig. 5a, BE ≈ 400 eV) and C–N–Hx (sp3-b, BE ≈ 401.5 eV) bonds. The resulting area ratios are shown in Fig. 5b, correlated with the H2 production yield (see later in the text). Although the samples with different precursors show small deviations of the N sp3 intensity, it is difficult to find a clear trend for the peaks area ratio, neither in relationship with the precursor nor with the preparation temperature. The fitting results are also affected by a large absolute error, due to the peak superpositions, up to 20% for peak areas. Even without considering the curve fitting, the simple spectra comparison already gives conflicting results: for instance, in the urea case the better performing (higher temperature) sample shows a lower sp3 degree as compared to the worst performing case; the opposite trend can be found in melamine based samples. By judging the preset data, it is then impossible to directly relate the photocatalytic performances to the sp2/sp3 ratio of the N 1s spectra, as it was proposed in the previous literature.17
The C 1s spectra for g-C3N4 samples are shown on Fig. 6a. Two distinct features are clearly visible in each spectrum: the main BE = 289 eV component correspond to NC–N bonds, while the lowest BE peak can be described by the sum of cyanide (CN, BE ≈ 286 eV) and C sp2 (C–C, BE ≈ 284.8 eV) contribution. In our case, the vast majority (more than 80%) of the photoemission intensity is ascribed to C–N bond, which is the only one expected in ideal g-C3N4. Such result is a further proof of the high quality of these samples and it is consistent with the extremely low variability of the overall N 1s peak shape. The C–C/CN features, whose intensity slightly changes among the samples, cannot be ascribed to environmental contamination only, which should appear with a similar amplitude in each case; it should then be ascribed to unpolymerized synthesis by-products. Unlike the case of N 1s sp2/sp3 ratio, it is possible to find a direct correlation between the C–C/NC–N area ratio and the photocatalytic performances. These results, evaluated through Voigt peak fitting, are shown in Fig. 6b. Among each sample precursor class, the H2 production is