
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient in situ N-heterocyclic carbene palladium(II) generated from Pd(OAc)2 catalysts for carbonylative Suzuki coupling reactions of arylboronic acids with 2-bromopyridine under inert conditions leading to unsymmetrical arylpyridine ketones: synthesis, characterization and cytotoxic activities†
Nedra Touja,
Abdullah S. Al-Ayedb,
Mathieu Sauthierc,
Lamjed Mansourb,
Abdel Halim Harrathb,
Jamil Al-Tamimib,
Ismail Özdemird,
Sedat Yaşard and
Naceur Hamdi *ab
*ab
aResearch Laboratory of Environmental Sciences and Technologies (LR16ES09), Higher Institute of Environmental Sciences and Technology, University of Carthage, Hammam-Lif, Tunisia. E-mail: naceur.hamdi@isste.rnu.tn; Fax: +966-6333-9351; Tel: +966-556-394-839
bChemistry Department, College of Science and Arts, Qassim University, Al-Rass, Kingdom of Saudi Arabia
cEcole Nationale Supérieure de Chimie de Lille, Unité de Catalyse et Chimie du Solide, UMR CNRS 8181, USTL, BP 90108, 59652 Villeneuve d’Ascq, France
dİnönü University, Faculty of Science and Art, Department of Chemistry, Malatya, Turkey
First published on 5th December 2018
Abstract
N,N-Substituted benzimidazole salts were successfully synthesized and characterized by 1H-NMR, 13C {1H} NMR and IR techniques, which support the proposed structures. Catalysts generated in situ were efficiently used for the carbonylative cross-coupling reaction of 2 bromopyridine with various boronic acids. The reaction was carried out in THF at 110 °C in the presence of K2CO3 under inert conditions and yields unsymmetrical arylpyridine ketones. All N,N-substituted benzimidazole salts 2a–i and 4a–i studied in this work were screened for their cytotoxic activities against human cancer cell lines such us MDA-MB-231, MCF-7 and T47D. The N,N-substituted benzimidazoles 2e and 2f exhibited the most cytotoxic effect with promising cytotoxic activity with IC50 values of 4.45 μg mL−1 against MDA-MB-231 and 4.85 μg mL−1 against MCF7 respectively.
1. Introduction
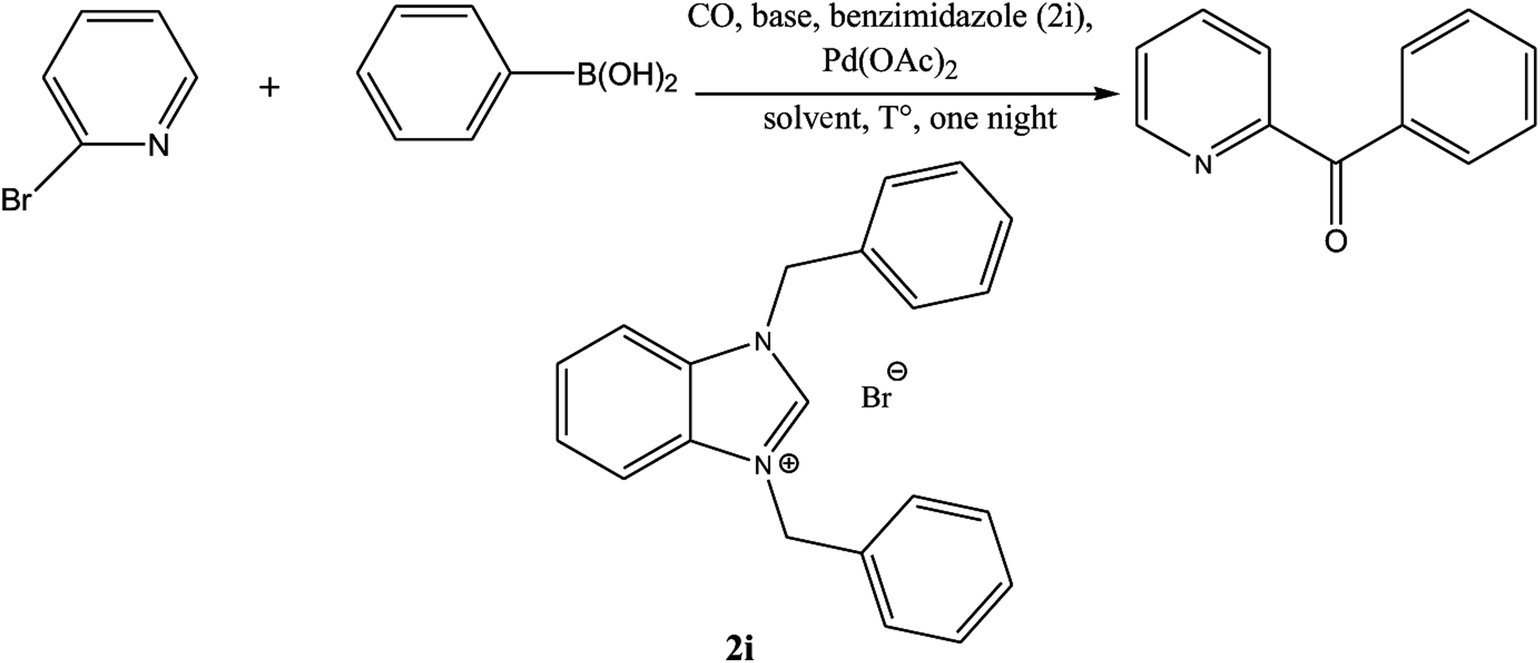
Ketones are vital building blocks in organic synthesis as well as an important functionality found in pharmaceutical compounds.1,2 The Friedel–Crafts (F–C) acylation of aromatic compounds is the most frequent route for the synthesis of aromatic ketones.2 However, it has intrinsic limitations such as the substituent-directing effects, reactive substrate requirements and the fact that recovery and recycling of the catalyst is seldom possible after aqueous work-up and a large amount of toxic wastes is generated. On the other hand, Pd-catalyzed cross-coupling reactions of acyl halides with organometallic reagents provides a direct procedure for the synthesis of isomeric ketones; a drawback is that Pd-catalysts are expensive and they are not recovered.3–8 In 1993, Suzuki's group9 reported a direct method (i.e., the carbonylative Suzuki coupling reaction) for the synthesis of aromatic ketones by the reaction of CO with an aryl halide and an arylboronic acid in the presence of PdCl2 and Pd(dba)2 (bis(dibenzylideneacetone)palladium). This reaction provides a versatile platform for the direct synthesis of aromatic ketones using boronic acids, which are generally non-toxic and stable to air and moisture. Ishiyama et al.10 used a variety of different catalysts to affect this carbonylative Suzuki reaction, including PdCl2 (PPh3)2 and PdCl2 (dppf) ([1,1′-bis(diphenylphosphino) ferrocene] dichloropalladium(II)). However, the conversion and selectivity of these reactions are low and require the presence of a ligand to proceed effectively.11,12 On the other hand, pyridine and its derivatives have a wide range of chemical and biological potentialities that are useful intermediates in the syntheses of various natural products and drugs.13 Several, different protocols have been developed for the synthesis of this important class of products.14 However, most of these protocols used unfriendly reagents and gave poor overall yields.The carbonyl coupling of bromopyridine with arylboronic acids (carbonyl version of the Suzuki reaction) has recently been shown to provide simple access to arylpyridine ketones with good yields after optimization of the operating conditions15 (Scheme 1).
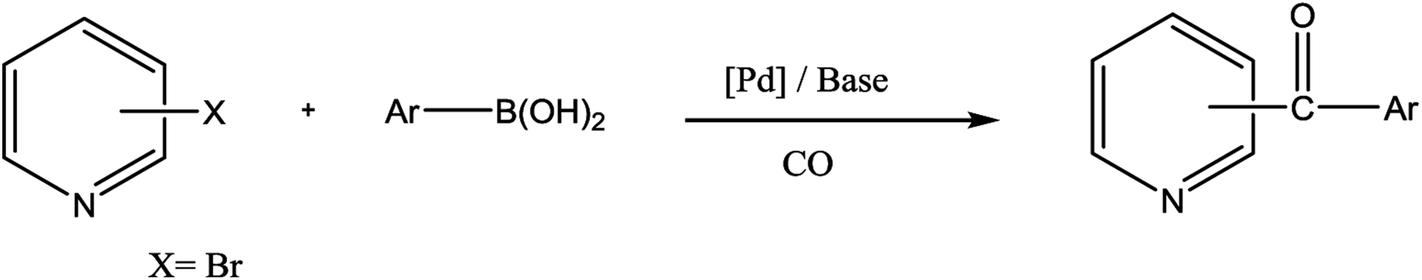 | ||
| Scheme 1 Carbonylative cross-coupling reaction of chloropyridines with arylboronic acids. | ||
Since the discovery of N-heterocyclic carbenes by Arduengo in 1991, NHCs have become one of the most versatile class of ligands for a wide range of transition metals16,17 and begun to replace phosphine ligands18–21 due to their unique proprieties.22–24 To date, various NHC complexes have been synthesised with different transition metals25 and have demonstrated unique activities as catalysts in different organic transformations.26
The palladium NHC complexes are one of the most interesting and widely investigated complexes in different catalytic transformations, especially C–C bond formation reactions such as Suzuki,27 Heck,28 and Sonogashira29 reactions. Thus, the use of N-heterocyclic carbenes associated to palladium has been reported as efficient catalysts for the carbonyl coupling under mild and varied conditions.30–35 Herein, we report the use of the two families of benzimidazolium salts already synthesized36–38 as a ligand in the carbonylative cross coupling of 2-bromopyridine with different boronic acids under inert condition to form unsymmetrical arylpyridine ketones (Scheme 2). In addition, the cytotoxic activities against the cancer human cell lines such us MDA-MB-231, MCF-7 and T47D of the compounds 2 and 4 were also determined.
