
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First use of a divalent lanthanide for visible-light-promoted photoredox catalysis†
Tyler C.
Jenks‡
 ,
Matthew D.
Bailey‡
,
Jessica L.
Hovey
,
Shanilke
Fernando
,
Gihan
Basnayake
,
Michael E.
Cross
,
Wen
Li
and
Matthew J.
Allen
*
,
Matthew D.
Bailey‡
,
Jessica L.
Hovey
,
Shanilke
Fernando
,
Gihan
Basnayake
,
Michael E.
Cross
,
Wen
Li
and
Matthew J.
Allen
*
Department of Chemistry, Wayne State University, 5101 Cass Avenue, Detroit, MI 48202, USA. E-mail: mallen@chem.wayne.edu
First published on 21st December 2017
Abstract
We report the first catalytic use of a divalent lanthanide in visible-light-promoted bond-forming reactions. Our new precatalyst uses europium in the +2 oxidation state and is active in the presence of blue light from light-emitting diodes. The use of low-energy visible light reduces the occurrence of potential side reactions that might be induced by higher-energy UV light. The system described here uses zinc metal as a sacrificial reductant and is tolerant to wet, protic solvents. The catalyst can be made in situ from relatively inexpensive and air-stable EuCl3·6H2O, and the ligand can be synthesized in large quantities in two steps. With 0.5% loading of precatalyst, an average of 120 turnovers was observed in six hours for reductive coupling of benzyl chloride. We expect that the results will initiate the study of visible-light-promoted photoredox catalysis using divalent europium in a variety of reactions.
Introduction
Metal-assisted photoredox catalysis uses light to promote the reactivity of metal-containing complexes in reactions such as halogen-atom abstractions, functional-group reductions, and carbon–carbon bond formations.1–3 Most reported metal-assisted photoredox systems rely on transition metals,2 with a small number of photoredox systems involving lanthanides that are either catalytic via the +3/+4 redox couple3–5 or noncatalytic starting from the +2 oxidation state.6–8 Among these metals, EuII is unique in that it is the mildest reducing agent of the divalent lanthanides. It can be handled in protic solvents including water; it can be produced from EuIII, which is inexpensive relative to second and third row transition metals commonly used in photoredox catalysis; and it undergoes metal–orbital-based electronic transitions that are not susceptible to photobleaching like organic dyes.9 Recently, we reported a luminescent, aqueous, EuII-containing complex that had a high quantum yield (26%) for a 5d–4f transition that occurred in the visible region of the electromagnetic spectrum using a ligand that can be prepared on large scale in two steps.10,11 We hypothesized that because this complex is luminescent and contains a redox-active metal, it could be employed in photoredox reactions with a sacrificial reducing agent to make the reaction catalytic in europium. Here, we report the first catalytic example of carbon–carbon bond formation using a europium-containing complex and visible light. Further, we evaluate the mechanism of the catalytic system.Results and discussion
Our photoredox system relies on azacryptand 1,4,7,10,13,16,21,24-octaazabicyclo[8.8.8]hexacosane, 1, to encapsulate EuII, inducing a bathochromic shift in the UV-visible absorption of EuII from the UV to the visible region of the electromagnetic spectrum (Fig. 1). This bathochromic shift arises from d-orbital splitting, caused by the nitrogen atoms of the cryptand, that results in a lower-energy 5d–4f transition relative to transitions induced by weaker field ligands.10b Upon absorption of blue light by EuII1, an electron is excited into an emissive state that has a luminescence lifetime of 0.98 ± 0.03 