systematically higher in samples with a lower C–C peak intensity. In this case, the fit error was much lower (<5% on peak areas) than in the case of the N 1s spectra, further strengthening our results. It should be noted that the intensity of this peak is also inversely related to the macroscopic disorder, since the higher H2 yields have been measured in the more porous materials; it appears then that the macroscopic disorder alone, due to gaseous product emission during polymerization, does not affect the local molecular ordering (local chemical environment), which improves at higher temperature synthesis. This result is also in agreement with the improved crystallinity of the samples observed by XRD and reported above.
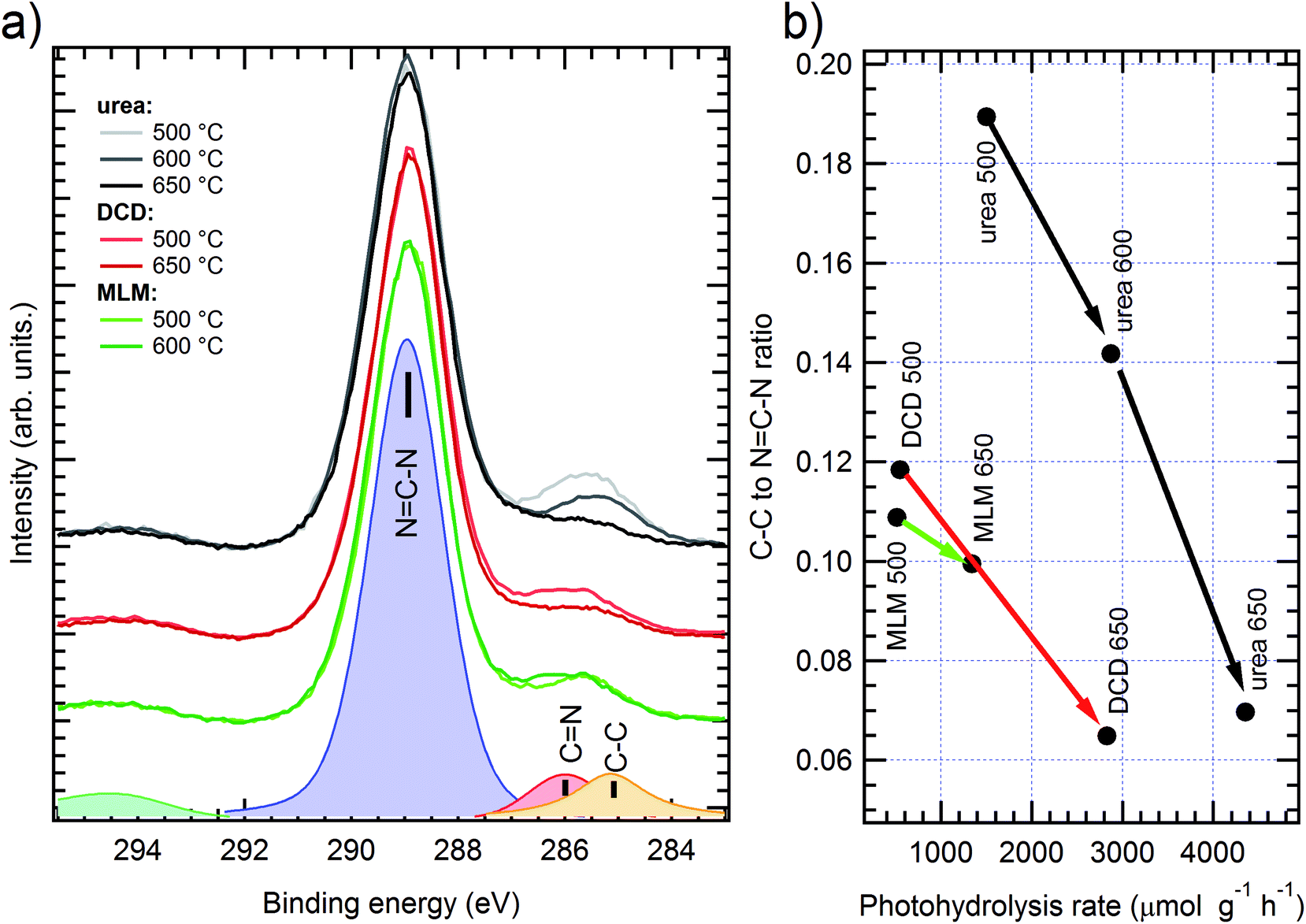 | ||
| Fig. 6 (a) XPS spectra of C 1s peak of g-C3N4 samples, obtained from urea (grey scale colors), DCD (red) and melamine (MLM, green); darker color corresponds to higher polymerization temperature. Peak fitting results for MM sample grown at 650 °C are shown on the bottom. (b) C–C/NC–N peaks area ratio dependence on photocatalytic activity; arrows connect samples grown with the same precursor with the raising synthesis temperature. | ||
3.6. Photocatalytic H2 production
The fourteen as-prepared samples have been tested in parallel for H2 production from aqueous TEOA (10%, v/v) under simulated solar light, in the same conditions (3 wt% Pt, 1 g L−1 catalyst, 6 h irradiation). Fig. 7 summarizes the results obtained in terms of HER (μmol g−1 h−1), reported as the mean of three independent irradiation tests.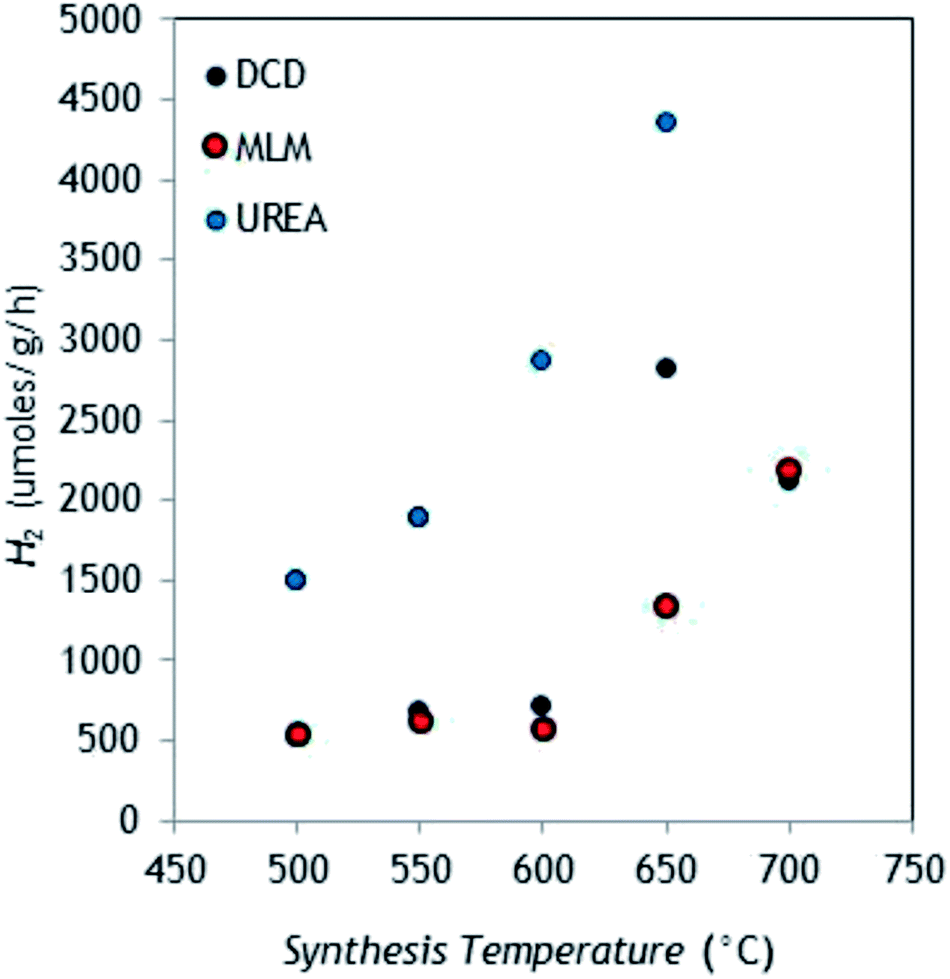 | ||
| Fig. 7 H2 evolution profiles obtained under simulated solar light using the g-C3N4 samples as function of polymerization temperature and precursor type (RSDs < 5%, n = 3). | ||
As it can be seen, H2 evolution turned out to be dependent on both polymerization temperature and g-C3N4 precursor. In particular, the catalysts produced from DCD provided increasingly enlarged HER as function of temperature up to 650 °C, with a decreased production for the sample obtained at 700 °C (this may be due to a possible detrimental effect of too high surface area35).
