
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient photoelectrochemical water oxidation using a TiO2 nanosphere-decorated BiVO4 heterojunction photoanode †
Wenchao Jiang,
Yi Jiang *,
Jing Tong,
Qian Zhang,
Siyuan Li,
Haili Tong and
Lixin Xia*
*,
Jing Tong,
Qian Zhang,
Siyuan Li,
Haili Tong and
Lixin Xia*
College of Chemistry, Liaoning University, Shenyang 110036, Liaoning, China. E-mail: jiangyi@lnu.edu.cn; lixinxia@lnu.edu.cn
First published on 12th December 2018
Abstract
Constructing heterojunctions by coupling dissimilar semiconductors is a promising approach to boost charge separation and charge transfer in photoelectrochemical (PEC) water splitting. In this work, we fabricated a highly efficient TiO2/BiVO4 heterojunction photoanode for PEC water oxidation via a simple hydrothermal method. The resulting heterojunction photoanodes show enhanced PEC performance compared to the bare BiVO4 due to the simultaneous improvements in charge separation and charge transfer. Under simulated sunlight illumination (AM 1.5G, 100 mW cm−2), a high photocurrent of 3.3 mA cm−2 was obtained at 1.23 V (vs. the reversible hydrogen electrode (RHE)) in a neutral solution, which exceeds those attained by the previously reported TiO2/BiVO4 heterojunctions. When a molecular Co–cubane catalyst was immobilized onto the electrode, the performance of the TiO2/BiVO4 heterojunction photoanode can be further improved, achieving a higher photocurrent density of 4.6 mA cm−2 at 1.23 V, an almost three-fold enhancement over that of the bare BiVO4. These results engender a promising route to designing an efficient photoelectrode for PEC water splitting.
Introduction
With gradual, increasing consumption resulting in the depletion of traditional fossil fuels, new forms of energy are being investigated. Sunlight has attracted more interest as it is the most abundant renewable energy source.1–3 Solar energy, a new type of renewable energy, has the advantages of being a clean and inexhaustible source of energy and is attracting research interest. Splitting water into hydrogen and oxygen using sunlight, via photoelectrochemical (PEC) devices, is a promising method of converting and storing solar energy as a chemical fuel.2–5 To date, an enormous effort has been invested in this field, however, the development of a highly efficient PEC water splitting cell for solar-to-hydrogen fuel conversion remains a challenge.6,7 And the PEC water splitting using semiconductors has received increasing attention, owing to the semiconductors have excellent light absorption characteristics, for example, bismuth vanadate (BiVO4) and TiO2. BiVO4 has appropriate band-edge position (2.4 eV) and a wide range of absorption of sunlight.8,9 However, the unmodified BiVO4 has significant electron–hole recombination and poor water oxidation kinetics.10 Various strategies have been implemented to address these restrictions, such as nanostructured control,11 elemental doping,12,13 loading cocatalysts such as Co–Pi,14–16 FeOOH17 and NiOOH,10,18 construction of heterojunction,19,20 and plasmonic enhancement.21–23In principle, constructing heterojunctions is the most effective and direct way to promote the efficiency of charge-separation in photoelectrodes. Many studies have investigated BiVO4-based heterojunction photoanodes, such as WO3/BiVO4,24–26 TiO2/BiVO4,27–29 etc. These composites have been demonstrated to have higher PEC performance compared to BiVO4. TiO2 has been intensively studied as a candidate photoanode due to its favourable band-edge positions, high resistance to photocorrosion, physical and chemical stability, nontoxicity and low cost.30,31 Recent studies have reported an improvement in PEC water oxidation using TiO2/BiVO4 heterojunctions.32 However, a high photocurrent density for this heterojunction electrode is scarce.
In this study, a facile hydrothermal method using modified nanoporous BiVO4 photoanodes with TiO2 nanospheres was proposed to develop TiO2/BiVO4 heterojunction photoanodes. The BiVO4 layers in this photoanode absorb sunlight and the TiO2 nanospheres facilitate the separation and transmission of the photogenerated charge in BiVO4. The shift of TiO2 and BiVO4 interfacial Fermi levels result in the formation of an n–n junction at the interface,33 promoting electron–hole separation. The proposed TiO2/BiVO4 heterojunction photoanodes had good PEC performance. A high photocurrent density of 3.3 mA cm−2 was achieved at 1.23 V, compared to a reversible hydrogen electrode (RHE) in a neutral electrolyte, which is much higher than that of BiVO4 and exceeds other TiO2/BiVO4 heterojunctions reported. To further optimise the kinetics of surface water oxidation of photoanodes, a molecular Co–cubane water oxidation catalyst (WOC) was immobilized on the electrode and an extremely high photocurrent density of 4.6 mA cm−2 was achieved at 1.23 V vs. RHE.