μs and a quantum yield of 37% in methanol. The quantum yield of EuII1 in methanol is 11% higher than the previously reported value for the same complex in a pH 12 aqueous solution,10a and the difference in the quantum yield is likely caused by the change of solvent. The luminescence lifetime of EuII1 is in the range of typical photoredox systems.12 Interestingly, both EuII and CeIII are known to be emissive through 5d–4f transitions with typical lifetimes on the order of 1 ns to 1 μs.3,7,13,14 This range of lifetimes for similar electronic transitions suggests that these lifetimes are largely dependent on ligand field and not necessarily intrinsic to the metal ions. The values for lifetime and quantum yield are toward the long and high end, respectively, of reports for solvated EuII.15,16 Due to the photophysical properties of EuII1, including the efficient conversion of visible light to a long-lived excited state, we hypothesized that EuII1 would be a good promoter of photoredox reactions.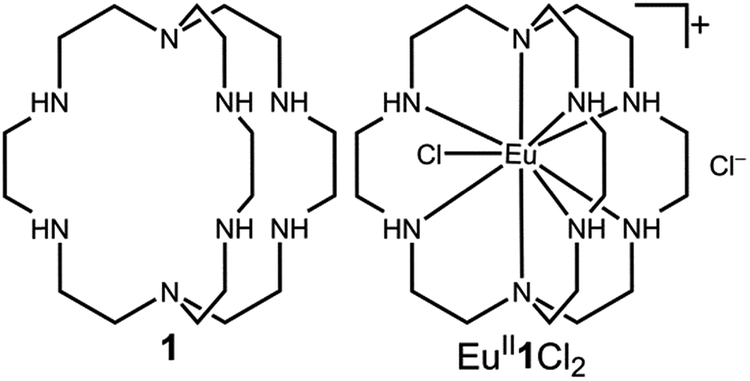 | ||
| Fig. 1 Structures of ligand 1 (left) and EuII1 (right). | ||
When a redox-active metal complex is excited to an emissive state, the E1/2 of the complex changes.1–6 To estimate the E1/2 of EuII1 in the emissive state, the excited-state potential 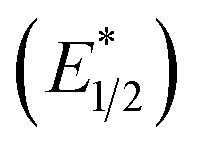 was calculated by means of the Rehm–Weller formalism (eqn (1)) using the ground-state potential (E1/2) and the energy of the emission band (E0,0), which is the energy of an electron in the excited state relative to the ground state as determined by the maximum emission wavelength (Fig. 2).17 There is an additional work-function term that has been omitted from eqn (1) because it was assumed to be negligibly small.4 To determine the ground-state potential of EuII1, cyclic voltammetry was performed with EuII1 in N,N-dimethylformamide. A reversible EuII/III1 couple was observed with an E1/2 of −0.90 V vs. Ag/AgCl, which represents a negative shift in the E1/2 potential relative to the solvated EuII/III couple, and the negative shift is consistent with other reported EuII complexes that contain nitrogen donors.18,19E0,0 was estimated to be 2.14 V by dividing the product of Planck's constant and the speed of light by the maximum emission wavelength (580 nm) in meters (hc/λ). Using these values for the ground-state potential and the emission-band energy, the
was calculated by means of the Rehm–Weller formalism (eqn (1)) using the ground-state potential (E1/2) and the energy of the emission band (E0,0), which is the energy of an electron in the excited state relative to the ground state as determined by the maximum emission wavelength (Fig. 2).17 There is an additional work-function term that has been omitted from eqn (1) because it was assumed to be negligibly small.4 To determine the ground-state potential of EuII1, cyclic voltammetry was performed with EuII1 in N,N-dimethylformamide. A reversible EuII/III1 couple was observed with an E1/2 of −0.90 V vs. Ag/AgCl, which represents a negative shift in the E1/2 potential relative to the solvated EuII/III couple, and the negative shift is consistent with other reported EuII complexes that contain nitrogen donors.18,19E0,0 was estimated to be 2.14 V by dividing the product of Planck's constant and the speed of light by the maximum emission wavelength (580 nm) in meters (hc/λ). Using these values for the ground-state potential and the emission-band energy, the 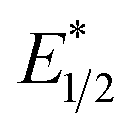 of EuII1 was calculated to be −3.0 V vs. Ag/AgCl. This calculated excited-state potential is among the most negative excited-state potentials reported to date for metal-based catalytic photoredox agents and is more negative than the potential of the potent reducing agent SmI2 in the presence of hexamethylphosphoramide.20,21 With a sense of the redox properties of EuII1 in hand, we were interested in probing the reactivity of EuII1. On the basis of a recent report from the Schelter group describing photocatalytic reductive couplings using a CeIII/IV system,3 we expected that EuII1 would display similar reactivity.