The trend was similar for MLM, but in this case the highest production point was observed at the highest synthesis temperature. Urea is the precursor that provided the higher hydrogen production, with a maximum HER of about 4400 μmol g−1 h−1 for the sample synthesized at 650 °C (the highest viable temperature for this precursor). This is in accordance with the results recently published by Oh et al.,36 who found higher HER from aqueous TEOA using urea rather than MLM and DCD. However, considering the low reaction yield of urea thermal condensation (ca. 5%), the semiconductor produced from DCD (reaction yield at 650 °C around 50%) seems most convenient. The control test confirmed that H2 production is effectively sustained by g-C3N4, indeed H2 evolution was negligible (0.4 μmol h−1) by omitting the catalyst. As well, the key role of Pt co-catalyst in catalyzing water hydrogen ions reduction was confirmed using pure g-C3N4 (from DCD 650 °C), observing a HER of 18 μmol g−1 h−1.
Overall, the best DCD-derived bulk catalyst (650 °C) coupled with Pt provided an apparent quantum yield (AQY)31 of 10.7% (the complete list of AQY and turn over number (TON) values are available in ESI†).
The irradiation experiments reproducibility was very good, with values of relative standard deviation (RSD) below 5% (n = 3), for all materials. The batch-to-batch reproducibility, evaluated by photoproduction tests on three independently prepared batches from DCD (650 °C), provided RSD not higher than 11%, accounting for good overall reproducibility, from g-C3N4 synthesis to the final H2 quantification.
For the interpretation of the H2 production data, it is useful to follow two distinct approaches. First we have to analyze the HERs within the same samples series (that is from the same precursor). The increased photocatalytic activity can be attributed to the combined effects of surface area, visible light harvesting ability, and crystallinity, which are factors that contribute to enlarge g-C3N4 HER.35,37 As it is shown in the characterization part, these three factors effectively increase along with polymerization temperature, thus reasonably standing for the enhanced photoreaction.