Experimental section
Fabricate of the BiVO4 photoanode
The BiVO4 electrodes were prepared by using an electrodeposition process as reported.10 Briefly, the electrodeposited solution was prepared as follow: 2.91 g Bi(NO3)3·5H2O (99.0%, AR) and 9.96 g KI (99.0%, AR) were dissolved in 150 mL deionized water respectively before its pH was adjusted to 1.7 by adding HNO3, then this solution was mixed with 20 mL of absolute ethanol contained 1.49 g p-benzoquinone (99%) by stirring for a few minutes vigorously. A three-electrode cell was used for electrodeposition, with an FTO as the working electrode, an Ag/AgCl (3.5 M KCl) reference, and a platinum counter electrode. A CHI660 was used for electrodeposition studies. Deposition was carried out potentiostatically at −0.1 V vs. Ag/AgCl for 3 min at 30 °C (equivalent to passing a total charge of 0.45C cm−2). All prepared films were rinsed by deionised water and blow-dried with nitrogen. 10 mL of a dimethyl sulfoxide (DMSO, AR) solution containing 1.06 g vanadyl acetylacetonate (VO(acac)2 98%) was placed on the BiOI electrode. Then the BiOI electrodes were annealed at 450 °C (ramping rate = 2 °C min−1) for 2 h. After annealing, the as deposited film was converted to crystalline BiVO4 and amorphous V2O5, and pure BiVO4 was obtained by dissolving the V2O5 in 1 M NaOH under stirring for several minutes.Fabricate of the TiO2/BiVO4 photoanode
The TiO2/BiVO4 electrode was developed using a hydrothermal method. The TiO2 nanospheres were formed using a hydrolysis reaction. 1 mL of TiCl4 solution was added to 50 mL deionized water, and centrifuged for 5 min at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm until the suspended precipitate was spun down into a pellet. The supernatant was removed and precipitate resuspended in deionized water. The TiO2 aqueous solution was subjected to ultrasonic treatment to promote the dispersion of TiO2 nanospheres throughout the solution. A BiVO4 electrode was immersed into the TiO2 aqueous solution for 10 min (10 min-TiO2/BiVO4), 30 min (30 min-TiO2/BiVO4) and 60 min (60 min-TiO2/BiVO4) at 60 °C. Finally, the electrode was washed using deionized water and then calcined at 500 °C for 2 h in air (2 °C min−1).
000 rpm until the suspended precipitate was spun down into a pellet. The supernatant was removed and precipitate resuspended in deionized water. The TiO2 aqueous solution was subjected to ultrasonic treatment to promote the dispersion of TiO2 nanospheres throughout the solution. A BiVO4 electrode was immersed into the TiO2 aqueous solution for 10 min (10 min-TiO2/BiVO4), 30 min (30 min-TiO2/BiVO4) and 60 min (60 min-TiO2/BiVO4) at 60 °C. Finally, the electrode was washed using deionized water and then calcined at 500 °C for 2 h in air (2 °C min−1).
Development of a TiO2 photoanode
A TiO2 electrode was prepared using a spin-coating method. TiO2 was deposited onto the FTO using spin coating at 1000 rpm for 20 s. The spin coating solution was the previously described solution in Section 2. After spin coating, the samples were annealed at 500 °C for 2 h with a temperature increase of 2 °C min−1.Synthesis of Co–cubane catalyst
The Co-catalyst was synthesised as below: Co(NO3)2·6H2O (2.90 g, 10 mmol) and CH3COONa·3H2O (2.70 g, 20 mmol) added in 30 mL methanol and heated to refluxing temperature, then 4-cyanopyridine (10 mmol) was added to the reaction mixture. 30% hydrogen peroxide (5 mL) was slowly added into the reaction mixture, which was held at reflux for a further 4 h. The reaction mixture was cooled and concentrated in a rotary evaporator, and the aqueous layer was separated by adding CH2Cl2. The light pink aqueous layer was discarded, and the CH2Cl2 layer was dried using anhydrous Na2SO4. An olive-green compound precipitated out on addition of petroleum ether. Fig S1† is the structure of the Co–cubane. Fig S2† is the 1H-NMR of the catalyst. 1H-NMR (500 MHz, CD3OD): 8.65 (d, 8H), 7.62 (d, 8H), 2.2 (d, 12H).Preparation of the Co–cubane/TiO2/BiVO4 photoanode
The catalyst solution is prepared by dissolving the Co–cubane catalyst into solution (CH3OH/CH2Cl2/Nafion mixture (7:2.5:0.5, v/v)) at a concentration of 1 mM. A 10 μL of the cobalt catalyst was then cast onto the surface of the TiO2/BiVO4 electrode and dried at room temperature.
Photoelectrochemical measurements
The PEC performance of the BiVO4 and TiO2/BiVO4 electrodes were tested in 0.1 M PBS solution and AM 1.5 G simulated sunlight illumination (100 mW cm−2). The active surface area of the BiVO4 and TiO2/BiVO4 heterojunctions were 1 cm2. A CHI660 was used for electrochemical PEC measurements which contained samples as the working electrode, an Ag/AgCl (3.5 M KCl) reference and a platinum counter electrode. EIS measurements was carried out at 0.6 V versus Ag/AgCl. Mott–Schottky measurements were performed under dark conditions, in the potential range −0.6 to +1.0 V versus Ag/AgCl with 500 Hz. All the potentials are presented against the reversible hydrogen electrode (RHE), and the conversion between the Ag/AgCl and RHE was performed using the equation:| ERHE = E Ag/AgCl + 0.059 pH + 0.197 V |
Incident photon-to-current efficiency (IPCE) at each wavelength was determined using illumination from a 300 W Xe arc lamp and neutral density filters, to imitate the light from the sun. Monochromatic light was produced using an Oriel Cornerstone 130 monochromator with a 10 nm bandpass, and the output was measured using a photodiode detector. IPCE was measured at 0.6 V vs. RHE using the same three-electrode setup described above for the photocurrent measurements.