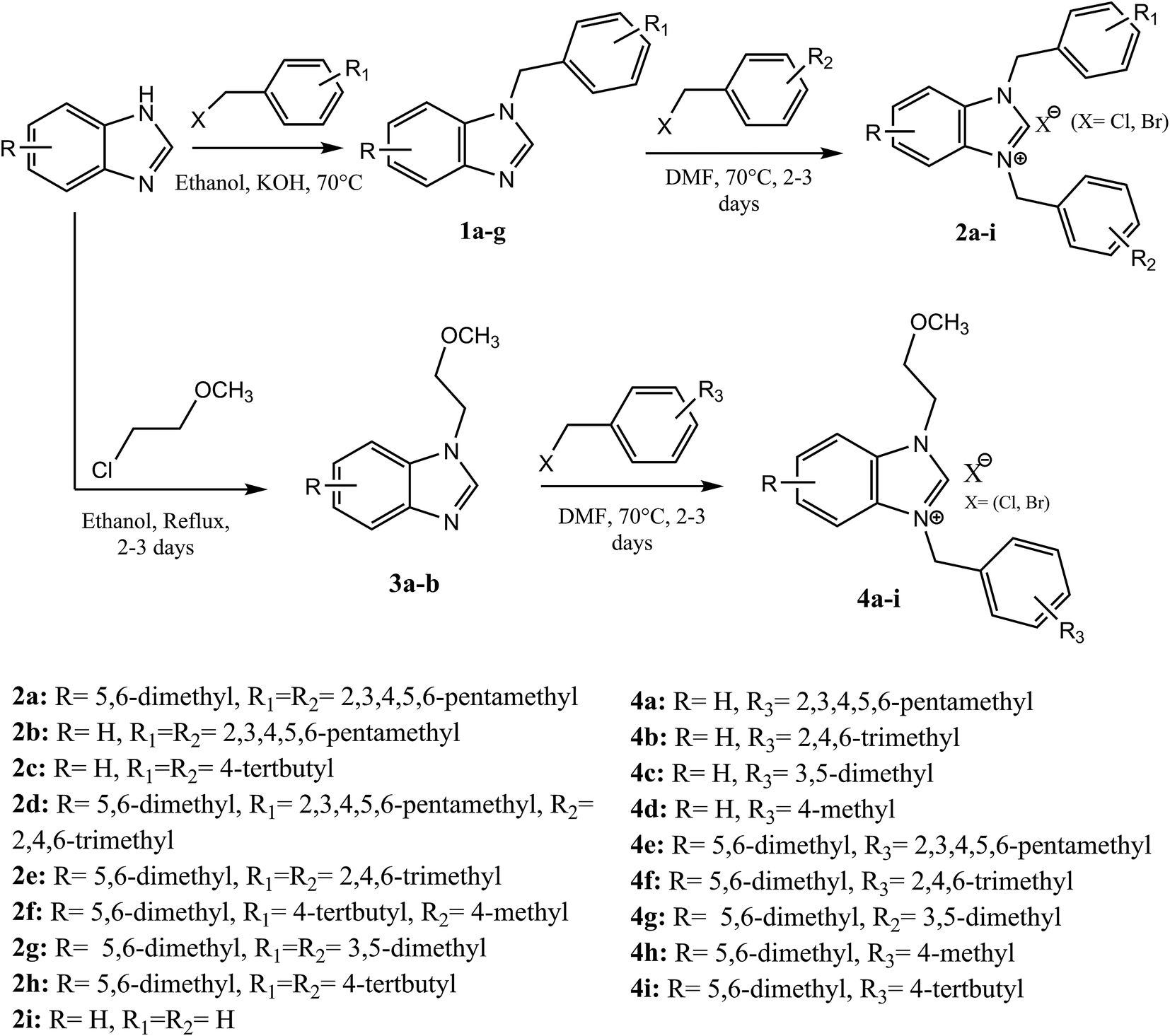 | ||
| Scheme 2 General preparation and structure formulae of benzimidazoles salts 2a–i and 4a–i | ||
2. Results and discussion
2.1 Preparation of benzimidazolium salts 2a–i and 4a–i
The formation of N,N-disubstituted azolium salts is mostly done through the introduction of alkyl/benzylhalide to an N-alkyl/benzylazole via quaternization reaction.In the present study, the precursors 2a–i and 4a–i were prepared by the quaternization of the intermediate 1 and 3 with a variety of benzyl chlorides or benzyl bromides in DMF at 70 °C (Scheme 2) as previously described36,37,39 in almost quantitatively yields. All compounds showed good solubility in water and common organic solvents, such as dichloromethane, chloroform, methanol, acetonitrile, and N,N-dimethylformamide.
The synthesized compounds were purified by crystallization/column chromatography and were identified by FTIR and NMR spectral data analysis (Experimental section) in particular NMR spectra of all the benzimidazolium salts 2a–i and 4a–i were recorded in CDCl3.
13C NMR chemical shifts were consistent with the proposed structure, the imino carbon appeared as a typical singlet in the 1H-decoupled mode at 141.3, 143.0, 143.0, 142.1, 142.7, 141.6, 141.8, 141.8 and 132.5 ppm respectively for imidazolinium salts 2a–i. The 1H NMR spectra of the benzimidazolium salts further supported the assigned structures; the resonances for C(2)-H were observed as sharp singlets in the 9.85, 10.34, 11.83, 10.41, 10.92, 11.51, 11.42, 11.63 and 11.47 ppm respectively for 2a–i.
The characteristic IR(CN) bond in the compounds was obtained as 1440.9, 1553.9, 1562.8, 1567.83, 1567.86, 1561.6, 1562.4, 1558.1 and 1559.02 cm−1 for 2a–i, respectively.
For example, the NCHN proton signal for the benzimidazole salt 2i was observed as singlet at 11.97 ppm that is in accordance with previous reports.36,37 The benzylic protons (N–CH2–Ar) appeared at 5.87 ppm (Fig. 1).
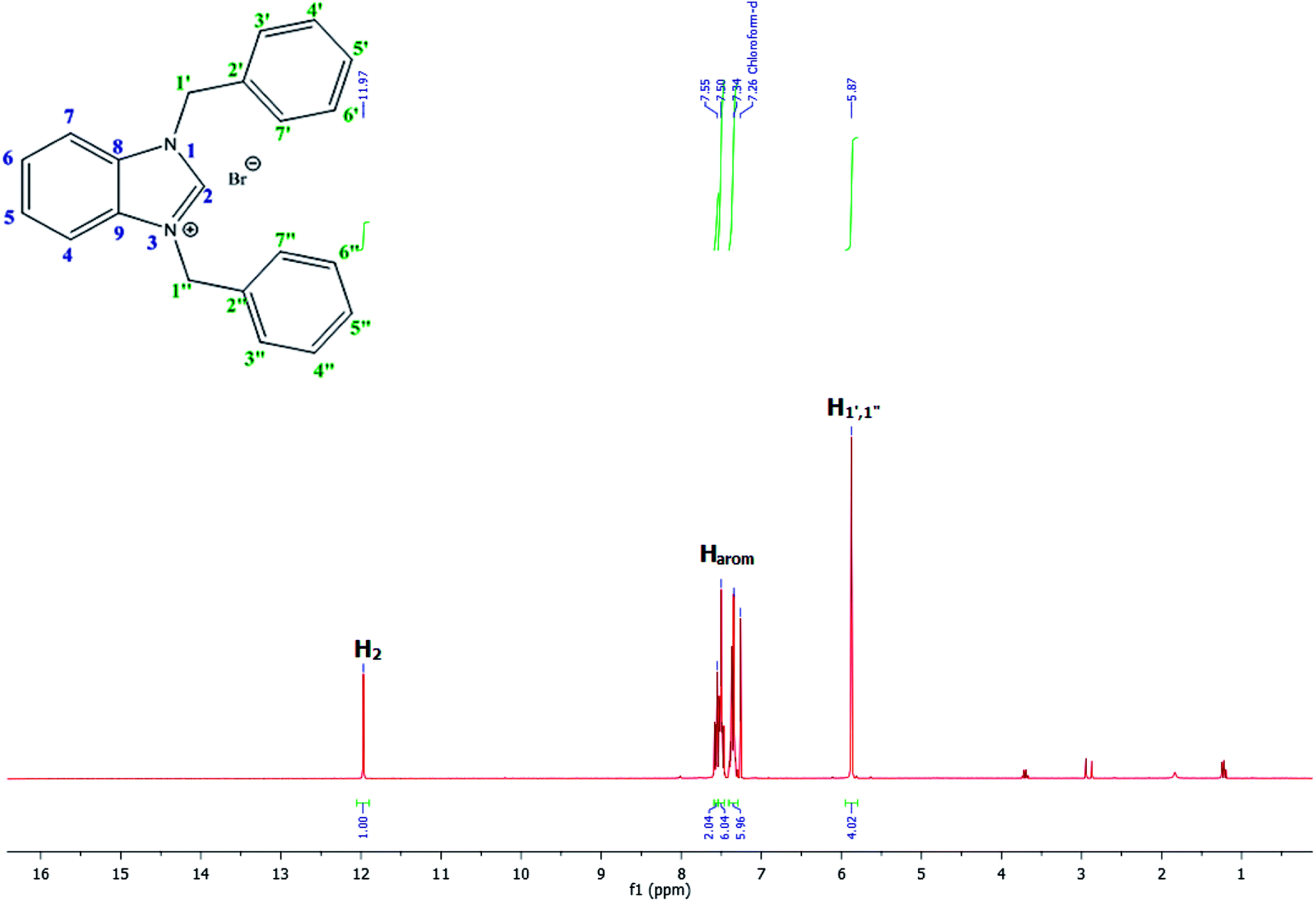 | ||
| Fig. 1 1H NMR spectra of benzimidazole salt 2i (CDCl3, 300 MHz). | ||
The NCHN carbone of the benzimidazole salts 2i in the 13C NMR spectra was obtained in low fields at δ = 132.5 ppm. Fig. 2 Palladium based N-heterocyclic carbene ligands have previously allowed the effective carbonylative coupling of different chloropyridines with phenylboronic acid40,41 but the results illustrate that commercial Pd-based catalyst is not effective towards this transformation, probably because the activity of the catalysts is strongly hindered by the strongly π-accepting CO ligand.42 Thus, we have studied the behavior of benzimidazolium salts 2a–i and 4a–i in the carbonylative cross coupling of 2-bromo pyridine with phenylboronic acid under standard conditions previously described (Scheme 3).16
 | ||
| Scheme 3 Carbonylative cross-coupling reaction of 2-bromopyridine with phenylboronicacid in presence of different benzimidazolium salts 2, 4. | ||
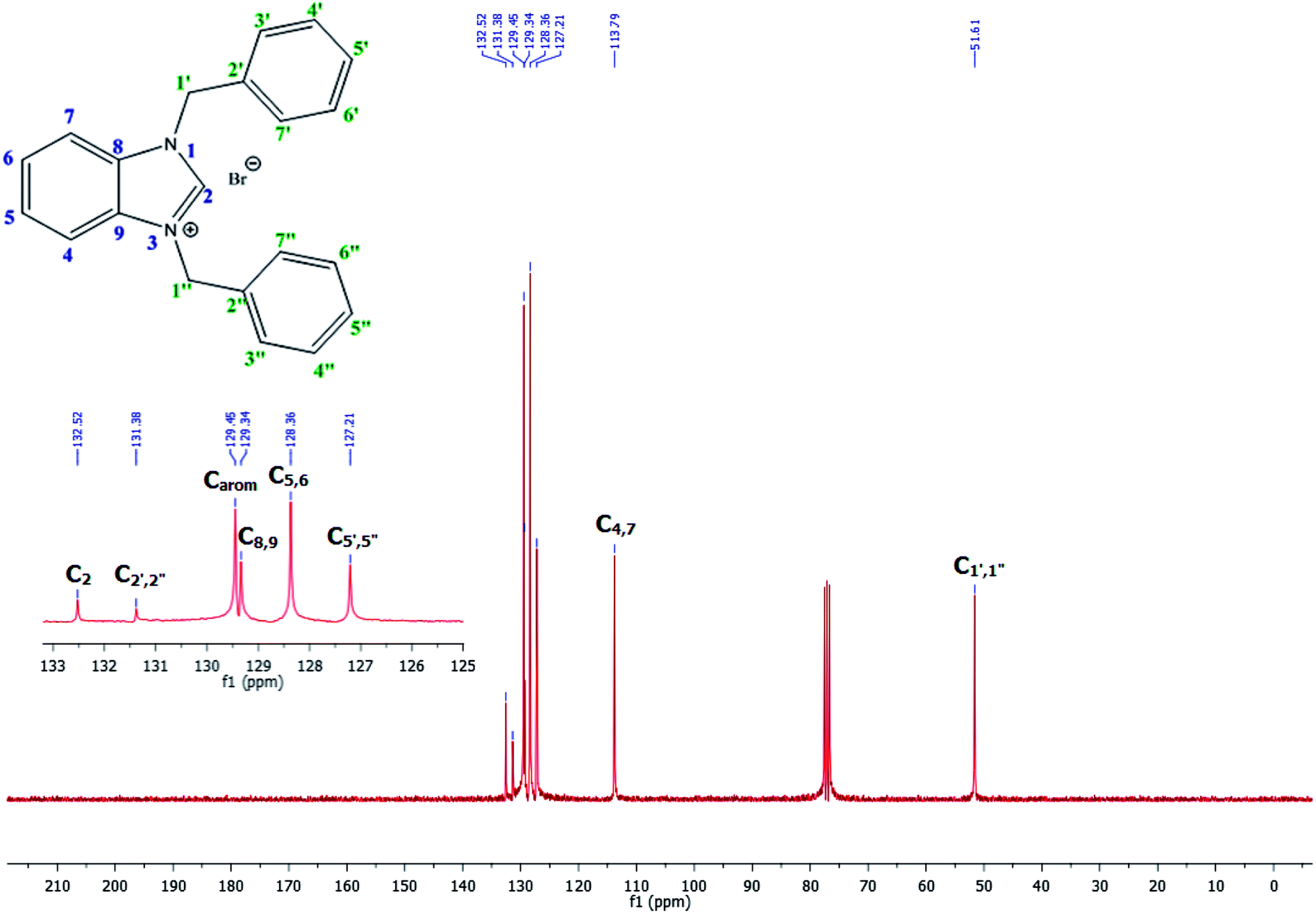 | ||
| Fig. 2 13C NMR spectra of benzimidazole salt 2i (CDCl3, 75 MHz). | ||
The corresponding results are summarized in the Table 1.