With regard to catalyst porosity, the H2 evolution profiles showed a superimposable correlation with the catalyst surface area, as it is apparent by comparing Fig. 7 with Fig. 8.
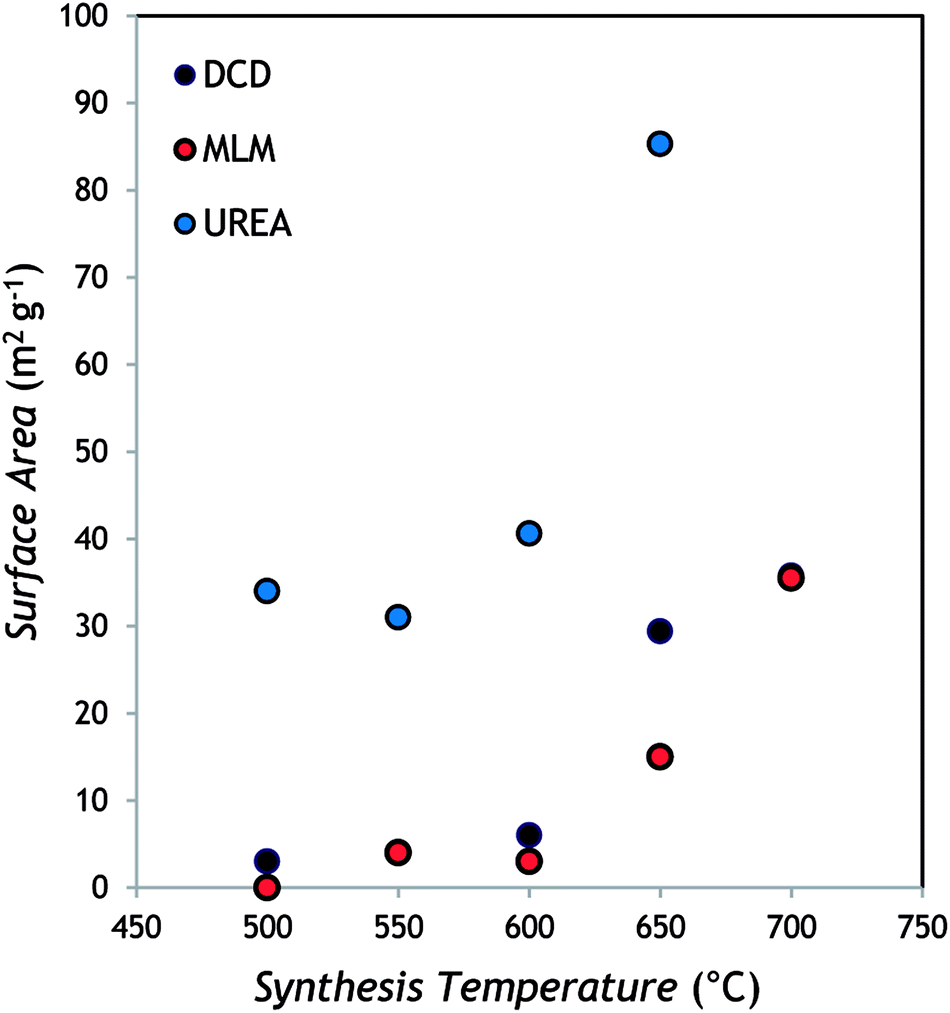 | ||
| Fig. 8 Correlation between surface area and polymerization temperature for the g-C3N4 catalysts. | ||
This clearly indicates that the photocatalytic activity of bulk g-C3N4 is strictly dependent on the available surface area of the material but also with the trend of the C–C/C–N ratio showed in the XPS results. In fact, the higher is the surface area the higher is the number of available reaction sites for the surface reactions and at the same time lower is recombination of the charge carriers.12,35,39,40 These considerations are further corroborated by comparing the HERs among the three precursors (polymerization at 650 °C, for instance), with the corresponding semiconductor physical–chemical properties (Table 2).