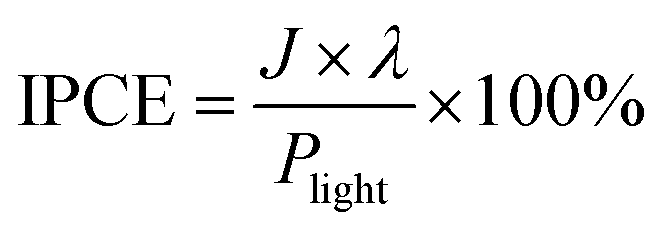
The applied bias photon-to-current efficiency (ABPE) was calculated from a J–V curve, where: J is the photocurrent density, Vbias is the applied bias and Pin is the incident illumination power density (AM 1.5 G, 100 mW cm−2):
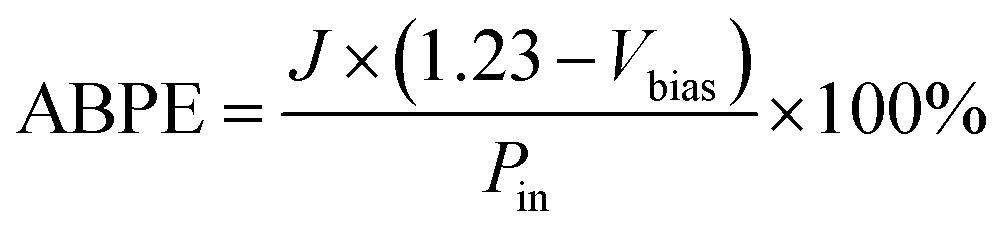
The ηtr was calculated using the equation:
| ηtr = Jwater/JNa2SO3 |
Results and discussion
A porous BiVO4 was produced using the previously reported method.10 Fig. 1a shows the microstructure of the porous BiVO4; the significant voids can be seen clearly. The porous structure of the BiVO4 enables sites for the TiO2 nanospheres to attach. TiO2 nanospheres, with a range of diameter size 200 to 500 nm (illustrated by DLS in Fig S3†) were attached onto the BiVO4 electrodes using a hydrothermal method. The SEMs of TiO2/BiVO4 electrodes with different hydrothermal times are shown in Fig. 1b–d, which show that the TiO2 nanospheres were attached onto the BiVO4 successfully and the quantity of TiO2 nanospheres increased with immersed time. The elemental mapping results shown in Fig. 1e–h suggest the existence of O, Ti, Bi, and V elements, indicating a uniform distribution of TiO2 nanospheres across the structure.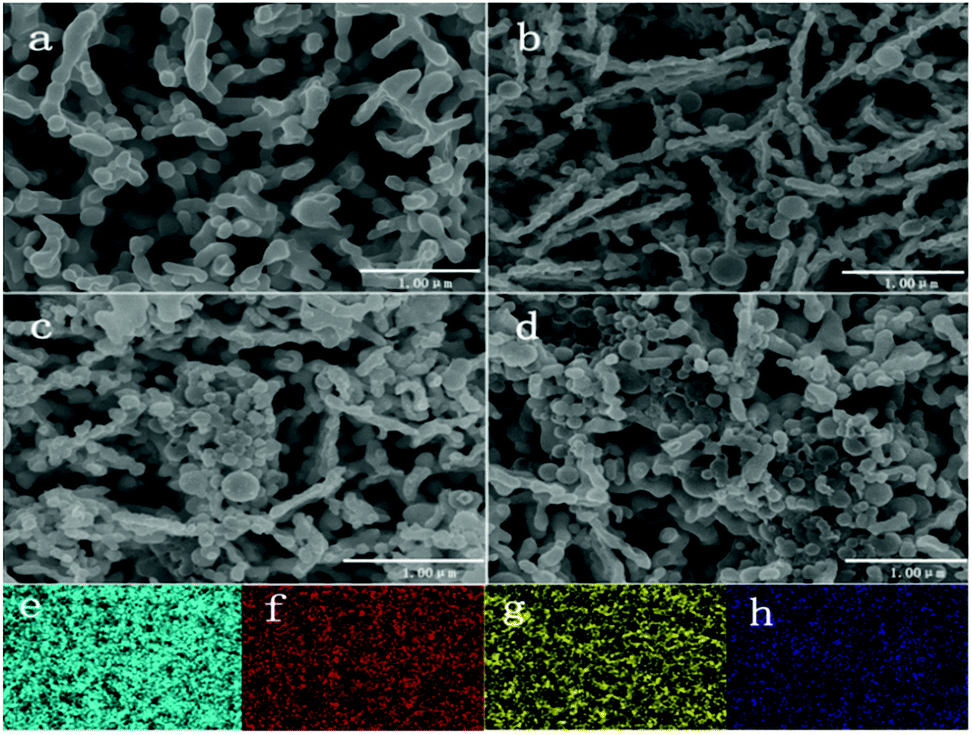 | ||
| Fig. 1 SEMs of different electrodes. (a) BiVO4, (b) 10 min-TiO2/BiVO4, (c) 30 min-TiO2/BiVO4, (d) 60 min-TiO2/BiVO4, and EDX-mapping of TiO2/BiVO4 are shown (e) Bi, (f) V, (g) O, (h) Ti. | ||
The crystalline structures of the FTO, BiVO4 and TiO2/BiVO4 samples were characterised using X-ray diffraction (XRD) patterns, as shown in Fig S4,† further confirming that TiO2 attached to the porous BiVO4 and that the crystalline structures of TiO2 are anatase.