of EuII1 was calculated to be −3.0 V vs. Ag/AgCl. This calculated excited-state potential is among the most negative excited-state potentials reported to date for metal-based catalytic photoredox agents and is more negative than the potential of the potent reducing agent SmI2 in the presence of hexamethylphosphoramide.20,21 With a sense of the redox properties of EuII1 in hand, we were interested in probing the reactivity of EuII1. On the basis of a recent report from the Schelter group describing photocatalytic reductive couplings using a CeIII/IV system,3 we expected that EuII1 would display similar reactivity.
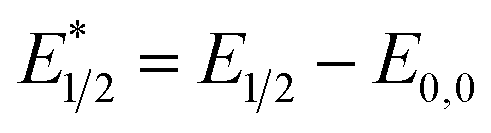 | (1) |
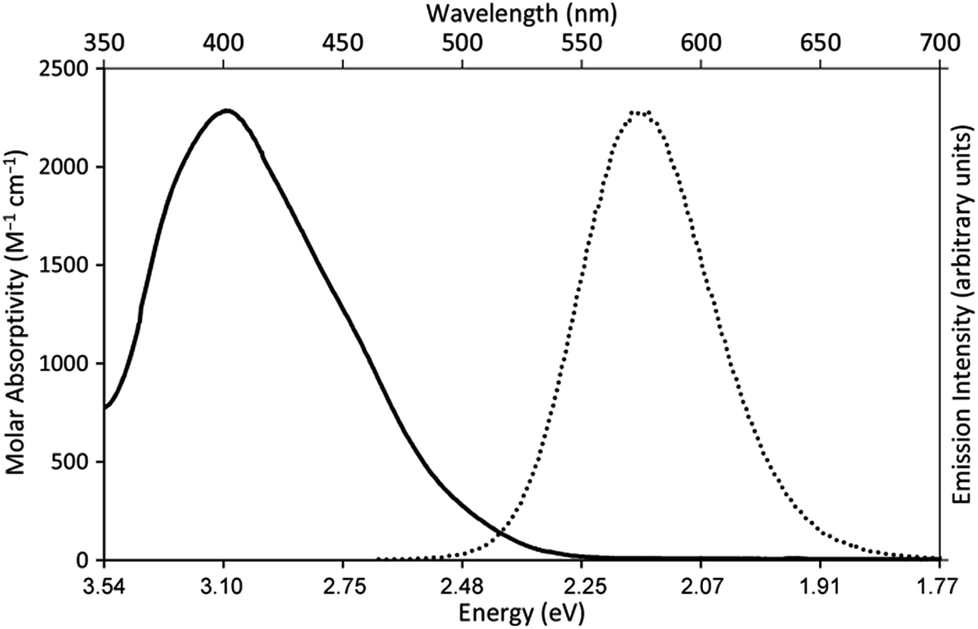 | ||
| Fig. 2 UV-visible absorption spectrum of EuII1Cl2 (—, left y-axis) and emission spectrum (λex = 460 nm, ε: 1044 M−1 cm−1) of EuII1Cl2 (••, right y-axis). Spectra were acquired in methanol. | ||
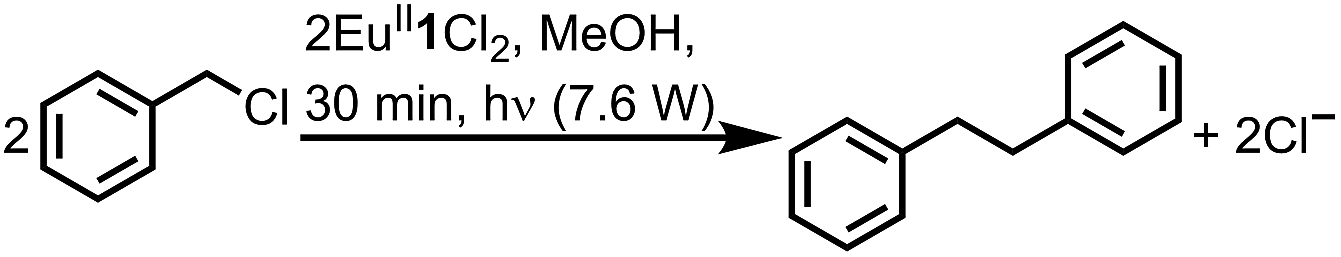
To study the reactivity of EuII1, we attempted to reductively couple alkyl halides to form carbon–carbon bonds. A solution containing EuCl2 (1 equiv.), 1 (1 equiv.), and benzyl chloride (1 equiv., 0.027 mmol) in methanol was illuminated with blue light (∼7.6 W, λem = 460 nm, Fig. S2†) using a strip of light-emitting diodes. We observed the formation of 1,2-diphenylethane (85 ± 2%) and toluene (4.7 ± 0.4%) within 30 minutes (Fig. 3A).22
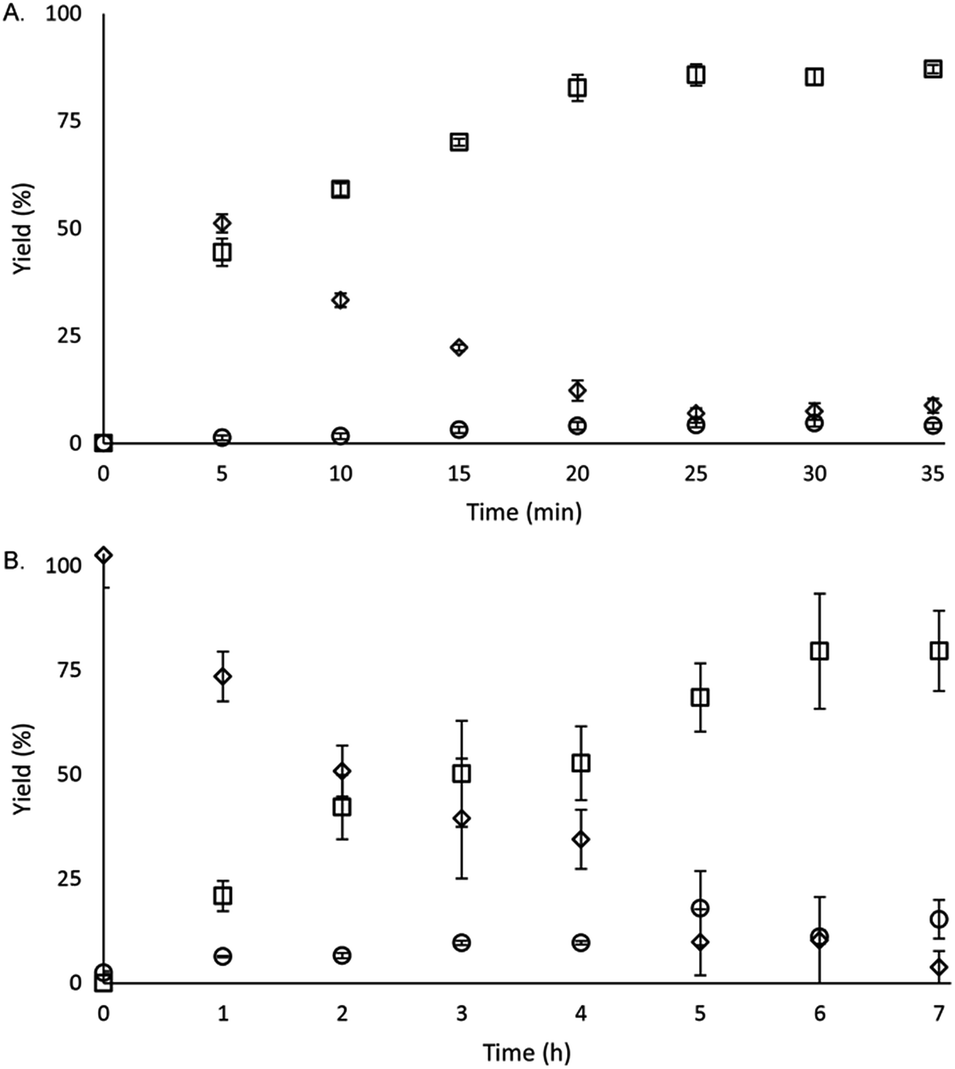 | ||
| Fig. 3 Formation of products and disappearance of starting material as a function of time for (A) stoichiometric and (B) catalytic (10 mol%) benzyl chloride coupling reactions (squares = 1,2-diphenylethane, diamonds = benzyl chloride, and circles = toluene). Each point is the mean of three independently prepared reactions, and the error bars represent the standard error of the means. | ||
To determine whether the reaction was promoted by the excited-state of EuII1, we performed three control reactions (Table 1). When the coupling of benzyl chloride was attempted in the absence of light, no product was observed. This observation indicated that for the reaction to proceed, light must be present, suggesting that the excited state of EuII1 was promoting the reaction and not the ground state of EuII1. When ligand 1 was omitted, no product was observed. This observation indicated that uncomplexed europium ions are incapable of performing the reductive coupling. When EuCl2 was omitted, no product was observed, indicating that europium is an active participant in the reduction of benzyl chloride. The control reactions demonstrate that light, ligand 1, and europium are all necessary to reduce benzyl chloride. To test for reactivity with methanol, fluorescence spectroscopy was performed before and after 12 h of light exposure on samples of EuII1 (Fig. S20†). Based on these studies, the excited state of EuII1 reacts with methanol, but no reaction with methanol was observed over the same time period in the dark. Despite the reactivity of the excited state of EuII1 with methanol, the observation of 1,2-diphenylethane in excellent yields in 30 min indicates that the reaction with methanol is relatively slow. To further understand how EuII1 promotes light-induced bond formation, we attempted to determine the mechanism of electron transfer.