From the data gathered in the table, it is clear that, despite the very similar UV-vis absorption (see Fig. 4) and structural properties, HERs are markedly different depending on the g-C3N4 precursor, and they are well correlated to surface area. This is in line with the results recently published by Oh et al.,36 who suggested that the photocatalytic activity is dominantly affected by surface area rather than light absorption ability. This can be explained not only in terms of available reaction sites,39,41 but also considering that the higher is the surface area the more uniform is the size distribution of the co-catalyst.36
Since from the N 1s XPS analysis it has been observed that the trend of sp2/sp3 carbon ratio is unrelated to the H2 photoproduction activity, it seems that the polymerization degree17 is not the key parameter in determining the overall H2 evolution activity of bulk g-C3N4; however, the C 1s peak analysis suggests that the lower presence of synthesis by-products (obtained with a higher reaction temperature) is positively correlated to a better photocatalysis process.
The present investigation showed that thermal condensation is a simple, reproducible way for mass production of structure-controlled bulk g-C3N4 semiconductors, which provide good performance for H2 evolution. As shown in Table 3, gathering some of the most recent applications of g-C3N4 (under UV/visible or simulated solar light), HERs and AQYs are competitive with those observed using bulk g-C3N4 (ref. 36, 39, 41 and 42) and also with those afforded by modified semiconductors.43–48
| Precursor | Photoproduction conditions | HER (μmol g−1 h−1) | AQY (%) | Ref. | |
|---|---|---|---|---|---|
| a n.a.: not available. | |||||
| Bulk g-C3N4 | Urea | 1 g L−1 catalyst, 3 wt% Pt, 10% TEOA, λ < 420 nm | 130 | n.a. | 36 |
| Urea | 1.4 g L−1 catalyst, 3 wt% Pt, 11% TEOA, UV/Vis | 4871 (λ < 400 nm) | n.a. | 41 | |
| 1405 (λ > 400 nm) | |||||
| MLM | 1 g L−1 catalyst, 3 wt% Pt, 15% TEOA, λ < 420 nm | 80 | n.a. | 39 | |
| MLM | 0.625 g L−1 catalyst, 1 wt% Au–Pt, 25% methanol, λ > 400 nm | 338 | n.a. | 42 | |
| Urea | 1 g L−1 catalyst, 3 wt% Pt, 10% TEOA, simulated solar light | 4353 | 13.4 | This work | |
| DCD | 2825 | 10.7 | |||
| MLM | 2196 | 8.3 | |||
| Modified g-C3N4 | Urea | 1 g L−1 catalyst, 2 wt% Pt, 10% methanol, simulated solar light | 2810 | 17.9 (λ 400 nm) | 44 |
| Urea | 0.625 g L−1 catalyst, 1 wt% Pt, 15% TEOA, λ > 420 nm | 213 | 0.9 (λ 420 nm) | 48 | |
| Urea | 1 g L−1 catalyst, 3 wt% Pt, 20% methanol, λ ≥ 420 nm | ∼420 | 6.25 | 43 | |
| MLM | 0.5 g L−1 catalyst, 4 wt% Pt, 10% TEOA, simulated solar light | 2040 | n.a. | 45 | |
| MLM | 0.5 g L−1 catalyst, 1 wt% Pt, 10% TEOA, simulated solar light | 8163 | n.a. | 46 | |
| DCD | 0.5 g L−1 catalyst, 3 wt% Pt, 10% TEOA, simulated solar light | 9578 | 9.01 (λ 420 nm) | 47 | |
4. Conclusions
This paper reported a systematic investigation of bulk g-C3N4 prepared from the three most common precursors used, and employing five different reaction temperatures. The investigation of the physico-chemical properties on each sample together with the hydrogen photogeneration activity allowed to clearly extract the key parameters affecting the catalytic activity for H2 production. In particular, while significant trends in the crystallinity, UV-Vis absorption and XPS data have been observed as a function of the precursor employed and the reaction temperature, it seems that the catalytic properties toward H2 evolution are dominated by the surface area of the samples. Moreover, we showed, by means of XPS, that the trend of sp2/sp3 carbon ratio is unrelated to the H2 photoproduction activity, indicating that the polymerization degree is not the key parameter, while the C 1s peak analysis suggests that the lower presence of synthesis by-products is positively correlated to a better photocatalysis process.Such conclusions, based on a huge analysis of a rich series of samples, provide a conclusive while simple correlation between the photoactivity and electronic, structural and morphological properties of bulk g-C3N4 allowing to focus the future research on the enhancement of surface area only. The possible real large-scale application of bulk g-C3N4 must take into account not only the catalytic activity but also “practical” aspects such as the polymerization yields and the balance between the amount of photogenerated hydrogen and viability of the synthetic procedure used.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by CATSTER Project within the framework of the Lombardy Region-INSTM Agreement (2015).References
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- S. Ye, R. Wang, M.-Z. Wu and Y.-P. Yuan, Appl. Surf. Sci., 2015, 358, 15–27 CrossRef CAS.
- Z. Zhao, Y. Sun and F. Dong, Nanoscale, 2015, 7, 15–37 RSC.
- Y. G. Gong, M. M. Li and Y. Wang, ChemSusChem, 2015, 8, 931–946 CrossRef CAS PubMed.
- A. Speltini, F. Maraschi, R. Govoni, C. Milanese, A. Profumo, L. Malavasi and M. Sturini, J. Chromatogr. A, 2017, 1489, 9–17 CrossRef CAS PubMed.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. A. Domen and M. A. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- K. Maeda, X. Wang, Y. Nishihara, D. Lu, M. Antonietti and K. Domen, J. Phys. Chem. C, 2009, 113, 4940–4947 CrossRef CAS.
- H. Ji, F. Chang, X. Hu, W. Qin and J. Shen, Chem. Eng. J., 2013, 218, 183–190 CrossRef CAS.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed.
- G. Zhang, J. Zhang, M. Zhang and X. Wang, J. Mater. Chem., 2012, 22, 8083–8091 RSC.
- F. Dong, Z. Wang, Y. Sun, W. K. Ho and H. Zhang, J. Colloid Interface Sci., 2013, 401, 70–79 CrossRef CAS PubMed.
- L. Yang, J. Huang, L. Shi, L. Cao, Q. Yu, Y. Jie, J. Fei, H. Ouyang and J. Ye, Appl. Catal., B, 2017, 204, 335–345 CrossRef CAS.
- A. B. Jorge, D. J. Martin, M. T. S. Dhanoa, A. S. Rahman, N. Makwana, J. Tang, A. Sella, F. Corà, S. Firth, J. A. Darr and P. F. McMillan, J. Phys. Chem. C, 2013, 117, 7178–7185 CrossRef CAS.
- Y. Zhang, J. Liu, G. Wu and W. Chen, Nanoscale, 2012, 4, 5300–5303 RSC.
- S. Martha, A. Nashim and K. M. Parida, J. Mater. Chem. A, 2013, 1, 7816–7824 RSC.
- Y. Zhong, Z. Wang, J. Feng, S. Yan, H. Zhang, Z. Li and Z. Zou, Appl. Surf. Sci., 2014, 295, 253 CrossRef CAS.
- D. J. Martin, K. Qiu, S. A. Shelvin, A. D. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 CrossRef CAS PubMed.
- Y. Cao, Z. Zhang, J. Long, J. Liang, H. Lin and X. Wang, J. Mater. Chem. A, 2014, 2, 17797–17807 RSC.
- W. Wu, J. Zhang, W. Fan, Z. Li, L. Wang, X. Li, Y. Wang, R. Wang, J. Zheng, M. Wu and H. Zeng, ACS Catal., 2016, 6, 3365–3371 CrossRef CAS.
- Q. Y. Lin, L. Li, S. J. Liang, M. H. Liu, J. H. Bi and L. Wu, Appl. Catal., B, 2015, 163, 135–142 CrossRef CAS.
- M. Shalom, S. Inal, C. Fettkenhauer, D. Neher and M. Antonietti, J. Am. Chem. Soc., 2013, 135, 7118–7121 CrossRef CAS PubMed.
- P. Niu, L. Zhang, G. Liu and H.-M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2012, 135, 18–21 CrossRef PubMed.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed.
- J. Tian, Q. Liu, C. Ge, Z. Xing, A. M. Asiri, A. O. Al-Youbi and X. Sun, Nanoscale, 2013, 5, 8921–8924 RSC.
- J. Tian, Q. Liu, A. M. Asiri, A. H. Qusti, A. O. Al-Youbi and X. Sun, Nanoscale, 2013, 5, 11604–11609 RSC.