In order to gain more insight into TiO2/BiVO4 heterojunctions, X-ray photoelectron spectroscopy (XPS) characterisation was performed, which showed signals from Ti, O, Bi and V elements (Fig. S5†). The HR-XPS spectra of the TiO2/BiVO4 are shown in Fig. 2. Two binding energy peaks for a typical Bi element in BiVO4 occurred at 158.9 and 164.2 eV, corresponding to the Bi 4f7/2 and Bi 4f5/2, suggesting that Bi is present as Bi3+ (Fig. 2a).34 The binding energies of the vanadium(V) peaks were centred at 516.5 and 524.5 eV, corresponding to V 2p3/2 and V 2p1/2 orbits, respectively, characteristic of the presence of the V5+ oxidation state (Fig. 2b).35 Two peaks for Ti 2p at 458.7 and 465.7 eV were assigned to Ti 2p3/2 and Ti 2p1/2, respectively, suggesting a Ti4+ oxidation state (Fig. 2c).36 The peak at binding energy of 529.8 eV in Fig. 2d, indicates that oxygen was present as surface lattice oxygen and free oxygen.37
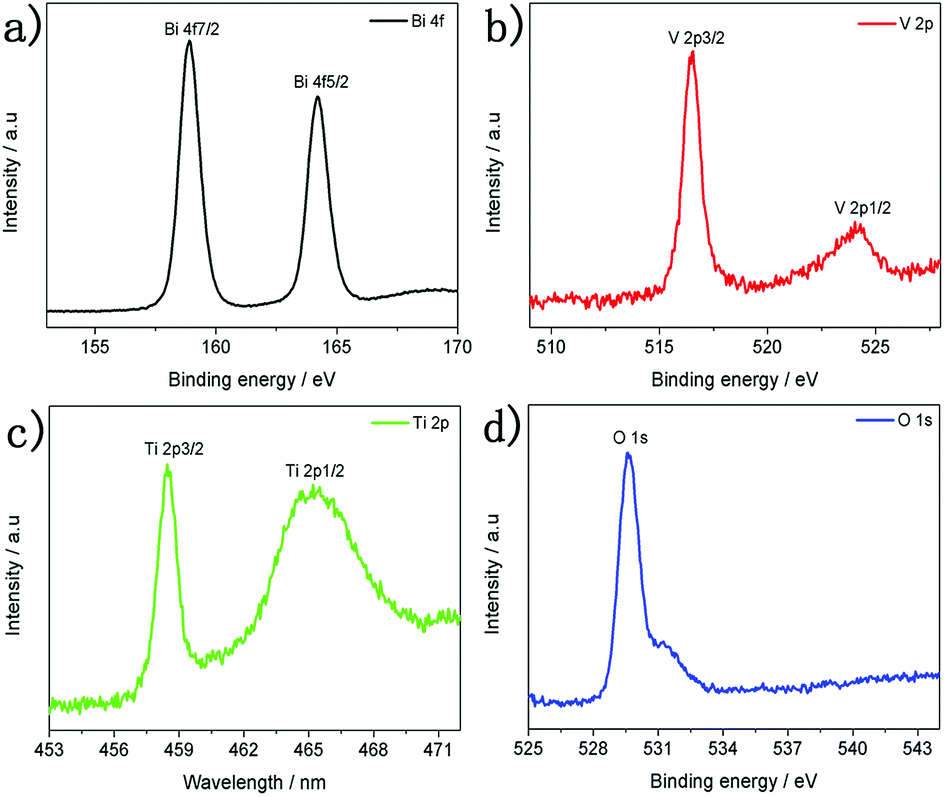 | ||
| Fig. 2 XPS spectra of the TiO2/BiVO4 electrode (a) Bi 4f, (b) V 2p, (c) Ti 2p, (d) O 1s. | ||
The PEC performance of the BiVO4 and TiO2/BiVO4 was measured using a three electrode system which included the proposed electrode as the working electrode, an Ag/AgCl (3.5 M KCl) reference and a platinum counter electrode. Fig. 3a shows the linear sweep voltammetry (LSV) curves of the BiVO4 and TiO2/BiVO4 electrodes. In the dark those electrodes show little photocurrent density. However, in the light, the differences in the TiO2 on the BiVO4 electrode resulted in a different photocurrent density, and TiO2/BiVO4 electrodes had a significantly enhanced photocurrent density compared to BiVO4 and TiO2 electrodes. The 30 min-TiO2/BiVO4 electrode had the highest photocurrent density, with a value of 3.3 mA cm−2 at 1.23 V vs. RHE, which was 2.5 times that of BiVO4. However, the 60 min-TiO2/BiVO4 electrode had lower photocurrent density compared to 30 min-TiO2/BiVO4, which may be a result of TiO2 hindering charge transfer. A significant cathodic shift of onset potential was observed. For the 30 min-TiO2/BiVO4 electrode, the potential at 0.5 mA cm2 was reduced greater than 500 mV, indicating an efficient charge separation or charge transfer originating from the formation of a heterojunction.