The emissive state of EuII1 is responsible for the observed reactivity, and it is unlikely that energy transfer occurs between the emissive state of EuII1 and benzyl chloride as shown by the lack of spectral overlap between the absorption of benzyl chloride and the emission of EuII1; therefore, the reductive coupling of benzyl chloride must occur through a photoinduced electron transfer, which would be expected to quench luminescence. We sought to investigate the mechanism of photoinduced electron transfer using substrates to quench luminescence with Stern–Volmer analyses.23 We measured the rate of quenching (kq) of the excited-state intensity (I) as function of concentration of substrates (Table 2). Additionally, we measured kq at three different temperatures for benzyl chloride and attempted to obtain lifetime quenching data. Entries 1 and 2 showed no detectable quenching of luminescence with EuII1, unlike entries 3 and 4 (Table 2). For entries 3 and 4, plots of I0/I versus concentration of quencher resulted in the observation of linear relationships (Fig. S15†). The linear relationships are indicative of well-behaved bimolecular quenching interactions that can be either collisional or static in nature.23 Furthermore, kq increased with increasing temperature, suggesting that the quenching is likely due to a diffusion-limited, collisional mechanism and is not static in nature (Fig. S16†). The collisional mechanism eliminates the possibility of the participation of a preorganized benzyl chloride adduct of EuII1 in the reaction. These results are consistent with the reaction of benzyl bromide with divalent europium in the presence of crown ethers.7 In both cases, the values of kq differ from the idealized collisional bimolecular quenching constant (1010 M−1 s−1).23 These differences are likely due to coordinative saturation of EuII, causing a lower frequency of productive collisions between EuII and substrates compared to idealized lumophores.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Quencher | E pc of quencher (V vs. Ag/AgCl) | k q (×107 M−1 s−1) | Product | Yielda (%) |
| a Determined by gas chromatography-mass spectrometry. b No quenching of the excited state was observed. | |||||
| 1 | (CH3)3CCl | −3.05 | 0b | [(CH3)3C]2 | 1.9 ± 0.1 |
| 2 | C6H5Cl | −2.93 | 0b | C6H6 | 5.4 ± 0.4 |
| 3 | CH2CHCH2Cl | −2.35 | 8.5 | (CH2CHCH2)2 | 46 ± 2 |
| 4 | C6H5CH2Cl | −2.34 | 73 | (C6H5CH2)2 | 85 ± 2 |
To explain the apparent selectivity observed in the Stern–Volmer analyses, cyclic voltammetry was performed for the complex and substrates (Table 2). The peak cathodic potentials of the substrates that showed no quenching of luminescence (Epc of entries 1 and 2 in Table 2) are close to or more negative than the calculated 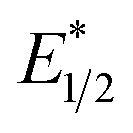 of EuII1. Because reliable cyclic voltammery of EuII1 could not be obtained in methanol, the E1/2 of EuII1 recorded in N,N-dimethylformamide might have resulted in a more negative value of E1/2 than would be present in methanol, propagating to a more negative estimation of
of EuII1. Because reliable cyclic voltammery of EuII1 could not be obtained in methanol, the E1/2 of EuII1 recorded in N,N-dimethylformamide might have resulted in a more negative value of E1/2 than would be present in methanol, propagating to a more negative estimation of 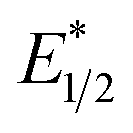 . However, the Epc of the substrates that quenched the luminescence of the excited state of EuII1 (entries 3 and 4 in Table 2) are between the calculated
. However, the Epc of the substrates that quenched the luminescence of the excited state of EuII1 (entries 3 and 4 in Table 2) are between the calculated 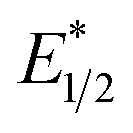 and ground-state E1/2 of EuII1, consistent with the difference in reactivity of EuII1 with benzyl chloride in the light and dark. Furthermore, allyl chloride, which has an Epc more positive than the
and ground-state E1/2 of EuII1, consistent with the difference in reactivity of EuII1 with benzyl chloride in the light and dark. Furthermore, allyl chloride, which has an Epc more positive than the 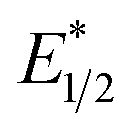 of EuII1, also shows expected product formation in the light (Table 2). Based on the cathodic potentials and lack of observed luminescence quenching, we would not expect chlorobenzene and 2-chloro-2-methylpropane to react with the excited state of EuII1; however, products were observed for these two substrates in yields of 1.9 and 5.4%, respectively. These data point toward a thermodynamic window of selectivity (−0.9 to approximately −3 V vs. Ag/AgCl) that is unique for EuII1*.
of EuII1, also shows expected product formation in the light (Table 2). Based on the cathodic potentials and lack of observed luminescence quenching, we would not expect chlorobenzene and 2-chloro-2-methylpropane to react with the excited state of EuII1; however, products were observed for these two substrates in yields of 1.9 and 5.4%, respectively. These data point toward a thermodynamic window of selectivity (−0.9 to approximately −3 V vs. Ag/AgCl) that is unique for EuII1*.