| Entry | Benzimidazolium salt | Yield |
|---|---|---|
| a Conditions: 2-Br-Py (1 mmol), Pd(OAc)2 (0.015 mmol), K2CO3 (2 mmol), ligand (0.03 mmol), PhB(OH)2 (1.1 mmol), CO (20 bars), THF (10 mL), 100 °C, one night. | ||
| 1 | 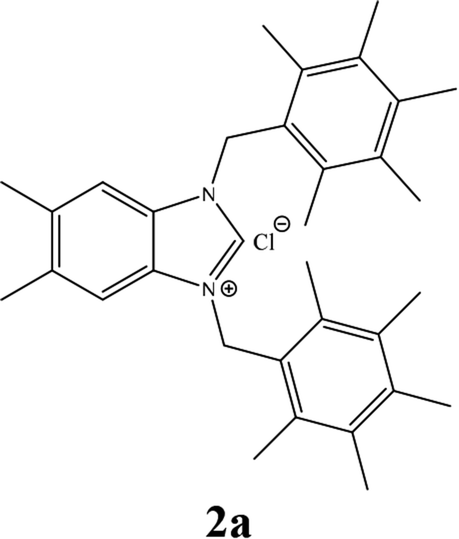 |
27% |
| 2 | 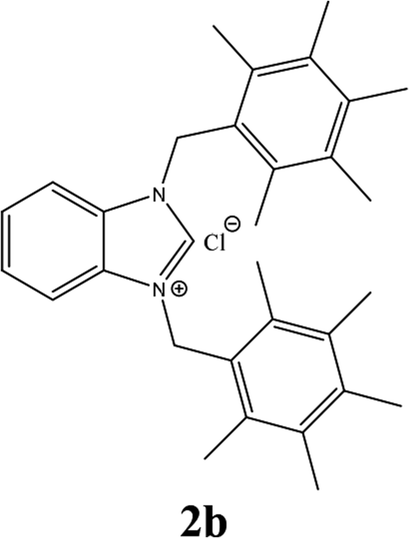 |
22% |
| 3 | 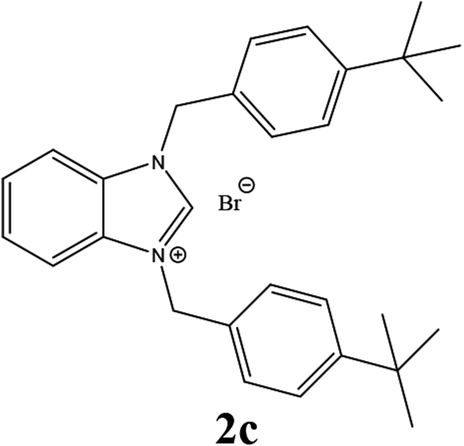 |
55% |
| 4 | 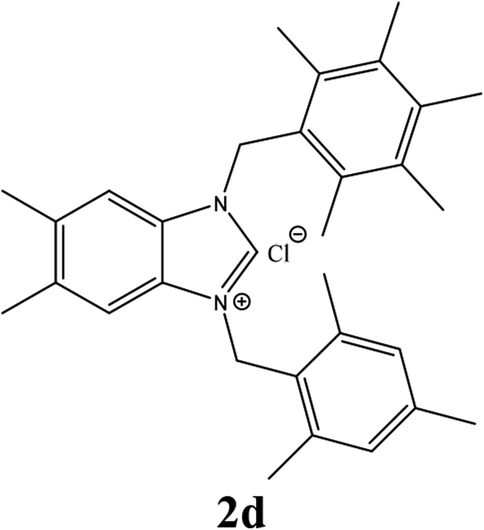 |
21% |
| 5 | 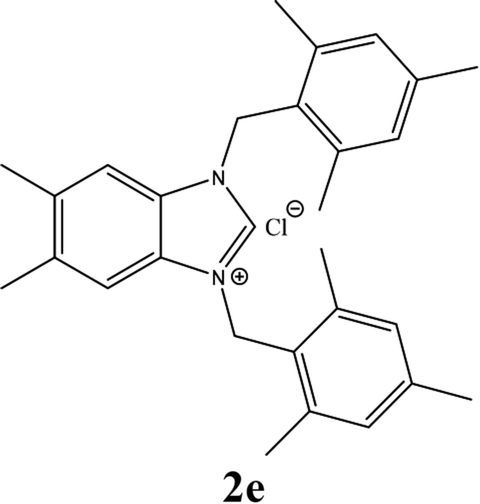 |
21% |
| 6 | 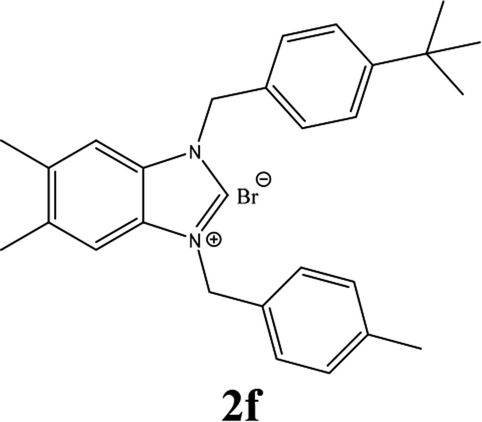 |
57% |
| 7 | 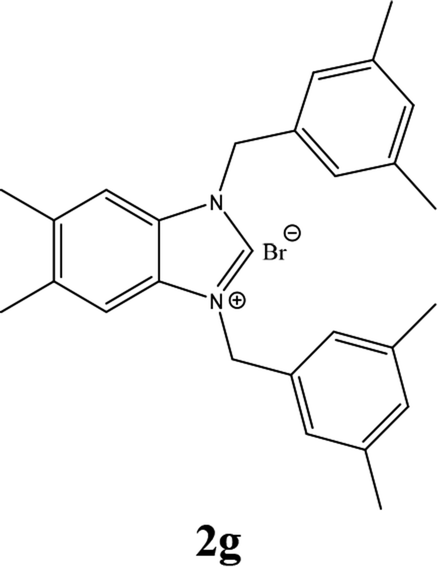 |
45% |
| 8 | 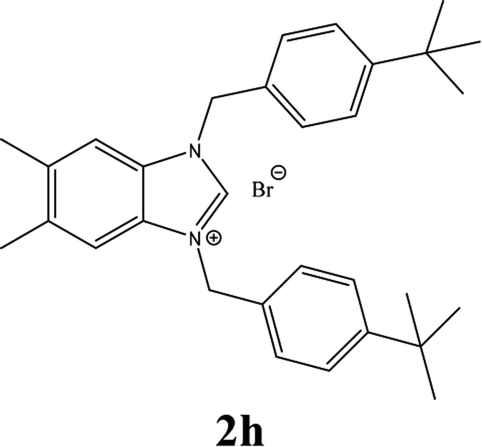 |
45% |
| 9 | 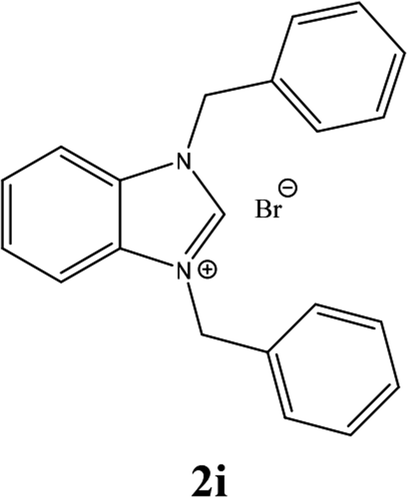 |
53.0% |
| 10 | 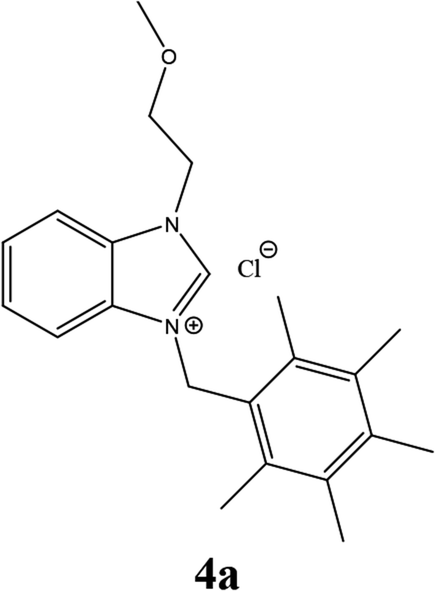 |
54% |
| 11 | 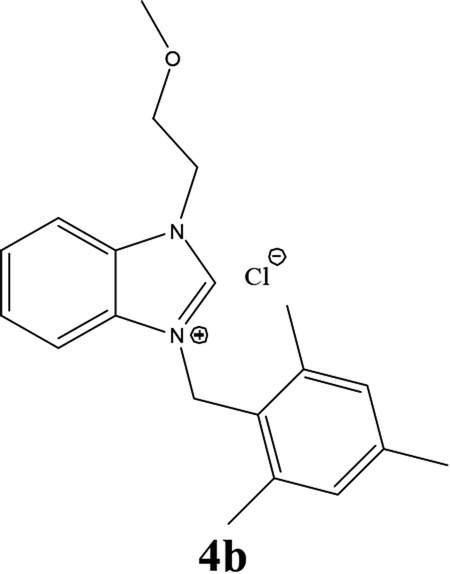 |
53% |
| 12 | 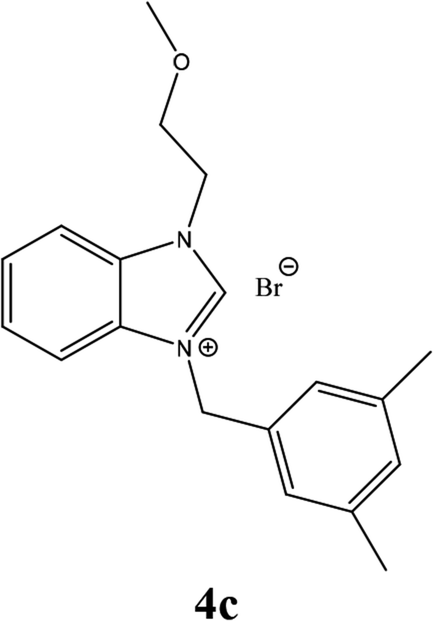 |
57% |
| 13 | 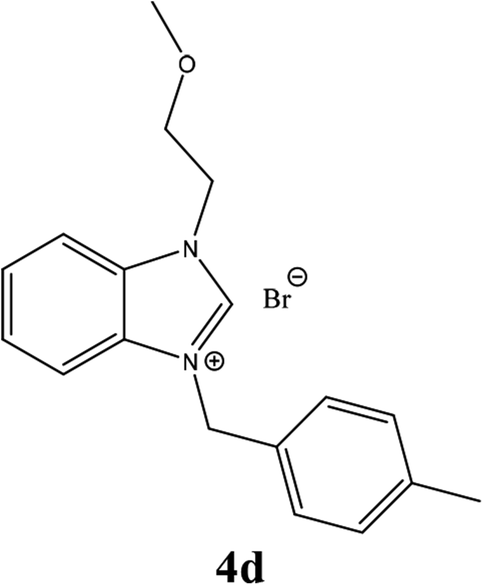 |
46% |
| 14 | 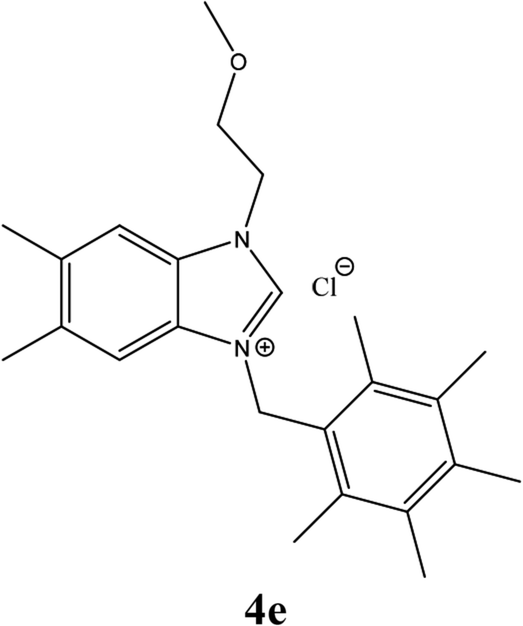 |
27% |
| 15 | 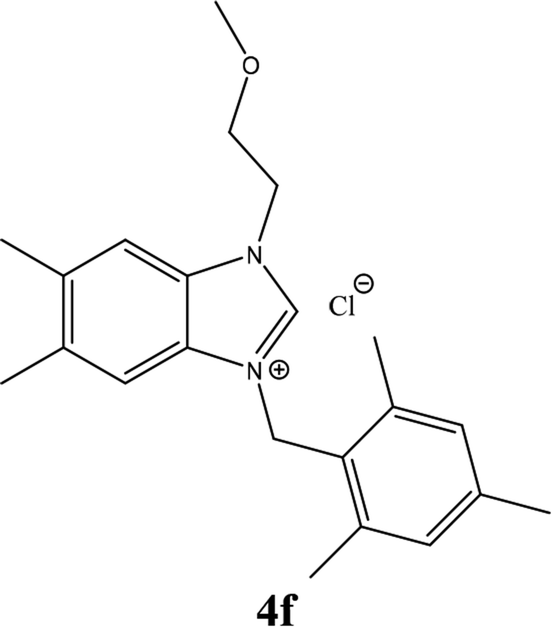 |
21% |
| 16 | 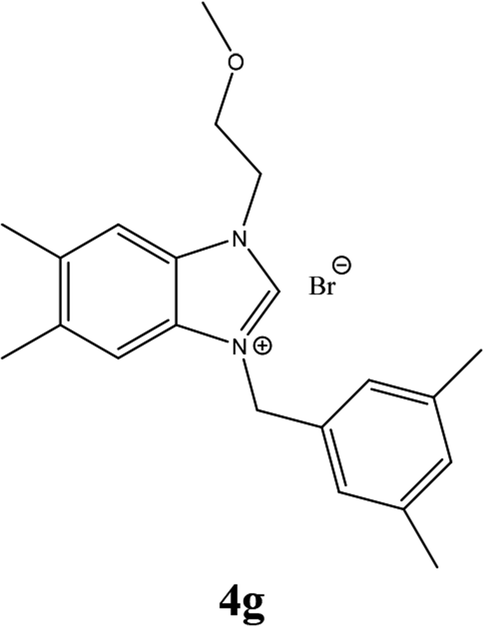 |
57% |