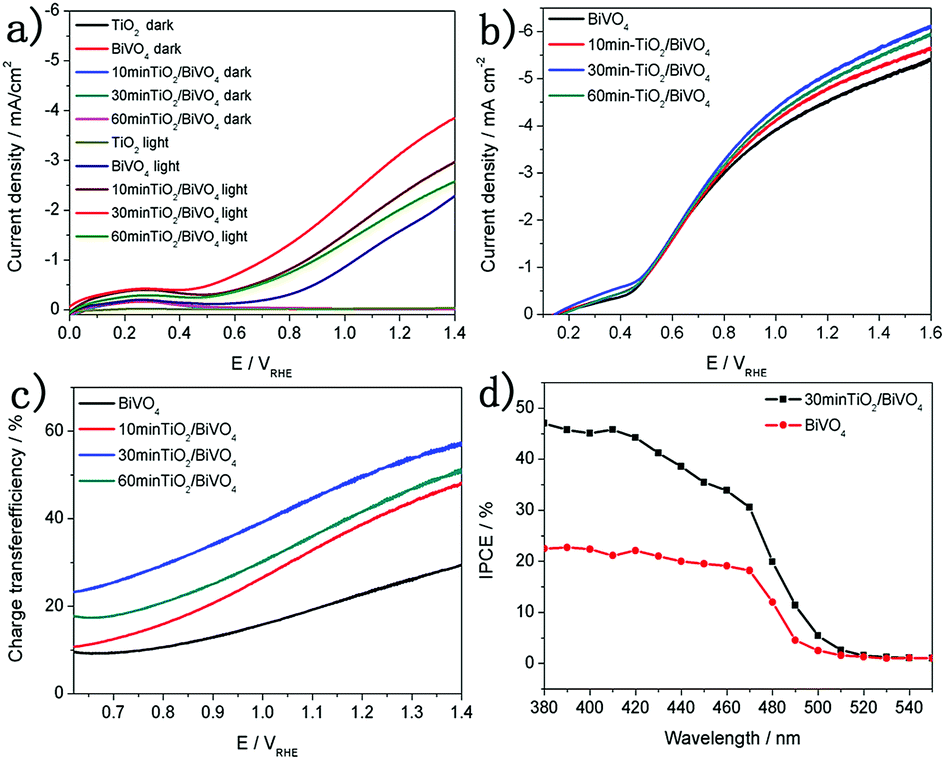 | ||
| Fig. 3 (a) LSV curves of different TiO2/BiVO4 electrodes. (b) LSV of curves BiVO4 and TiO2/BiVO4 in 0.1 M PBS containing 0.1 M Na2SO3. (c) The charge transfer efficiencies of different TiO2 content on BiVO4. (d) IPCEs of BiVO4 and TiO2/BiVO4 electrodes. | ||
To test the charge recombination behaviour of the TiO2/BiVO4 electrodes, 0.1 M Na2SO3 solution was added to the electrolyte as a hole scavenger for the PEC measurements. The LSV curves of TiO2/BiVO4 electrodes are shown in Fig. 3b. TiO2/BiVO4 electrodes show higher photocurrent density compared to the BiVO4 electrode, demonstrating a heterojunction was formed and better charge separation occurred. The 30 min-TiO2/BiVO4 electrode had the highest photocurrent density, 5.4 mA cm−2 at 1.23 V vs. RHE. The photocurrent can be described as: J = Jabs × ηsep × ηtr38 the ηr values can be calculated as Jwater/JNa2SO3. As shown in Fig. 3c, all TiO2/BiVO4 electrodes had improved ηtr compared to the BiVO4 electrode, over the range tested in this study. The 30 min-TiO2/BiVO4 electrode had the highest ηtr, 60% at 1.23 V vs. RHE, 3 times that of the BiVO4 electrode. These results demonstrate that the construction of a TiO2/BiVO4 heterojunction could promote charge separation and charge transfer efficiencies. The incident-photon-to-current-conversion efficiency (IPCE) values are shown in Fig. 3d. The TiO2/BiVO4 was higher than BiVO4 across the visible spectrum and at 520 nm the two electrodes reached 0, which is in agreement with the light absorbance of BiVO4 and TiO2/BiVO4 electrodes (Fig S6†).