With an understanding of the electron transfer mechanism of EuII1, we were interested in moving from reactions that were stoichiometric in Eu to reactions that were catalytic in Eu. To enable catalysis, a sacrificial reducing agent was needed, and it is known that EuIII can be reduced to EuIIin situ with Zn0.19,24 To ensure that EuII1 could be assembled in situ from EuIII, 1, and Zn0, UV-visible and fluorescence spectroscopies were performed on a mixture of EuCl3, Zn0, and 1. Absorption at wavelengths >400 nm and a broad emission between 500 and 700 nm, which are both characteristic of EuII1, indicated that EuII1 can be assembled in situ (Fig. S18 and S19†). Furthermore, X-ray diffraction of material nucleated from a mixture of EuCl3, Zn0, and 1 in methanol provides direct evidence that EuII1, as well as oxidized zinc species, are formed under the reaction conditions (Fig. 4). The crystal structure in Fig. 4 is from a crystal isolated from the reaction mixture. Although several crystals formed, a yield was not determined. However, because it nucleated from a reaction mixture in which EuII was not directly added, this structure demonstrates that Zn0 is able to complete the catalytic cycle by either reducing EuCl3 followed by metalation with 1 or by reducing EuIII1 to EuII1. Direct evidence of the reduction of EuIII to EuII can be found in the Eu–N bond distances between Eu and the ligand [2.7116(10)–2.7484(10) Å for secondary amines and 2.8030(11)–2.8333(10) Å for tertiary amines] that are in the expected range for EuII–N bonds.10a,25 In the structure in Fig. 4, unlike with the previously reported structure of EuII1, there was no inner-sphere chloride, and the associated anion was ZnCl42− instead of two equivalents of Cl−, indicating oxidation of Zn0 and demonstrating the formation of EuII1via reduction of EuIII by Zn0.
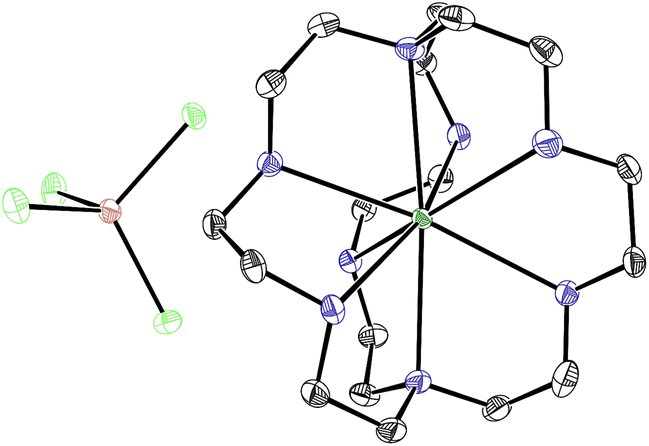 | ||
| Fig. 4 Crystal structure of [EuII1][ZnCl4] generated from a mixture of EuCl3, Zn0, and 1 in methanol. Thermal ellipsoids are drawn at 50% probability. Final refinement indicators: R1 = 2.89%; wR2 = 6.25%; resolution = 0.4929 Å; Rint = 4.91%; and Rsigma = 3.05%. Crystallographic data for this structure has been deposited at the Cambridge Crystallographic Data Centre under deposition number CCDC 1539923. An outer-sphere molecule of methanol has been omitted for clarity. Grey = C; blue = N; seagreen = Eu; green = Cl; and brown = Zn. | ||
To ensure that Zn0 could not promote the reductive coupling of benzyl chloride, a control experiment was performed with Zn0, light, and benzyl chloride. Only the formation of toluene was observed after 6 h, indicating that Zn0 does not promote the reductive coupling of benzyl chloride. To probe whether Zn0 promoted the formation of toluene, another control experiment was performed that only included benzyl chloride, methanol, and light. This experiment showed no formation of toluene, indicating that Zn0 induces the reduction of benzyl chloride to toluene.
Knowing that EuII1 can be formed in situ and that Zn0 does not promote the reductive coupling of benzyl chloride, we wanted to probe the catalytic activity of EuII1. A benzyl chloride coupling reaction was performed starting from EuCl3 (10 mol%) and 1 (10 mol%). This reaction yielded 1,2-diphenylethane (80 ± 10%) and toluene (11 ± 2%) in six hours (Fig. 3B). The variation in yields is likely due to the heterogeneity of the reaction mixture and small differences in stir rate, causing a variability in light penetration. These experiments demonstrate that the photoredox reaction can be rendered catalytic (∼8 turnovers) in europium.