- G. Zhang, L. Lin, G. Li, Y. Zhang, A. Savateev, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2018, 57, 9372–9376 CrossRef CAS PubMed.
- M. K. Bhunia, K. Yamauchi and K. Takanabe, Angew. Chem., Int. Ed., 2014, 53, 11001–11005 CrossRef CAS PubMed.
- L. Lin, H. Ou, Y. Zhang and X. Wang, ACS Catal., 2016, 6, 3921–3931 CrossRef CAS.
- G. Drera, G. Salvinelli, J. Åhlund, P. G. Karlsson, B. Wannberg, E. Magnano, S. Nappini and L. Sangaletti, J. Electron Spectrosc. Relat. Phenom., 2014, 195, 109–116 CrossRef CAS.
- A. Speltini, M. Sturini, F. Maraschi, D. Dondi, G. Fisogni, E. Annovazzi, A. Profumo and A. Buttafava, Int. J. Hydrogen Energy, 2015, 40, 4303–4310 CrossRef CAS.
- H. Sun, X. Zhou, H. Zhang and W. Tu, Int. J. Hydrogen Energy, 2017, 42, 7930–7937 CrossRef CAS.
- K. L. Willett and R. A. Hites, J. Chem. Educ., 2000, 77, 900–902 CrossRef CAS.
- A. Speltini, M. Sturini, D. Dondi, E. Annovazzi, F. Maraschi, V. Caratto, A. Profumo and A. Buttafava, Photochem. Photobiol. Sci., 2014, 13, 1410–1419 RSC.
- X. Li, A. F. Masters and T. Maschmeyer, Chem. Commun., 2017, 53, 7438–7446 RSC.
- J. Oh, J. M. Lee, Y. Yoo, J. Kim, S. J. Hwang and S. Park, Appl. Catal., B, 2017, 218, 349–358 CrossRef CAS.
- H. Zhang and A. Yu, J. Phys. Chem., 2014, 118, 11628–11635 CAS.
- Y. Kang, Y. Yang, L. C. Yin, X. Kang, L. Wang, G. Liu and H. M. Cheng, Adv. Mater., 2016, 28, 6471–6477 CrossRef CAS PubMed.
- Q. Gu, Z. Gao, H. Zhao, Z. Lou, Y. Liao and C. Xue, RSC Adv., 2015, 5, 49317–49325 RSC.
- J. Wen, J. Xie, X. Chen and X. Li, Appl. Surf. Sci., 2017, 391, 72–123 CrossRef CAS.
- Z. Zhang, Y. Zhang, L. Lu, Y. Si, S. Zhang, Y. Chen, K. Dai, P. Duan, L. Duan and J. Liu, Appl. Surf. Sci., 2017, 391, 369–375 CrossRef CAS.
- J. Jiang, J. Yu and S. Cao, J. Colloid Interface Sci., 2016, 461, 56–63 CrossRef CAS PubMed.
- Y. A. Haleem, Q. He, D. Liu, C. Wang, W. Xu, W. Gan, Y. Zhou, W. Chuangqiang, Y. Ding and L. Song, RSC Adv., 2017, 7, 5390–5396 RSC.
- V. W. Lau, V. W. Yu, F. Ehrat, T. Botari, I. Moudrakovski, T. Simon, V. Duppel, E. Medina, J. K. Stolarczyk, J. Feldmann, V. Blum and B. V. Lotsch, Adv. Energy Mater., 2017, 7, 1602251 CrossRef.
- N. Ding, L. Zhang, M. Hashimoto, K. Iwasaki, N. Chikamori, K. Nakata, Y. Xu, J. Shi, H. Wu, Y. Luo, D. Li, A. Fujishima and Q. Meng, J. Colloid Interface Sci., 2018, 512, 474–479 CrossRef CAS PubMed.
- L. Zhang, Q. Liu, Y. Chai and W. L. Dai, Int. J. Hydrogen Energy, 2018, 43, 5591–5602 CrossRef CAS.
- W. Iqbal, B. Qiu, Q. Zhu, M. Xing and J. Zhang, Appl. Catal., B, 2018, 232, 306–313 CrossRef CAS.
- Q. Xu, B. Cheng, J. Yu and G. Liu, Carbon, 2017, 118, 241–249 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08880b |
| This journal is © The Royal Society of Chemistry 2018 |