The photocurrent transient behaviours of the TiO2/BiVO4 electrodes were measured in 0.1 M PBS. A photocurrent spike was obtained using sudden illumination due to capacitive charging of the interface, and the spike decay due to the recombination of the electrons and holes that is associated with holes getting trapped at the electrode surface. A stabilised photocurrent was achieved when the charge generation and recombination rates were equal. Furthermore, the recombination rate could be reflected by longer transient decay times, which can be analysed on a logarithmic plot of parameter D, using the following equation:39
| D = (It − Is)/(Im − Is) |
D = −1. Fig. 4a shows the logarithmic plots of parameter D of the BiVO4 and TiO2/BiVO4 electrodes. The transient decay time for TiO2/BiVO4 was 4.1 s, longer than that of BiVO4 (1.5 s), suggesting a lower rate of electron and hole recombination in TiO2/BiVO4, which could be attributed to the TiO2 nanospheres acting as hole resistors, resulting in increased efficiency of charge separation and prolonging the lifetime of the holes. As a result, abundant photogenerated charge carriers in the TiO2/BiVO4 electrode were effectively collected, leading to an improved PEC performance.
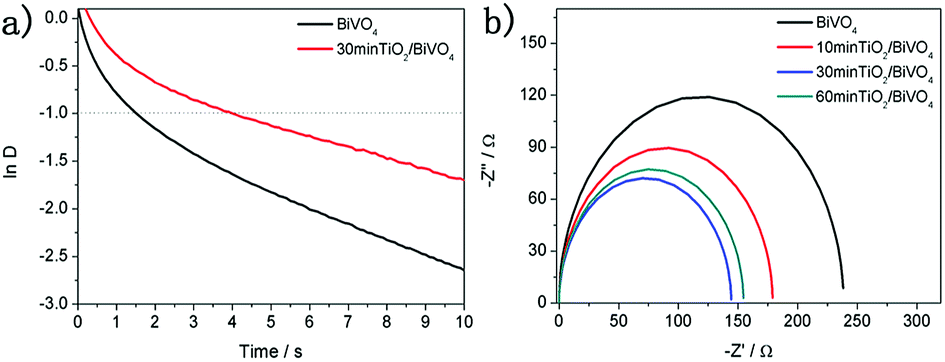 | ||
| Fig. 4 (a) The logarithmic plots of the parameter D of the BiVO4 and TiO2/BiVO4 electrodes. (b) Electrochemical impedance spectroscopy (EIS) of the TiO2 under different soaking times. | ||
To characterise the kinetics of the charge transfer process of the TiO2/BiVO4 and BiVO4 electrodes, electrochemical impedance spectroscopy (EIS) tests were performed at 1.23 V vs. RHE in 0.1 M PBS under simulated solar light illumination. Fig. 4b shows Nyquist at frequencies from 1 Hz to 100 kHz. The BiVO4 electrode had the greatest resistance to charge transfer and the 30 min-TiO2/BiVO4 had the lowest charge transfer resistance. However, the 60 min-TiO2/BiVO4 electrode had a lower charge transfer resistance, which may be a result of the TiO2 impeding the transfer of surface charge, further indicating the optimum amount of TiO2 was achieved in the 30 min-TiO2/BiVO4 electrode. The above results suggest that the TiO2 facilitates the BiVO4 electrode charge separation and transfer.
In order to further investigate the PEC performance, a molecular Co–cubane water oxidation catalyst was immobilised onto the TiO2/BiVO4 electrode. As the LSV curves show in Fig. 5a, the Co–cubane/TiO2/BiVO4 photoanode exhibits a high photocurrent density of 4.6 mA cm−2 at 1.23 V vs. RHE, 3.1 times higher than that of BiVO4, indicating that the Co–cubane was acting as an efficient OER catalyst, further enhancing the charge transfer. Furthermore, after 400 s photoelectrolysis, the photocurrent density of Co–cubane/TiO2/BiVO4 could maintain more than 2.6 mA cm−2, which is 2.6 times than that of the bare BiVO4 (Fig S7†). The half-cell photoconversion efficiencies of BiVO4 and Co–cubane/TiO2/BiVO4 electrodes were calculated using the LSV results, (Fig. 5b). The maximum value of Co–cubane/TiO2/BiVO4 achieved 1.48% at 0.72 V, approximately 6 fold compared to BiVO4. The XPS spectra of the Co–cubane/TiO2/BiVO4 electrode was shown in Fig S8,† which indicated the element of Co was exited.
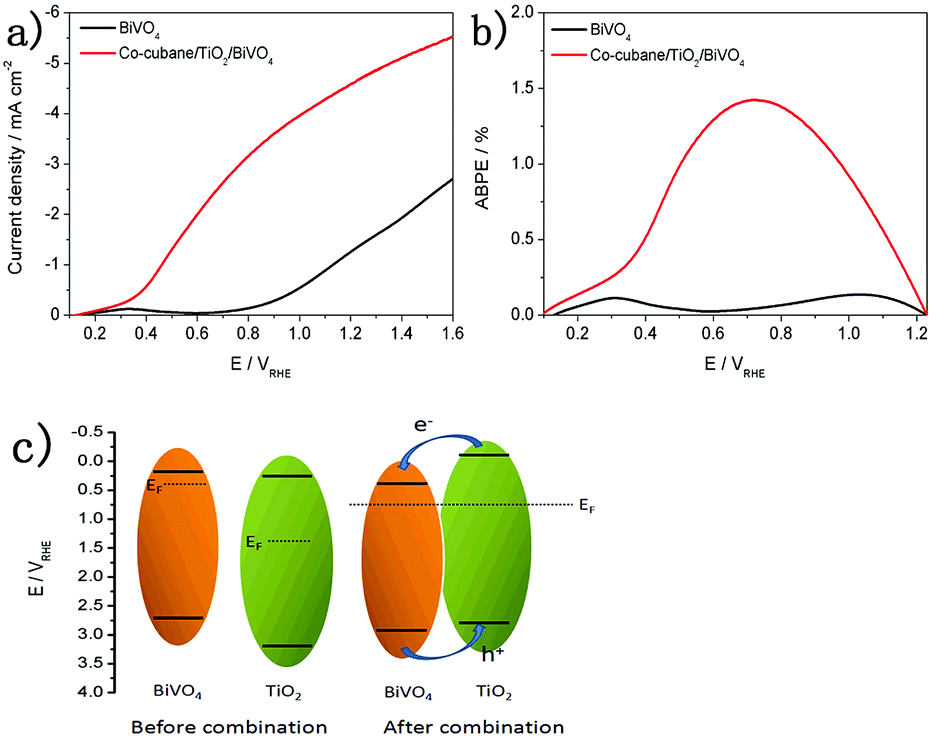 | ||
| Fig. 5 (a) LSV curves of BiVO4 and Co–cubane/TiO2/BiVO4 electrodes. (b) ABPEs of BiVO4 and Co–cubane/TiO2/BiVO4 electrodes. (c) The charge transfer mechanism of TiO2/BiVO4 electrode. | ||
To explore the charge transfer mechanism between the BiVO4 and TiO2, Mott–Schottky plots of the BiVO4 and TiO2 were performed under dark conditions.40 In Fig S9b,† the linear part of Mott–Schottky plots of TiO2/BiVO4 rather than BiVO4 show the gentlest slope and the more negative flatband potential. The gentle slope means the modifying with TiO2 can obviously increase the density of the carriers.41 From Fig S9a and c,† the CB position of BiVO4 and TiO2 were 0.12 eV and 0.2 eV. And from Fig S10,† the band gap energies of BiVO4 and TiO2 were 2.42 eV and 3.09 eV. Therefore, the VB position of BiVO4 and TiO2 were 2.54 eV and 3.29 eV, calculated using ECB = EVB − Eg. Compared the BiVO4, the TiO2 approach to the intrinsic semiconductor, So we suppose the charge transfer mechanism shown the Fig. 5c, When these two types of semiconductor materials are closely joined together, the heterojunction structure was formed. At this moment, the two semiconductors have a uniform Fermi level and the system is in equilibrium. An electron transfer from TiO2 to BiVO4 possibly occurred, providing evidence for the efficient charge transfer between BiVO4 and TiO2.
Conclusions
In summary, a simple hydrothermal method was proposed to produce a TiO2/BiVO4 heterojunction electrode. The proposed TiO2/BiVO4 electrode provided a high performance for the oxidation of PEC water. The TiO2/BiVO4 heterojunction results indicate good charge separation and transfer efficiency. When a molecular Co–cubane catalyst was attached onto the TiO2/BiVO4 electrode, a further improvement in the PEC performance was achieved, with a photocurrent of photocurrent density of 4.6 mA cm−2 at 1.23 V, more than 3 times for BiVO4 alone. This study provides a simple and effective approach in the production of heterojunction photoelectrodes for increasing the performance of PEC, which could be used to design efficient photocatalytic systems in future studies.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21401092, 21671089, 21773100), the Shenyang Natural Science Foundation of China (F16-103-4-00), Scientific Research Found of Liaoning Province (LT2017010, 20170540409, 20180510011).References
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- O. Khaselev, Science, 1998, 280, 425–427 CrossRef CAS.
- Z. Li, W. Luo, M. Zhang, J. Feng and Z. Zou, Energy Environ. Sci., 2013, 6, 347–370 RSC.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- K. O. Akihiko Kudo and H. Kato, J. Am. Chem. Soc., 1999, 121, 11459–11467 CrossRef.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- Z. Yin, Z. Wang, Y. Du, X. Qi, Y. Huang, C. Xue and H. Zhang, Adv. Mater., 2012, 24, 5374–5378 CrossRef CAS PubMed.
- H. K. Saimi Tokunaga and A. Kudo, Chem. Mater., 2001, 13, 4624–4628 CrossRef.
- M. Zhong, T. Hisatomi, T. Minegishi, H. Nishiyama, M. Katayama, T. Yamada and K. Domen, J. Master. Chem. A, 2016, 4, 9858–9864 RSC.
- T. W. Kim and K. S. Choi, Science, 2014, 343, 990–994 CrossRef CAS.
- G. Wang, Y. Ling, X. Lu, F. Qian, Y. Tong, J. Z. Zhang, V. Lordi, C. Rocha Leao and Y. Li, J. Phys. Chem. C, 2013, 117, 10957–10964 CrossRef CAS.
- Y. Park, D. Kang and K. S. Choi, Phys. Chem. Chem. Phys., 2014, 16, 1238–1246 RSC.
- M. Huang, J. Bian, W. Xiong, C. Huang and R. Zhang, J. Master. Chem. A, 2018, 6, 3602–3609 RSC.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert and T. W. Hamann, J. Am. Chem. Soc., 2012, 134, 16693–16700 CrossRef CAS.
- C. Zachaus, F. F. Abdi, L. M. Peter and R. van de Krol, Chem. Sci., 2017, 8, 3712–3719 RSC.
- F. F. Abdi and R. van de Krol, J. Phys. Chem. C, 2012, 116, 9398–9404 CrossRef CAS.
- B. Zhang, L. Wang, Y. Zhang, Y. Ding and Y. Bi, Angew. Chem., Int. Ed. Engl., 2018, 57, 2248–2252 CrossRef CAS PubMed.
- H. Zhang, Y. Yu, L. Zhang and S. Dong, Angew. Chem., Int. Ed. Engl., 2018, 57, 1547–1551 CrossRef CAS PubMed.
- S. Gu, W. Li, F. Wang, S. Wang, H. Zhou and H. Li, Appl. Catal., B, 2015, 170–171, 186–194 CrossRef CAS.
- A. P. Singh, N. Kodan, B. R. Mehta, A. Held, L. Mayrhofer and M. Moseler, ACS Catal., 2016, 6, 5311–5318 CrossRef CAS.
- S.-W. Cao, Z. Yin, J. Barber, F. Y. C. Boey, S. C. J. Loo and C. Xue, ACS Appl. Mater. Interfaces, 2011, 4, 418–423 CrossRef PubMed.
- L. Zhang, L. O. Herrmann and J. J. Baumberg, Sci. Rep., 2015, 5, 16660 CrossRef CAS PubMed.
- H. Hirakawa, S. Shiota, Y. Shiraishi, H. Sakamoto, S. Ichikawa and T. Hirai, ACS Catal., 2016, 6, 4976–4982 CrossRef CAS.
- J. Su, L. Guo, N. Bao and C. A. Grimes, Nano Lett., 2011, 11, 1928–1933 CrossRef CAS.
- P. Chatchai, Y. Murakami, S.-y. Kishioka, A. Y. Nosaka and Y. Nosaka, Electrochim. Acta, 2009, 54, 1147–1152 CrossRef CAS.
- Y. Pihosh, I. Turkevych, K. Mawatari, T. Asai, T. Hisatomi, J. Uemura, M. Tosa, K. Shimamura, J. Kubota, K. Domen and T. Kitamori, Small, 2014, 10, 3692–3699 CrossRef CAS.
- S. Ho-Kimura, S. J. A. Moniz, A. D. Handoko and J. Tang, J. Master. Chem. A, 2014, 2, 3948–3953 RSC.
- H. Zhang and C. Cheng, ACS Energy Lett., 2017, 2, 813–821 CrossRef CAS.
- M. Xie, X. Fu, L. Jing, P. Luan, Y. Feng and H. Fu, Adv. Energy Mater., 2014, 4, 1300995 CrossRef.
- J. S. Yang, W. P. Liao and J. J. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 7425–7431 CrossRef CAS.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- D. Lee, A. Kvit and K.-S. Choi, Chem. Mater., 2018, 30, 4704–4712 CrossRef CAS.
- S. S. Kalanur, I.-H. Yoo, J. Park and H. Seo, J. Master. Chem. A, 2017, 5, 1455–1461 RSC.
- L. Dong, S. Guo, S. Zhu, D. Xu, L. Zhang, M. Huo and X. Yang, Catal. Commun., 2011, 16, 250–254 CrossRef CAS.
- Y. Guo, X. Yang, F. Ma, K. Li, L. Xu, X. Yuan and Y. Guo, Appl. Surf. Sci., 2010, 256, 2215–2222 CrossRef CAS.
- N. Myung, S. Ham, S. Choi, Y. Chae, W.-G. Kim, Y. J. Jeon, K.-J. Paeng, W. Chanmanee, N. R. de Tacconi and K. Rajeshwar, J. Phys. Chem. C, 2011, 115, 7793–7800 CrossRef CAS.
- N. J. Bell, Y. H. Ng, A. Du, H. Coster, S. C. Smith and R. Amal, J. Phys. Chem. C, 2011, 115, 6004–6009 CrossRef CAS.
- D. K. Zhong, S. Choi and D. R. Gamelin, J. Am. Chem. Soc., 2011, 133, 18370–18377 CrossRef CAS PubMed.
- A. L. Hagfeldt, H. Lindström, S. Soedergren and S.-E. Lindquist, J. Electroanal. Chem., 1995, 381, 39–46 CrossRef.
- J. A. T. G. Cooper and A. J. Nozik, J. Electrochem. Soc., 1982, 129, 1973–1977 CrossRef.
- F. Ning, M. Shao, S. Xu, Y. Fu, R. Zhang, M. Wei, D. G. Evans and X. Duan, Energy Environ. Sci., 2016, 9, 2633 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09072f |
| This journal is © The Royal Society of Chemistry 2018 |