To determine how catalyst loading influenced product formation, the loading of EuCl3 and 1 were systematically varied, keeping ten equivalents of Zn0 relative to benzyl chloride constant, and yields were compared at six hours. Benzyl chloride coupling reactions were performed at catalyst loadings of 5, 1, and 0.5 mol%. Yields of 1,2-diphenylethane of 71 ± 5% (∼14 turnovers), 70 ± 5% (∼70 turnovers), and 60 ± 3% (∼120 turnovers), respectively, were observed. Toluene was also formed at yields of 12 ± 2, 21 ± 2, and 26 ± 1% for 5, 1, and 0.5% catalyst loadings, respectively. This trend demonstrates that decreased catalyst loading correlates to increased toluene production. At a much lower catalyst loading (0.005%), only toluene formation was observed. These results indicate that the precatalyst operates efficiently at low concentrations but is likely in competition with zinc for reduction versus reductive coupling.
After examining the catalytic utility of EuII1, we were interested in examining the effect of water on the system because all of the reactions to this point were performed under anhydrous conditions. To introduce water into the system, EuCl3·6H2O was used as the EuIII source and the samples were prepared in a wet glovebox (water allowed but no molecular oxygen). Reactions of the catalytic reductive coupling of benzyl chloride under these wet conditions were prepared at 10 mol% catalyst loading, and the formation of 1,2-diphenylethane in yields of 80 ± 3% was observed. These yields are not different from those of reactions performed under anhydrous conditions, indicating that small amounts of water have no significant effect on the performance of the precatalyst.
To determine if EuIII remains complexed after the oxidation of EuII, luminescence intensities were compared of solutions containing EuCl3, EuCl3 in the presence of 1, and EuII1 that was opened to air to oxidize (Fig. S17†). The spectra were normalized to the 5D0 → 7F1 transition at 591 nm that is insensitive to ligand environment, and the emission intensities of the spectra were compared at the 5D0 → 7F2 transition (610–630 nm) that is hypersensitive to ligand environment.26 The change in spectral profile of the 5D0 → 7F2 transitions indicates that there is an interaction between EuIII and 1, but the exact nature of this interaction is ambiguous.
Based on the data presented here, we propose that the photocatalytic reductive coupling of benzyl chloride using EuII1 proceeds through the catalytic cycle shown in Scheme 1. From luminescence experiments, EuII1 is excited by blue light into an excited state (EuII1*). Two molecules of EuII1* reduce two molecules of substrate through a collisional electron transfer based on Stern–Volmer analyses, followed by reductive coupling of substrate molecules. The electron transfer also generates EuIII that interacts with 1 to some extent. Zn0 reduces EuIII to EuII either as the complex or the uncomplexed ion. Spectroscopic evidence (Fig. S17†) supports the presence of interactions between EuIII and 1, but this evidence is not conclusive with respect to the nature of speciation of the trivalent ion. Regardless of the extent of encapsulation of EuIII by 1, reduction by Zn0 regenerates EuII1, evidenced by spectroscopy and the crystal structure in Fig. 4, restarting the catalytic cycle.
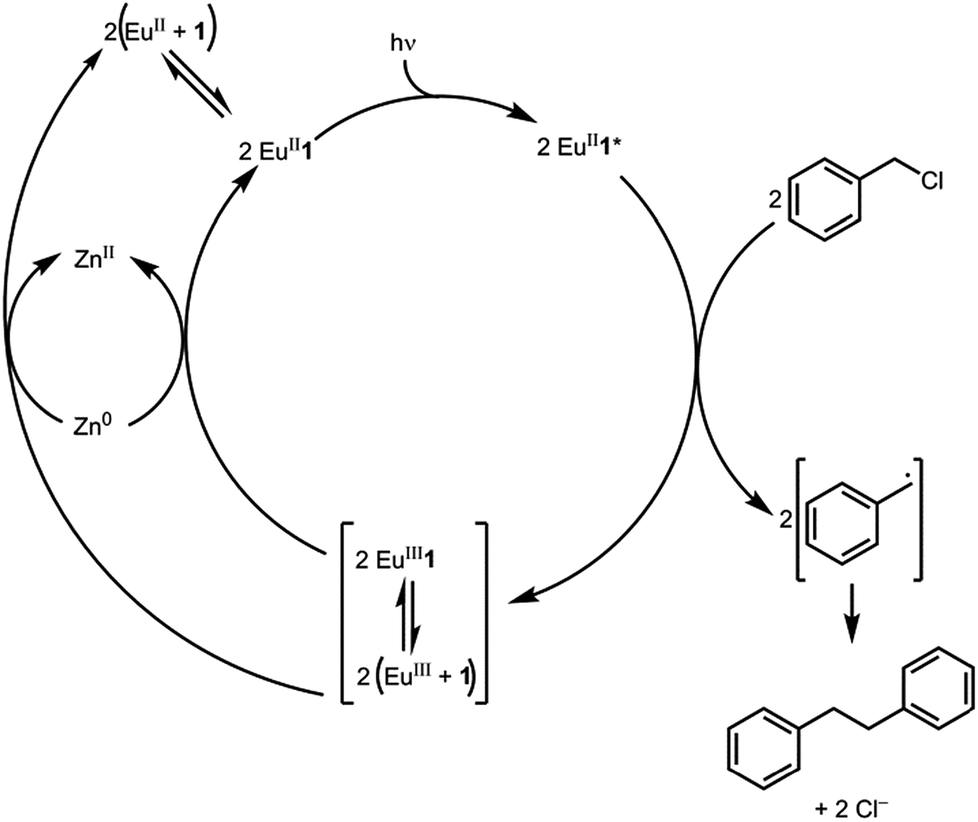 | ||
| Scheme 1 Proposed catalytic cycle. | ||
Conclusions