| 17 | 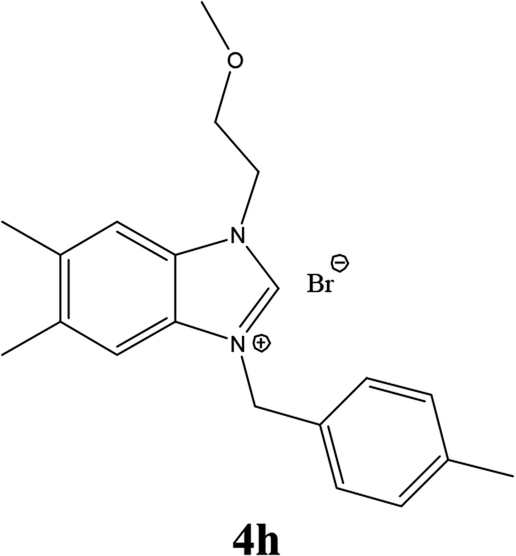 |
49% |
| 18 | 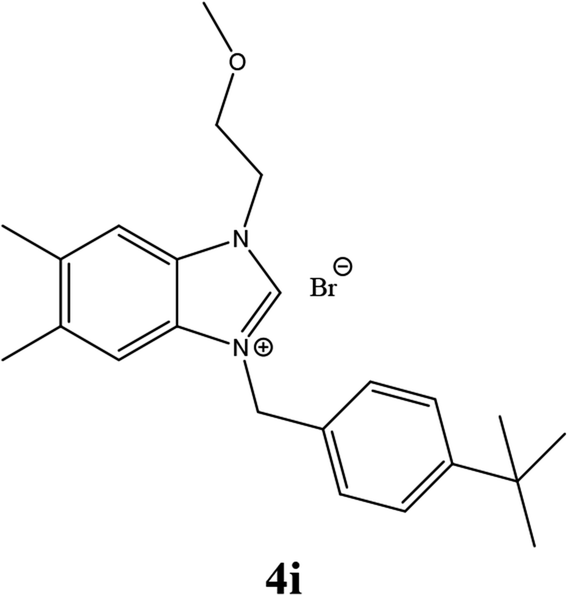 |
55% |
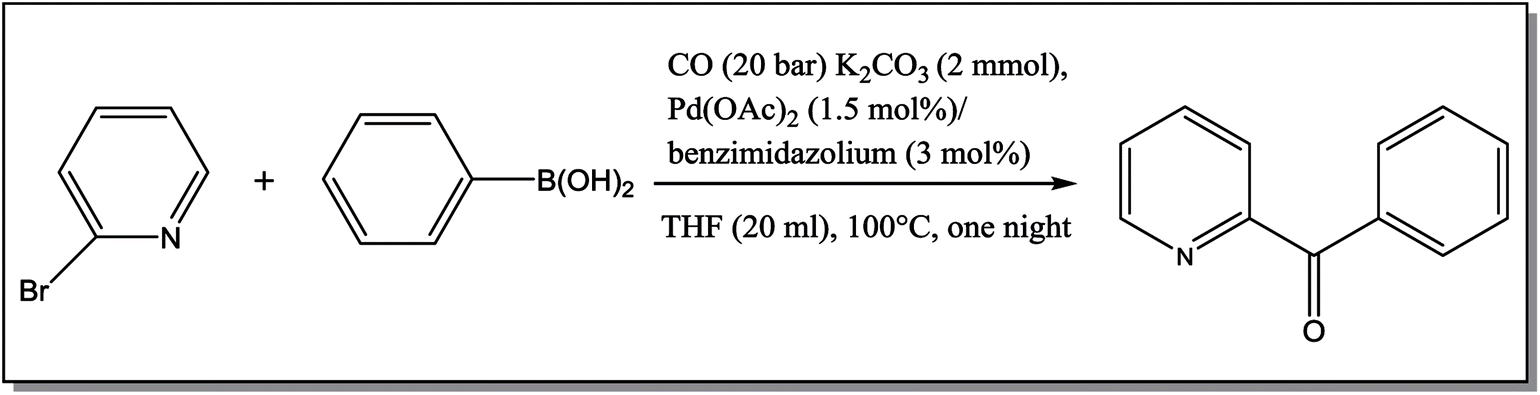
From Table 1 all the complexes gave the corresponding carbonyl product with moderate yields. Good yields were obtained for the less sterically hindered benzimidazoles salts (Table 1, entries 3, 6–13, 16–18). However, lower yields were obtained for the rest of benzimidazoles having most sterically hindered groups (Table 1, entries 1–2, 4–5, 14–15). These results can be explained by the difference in the steric effects stem from the bulky groups on the carbene ring. Based on these results, we tried to improve the selectivity of the reactions by optimizing the reaction conditions. We chose 2-bromopyridine and phenylboronic acid as model substrates to explore the viability of the process. Various solvents, bases and CO pressures were tested in combination with Pd(OAc)2 precursor and benzimidazole 2i as ligand (Scheme 4).
 | ||
| Scheme 4 Carbonylative cross-coupling reaction of 2-bromopyridine with phenylboronic acid. | ||
Selected results are shown in Table 2.
Solvents play a significant role in determining the conversion. Thus, a series of solvents were tested, such as 1,4-dioxane, THF and DMF, (Table 2, entries 1–3). With polar aprotic solvents such as DMF, no reaction took place (Table 2, entry 1). Even, the direct coupling is working in DMF, the presence of CO in the reaction medium could so be the cause. The absence of reaction can be explained by the fact that, in our reaction conditions, the coordination of the combination DMF-CO on the palladium caused the delay of its nucleophilic attack on the halogenated derivative. On the other hand, the use of the ethereal solvent 1,4-dioxane, afforded the carbonylative product in 42% yield (Table 2, entry 2) when we use 1,4-dioxane. However, when the reaction was carried out in tetrahydrofuran (THF), the selectivity was increased and the desired product was obtained with a yield of 53% (Table 2, entry 3).
| Entry | Benzimidazolium salt | Ligand (mol%) | Base | Temperature | CO pressure (bar) | Solvent | Yield |
|---|---|---|---|---|---|---|---|
| a Conditions: 2-Br-Py (1 mmol), Pd(OAc)2, base (2 mmol), 2i, PhB(OH)2 (1.1 mmol), CO, solvent (10 mL), T°, one night. | |||||||
| 1 | 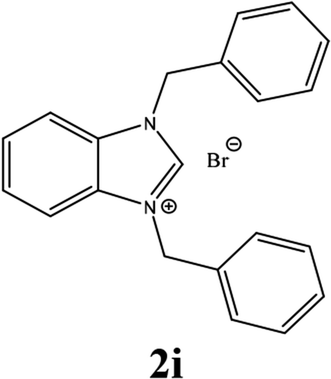 |
3 mol% | K2CO3 | 100 °C | 20 | DMF | — |
| 2 | 3 mol% | K2CO3 | 100 °C | 20 | 1,4-Dioxane | 42% | |
| 3 | 3 mol% | K2CO3 | 100 °C | 20 | THF | 53% | |
| 4 | 3 mol% | t-BuOK | 100 °C | 20 | THF | — | |
| 5 | 3 mol% | Cs2CO3 | 100 °C | 20 | THF | 55% | |
| 6 | 3 mol% | K2CO3 | 100 °C | 10 | THF | 26% | |
| 7 | 3 mol% | K2CO3 | 100 °C | 40 | THF | 34% | |
| 8 | 3 mol% | K2CO3 | 110 °C | 20 | THF | 56% | |
| 9 | 3 mol% | K2CO3 | 120 °C | 20 | THF | 41% | |
| 10 | 5 mol% | K2CO3 | 110 °C | 20 | THF | 70% | |
| 11 | 5 mol% | K2CO3 | 120 °C | 20 | THF | 56% | |
| 12 | 6 mol% | K2CO3 | 110 °C | 20 | THF | 68% | |
Next, the base-screening study was commenced. With the t-BuOK no reaction was observed (Table 2, entry 4). However, alkaline carbonates such as K2CO3 and CsCO3 gave good yields (Table 2, entries 3 and 5). The impact of the reaction temperature for reaction efficiency was investigated. The best yield was obtained for a temperature of 110 °C (Table 2, entry 8). An increase of the temperature to 120 °C, caused a decrease in the yield to 41.11% (Table 2, entry 9) 110 °C was thus chosen as the optimal temperature for the rest of the study.
Various ligand concentrations were also investigated (Table 2, entries 10–12). Optimum ligand loading was found to be 5 mol% (Table 2, entry 10). The increase of the ligand concentration to 6 mol% has caused a slightly drop in the activity (Table 2, entry 12).
The coupling reaction being carried out under a flow of CO, the influence of this parameter on the yield of the reaction was studied. According to Table 1, low yields were obtained using low and very high carbon monoxide pressure (Table 1, entries 6 and 7) while the best yield was obtained using a pressure of 20 bar (Table 2, entry 3).
Based on these results, the optimized reaction conditions were determined to be as follows: 2-bromopyridine (1.0 mmol), arylboronic acid (1.1 mmol), ligand (5 mol%) Pd(OAc)2 (0.025 mmol) and K2CO3 (2.0 mmol) in THF at 110 °C under CO atmosphere (20 bar).
In order to generalize this reaction, a range of arylboronic acids were subjected to the standard conditions (Table 3), regarding their electronic properties and steric effects these reactions were executed smoothly, giving the corresponding products in good yields (Scheme 5).
| Entry | Benzimidazolium salt | Boronic acid | Carbonyl product | Yield |
|---|---|---|---|---|
| a Conditions: 2-Br-Py (1 mmol), Pd(OAc)2 (0.025 mmol), K2CO3 (2 mmol), ligand (5 mol%), arylboronic acid (1.1 mmol), CO (20 bars), 110 °C, THF (10 mL), one night. | ||||
| 1 | 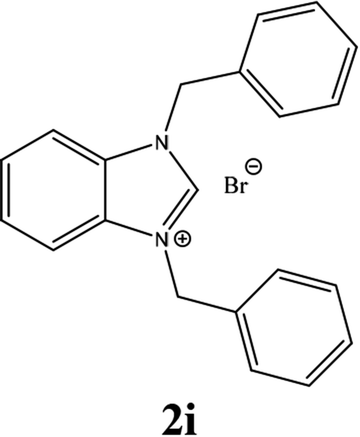 |
 |
 |
70% |
| 2 | 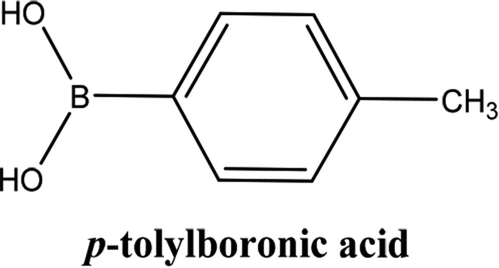 |
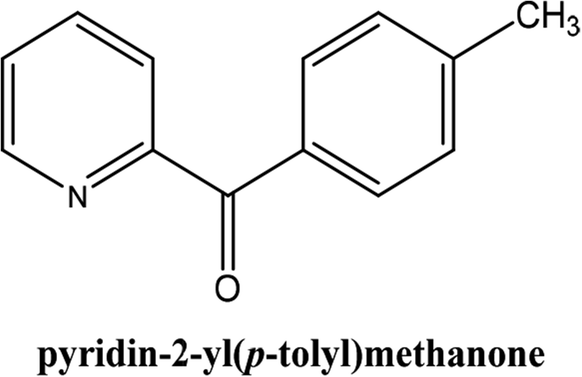 |
51% | |
| 3 | 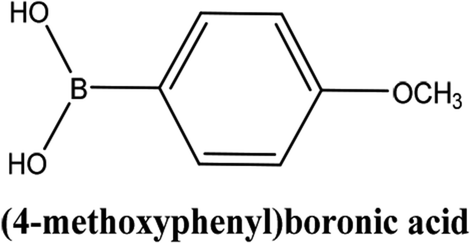 |
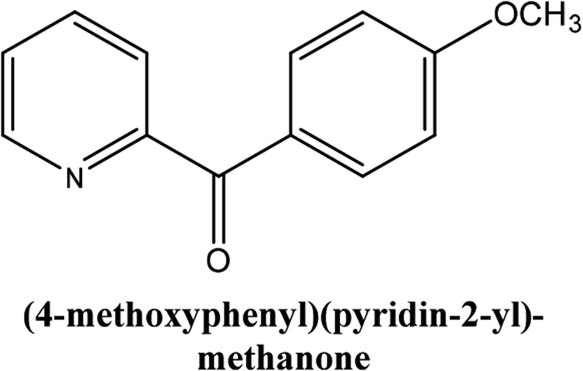 |
51% | |
| 4 | 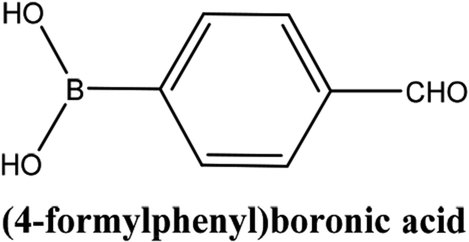 |
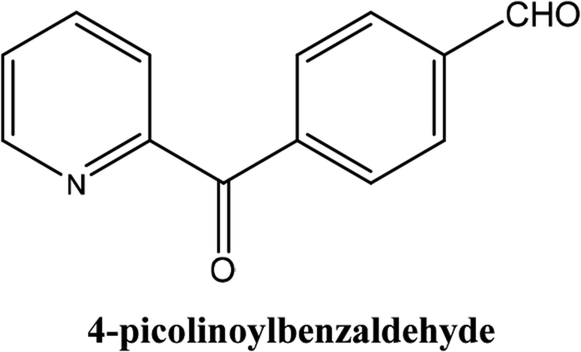 |
10% | |
| 5 | 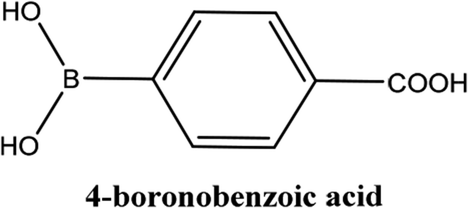 |
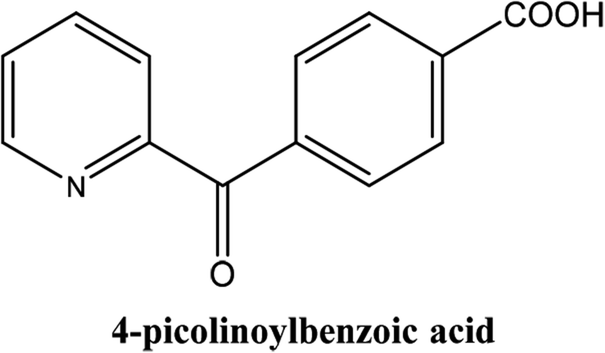 |
Trace | |
 | ||
| Scheme 5 Carbonylative cross-coupling reaction of 2-bromopyridine with different arylboronic acids. | ||
The results are summarized in Table 3.
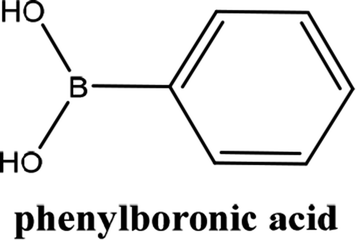
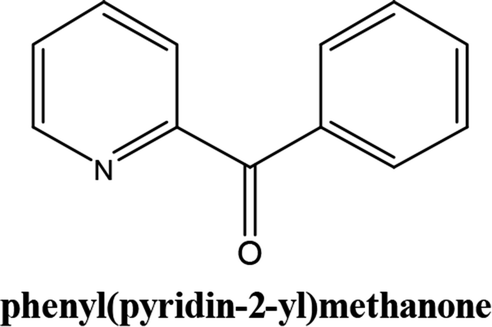
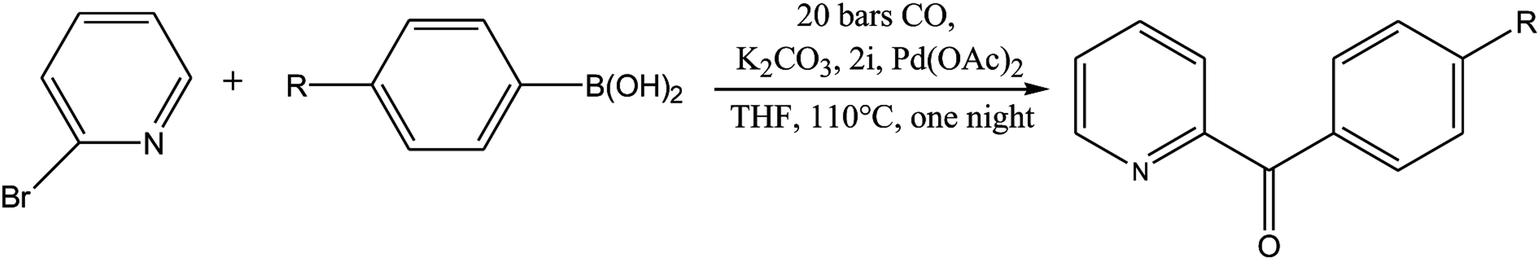
As shown in Table 3, the yield of arylpyridine ketones was depending on the nature of the used arylboronic acid. The best result was obtained with phenylboronic acid giving carbonylated coupling product with a high yield of 70% (Table 3, entry 1). When we used p-tolylboronic acid and 4-carboxyphenylboronic acid, having a strong electrodonor effects, very similar results were obtained (Table 3, compare entries 2 and 3) and an important amount of by-product was found in the reaction medium for the two reactions resulting from the deboronation of the corresponding boronic acid under basic conditions and high temperatures. Thus, the modest conversion obtained in this case was probably due to the lack of reagent. Other boronic acids with electron-withdrawing groups (–CHO, –COOH) were also tested on the carbonylative cross-coupling reaction of 2-bromopyridine with arylboronic acids. A low yield of 10% was obtained for the 4-formylphenylboronic acid (Table 3, entry 4) whereas, no carbonyl product was formed using 4-bromobenzoic acid only a small amount of direct coupling product (Table 3, entry 5) was detected. Thus, we can conclude that the differences in the activity for the arylboronic acids are presumably due to the difference in the electronic properties of the substituents. The chemical structures of unsymmetrical arylpyridine ketones were established using spectroscopic methods including FTIR, 1H-NMR and 13C-NMR. For example 1H-NMR spectra of phenyl(pyridin-2-yl)methanone revealed aromatic protons between 7.26 ppm and 8.74 ppm (Fig. 3).
 | ||
| Fig. 3 1H NMR spectra of phenyl(pyridin-2-yl)methanone (CDCl3, 300 MHz). | ||
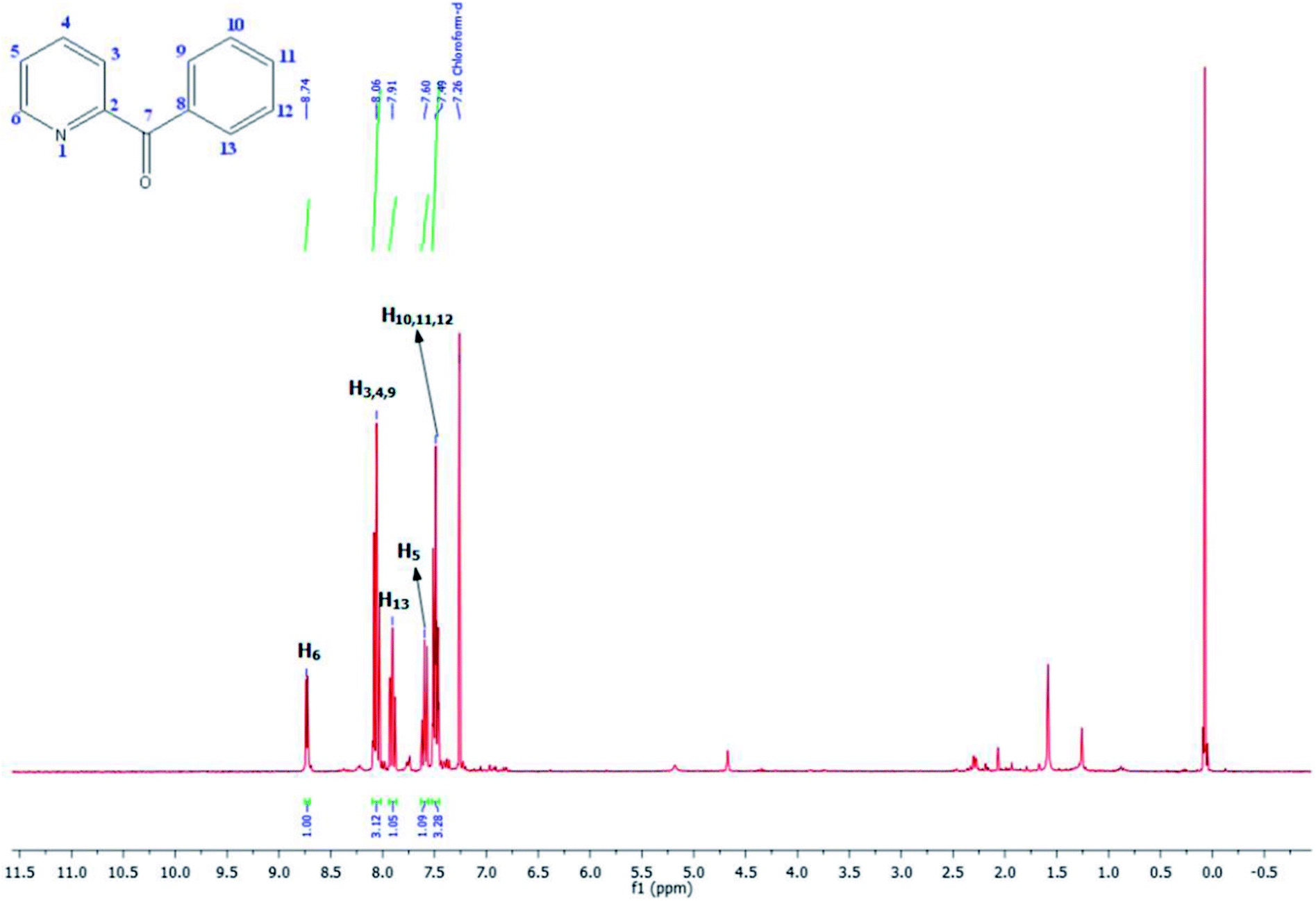
Its 13C-NMR spectrum of phenyl(pyridin-2-yl)methanone revealed C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at δ 192.8 ppm, aromatic carbons between 123.8 ppm and 154.1 ppm (Fig. 4).
O at δ 192.8 ppm, aromatic carbons between 123.8 ppm and 154.1 ppm (Fig. 4).
 | ||
| Fig. 4 13C NMR spectra of phenyl(pyridin-2-yl)methanone (CDCl3, 75 MHz). | ||
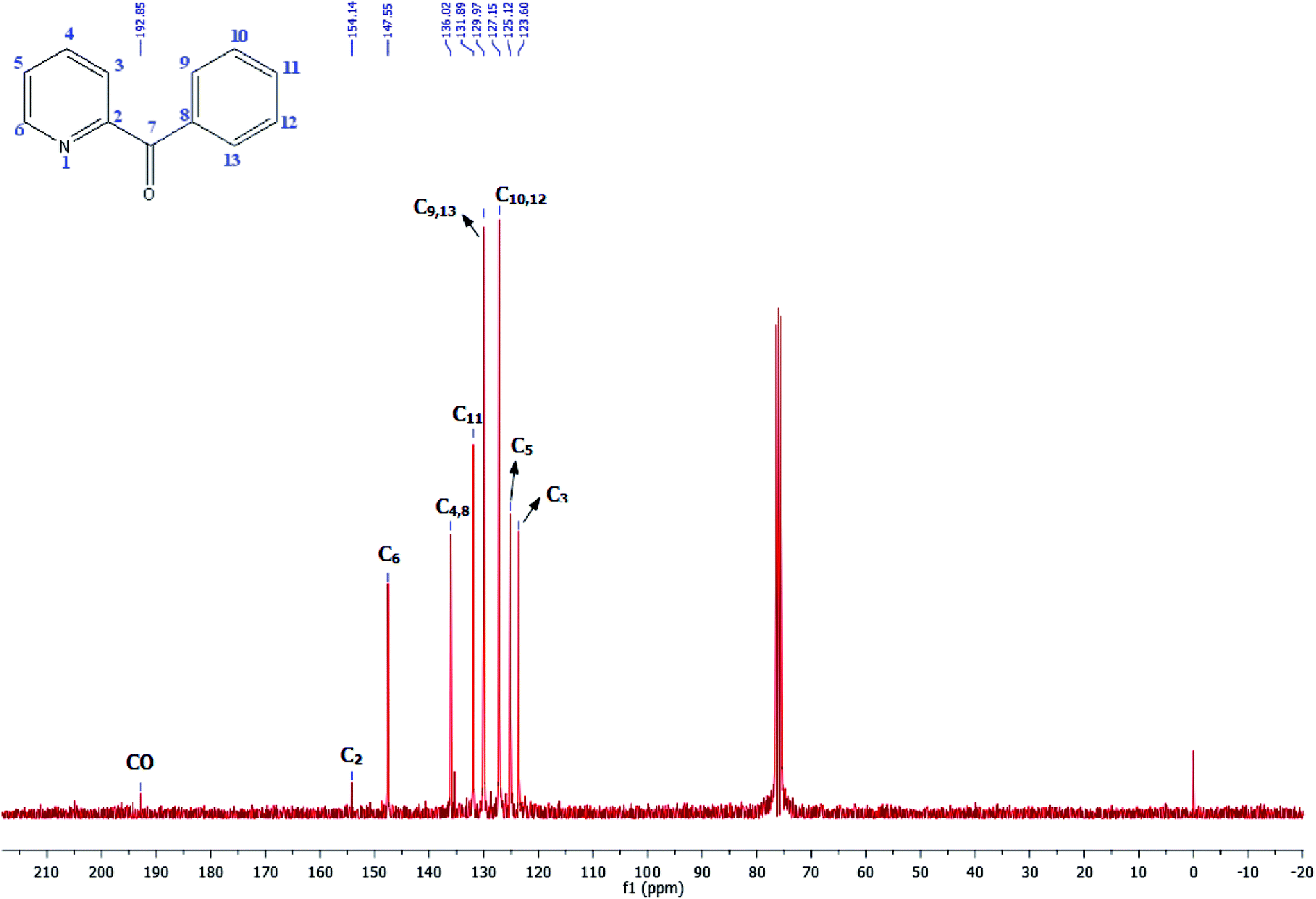
3. Reaction mechanism for the carbonylative Suzuki–Miyaura coupling reaction
A plausible mechanism is given in the following Scheme 6. | ||
| Scheme 6 The fundamental steps involved in palladium-catalyzed carbonylative cross-coupling. | ||
The first step of the catalytic cycle consists of an oxidative addition of the halide on the palladium species leading to the formation of palladium pyridine complex. The next step is the insertion and coordination of CO to palladium pyridine complex. The step after insertion is transmetallation. The final step of the catalytic cycle is reductive elimination. This step proceeds through a three-centered transition state in order to form a new C–C bond between the electrophilic acyl group and the nucleophile leading to the formation of the expected unsymmetrical arylpyridine ketones.
In order to compare the efficiency of our catalysts (NHC–Pd complex) generated in situ from benzimidazolium salts 2 and 4 with literature further tests were established. The results are given in Table 4.
| Entry | Catalyst | Arylbromide | Arylboronic acid | Direct coupling yield | Carbonylative coupling yield |
|---|---|---|---|---|---|
| a Conditions: 2-Br-Py (1 mmol), Pd(OAc)2 (0.025 mmol), K2CO3 (2 mmol), ligand (5 mol%), PhB(OH)2 (1.1 mmol), CO (20 bars), 110 °C, THF (10 mL), one night.b PhB(OH)2 (1.5 mmol). | |||||
| 1 | 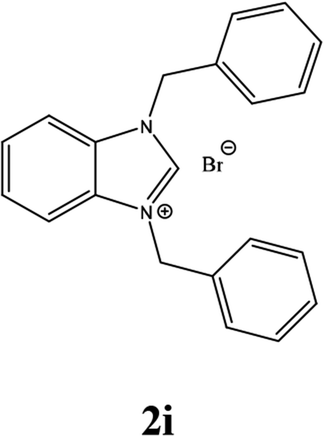 |
 |
 |
4.8% | 69.9% |
| 2b |  |
3.9% | 27.7% | ||
| 3 | 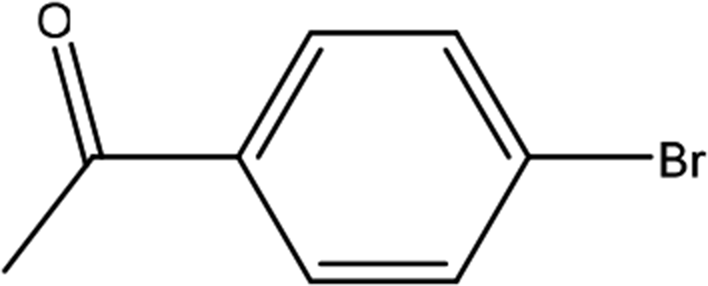 |
20.2% | — | ||
| 4 | 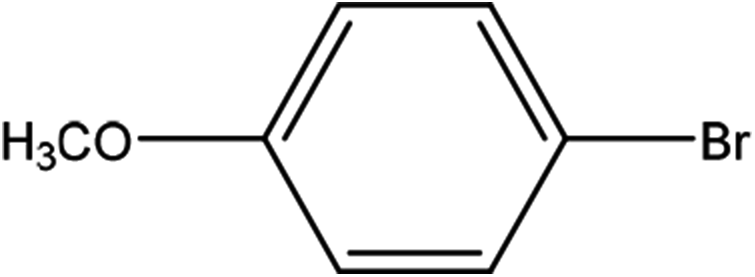 |
4.1% | — | ||
| 5 | 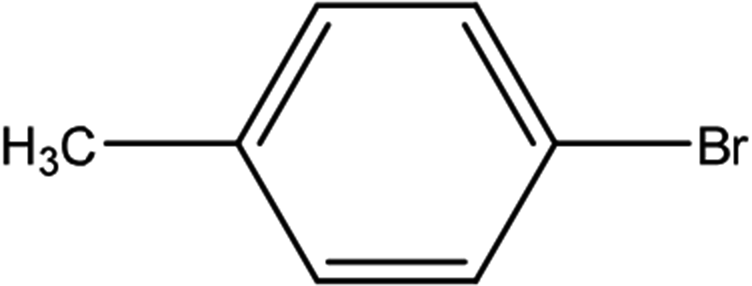 |
3.7% | — | ||
| 6 | 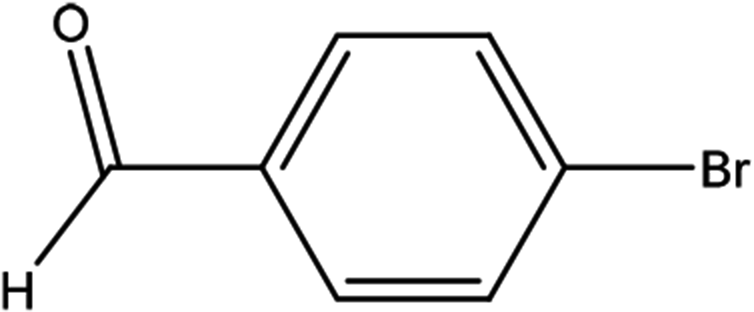 |
17.7% | 35.17% | ||
First, we thought that the obtained moderate yields are due to the lack of boronic acid in the medium caused by decomposition of boronic acid at high temperatures. In this way by increasing the amount of boronic acid the yields of the obtained products decrease (Table 4, entries 1 and 2) so we conclude that the lack of boronic acid not affect the yields. Then we tried to test benzimidazole salt 2i with others aryl bromides used in Suzuki couplings. As shown in Table 4, using bromoacetophenone, bromoanisole and bromotoluene, only the direct coupling product was obtained (Table 4, entries 3–5). While in the case of bromobenzaldehyde low yields of carbonyl product and direct coupling product were obtained.
Previous researchers have reported induction periods for Suzuki reactions promoted by Pd(OAc)2/imidazolium salts. It was proposed that during these induction periods Pd(II)/NHC complexes were formed and were then slowly reduced to catalytically active Pd(0)/NHC complexes.43 It is important to note that these induction periods could be avoided with our present catalyst system.
4. Anticancer cytotoxic activity
The cytotoxic activity of the synthesized compounds 2a–i and 4a–i was tested in vitro against cancer human cell lines such us MDA-MB-231, MCF-7 and T47D.44,45 The results are summarized in Table 5.| Benzimidazoles salts 2 | MDA-MB-231 | MCF-7 | T47D |
|---|---|---|---|
| a The IC50 values represent an average of three independent experiments (mean ± SD). | |||
| 2a | 18 ± 4 | 17 ± 2 | 20 ± 4 |
| 2b | 19.37 ± 0.85 | 18.36 ± 0.85 | 20.38 ± 0.85 |
| 2c | 8.45 ± 3 | 10.55 ± 3 | 11.55 ± 5.75 |
| 2d | 10.75 ± 2 | 10.75 ± 2 | 10.75 ± 2 |
| 2e | 5.95 ± 1 | 4.85 ± 1 | 14.96 ± 1 |
| 2f | 4.45 ± 0.95 | 8.08 ± 0.95 | 14.42 ± 0.95 |
| 2g | 19.30 ± 2 | 20.35 ± 2 | 25.32 ± 1 |
| 2h | 20.25 ± 1 | 20.25 ± 2 | 20.25 ± 3.10 |
| 2i | 18.45 ± 2 | 16.42 ± 2 | 22.43 ± 2 |
| 4a | 16 ± 4 | 15 ± 2 | 18 ± 4 |
| 4b | 18 ± 0.85 | 17 ± 0.85 | 19 ± 0.85 |
| 4c | 9.23 ± 2 | 13.55 ± 3 | 14.55 ± 4 |
| 4d | 14.75 ± 3 | 15.75 ± 2 | 13.75 ± 2 |
| 4e | 7.95 ± 1 | 6.85 ± 1 | 13.96 ± 1 |
| 4f | 7.45 ± 0.95 | 10.08 ± 0.95 | 15.42 ± 0.95 |
| 4g | 18.30 ± 2 | 22.35 ± 2 | 25.32 ± 1 |
| 4h | 20.25 ± 1 | 20.25 ± 2 | 24.25 ± 2 |
| 4i | 19.45 ± 2 | 17.42 ± 2 | 23.43 ± 3 |
All synthesized compounds showed good cytotoxic activity against MDA-MB-231, MCF-7 and T47D cell lines. The most actives compounds are 2e and 2f against MDA-MB-231 and MCF-7.
5. Conclusion
In conclusion, two families of benzimidazolium salts 2a–i and 4a–i have been prepared and characterized. We have demonstrated that carbonylative cross-coupling reaction of arylboronic acids with 2-brompyridine is a versatile route to prepare unsymmetrical arylpyridine ketones in the presence of a catalyst formed in situ from Pd(OAc)2 and K2CO3. The efficiency of the reaction was especially related to the nature of the arylboronic acid. The cytotoxic activity of the compounds (2–4) were also determined against a panel of cell lines such us MDA-MB-231, MCF-7 and T47D. The most actives compounds are 2e, 2f, 4e and 4f against MDA-MB-231 and MCF-7.6. Experimental section
6.1 General information
All manipulations were performed using Standard Schlenck techniques under Argon atmosphere. Chemicals were purchased from Sigma Aldrich and used without further purification. All solvents were purified and dried by MBraun SPS 800 solvent purification system. Column chromatography was performed using silica gel 60 (70–230 mesh). NMR spectra were recorded at 300 and 400 MHz for 1H NMR and 75 and 100 MHz for 13C NMR. Chemical shifts, δ, are reported in ppm relative to the internal standard TMS for both 1H and 13C NMR. The products were characterized by GC (gas chromatography). Quantitative GC analyses were performed gas chromatography (CHROMPACK CP-9002, ENSCL, France). The NMR studies were carried out in high-quality 5 mm NMR tubes. Signals are quoted in parts per million as δ downfield from tetramethylsilane (δ = 0.00) as an internal standard. NMR multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, m = multiplet signal. IR spectra were recorded on a 398 spectrophotometer (Perkin-Elmer, Malatya University,Turkey). Melting points were determined with Stuart automatic melting point apparatus (SMP-40) at Malatya University, Turkey.6.2 Synthesis of compounds 1a–g and benzimidazoles 2a–i
Compounds 1a–f were synthesized according to our previous works.36,39Benzimidazoles 2a–h were synthesized according to the literature36,37
6.3 Compounds 3a-b and benzimidazoles 4a–i were synthesized according to our previous works38
6.4 Synthesis of benzimidazoles (4a–i)
A mixture of benzimidazolium salt (3a-b) (1 g) and the corresponding benzyl bromide or chloride (1 eq.) in DMF (2 mL) was stirred at 70 °C for 2–3 days. After that time, the white solid formed was washed with diethyl ether (20 mL) and stirred for couple hours. Then the reaction mixture was filtered through filter paper and the white solid was dried under vaccum.6.4 General procedure for carbonylative coupling
A stainless steel autoclave equipped with a magnetic stirring bar was charged with benzimidazolium salt (0.05 mmol), K2CO3 (2 mmol) and Pd(OAc)2 (0.025 mmol). After flushing the atmosphere with N2, a solution of 2-bromopyridine (1 mmol) in 5 mL of dry THF was added under N2. The reaction mixture was stirred for 1 h at 80 °C. After this time, the autoclave was cooled down to room temperature and a solution of boronic acid (1.1 mmol) in 5 mL of dry THF was added under N2. The autoclave was flushed with CO, pressurized to 20 bar and heated to 120 °C. At the end of the reaction and after cooling down to room temperature, the solution was filtred and the solvent was removed under vacuum. The residue was purified by silica gel chromatography (EtOAc/hexane) to give pure product.The cytotoxic activity was assessed according to the literature.44,45
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to extend their sincere appreciation to the Deanship of Scientific Research at King Saud University for its funding this research group (No. RG-1435-023).References
- X. J. Wang, L. Zhang, X. F. Sun, Y. B. Xu, D. Krishnamurthy and C. H. Senanayake, Org. Lett., 2005, 7, 5593 CrossRef CAS PubMed.
- B. Hatano, J. Kadokaw and H. Tagaya, Tetrahedron Lett., 2002, 43, 5859 CrossRef CAS.
- S. Gmouh, H. L. Yang and M. Vaultier, Org. Lett., 2003, 5, 2219 CrossRef CAS PubMed.
- T. Yamamoto, T. Kohara and A. Yamamoto, Chem. Lett., 1976, 11, 1217 CrossRef.
- Y. Hatanaka, S. Fukushima and T. Hiyama, Tetrahedron, 1992, 48, 2113 CrossRef CAS.
- J. J. Brunet and R. Chauvin, Chem. Soc. Rev., 1995, 24, 89 RSC.
- E. Fillion, D. Fishlock, A. Wilsily and J. M. Goll, J. Org. Chem., 2005, 70, 1316 CrossRef CAS PubMed.
- D. O. Jang, K. S. Moon, D. H. Cho and J. G. Kim, Tetrahedron Lett., 2006, 47, 6063 CrossRef CAS.
- T. Ishiyama, H. Kizaki, N. Miyaura and A. Suzuki, Tetrahedron Lett., 1993, 34, 7595 CrossRef CAS.
- T. Ishiyama, H. Kizaki, T. Hayashi, A. Suzuki and N. Miyaura, J. Org. Chem., 1998, 63, 4726 CrossRef CAS.
- M. V. Khedkar, T. Sasaki and B. M. Bhanage, RSC Adv., 2013, 3, 7791 RSC.
- J. R. Niu, M. M. Liu, P. Wang, Y. Long, M. Xie, R. Li and J. T. Ma, New J. Chem., 2014, 38, 1471 RSC.
- (a) F. S. Yates, Comprehensive Heterocyclic Chemistry, ed. A. J. Boulton and A. Mckillop, Pergamon, Oxford, 1984, vol. 2, ch. 2.08 Search PubMed; (b) M. Balasubramanian and J. G. Keay, Comprehensive Heterocyclic Chemistry II, ed. A. Mckillop, Pergamon, Oxford, 1995, vol. 5, ch. 5.06 Search PubMed.
- (a) D. J. Hlasta and J. J. Court, Tetrahedron Lett., 1989, 30, 1773–1776 CrossRef CAS; (b) C. K. Herrmann, Y. P. Sachdeva and J. F. Wolfe, J. Heterocycl. Chem., 1987, 24, 1061–1065 CrossRef; (c) T. Sakamoto, Y. Kondo, N. Murata and H. Yamanaka, Tetrahedron, 1993, 49, 9713–9720 CrossRef CAS; (d) Y. Yamamoto, H. Ouchi and T. Tanaka, Chem. Pharm. Bull., 1995, 43, 1028–1030 CrossRef CAS; (e) F. Trécourt, G. Breton, V. Bonnet, F. Mongin, F. Marsais and G. Queguiner, Tetrahedron Lett., 1999, 40, 4339–4342 CrossRef.
- Samuel COUVE-BONNAIRE, Synthesis of functionalized pyridine derivatives-ketones, ketoamides and ketoesters-by carbonylation reaction of halogenated precursors, PhD thesis, thesis no. 3015, Université de Lille, 1, 2001.
- (a) S. Couve-Bonnaire, J.-F. Carpentier, A. Mortreux and Y. Castanet, Tetrahedron Lett., 2001, 42, 3689–3691 CrossRef CAS; (b) S. Couve-Bonnaire, J.-F. Carpentier, A. Mortreux and Y. Castanet, Tetrahedron, 2003, 59, 2793–2799 CrossRef CAS.
- (a) A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS; (b) G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC; (c) H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC.
- W. A. Herrmann, M. Elison, J. Fisher, C. Köcher and G. R. Artus, Angew. Chem., Int. Ed., 1995, 34, 2371 CrossRef CAS.
- N. M. Scott and S. P. Nolan, Eur. J. Inorg. Chem., 2005, 1815 CrossRef CAS.
- H. W. Wanzlick and E. Schikora, Angew. Chem., 1960, 72, 494 CrossRef CAS.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef CAS PubMed.
- S. Díez-González and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874 CrossRef.
- W. A. Hermann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef.
- G. A. Grasa, M. S. Viciu, J. Huang, C. Zhang, M. L. Trudell and P. Nolan, Organometallics, 2002, 21, 2866 CrossRef CAS.
- G. A. Grasa, M. S. Viciu, J. Huang, C. Zhang, M. L. Trudell and S. P. Nolan, Organometallics, 2002, 21(14), 2866–2873 CrossRef CAS.
- (a) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed; (b) N-Heterocyclic Carbenes in Transition Metal Catalysis, ed. F. Glorius, Springer, Berlin Heidelberg, 2007 Search PubMed; (c) N-Heterocyclic Carbenes From Laboratory Curiosities to Efficient Synthetic Tools, ed. S. Díez-González, RSC Publishing, Cambridge, UK, 2011 Search PubMed; (d) E. L. Kolychev, A. F. Asachenko, P. B. Dzhevakov, A. A. Bush, V. V. Shuntikov, V. N. Khrustalev and M. S. Nechaev, Dalton Trans., 2013, 42, 6859–6866 RSC; (e) A. R. Leverett, A. I. McKay and M. L. Cole, Dalton Trans., 2015, 44, 498–500 RSC.
- (a) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS PubMed; (b) E. Colacino, J. Martinez and F. Lamaty, Coord. Chem. Rev., 2007, 251, 726–764 CrossRef CAS; (c) C. Samojlowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708–3742 CrossRef CAS PubMed; (d) M. Yus and I. M. Pastor, Chem. Lett., 2013, 42, 94–108 CrossRef CAS; (e) L. A. Schaper, S. J. Hock, W. A. Herrmann and F. E. Kuhn, Angew. Chem., Int. Ed., 2013, 52, 270–289 CrossRef CAS PubMed; (f) M. J. Ingleson and R. A. Layfield, Chem. Commun., 2012, 3579–3589 RSC; (g) N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440–1449 CrossRef CAS PubMed; (h) S. Çekirdek, S. Yaşar and İ. Özdemir, Appl. Organomet. Chem., 2014, 28, 423–431 CrossRef; (i) S. Yaşar, K. J. Cavell, B. D. Ward and B. Kariuki, Appl. Organomet. Chem., 2011, 25, 374–382 CrossRef; (j) S. Yaşar, S. Çekirdek and İ. Özdemir, J. Coord. Chem., 2014, 67, 1236–1248 CrossRef; (k) E. . Ö. Karaca, M. Akkoç, E. Öz, S. Altin, V. Dorcet, T. Roisnel, N. Gürbüz, Ö. Çelik, A. Bayri, C. Bruneau, S. Yaşar and I. Özdemir, J. Coord. Chem., 2017, 70, 1270–1284 CrossRef CAS; (l) S. Yaşar, E. Ö. Karaca, Ç. Şahin, İ. Özdemir, O. Şahin and O. Büyükgüngör, J. Organomet. Chem., 2015, 789–790, 1–7 CrossRef.
- (a) H. Ren, Y. Xu, E. Jeanneau, I. Bonnamour, T. Tu and U. Darbos, Tetrahedron, 2014, 70, 2829–2837 CrossRef CAS; (b) R. Zhong, A. Pothig, Y. Feng, K. Riener, W. A. Herrmann and F. E. Kuhn, Green Chem., 2014, 16, 4955–4962 RSC; (c) H. Turkmen, L. Gok, I. Kani and B. Cetinkaya, Turk. J. Chem., 2013, 37, 633–642 CAS; (d) M. Teci, E. Brenner, D. Matt, C. Gourlaouen and L. Toupet, Dalton Trans., 2014, 43, 1251–1262 RSC; (e) M. Teci, E. Brenner, D. Matt and L. Toupet, Eur. J. Inorg. Chem., 2013, 2841–2848 CrossRef CAS; (f) R. L. Sutar, V. K. Kumar, R. D. Shingare, S. Thorat, R. Gonnade and D. S. Reddy, Eur. J. Org. Chem., 2014, 4482–4486 CrossRef CAS.
- (a) H. Valdes, M. Poyatos, G. Ujaque and E. Peris, Chem.–Eur. J., 2014, 20, 1–12 CrossRef; (b) Y. Li, L. Yang, Q. Chen, C. Cao, P. Guan, G. Pang and Y. Z. Shi, Z. Anorg. Allg. Chem., 2013, 639, 575–581 CrossRef CAS; (c) Y. C. Lin, H. H. Hsueh, S. Kanne, L. K. Chang, F. C. Liu, I. J. B. Lin, G. H. Lee and S. M. Peng, Organometallics, 2013, 32, 3859–3869 CrossRef CAS.
- (a) R. A. Batey, M. Shen and J. Lough, Org. Lett., 2002, 4, 1411–1414 CrossRef CAS; (b) L. Ray, S. Barman, M. M. Shaikh and P. Ghosh, Chem.–Eur. J., 2008, 14, 6646–6655 CrossRef CAS PubMed; (c) F. T. Luo and H. K. Lo, J. Organomet. Chem., 2011, 696, 1262–1265 CrossRef CAS.
- (a) H. V. Huynh and C. S. Lee, Dalton Trans., 2013, 42, 6803–6809 RSC; (b) L. Yang, Y. Li, Q. Chen, Y. Du, C. Cao, Y. Shi and G. Pang, Tetrahedron, 2013, 69, 5178–5184 CrossRef CAS; (c) A. Biffis, M. Cipani, E. Bressan, C. Tubaro, C. Graiff and A. Venzo, Organometallics, 2014, 33, 2182–2188 CrossRef CAS; (d) H. Lu, L. Wang, F. Yang, R. Wu and W. Shen, RSC Adv., 2014, 4, 30447–30452 RSC.
- R. Miri, O. Firuzi, P. Peymani, Z. Nazarian and A. Shafiee, Synthesis and Cytotoxicity Study of New Cyclopenta [b] quinoline-1,8-dione Derivatives, Iran. J. Pharm. Res., 2011, 10, 489–496 CAS.
- R. Miri, K. Javidnia, Z. Amirghofran, S. H. Salimi, Z. Sabetghadam, S. Meilia and A. R. Mehdipour, Cytotoxic Effect of Some 1,4-Dihydropyridine Derivatives Containing Nitroimidazole Moiety, Iran. J. Pharm. Res., 2011, 10, 497–503 CAS.
- M. Vosooghi, H. Yahyavi, K. Divsalar, H. Shamsa, A. Kheirollahi, M. Safavi, S. K. Ardestani, S. Sadeghi-Neshat, N. Mohammadhosseini, N. Edraki, M. Khoshneviszadeh, A. Shafiee and A. Foroumadi, Synthesis and in vitro cytotoxic activity evaluation of (E)-16-(substituted benzylidene) derivatives of dehydroepiandrosterone, Daru, J. Pharm. Sci., 2013, 21, 34 CrossRef CAS PubMed.
- B. Lamia, A. Chakchouk-Mtibaa, B. Hallouma, S. Yasser, I. Ozdemir, L. Mansour, L. Mellouli and N. Hamdi, Molecules, 2017, 22(3), 420 CrossRef PubMed.
- N. Touj, A. Chakchouk-Mtibaa, L. Mansour, A. H. Harrath, J. H. Al-Tamimi, I. Özdemir, L. Mellouli, S. Yaşar and N. Hamdi, J. Organomet. Chem., 2017, 853, 49–63 CrossRef CAS.
- N. Touj, I. Özdemir, S. Yasar and N. Hamdi, Inorganica Chim. Acta, 2017, 467, 21–32 CrossRef CAS.
- N. Touj, N. Gürbüz, N. Hamdi, S. Yaşar and İ. Özdemir, Palladium PEPPSI complexes: Synthesis and catalytic activity on the Suzuki-Miyaura coupling reactions for aryl bromides at room temperature in aqueous media, Inorganica Chim. Acta, 2018, 478, 187–194 CrossRef CAS.
- L. Boubakri, L. Mansour, A. H Harrath, I. Özdemir, S. Yaşar and N. Hamdi, N-heterocyclic carbene-Pd (II)-PPh3 complexes a new highly efficient catalyst system for the Sonogashira cross-coupling reaction: Synthesis, characterization and biological activities, J. Coord. Chem., 2018, 71, 183–199 CrossRef CAS.
- E. Maerten, M. Sauthier, A. Mortreux and Y. Castanet, Tetrahedron, 2007, 63, 682–689 CrossRef CAS.
- A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef PubMed.
- D.-J. Chen, Z.-Y. Zhou, Q. Wang, D.-M. Xiang, N. Tian and S.-G. Sun, Chem. Commun, 2010, 46, 4252–4254 RSC.
- D. S. McGuinness and K. J. Cavell, Organometallics, 2000, 19, 741 CrossRef CAS.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- A. Zonouzi, R. Mirzazadeh, M. Safavi, S. Kabudanian Ardestani, S. Emami and A. Foroumadi, Iran. J. Pharm. Res., 2013, 12(4), 679–685 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08897g |
| This journal is © The Royal Society of Chemistry 2018 |