We have described the first report of photoredox catalysis based on europium. Exposure of EuII1 to visible light forms an excited state with a calculated electrochemical potential that rivals SmI2 in the presence of hexamethylphosphoramide, has a long luminescence lifetime, is tolerant of protic solvents and some H2O, and can be assembled in situ starting from air-stable and relatively inexpensive EuCl3·6H2O. We expect that the mechanistic insight provided here will open the door for the study of visible-light-promoted photoredox catalysis using EuII1 in reactions that require large negative electrochemical potentials between −0.9 and approximately −3 V vs. Ag/AgCl, including challenging systems like unactivated halides such as aryl bromides. Furthermore, studies from our laboratory have shown that ligand modifications to EuII1 can influence its spectroscopic properties,25a and these modifications are likely to impact excited-state redox properties. Studies exploring ligand modifications and the scope of reactivity of EuII1 are underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Science Foundation (CHE-1564755). W. L. thanks the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES), under Award # DE-SC0012628 for financial support. We thank Jennifer Stockdill for helpful conversations and use of her gas chromatograph, and we thank Jeremy Kodanko for use of his spectrophotometer. The authors thank Duke Debrah and Lin Fan for help with experimental setup for lifetime measurements.Notes and references
- (a) A. G. Amador and T. P. Yoon, Angew. Chem., Int. Ed., 2016, 55, 2304 CrossRef CAS PubMed; (b) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS PubMed; (c) Y. Slutskyy and L. E. Overman, Org. Lett., 2016, 18, 2564 CrossRef CAS PubMed; (d) A. Singh, C. J. Fennell and J. D. Weaver, Chem. Sci., 2016, 7, 6796 RSC; (e) J. J. Douglas, M. J. Sevrin and C. R. J. Stephenson, Org. Process Res. Dev., 2016, 20, 1134 CrossRef CAS; (f) J. A. Terrett, J. D. Cuthbertson, V. W. Shurtleff and D. W. C. MacMillan, Nature, 2015, 524, 330 CrossRef CAS PubMed; (g) C. C. Nawrat, C. R. Jamison, Y. Slutskyy, D. W. C. MacMillan and L. E. Overman, J. Am. Chem. Soc., 2015, 135, 11270 CrossRef PubMed; (h) A. Arora, K. A. Teegardin and J. D. Weaver, Org. Lett., 2015, 17, 3722 CrossRef CAS PubMed; (i) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433 CrossRef CAS PubMed; (j) K. Singh, S. J. Staig and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 5275 CrossRef CAS PubMed; (k) G. Bergonzini, C. S. Schindler, C.-J. Wallentin, E. N. Jacobsen and C. R. J. Stephenson, Chem. Sci., 2014, 5, 112 RSC; (l) Y. Xi, H. Yi and A. Lei, Org. Biomol. Chem., 2013, 11, 2387 RSC; (m) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (n) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed; (o) E. R. Welin, C. Le, D. M. Arias-Rotondo, J. K. McCusker and D. W. C. MacMillan, Science, 2017, 355, 380 CrossRef CAS PubMed.
- (a) W. Sattler, L. M. Henling, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2015, 137, 1198 CrossRef CAS PubMed; (b) S. B. Harkins and J. C. Peters, J. Am. Chem. Soc., 2005, 127, 2030 CrossRef CAS PubMed; (c) H. Huo, K. Harms and E. Meggers, J. Am. Chem. Soc., 2016, 138, 6936 CrossRef CAS PubMed; (d) L. A. Büldt, X. Guo, A. Prescimone and O. S. Wenger, Angew. Chem., Int. Ed., 2016, 55, 11247 CrossRef PubMed; (e) D. Li, C.-M. Che, H.-L. Kwong and V. W.-W. Yam, J. Chem. Soc., Dalton Trans., 1992, 23, 3325 RSC; (f) S. E. Creutz, K. J. Lotito, G. C. Fu and J. C. Peters, Science, 2012, 338, 647 CrossRef CAS PubMed; (g) W. Sattler, M. E. Ener, J. D. Blakemore, A. A. Rachford, P. J. LaBeaume, J. W. Thackeray, J. F. Cameron, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2013, 135, 10614 CrossRef CAS PubMed; (h) J.-M. Kern and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1987, 8, 546 RSC.
- (a) H. Yin, P. J. Carroll, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2015, 137, 9234 CrossRef CAS PubMed; (b) H. Yin, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 5984 CrossRef CAS PubMed.
- H. Yin, Y. Jin, J. E. Hertzog, K. C. Mullane, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 16266 CrossRef CAS PubMed.
- (a) K. Suzuki, F. Tang, Y. Kikukawa, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2014, 53, 5356 CrossRef CAS PubMed; (b) J.-J. Guo, A. Hu, Y. Chen, J. Sun, H. Tang and Z. Zuo, Angew. Chem., Int. Ed., 2016, 55, 15319 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, S. P. Ellery and J. S. Chen, Angew. Chem., Int. Ed., 2009, 48, 7140 CrossRef CAS PubMed; (b) W. G. Skene, J. C. Scaiano and F. L. Cozens, J. Org. Chem., 1996, 61, 7918 CrossRef CAS PubMed; (c) A. Ogawa, Y. Sumino, T. Nanke, S. Ohya, N. Sonoda and T. Hirao, J. Am. Chem. Soc., 1997, 119, 2745 CrossRef CAS; (d) G. A. Molander and C. N. Wolfe, J. Org. Chem., 1998, 63, 9031 CrossRef CAS; (e) G. A. Molander and C. Alonso-Alija, J. Org. Chem., 1998, 63, 4366 CrossRef CAS; (f) C. N. Rao and S. Hoz, J. Org. Chem., 2012, 77, 4029 CrossRef CAS PubMed; (g) C. N. Rao and S. Hoz, J. Org. Chem., 2012, 77, 9199 CrossRef CAS PubMed; (h) Y. Sumino, N. Harato, Y. Tomisaka and A. Ogawa, Tetrahedron, 2003, 59, 10499 CrossRef CAS; (i) S. Maity, K. A. Choquette, R. A. Flowers II and E. Prasad, J. Phys. Chem. A, 2012, 116, 2154 CrossRef CAS PubMed; (j) E. Prasad, B. W. Knettle and R. A. Flowers II, Chem.–Eur. J., 2005, 11, 3105 CrossRef CAS PubMed; (k) A. Nomoto, Y. Kojo, G. Shiino, Y. Tomisaka, I. Mitani, M. Tatsumi and A. Ogawa, Tetrahedron Lett., 2010, 51, 6580 CrossRef CAS; (l) Y. Tomisaka, A. Nomoto and A. Ogawa, Tetrahedron Lett., 2009, 50, 584 CrossRef CAS.
- S. Maity and E. Prasad, J. Photochem. Photobiol., A, 2014, 274, 64 CrossRef CAS.
- P. L. Watson, T. H. Tulip and I. Williams, Organometallics, 1990, 9, 1999 CrossRef CAS.
- (a) M. Neumann and K. Zeitler, Org. Lett., 2012, 14, 2658 CrossRef CAS PubMed; (b) Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, J. Mater. Chem. A, 2014, 2, 18720 RSC.
- (a) A. N. W. Kuda-Wedagedara, C. Wang, P. D. Martin and M. J. Allen, J. Am. Chem. Soc., 2015, 137, 4960 CrossRef CAS PubMed; (b) B. A. Corbin, J. L. Hovey, B. Thapa, H. B. Schlegel and M. J. Allen, J. Organomet. Chem., 2017 DOI:org/10.1016/j.jorganchem.2017.09.007.
- (a) P. H. Smith, M. E. Barr, J. R. Brainard, D. K. Ford, H. Freiser, S. Muralidharan, S. D. Reilly, R. R. Ryan, L. A. Silks III and W.-h. Yu, J. Org. Chem., 1993, 58, 7939 CrossRef CAS; (b) M. Y. Redko, R. Huang, J. L. Dye and J. E. Jackson, Synthesis, 2006, 5, 759 CrossRef.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- J. Jiang, N. Higashiyama, K.-i. Machida and G.-y. Adachi, Coord. Chem. Rev., 1998, 170, 1 CrossRef CAS.
- P. N. Hazin, J. W. Bruno and H. G. Brittain, Organometallics, 1987, 6, 913 CrossRef CAS.
- C. A. P. Goodwin, N. F. Chilton, L. S. Natrajan, M.-E. Boulon, J. W. Ziller, W. J. Evans and D. P. Mills, Inorg. Chem., 2017, 56, 5959 CrossRef CAS PubMed.
- L. Raehm, A. Mehdi, C. Wickleder, C. Reyé and R. J. P. Corriu, J. Am. Chem. Soc., 2007, 129, 12636 CrossRef CAS PubMed.
- (a) J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617 CrossRef CAS PubMed; (b) D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259 CrossRef CAS.
- (a) S. Seibig, É. Tóth and A. E. Merbach, J. Am. Chem. Soc., 2000, 122, 5822 CrossRef CAS; (b) L. Burai, É. Tóth, S. Seibig, R. Scopelliti and A. E. Merbach, Chem.–Eur. J., 2000, 6, 3761 CrossRef CAS PubMed; (c) M. Regueiro-Figueroa, J. L. Barriada, A. Pallier, D. Esteban-Gómez, A. de Blas, T. Rodríguez-Blas, É. Tóth and C. Platas-Iglesias, Inorg. Chem., 2015, 54, 4940 CrossRef CAS PubMed.
- L. A. Ekanger, D. R. Mills, M. M. Ali, L. A. Polin, Y. Shen, E. M. Haacke and M. J. Allen, Inorg. Chem., 2016, 55, 9981 CrossRef CAS PubMed.
- H. Yin, Y. Jin, J. E. Hertzog, K. C. Mullane, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 16266 CrossRef CAS PubMed.
- R. A. Flowers II, Synlett, 2008, 10, 1427 CrossRef.
- Yields are reported as the mean ± standard error of the mean of three independently prepared reactions.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer Science+Business Media, LLC, New York, 3rd edn, 2006, pp. 278–327 Search PubMed.
- S. Shinoda, M. Nishioka and H. Tsukube, J. Alloys Compd., 2009, 488, 603 CrossRef CAS.
- (a) G.-X. Jin, M. D. Bailey and M. J. Allen, Inorg. Chem., 2016, 55, 9085 CrossRef CAS PubMed; (b) C. U. Lenora, F. Carniato, Y. Shen, Z. Latif, E. M. Haacke, P. D. Martin, M. Botta and M. J. Allen, Chem.–Eur. J., 2017, 23, 15404 CrossRef CAS PubMed.
- (a) D. J. Averill and M. J. Allen, Inorg. Chem., 2014, 53, 6257 CrossRef CAS PubMed; (b) A. de Bettencourt-Dias, in Luminescence of Lanthanide Ions in Coordination Compounds and Nanomaterials, ed. A. de Bettencourt-Dias, John Wiley and Sons, Ltd., West Sussex, 2014, pp. 38–40 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1539923. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc02479g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |