
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeciphering the incognito role of water in a light driven proton coupled electron transfer process†
Senthil Kumar
Thiyagarajan
 ,
Raghupathy
Suresh
,
Vadivel
Ramanan
and
Perumal
Ramamurthy
*
,
Raghupathy
Suresh
,
Vadivel
Ramanan
and
Perumal
Ramamurthy
*
National Centre for Ultrafast Processes, University of Madras, Taramani Campus, Chennai – 600 113, India. E-mail: prm60@hotmail.com
First published on 10th November 2017
Abstract
Light induced multisite electron proton transfer in two different phenol (simple and phenol carrying an intramolecularly hydrogen bonded base) pendants on acridinedione dye (ADD) and an NADH analogue was studied by following fluorescence quenching dynamics in an ultrafast timescale. In a simple phenol derivative (ADDOH), photo-excited acridinedione acquires an electron from phenol intramolecularly, coupled with the transfer of a proton to solvent water. But in a phenol carrying hydrogen bonded base (ADDDP), both electron and proton transfer occur completely intramolecularly. The sequence of this electron and proton transfer process was validated by discerning the pH dependency of the reaction kinetics. Since photo-excited ADDs are stronger oxidants, the sequential electron first proton transfer mechanism (ETPT) was observed in ADDOH and hence there is no change in the PCET reaction kinetics kETPT ∼ 6.57 × 109 s−1 in the entire pH range (pH 2–12). But the phenol carrying hydrogen bonded base (ADDDP) unleashes concerted electron proton transfer where the PCET reaction rate decreases upon decreasing the pH below its pKa. Noticeably, the concerted EPT process in ADDDP mimics the donor side of photosystem II and it occurs by two distinct pathways: (i) through direct intramolecular hydrogen bonding between the phenol and amine, kDEPT ∼ 12.5 × 1010 s−1 and (ii) through the bidirectional hydrogen bond extended by the water molecule trapped in between the proton donor and acceptor, which mediates the proton transfer and serves as a proton wire, kWMEPT ∼ 2.85 × 1010 s−1. These results unravel the incognito role played by water in mediating the proton transfer process when the structural elements do not favor direct hydrogen bonding between the proton donor and acceptor in a concerted PCET reaction.
Introduction
The fundamental yet mechanistically intricate proton coupled electron transfer process (PCET) is a key for annual energy storage of ∼1018 kJ via photosynthesis.1 By avoiding high energy intermediates and charge build up, it helps to store solar energy in chemical bonds.2 Photosystem II is a beacon that represents the significance of the PCET process, and it is prevalent in various vital enzymatic reactions,1,3 catalytic oxidation,1,4 the production of molecular hydrogen,5 artificial photosystems6 and most chemical and biological energy conserving reactions. Hence, exploring various factors that influenc the PCET process and its energetics,7,8 dynamics9–15 and mechanistic pathways1,11,16–19 is necessary to design an efficient artificial photosystem which can harness solar energy in chemical bonds for the growing global energy need.Based on energetics, molecularity, number of mechanistic steps involved and degree of adiabaticity, PCET processes are classified into different types.1,19–21 Among them, multisite electron proton transfer (MSEPT) is a class where the translocation of an e−/H+ pair occurs from a common donor to a different acceptor or from different donors to a common acceptor.1 Photosystem II is the quintessential example of the MSEPT process, where photo-excited chlorophyll (P680+) acquires an electron (Ae) from tyrosine (De−/H+), which decreases its pKa value drastically (pKa = −2) and facilitates the transfer of the proton from tyrosine to the base histidine (AH+) via the intramolecular hydrogen bond.22 Otherwise, the neutralization reaction between tyrosine (pKa = 10) and histidine (pKa = 7) under biological conditions is not favoured.1 According to the pKa slider rule,23 intramolecular hydrogen bonding between tyrosine and histidine is expected to be very strong (ΔpKa ∼ 3), provided these two molecular species are at a communicable distance. The high resolution (1.9 Å) crystal structure of photosystem II (extracted from thermophilic cyanobacterium)24 estimates that the distance between the phenolic oxygen of tyrosine and the imidazole nitrogen of histidine is ∼2.49 Å, short enough for the direct hydrogen bonding interaction. Eventually the shorter and stronger hydrogen bonding interaction facilitates the translocation of electron and proton to occur in a concerted fashion.
Although the MSEPT process can happen in several distinct pathways, the concerted transfer of electron and proton (CPET) has a large thermochemical bias conducive for energy harvesting, but with an intrinsic kinetic barrier imposed by the larger reorganisation energy for the proton transfer process.11 However, the hydrogen bonding interaction between the proton donor and acceptor effectively decreases the reorganization energy by increasing the proton vibrational wavefunction overlap and it accelerates the PCET reaction rate.25 Moreover, the same non-covalent interaction considerably decreases the oxidation potential of the phenol which makes ΔGPET more negative. Thus, the hydrogen bonding interaction navigates the PCET reactions by accelerating the reaction kinetics and also by altering the energetics. Successive publications on this hydrogen bonding interaction unveil that by altering the relative orientation of the hydrogen bond, proton transfer distance and π-conjugation, the PCET reaction dynamics can be tuned, which will enable us to control the associated energy conservation process.26–33
Knowledge of these different structural elements allows researchers to design a plethora of model systems to biomimic the ‘oxygen evolving complex’ (OEC) of photosystem II,34–36 yet duplicating this intricate process to efficiently harvest solar energy is still challenging. Most of the literature that addresses the biomimetics of the PCET phenomenon in photosystem II is focused on ruthenium complexes carrying a tyrosine/tryptophan moiety.9–11,17,19,21,32,34–42 It is because these complexes imitate the energetics of OEC of photosystem II (E0 of P680+ is ∼1.1 V vs. NHE)22 and at the same time, the photooxidation of Ru(II) complexes occur both reversibly and irreversibly (using sacrificial electron acceptors), which facilitate researchers to monitor the PCET reaction kinetics in a broader time scale (ps to 5 ms).9–11,19 Meanwhile, reports on pure organic compounds which resemble the PCET process in photosystem II are very sparse and in all cited works intermolecular fluorescence quenching dynamics exhibited by hydrogen bonded phenol base pairs in non-aqueous media were routinely studied.43–46 Organic compounds exhibiting intramolecular charge transfer (ESICT) coupled with proton transfer (ESIPT) in the excited state were designed and their excited state dynamics in an ultrafast timescale were unravelled. These systems possess strong ground state interactions between the electron donor and acceptor (π-conjugation) and also between the proton donor and acceptor (short and strong hydrogen bond). Thus, these artificial organic systems failed to practically mimic photosystem II and are usually referred to as PCET*.47,48 On the whole, no attempts were made until now to design a pure organic compound which mimics the role of tyrosine and histidine in photosystem II and to study its intra- and intermolecular PCET behaviour in an aqueous environment.
Similarly, the indigenous role of solvent water in a PCET reaction is not well understood. Strenuous effort from various research groups convinces one both experimentally and theoretically of the vital role of solvent water as a proton acceptor in a PCET reaction. Yet the factors influencing the reaction dynamics and the sequence of the process are still a subject of much debate.37–42 Even recently, while reporting a theoretically conflicting result (where a tryptophan residue underwent a concerted MSEPT process with water), Hammarström et al., mentioned that our current understanding of the PCET process in water (after two decades of research) is still incomplete.42 At the same time, restricting the role of solvent water only as a proton acceptor in the PCET reaction is ingenuous. Being both a hydrogen bond donor and acceptor facilitates the water molecule to build a bridge between the proton donor (acid) and acceptor (base). Transfer of the proton across this water mediated bridge is observed in many excited state intra- and intermolecular proton transfer processes, photochemical reactions49–52 and even in the case of photoacids.1 For example, in photoacids the solvent mediated proton transfer bridge facilitates the exchange of proton between the acid and base by adopting a Von Grotthus type proton hopping mechanism.53 It was experimentally demonstrated in real time by O. F. Mohammed and his co-workers using ultrafast infrared spectroscopy.54 But for the analogous MSEPT process, water was considered only as a proton acceptor and the role of solvent water as a proton wire was unfathomed except for a few times.55,56
In this work, we tailored a first ever organic molecular triad (ADDDP), which biomimics the donor side of photosystem II, in which the role of P680+, tyrosine and histidine is played by the ADD*, phenol and DPA unit respectively. To comprehend the essence of each molecular entity, preliminary investigations were carried out with the model compound ADDOH, an organic molecular dyad, in which selectively photo-exciting the ADD unit dislodges the e−/H+ pair from phenol only in aqueous media and quenches the emission. The influence of isotopic substitution and the pH of the medium in ultrafast fluorescence quenching dynamics was used as the experimental marker to identify the reaction mechanism and the factors controlling the reaction kinetics. Applying the knowledge acquired from this preliminary study and enacting similar investigations with the triad ADDDP allow us to disentangle the role of water in mediating the MSEPT process in concerted MSEPT reactions.
Results and discussion
Similar to NADH, acridinediones are 1,4 dihydropyridines57 having a longer wavelength absorption maximum at ∼350 nm due to the intramolecular charge transfer transition from the ring nitrogen to the carbonyl moiety, and the corresponding emission is observed at ∼450 nm. Investigations on various ADD derivatives unveil that ADD is a good electron donor as well as acceptor in the excited state.57–60 Noticeably, ADD bearing an electron donor moiety at the 9th position has a tendency to undergo a ‘through space intramolecular photoinduced electron transfer process’ (PET) after photoexcitation, despite there being only a negligible electronic coupling between the electron donor and acceptor in the ground state. Hence, unlike other organic model systems (having strong electronic coupling in the ground state) ADD can act as a suitable template for mimicking the donor side of photosystem II. Accordingly, two different ADD derivatives, ADDOH and ADDDP, bearing a different phenolic moiety at the 9th position were synthesised following the synthetic shown in Scheme S1.† It is shown in Fig. S1† that the summation of indigenous absorption spectrum of phenols used (both PC and PCDPA) and basic ADD dye is similar to the absorption spectrum of ADDOH and ADDDP. This indicates that there is no ground state interaction or electronic coupling between the phenolic group and the ADD fluorophore.Unravelling the bimolecular PCET process in an aqueous solution of ADDOH
The steady state absorption and emission spectra of ADD1 and ADDOH in different solvents are shown in Fig. S2† and the corresponding data are given in Tables S1 and S2.† From the table, it is clear that there is not much difference in the absorption and emission maxima after phenolic substitution. However, there is a significant difference in the fluorescence quantum yield and lifetime value in all of the solvents, especially in protic solvents (Tables S1 and S2†). Although there is no ground state electronic coupling between phenol and the ADD moiety, being a good electron acceptor in the excited state may facilitate the ADD moiety to acquire the electron from phenol intramolecularly, which can quench the emission significantly in all of the solvents. However, the extent of quenching observed is enormous in protic solvents (40 times in water) when compared to that in polar aprotic solvents (2 times). The observed bias in the quenching process may be due to the specific solvent effect. To unravel this, we carried out time resolved fluorescence studies. The fluorescence decay curve of ADDOH fits a single exponential in non-polar and polar aprotic solvents, whereas it becomes biexponential in protic solvents with an ultrashort component (τf < 300 ps) in a larger proportion (>90%) (Table S2†). In general, the inverse of the lifetime is the sum of the rates that depopulate the excited state (kf + knr)−1, where kf and knr are the emissive and non-radiative decay rate constants. From Table S2† it is deduced that in protic solvents the rate of the non-radiative decay process is significantly faster than the radiative process which in turn dictates the (total) excited state decay rate. So, the observed decay rate (kobs = τ−1) in polar protic solvents is tantamount to the rate of the non-radiative deactivation process induced by the protic solvents. Thus, there is an involvement of specific solute–solvent interaction in protic solvents that quenches the excited state of the ADD moiety. Some of the common non-radiative channels induced by the protic solvents are (1) excited state hydrogen bond dynamics;61 which are not possible in this case because the hydrogen bonded phenol is in the ground state, (2) the hydrogen bond facilitated photoinduced electron transfer (PET) process;61 weakly acidic phenolic –OH group can participate in hydrogen bonding interaction with the protic solvents which can lower the oxidation potential of the phenol and make the ΔGPET value more negative and (3) the proton coupled electron transfer process; it is well known that the phenol radical cation formed during the PET process is highly acidic with a pKa value of −2 and hence it may undergo deprotonation and quench the emission. Among these different decay channels, the possible non-radiative decay process/processes that quench the excited state can be determined experimentally by following the bimolecular quenching rate at both steady state and time resolved conditions.ADDOH exhibits a significantly higher fluorescence quantum yield in polar aprotic solvent acetonitrile (τf = 0.28). While adding water to this acetonitrile solution, the emission intensity decreases as shown in Fig. S3.† The Stern–Volmer (SV) plot of the ratio of emission intensity as a function of quencher concentration (water) exhibits a linear relation as shown in Fig. 1 and the bimolecular quenching rate constant (kq)I was determined to be 6.50 × 107 M−1 s−1. Similarly, the fluorescence lifetime value of ADDOH also gets quenched as a function of water percentage and the corresponding SV plot (Fig. S4, S5 and Table S3†) estimates the bimolecular quenching rate constant (kq)τ as 6.37 × 107 M−1 s−1, which is almost equivalent to (kq)I. This indicates that the quenching may be completely dynamic,62i.e. due to diffusive encounters between the excited state of the ADD fluorophore and the water molecule present in relatively large excess.
 | ||
| Fig. 1 S–V plot showing the quenching of ADDOH fluorescence by H2O/D2O. | ||
But the bare acridinedione dye ADD1 exhibits a relatively higher fluorescence quantum yield and lifetime in water than in acetonitrile, which is quite common for a fluorophore exhibiting charge transfer transition62 (even in the case of ADDOH, increasing the water percentage increases the absorptivity and red shifts the CT band). Hence the presumption that collisional encounters between the photoexcited ADDOH and water quench the emission is inappropriate.
In the case of ADDOH, the actual quenching species is the phenol, an intramolecular PET donor, and not water. So, any factor that significantly influences the intramolecular PET process i.e. ΔGPET will severely affect the fluorescence quantum yield and lifetime (bimolecular quenching due to PET from the phenol of one molecule to the ADD fluorophore of another molecule is negligible at this dye concentration). While applying the classical equation for Gibbs energy of photoinduced electron transfer,63 it is understood that ΔGPET is a function of the oxidation potential of the phenol (donor) and the excited state reduction potential of the ADD (acceptor).
| ΔGPET = E0(PhOH/PhOH+˙) − {E0(ADD/ADD−˙) + E0−0} |
It has already been reported that the addition of water in acetonitrile solution of phenol will shift its Eoxp to a less positive potential due to the relative ease of oxidation of phenol hydrogen bonded to water. The addition of even 2% water to a dry ACN solution of mesitol (sterically hindered phenol) shifts its Eoxp value ∼125 mV towards a less positive region.64 The concentration of the dye used in our study was 25 μM and the minimum amount of water added was 1.38 M (5%), hence there is a sufficient amount of water molecule to interact with ADDOH through hydrogen bonding in the ground state itself, which can significantly reduce the oxidation potential of the phenol (∼350 mV). Moreover, the excited state reduction potential of acridinedione E0(ADD*/ADD−˙) becomes more positive (discussion D1 given in ESI†) in an aqueous environment i.e., easily reducible, which in turn brought down the ΔGPET to a more negative value in water. Thus it seems apparent that the hydrogen bond assisted intramolecular photoinduced electron transfer process is responsible for the observed quenching in both the fluorescence intensity and lifetime of the ADD moiety.
Since it is dubious that the phenols are prone to undergo deprotonation during the photoinduced electron transfer process, we intended to investigate the influence of isotopic substitution on electron transfer kinetics. From 1H NMR titration, it is identified that the phenolic hydrogen of ADDOH in CD3CN is involved in deuterium exchange in the presence of D2O (10%). And if this deuterated phenol undergoes deprotonation during the electron transfer process, it will significantly affect the excited state quenching dynamics.
Accordingly, increasing the percentage of D2O quenches both the steady state emission intensity (Fig. S8†) and fluorescence lifetime (Fig. S9 and Table S5†) with a bimolecular quenching rate constant of ∼2.97 × 107 M−1 s−1 which gives a kinetic isotope value (kH/kD) of ∼2.1. Such a significant isotopic effect (Fig. S5†) implies that the intermolecularly hydrogen bonded water must have acted as the proton acceptor and drives the electron transfer process. But there are also cases where kinetic isotopic values up to ∼1.5 are observed even for the simple electron transfer process.65–67 Hence, to subdue such arguments similar investigations were carried out for other ADD derivatives as shown in Fig. S10,† but they show no such kinetic isotopic effect. Thus, the quenching dynamics observed in the presence of D2O have conclusively proven that it is not simply the hydrogen bond assisted PET process from phenol that quenches the emission intensity. But, there is a translocation of electron and proton at two different sites i.e., a multisite electron and proton transfer process, where the ADD fluorophore acts as the electron acceptor and the solvent water as the proton acceptor as shown in Scheme 1, and this quenches both the fluorescence intensity and lifetime.
 | ||
| Scheme 1 Light driven PCET processes in ADDOH. | ||
Is the PCET process in ADDOH a sequential or concerted process?
Light driven multisite electron proton transfer can be coupled in many different ways. A simple and useful mechanistic tool to spot the sequence of electron proton transfer is following the pH dependent reaction kinetics. While investigating the MSEPT process in a Ru(III)–tyrosine system, Hammarstörm and co-workers have identified the existence of four different mechanisms that dominate in different pH regions/zones.19 However, some of the mechanistic regimes are absent or shifted to a different pH region as a function of the oxidizing power of different Ru(III) derivatives. So, to probe the mechanism that operates the PCET process in the ADDOH–H2O system, it is expected that the basic ADD fluorophore must be insensitive to pH for a broader range. It has already been established that the photophysical behaviour of acridinedione bearing N-alkyl substitution is insensitive for a broader pH range (2–13)68 and hence any change in the steady state and time resolved photophysical behaviour of ADDOH within this pH range will be primarily due to modulation in the PCET reaction kinetics.The absorption and emission spectra of ADDOH at different pH values are shown in Fig. S11.† No change in the longer wavelength absorption band at pH 2–9.2 indicates that there is no significant ground state reaction within this pH range. At pH > 9.2, the pendant phenol undergoes deprotonation (pKa ∼ 10) and the equilibrium shifts more towards phenolate anion formation which in turn intensifies and elongates the longer wavelength absorption band. Similarly, the emission intensity remains the same for a broader pH range (ϕf = 1.5%) but from pH 9.2, the intensity starts to decline as shown in Fig. 2. At pH > 12, ADDOH exhibits very weak emission even though it has a stronger visible absorption; this implies that the deprotonated form of ADDOH might be non-emissive. According to Harriman,69 the oxidation potential of the phenol in aqueous medium varies as a function of pH and it undergoes oxidation much easier at higher pH. Also, Hammarstörm19,21 and co-workers have established that the rate of oxidation of phenol increases with the increase in pH. Hence, the very fast intramolecular PET process from the pendant phenolate anion to the highly oxidizing photoexcited acridinedione might lead the deprotonated form of ADDOH to be non-emissive. This is further supplemented by the work of Ashokkumar et al., where they have demonstrated the existence of the non-emissive nature of the deprotonated form of ADDOH in acetonitrile.70
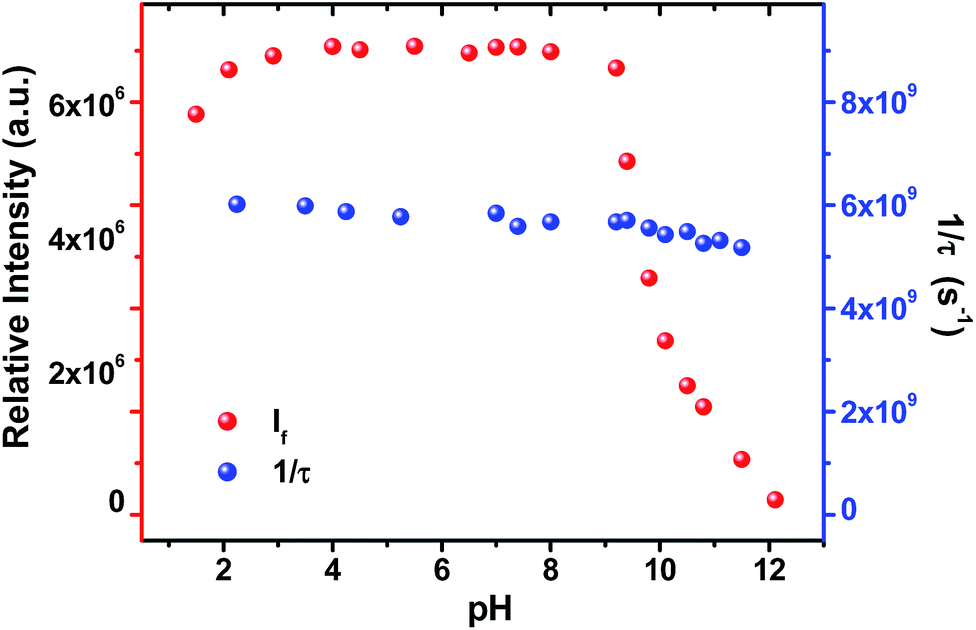 | ||
| Fig. 2 Variation in the emission intensity and inverse of the fluorescence lifetime of ADDOH at different pH values. | ||
To identify the presence of pH dependent mechanistic zones, the excited state decay rates (kobs = τ−1) were monitored at different pH values as shown in Fig. S12 and S13.† At neutral pH, the fluorescence decay profile recorded using TCSPC fits a biexponential with an ultrashort component present in the larger proportion (>95%) having a lifetime value of ∼160 ps, but it is less than the IRF (∼550 ps). Hence, the fluorescence decay profiles were recorded using a femtosecond upconversion technique (IRF ∼ 200 fs) and they fit a single exponential with a lifetime value of ∼170 ps. Similar analysis on the fluorescence decay monitored at different pH values (Tables S6 and S7†) has shown that there is not much variation in the kobs in the entire pH range (Fig. 2). So, it appears that there is only one type of PCET mechanism that operates and dominates in the entire pH range. According to Irebo et al., by tuning the pH and oxidation strength of the acceptor, different mechanisms can be enhanced or suppressed19 but in our case tuning the pH has proven futile. This may be due to the very high oxidizing strength of the electron acceptor. In an analogous system,11 Ru(III)-dce with a stronger oxidant (1.53 V vs. NHE), the pH independent region was observed at pH < 8. Similarly, in our case photoexcited acridinedione in water is a stronger oxidant  and under such highly oxidizing conditions, the influence of the pH dependent oxidation potential of phenol (∼300 mV) in deciding the reaction kinetics will be negligible (discussion D2 given in the ESI†). Hence, the electron transfer limited ETPT mechanism governs the acidic regime (pH 2–7). In the basic region, PT limited electron transfer or pure electron transfer from the deprotonated species is expected to quench the fluorescence lifetime further. But to our dismay, there is no change in the fluorescence decay profile even under basic conditions. The deprotonated form of ADDOH is the predominant species (ADDO−) that has formed at higher pH due to either the ground state or excited state (PTET) deprotonation process.
and under such highly oxidizing conditions, the influence of the pH dependent oxidation potential of phenol (∼300 mV) in deciding the reaction kinetics will be negligible (discussion D2 given in the ESI†). Hence, the electron transfer limited ETPT mechanism governs the acidic regime (pH 2–7). In the basic region, PT limited electron transfer or pure electron transfer from the deprotonated species is expected to quench the fluorescence lifetime further. But to our dismay, there is no change in the fluorescence decay profile even under basic conditions. The deprotonated form of ADDOH is the predominant species (ADDO−) that has formed at higher pH due to either the ground state or excited state (PTET) deprotonation process.
But, ADDO− exhibits an ultrafast intramolecular electron transfer process and hence its excited state decay kinetics fall well within the IRF (200 fs) which results in no significant modulation in the fluorescence decay profile. However, a significant loss in the measurement counts/intensity while recording the lifetime decay using the femtosecond upconversion technique has shown its existence (Fig. S14†). Hence, it can be concluded that the sequential ETPT mechanism supersedes the CPET process due to the very high oxidizing strength of the photoexcited acridinedione and it dominates the acidic region. Whereas, the PTET or pure electron transfer that occurs in the basic region doesn’t bring any significant modulation in the fluorescence decay profile because of the ultrafast decay kinetics which fall short of the IRF. Hence, the persistence of the ETPT mechanism (and no CPET) was felt even under highly basic conditions.
To the best of our knowledge, this is the first report where a detailed investigation on the MSEPT process exhibited by a neat organic system with water as the proton acceptor was established. This preliminary investigation with the molecular dyad ADDOH unequivocally establishes the role of phenol and the photoexcited ADD moiety towards the PCET reaction and the factor that controls the bimolecular PCET reaction kinetics in aqueous media.
Impact of intramolecular hydrogen bonding in the molecular triad ADDDP
ADDDP carries an intramolecularly hydrogen bonded phenol covalently linked at the 9th position; two di-2-picolyl amine units (DPA) at both ortho positions act as the hydrogen bond acceptor. The presence of the intramolecular hydrogen bond between the phenolic –OH and DPA unit in the solid state is indubitably established by the broad IR signal observed at ∼3150 cm−1 corresponding to the hydrogen bonded O–H stretching (Fig. S15†). However, the existence of intramolecular hydrogen bonding interaction even in the solution state has to be validated. Hence, to explore this the 1H NMR spectra of ADDOH and ADDDP were recorded in three different deuterated solvents and are shown in Fig. S16.† The singlet at 6.8 ppm in the 1H NMR spectrum of ADDOH in CD3CN is exchangeable with D2O, this indicates that it is due to phenolic –OH. But it is shifted downfield to 9.0 ppm in DMSO-d6 due to the facile intermolecular hydrogen bonding interaction with solvent DMSO. Whereas in the case of ADDDP, the proton exchangeable with D2O appears at 10.75 ppm in both CD3CN and DMSO-d6; this indicates that the phenolic –OH is involved in the intramolecular hydrogen bonding interaction with the di-2-picolyl amine unit and hence it is shifted to downfield in both the solvents. This is further supplemented by the cyclic voltammogram of ADDDP in ACN (Fig. S17†) which shows that the oxidation potential of phenol shifts towards a less positive region after picolyl amine substitution and is due to the efficient intramolecular hydrogen bonding interaction between phenol and amine even in the solution state. Hence, it is envisaged that this organic molecular triad may function as an organic mimetic for photosystem II by performing a completely intramolecular MSEPT process.Before studying the PCET process in this molecular triad, the possibility of direct PET from the DPA unit to the ADD fluorophore needs to be validated because the photoexcited acridinedione in aqueous medium is a stronger oxidant and capable enough to oxidize even DPA. The PET process from DPA can be either intra- or intermolecular. On adding up to 10 equivalents of DPA to the aqueous solution of ADDOH, there is no change in the longer wavelength absorption band, emission band, emission intensity, and lifetime as shown in Fig. S18† which indicates that there is no intermolecular PET process from the DPA unit even in such a high concentration. Hence, ADDDP carrying only two equivalents of DPA involved in the intermolecular PET process is not feasible. Similarly, to validate the absence of the direct intramolecular PET process from the DPA to ADD fluorophore, a new ADD derivative bearing DPA at the phenolic oxygen (an ether functionality) was synthesized, ADDODP (Scheme S2†). The photophysical behaviour of this compound is almost similar to that of ADDOMe, as shown in Fig. S19,† which indicates that there is no direct PET process from the DPA unit. Hence, in the presence of a stronger reductant like phenol, the direct intramolecular PET process from the DPA unit in ADDDP is non-viable. These studies allow us to conclude that any difference in the photophysical behaviour of ADDDP relative to ADDOH would be primarily due to the intramolecular hydrogen bonding between the phenolic –OH and the DPA unit.
The photophysical behaviour of ADDDP in different solvents is shown in Fig. S20† and the corresponding data are given in Tables S8 and S9.† From the data it is apparent that the DPA substitution has not appreciably altered the absorption and emission maxima but as expected there is significant quenching in both the fluorescence quantum yield and lifetime due to the completely intramolecular MSEPT process. The fluorescence quantum yield values of ADDDP in all of the solvents under investigation were <2%. This indicates that the intramolecular hydrogen bonding interaction with the pendant base paves the way for MSEPT even in a polar aprotic solvent like acetonitrile and hence the fluorescence quantum yield (∼1.8%) of ADDDP in acetonitrile is almost equivalent to that of ADDOH in water. Similarly, the fluorescence decay profile becomes biexponential (after DPA substitution) in all of the solvents with an ultra-short component having a lifetime of <400 ps, which exists in a larger proportion. Since the translocation of electron and proton occurs completely intramolecularly, it depopulates the excited state through a non-radiative deactivation pathway and leads the fluorescence quenching dynamics to proceed in a sub nanosecond timescale i.e. the non-radiative decay rate (knr) dictates the observed decay rate (kobs). These preliminary observations have convincingly proven the occurrence of a completely intramolecular MSEPT process in ADDDP in almost all of the solvents under investigation. However, to precisely determine the MSEPT reaction kinetics, fluorescence lifetime measurements were carried out at higher time resolution. The completely intramolecular PCET process in ADDDP delineates an ultrashort component in the polar aprotic solvents (<250 ps) as shown in Fig. S21† whereas we observed two such ultrashort decay components (Table S10†) for ADDDP only in protic solvents. The role played by the protic solvents in altering this PCET reaction kinetics requires an elaborate photophysical investigation, which is described below.
Deciphering the role of protic solvents in modulating MSEPT reaction kinetics
On adding water to the solution of ADDDP in ACN, the CT band in the absorption spectrum undergoes a red shift with an enhancement in its absorptivity due to the charge transfer nature of the locally excited state in high polar solvents (Fig. S22a†). However, the added water quenches the emission intensity (Fig. S22b†) and the fluorescence quantum yield of ADDDP becomes 0.2% in 90% water. This indicates that the solvent water accompanies the pendant amine in quenching the emission intensity of ADDDP. But aberrantly, the S–V plot of the ratio of emission intensity as a function of the quencher concentration (water) follows a non-linear behaviour as shown in Fig. S22c.† The fluorescence decay curves recorded using TCSPC are shown in Fig. S22d.† The fit is biexponential up to 50% of water, where τ1 (<200 ps) accounts for the MSEPT process and τ2 (∼3 ns) for the locally excited state (Table S11†). Whereas at a higher percentage of water, an anomalous longer decay component appears (τm ∼ 1 ns) and its relative amplitude increases with the increase in the percentage of water (the origin of this new decay component will be discussed later).Increasing the time resolution in lifetime measurements (Fig. 3a) reveals that an ultrashort component observed in pure acetonitrile (τ1 ∼ 115 ps) is divided into two ultrashort components (τu and τ1) upon adding water (Table S12†). The lifetime values of these two decay components decrease (τu ∼ 15 to 5 ps and τ1 ∼ 80 to 35 ps) with the increase in the addition of water as shown in Fig. 3b and S23.† This indicates that both decay components are responsible for the observed quenching in the steady state emission intensity. Similar investigations were carried out by adding D2O and we observed an analogous behaviour. However, the lifetime values of the two ultrashort components in D2O are twice that of those in water in all proportions as shown in Fig. S24 and S25 and Tables S13 and S14.† So, the ratio of the observed decay rate measured using the two ultrashort components in water and D2O has given a kinetic isotopic value of ∼2.1. These results implicate the involvement of two independent non-radiative deactivation processes (with a significant kinetic isotopic effect) in the observed fluorescence quenching dynamics. Applying the knowledge acquired from our earlier investigations with the dyad ADDOH, it is assumed that these two ultrashort components must be due to two independent MSEPT processes with a significant difference in the fluorescence quenching dynamics. The difference in the reaction kinetics may be due to the different proton acceptors or the difference in the hydrogen bond strength. In the case of ADDDP, there are only three possible different pathways for the MSEPT process to occur with ADD as the electron acceptor and (1) tertiary amine, (2) water, and (3) pyridine as the proton acceptors.
 | ||
| Fig. 3 (a) Variation in the fluorescence decay profile of ADDDP in acetonitrile with an increase in the addition percentage of water recorded using the femtosecond upconversion technique. (b) Fluorescence lifetime values of the ultrashort components of ADDDP with an increase in the addition percentage of water. | ||
ADDDP exhibits an intramolecular hydrogen bond with moderate strength in acetonitrile (ΔpKa ∼ 10, discussion D3 given in ESI†). Upon adding even a minimum amount of water (5%), it is sufficient enough (1.5 × 104 equivalents) to interact with the ADDDP and to break the intramolecular hydrogen bond between the phenol and amine with the concomitant formation of intermolecular hydrogen bonding with water. So, there is a greater chance for the solvent water to be the proton acceptor. But, the lifetime of the two ultrashort components is significantly less than the lifetime value established (∼160 ps) for the direct proton transfer to the solvent water (ADDOH in water). Hence, it is apparent that the proton transfer to the solvent water is not likely to happen. This is further supplemented by the pH dependent PCET reaction kinetics which will be discussed later. Similarly, the pendant pyridine functioning as the proton acceptor (both intra- and intermolecular) is also less probable due to the larger proton transfer distance as well as steric reasons. If it occurs, then these two ultrashort components are also warranted in other polar aprotic solvents. But these two ultrashort components exist only in protic solvents. This clearly indicates that the direct proton transfer to the pendant base pyridine is also not feasible. Due to its proximity to the phenolic –OH group, tertiary amine can form a stable six membered cyclic structure during hydrogen bond formation. Hence, direct proton transfer along the intramolecular hydrogen bond is possible (only) with the tertiary amine unit. There are a plethora of reports endorsing the direct proton transfer between the phenol and hydrogen bonded amine by following the kinetics of electro- and photochemical oxidation.62 For an intramolecularly hydrogen bonded system, it is already established that the rate of the MSEPT process depends upon the strength of the hydrogen bond,27–33 the distance between the hydrogen bond donor and acceptor32,71 and a resonance assisted hydrogen bond as a special case.31 Since no other species in ADDDP can form a shorter and stronger hydrogen bond than the amine moiety, the ultrashort decay components τu have been assigned to the MSEPT process with the directly hydrogen bonded amine as the proton acceptor.
The other decay component τ1 (observed only in protic solvents) is not involved in direct proton transfer with any of the other proton acceptors but exhibits a significant kinetic isotopic effect. The possible mechanistic pathway followed by this acid base neutralization reaction in protic solvent is uncertain. But the situation is very much similar to photoacids, in which the bimolecular neutralization reaction between the (photoexcited) acid and base occurs in a much faster time scale, which is facilitated by a (solvent) water mediated proton transfer bridge constructed using the bidirectional hydrogen bond of a water molecule locked in between the acid and base. A similar kind of water mediated proton transfer process is also viable in ADDDP. As a precedent, Akermark and co-workers have synthesised a similar model compound (phenol carrying an imidazole unit at its ortho position) to biomimic water binding in enzymes,55 and have shown the existence of a water mediated hydrogen bond between the phenolic –OH and the pendant imidazole unit in the crystalline state using XRD and deuterium solid state NMR studies, in which the strength of the hydrogen bond between phenol and water is almost equivalent to direct hydrogen bonding with the imidazole unit. Hence, it is envisaged that a single water molecule can form a stronger hydrogen bond between the phenolic –OH and amine/pyridine, such that the strength of the hydrogen bond between the phenol and water is almost equivalent to the direct hydrogen bond between the phenol and the basic amine/pyridine (Scheme 2 and S3†). Under such conditions the water mediated MSEPT process is feasible with these bases as the proton acceptor. Accordingly, τ1 has been assigned to the MSEPT process with amine/pyridine as the proton acceptor which is inherently mediated by the hydrogen bonded water. Reports for the excited state intramolecular proton transfer processes mediated by the water molecule without any participation from electrons are available.49–52 However, no direct evidence for the involvement of a water molecule in transferring the proton from the donor to the acceptor during the electron transfer process was established until now.
 | ||
| Scheme 2 Light driven intramolecular PCET process in ADDDP. | ||
Table S12† and Fig. 3b show that the lifetime value of both the shorter components decreases, but the relative amplitude of τu increases and τ1 decreases with the increase in water percentage i.e., the relative distribution of the lifetime component corresponding to the MSEPT process assisted by direct hydrogen bonding increases with the increase in water percentage (Fig. S23†). This indicates that the phenol prefers to form direct hydrogen bonding with the tertiary amine (rather than a water mediated hydrogen bond) in the presence of a large excess of water. This hypothesis will be true only when the strengthening of the hydrogen bond occurs with the increase in water percentage. Hence, to probe the strength of the hydrogen bond, the pKa slider rule was applied under aqueous conditions. The pKa value of phenol in water is ∼10 and for analogous TEA it is 10.8 and the ΔpKa value is estimated to be ∼1, a very strong hydrogen bond. Whereas in acetonitrile the ΔpKa is ∼10, a hydrogen bond with moderate strength. Hence, while adding a minimum amount of water (5%) to the acetonitrile solution of ADDDP, it cleaves the intramolecular hydrogen bond between the phenol and amine and the water molecule resides in between the phenol and DPA unit through the bidirectional hydrogen bond. Whereas upon increasing the water percentage there is a substantial decrease in the ΔpKa value, which facilitates the formation of the direct hydrogen bond between the phenol and amine. Under this condition, the bonded hydrogen is expected to exist as a 3-center-2-electron bond. Thus, the observed variation in the relative amplitude of τu and τ1 correlates well with the strengthening of the direct hydrogen bond in the presence of a large excess of water. From all of these studies in ACN–H2O/D2O mixture it is clear that ADDDP exhibits only a direct MSEPT process in polar aprotic solvents like acetonitrile whereas on adding a minimum amount of water to the acetonitrile solution, ADDDP prefers to exhibit a water (solvent) mediated MSEPT process and at a higher water percentage it exhibits both direct and water mediated electron proton transfer in almost similar proportions. The transients formed due to the MSEPT process in ADDDP and ADDOH have been validated using femtosecond transient absorption spectral studies and they are described briefly in discussion D4 (ESI†).
Sequence of e−/H+ transfer in a completely intramolecular PCET process
Analogous to ADDOH, monitoring the impact of the pH of the medium on the EPT reaction rate can be used as an experimental marker to explore the sequence of the electron and proton transfer process in ADDDP.Our preliminary investigations with ADDOH have shown that ADDOH in aqueous medium exists in two different forms viz., neutral and deprotonated forms (at pH 2–13), and in the ground state ADDOH exhibits equilibrium between these two forms as a function of the pH of the medium. But in the case of ADDDP, three different pH sensitive groups viz., phenol, tertiary amine and pyridine, exist and hence more than one equilibrating pair may be available as a function of pH (that depends on the pKa value of the pH sensitive groups). So, identifying the different pH dependent ground state forms of ADDDP is a prerequisite to properly assign the excited state deactivation kinetics observed at different pH values. To realize that, steady state and time resolved photophysical studies of ADDDP were carried out at different pH values.
The absorption and emission spectra of ADDDP at different pH values are shown in Fig. S26 and S27† and they show a significant difference in the entire pH range of interest (pH 2–12). While tuning the pH from neutral to acidic, the absorption spectrum shows a decrease in absorbance at ∼350 nm (corresponding to the deprotonated or the strongly hydrogen bonded phenolic unit) with a concomitant increase in absorbance at ∼300 nm (corresponding to the bare/free phenol). This is contrary to ADDOH where there is no change in the absorption spectrum in the entire acidic regime. The change in the absorption spectrum observed in ADDDP may be due to the following: (1) a small fraction of phenol may get deprotonated in the ground state itself due to the basic amine/pyridine which may get protonated while decreasing the pH, or/and (2) the protonation of the basic DPA unit may split apart the phenol from hydrogen bonding and increase the absorbance at ∼300 nm. Hence, from the absorption spectrum it is clear that protonation of either the phenoxide ion or the DPA unit leads to a free phenol analogous to ADDOH, which in turn is expected to increase the emission intensity (Fig. S26c†). Accordingly, there is an enhancement in the emission intensity but to our surprise the fluorescence quantum yield of ADDDP at pH 2.5 is five times greater than that of ADDOH at the same pH and concentration as shown in the photographs in Fig. S26e and S27d.† Decreasing the pH from neutral to acidic shifts the equilibrium from the deprotonated to neutral form of the phenol and the neutral to protonated form of the bases. At pH 2.5, the whole of the DPA unit might get protonated and the bare phenolic moiety is expected to behave analogous to ADDOH. Instead, there is a very large increase in the emission intensity and fluorescence quantum yield value, which indicates the formation of some other new species in the acidic regime. Whereas under basic conditions, the steady state photophysical behaviour of ADDDP is much similar to that of ADDOH (Fig. S27†), where there is an increase in absorbance and decrease in emission intensity due to the ground state deprotonation of the phenolic group (Fig. 4).
 | ||
| Fig. 4 Emission intensity of ADDDP as a function of pH. | ||
To probe this further, time resolved fluorescence studies were carried out at different pH values. Unlike ADDOH, the fluorescence decay curve of ADDDP recorded using TCSPC fits a tri-exponential at all pH values (Fig. S28†). The ultrashort component τ1 has a lifetime value (<100 ps) less than the IRF (550 ps), which traces the kinetics of the MSEPT process and the longer lifetime component τ3 is due to the LE state (similar to ADDOH). Another longer component τ2 (which is not observed in ADDOH) has a relative amplitude that increases with the decrease in pH and it exists predominantly ∼70% at pH 2.5 (Table S15†). This new/predominant emissive species might be responsible for the exponential increase in the emission intensity observed in the acidic regime. Based on the pKa value of the pH sensitive groups, it can be predicted that the predominant species that can possibly exist at this pH is ADDDP carrying a neutral phenolic and protonated DPA unit. If so, then the fluorescence decay profile of ADDDP at pH ∼ 2.5 is expected to duplicate the fluorescence decay profile of ADDOH. But instead, a longer decay component (τ2) with a lifetime value of ∼1 ns exists predominantly. This indicates that at a lower pH some specific interaction in ADDDP blocks the electron transfer process and enhances the emission. Under this condition, the possible specific interaction which can hinder the PET process from phenol is the intramolecular hydrogen bonding between the phenol and the protonated DPA. Because of its closer vicinity with the phenolic group, the protonated amine can act as a hydrogen bond donor and establishes an intramolecular hydrogen bond with the lone pair of electrons available on the phenolic oxygen which in turn hinders the PET process and enhances the emission. This is further supplemented by the time resolved fluorescence studies of ADDDP in other solvents and especially in ACN–H2O/D2O mixture. The fluorescence decay curve of ADDDP in ACN–H2O mixture (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 v/v) recorded using TCSPC exhibits triexponential behaviour which accommodates a longer decay component (τ2) with a lifetime value of 1 ns and a relative amplitude of 26%. Whereas in ACN–D2O mixture, the decay fits only biexponential and there is no such anomalous longer lifetime component. It is well known that the autoprotolysis constant of D2O is much less than that of water (Kw(H2O) = 7.5 Kw(D2O))19 and hence protonating the DPA unit is reluctant in D2O. Similarly, the fluorescence decay profiles of ADDDP in other polar protic and aprotic solvents fit only biexponentially.
:90 v/v) recorded using TCSPC exhibits triexponential behaviour which accommodates a longer decay component (τ2) with a lifetime value of 1 ns and a relative amplitude of 26%. Whereas in ACN–D2O mixture, the decay fits only biexponential and there is no such anomalous longer lifetime component. It is well known that the autoprotolysis constant of D2O is much less than that of water (Kw(H2O) = 7.5 Kw(D2O))19 and hence protonating the DPA unit is reluctant in D2O. Similarly, the fluorescence decay profiles of ADDDP in other polar protic and aprotic solvents fit only biexponentially.
Thus these results show that an intramolecular hydrogen bond donor at the closer proximity of the phenolic –OH group can hinder the electron transfer process more effectively through hydrogen bonding than adding the acid externally as was done in ADDOH. So, the anomalous longer decay component (τ2) is due to ADDDP carrying a protonated DPA unit involved in the hydrogen bonding interaction with the phenol, a new emissive species that has formed in the ground state.
The fluorescence decay profiles recorded using the femtosecond upconversion technique are shown in Fig. 5 and were fitted triexponentially by fixing the lifetime of the longer component (τ2) obtained from the TCSPC technique, and the corresponding lifetime data are given in Table S16.† While lowering the pH from 11 to 2, the lifetime of the two shorter components (τu, τ1) increases with a continuous decrease in relative amplitude as shown in Fig. 6. Noticeably, Fig. 6b shows that the lowering of pH increases the relative amplitude of the longer component τ2 by consuming the relative amplitude of both τu and τ1. This indicates that neither of these two ultrashort components belong to ADDDP carrying a protonated DPA unit. Thus, the two ultrashort components are from the neutral ADDDP existing in equilibrium with the protonated ADDDP in the ground state. So, ADDDP at neutral pH exists in four different forms in the ground state (Fig. S30†): (i) deprotonated form (DF) where the phenolic unit is in the deprotonated state, and it is non-emissive, (ii) protonated form (PF) in which the pendant bases are protonated and are involved in hydrogen bonding with the lone pair of electrons available on the phenolic oxygen, and it is moderately emissive, (iii) neutral form with direct hydrogen bonding between the phenol and amine (NFD) and (iv) neutral form with water mediated hydrogen bonding between the phenol and amine/pyridine (NFW). The latter two cases are weakly emissive.
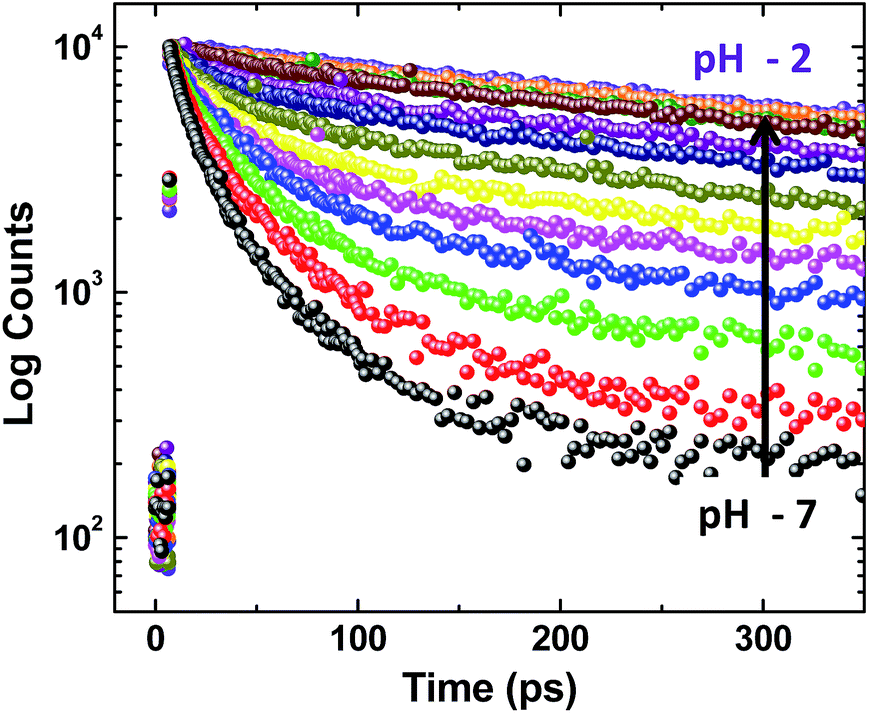 | ||
| Fig. 5 Fluorescence decay curve of ADDDP as a function of pH. | ||
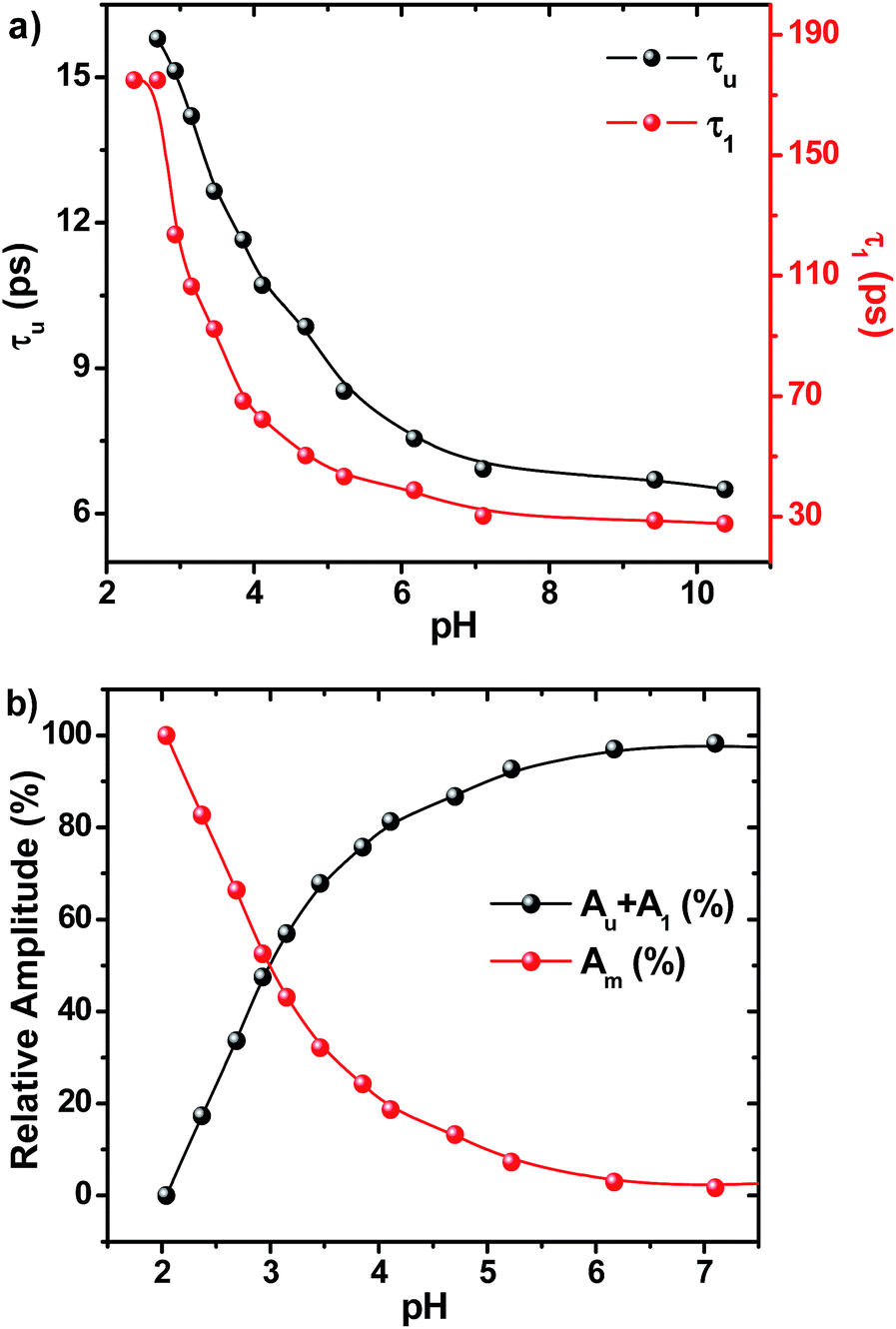 | ||
| Fig. 6 (a) Plot showing the change in the fluorescence lifetime value of τu and τ1 (and hence the rate of the MSEPT process) as a function of pH and (b) upon decreasing the pH, the relative amplitude of A2 increases with the consumption of the sum of the relative amplitude of Au and A1. | ||
On altering the pH, there must be a shift in this acid base equilibrium and it will bring variation in the relative population of the above four components in the ground state. Under such conditions, it is expected that there must be a variation only in the relative amplitude of the respective lifetime components and not in the lifetime value.
Instead, there is a continuous increase in the fluorescence lifetime value of both τu and τ1 as shown in Fig. 6a while decreasing the pH. Our earlier investigation in ACN–H2O/D2O mixture clearly demonstrates that the factor which governs the rate of the MSEPT process in ADDDP is the strength of the intramolecular hydrogen bond. The stronger the hydrogen bond, the faster the MSEPT reaction rate. If so, then the continuous increase in the fluorescence lifetime value of both τu and τ1 must be due to the weakening of the intramolecular hydrogen bond at lower pH. As per the pKa slider rule, the hydrogen bonds in ADDDP are much stronger. However, the decrease in the PCET reaction kinetics indicates that the bonding becomes weaker at lower pH. These results indicate that the ΔpKa value closer to zero may indicate a stronger intramolecular hydrogen bond but it depends upon the pH of the medium. In this case, while decreasing the pH towards ΔpKa (= 0), it weakens the hydrogen bond exponentially. At acidic pH, the interaction between the large excess of H3O+ ion and the basic tertiary amine/pyridine unit weakens the intramolecular hydrogen bonding interaction, which in turn shifts the oxidation potential of phenol to a more positive value and detrimentally alters the energetics of the PCET process. Moreover, the weakening of the hydrogen bond will slow down the PCET reaction kinetics by increasing the reorganization energy corresponding to the proton transfer process. Thus, altering the pH of the medium alters the strength of the intramolecular hydrogen bond which in turn tunes the oxidation potential of phenol and also the PCET reaction rate. Thus, the variation in the PCET reaction rate as a function of pH implicates that the translocation of the electron and proton occurs concertedly. This appears contradictory to what we have observed in ADDOH where no such influence from the pH dependent oxidation potential of the phenol is observed in the entire acidic regime. In the case of ADDOH, the hydrogen bonding interaction is intermolecular and also relatively weaker; hence the rate of EPT is completely dictated by the oxidation strength of the acridinedione moiety. Whereas in ADDDP, the strength of the intramolecular hydrogen bond (both direct and water mediated) is very strong which significantly alters the oxidation potential of the phenol as a function of pH and plays a prime role in tuning the rate of the MSEPT process.
Summary and conclusion
The dual role of solvent water in a multisite electron proton transfer process was unravelled by engaging two different phenolic derivatives of acridinedione dye. Photo-exciting the simple phenol derivative ADDOH triggers a ‘through space electron transfer’ from a non-conjugative pendant phenol to the highly oxidizing excited state of an ADD fluorophore. But in protic solvents like water, this light induced redox process was coupled with the transfer of proton from the phenolic unit to the intermolecularly hydrogen bonded water, as revealed by the significant kinetic isotopic effect (kH/kD ∼ 2.1) on the water induced fluorescence quenching dynamics. In the case of the molecular triad ADDDP, it is an outright organic molecular system that mimics the donor side of photosystem II, in which the phenol (e−/H+ donor) carries the acridinedione (e− acceptor) at its para position and two di-2-picolylamine units at both the ortho positions (H+ acceptor). Hence, after photo-exciting the ADD unit, the unimolecular MSEPT process occurs even in polar aprotic solvents which eventually quenches the emission through a non-radiative deactivation pathway. But in protic solvents like water, two such deactivation channels with a significant kinetic isotopic effect were observed. From a thorough kinetic analysis, it was established that these two excited state deactivation processes are due to two independent MSEPT processes which occur through direct and water mediated hydrogen bonding between the phenol and the DPA unit. Thus, apart from functioning as a proton acceptor, the solvent water can also serve as a proton wire and mediates the multisite electron proton transfer process.Conspicuously, the rate of the MSEPT process exhibited by ADDOH with the solvent water as proton acceptor is unaffected by the pH of the medium. It indicates that being a stronger oxidant facilitates the photoexcited acridinedione to unanimously decide the PCET bimolecular reaction kinetics, without any influence from the pH dependent oxidation potential of the phenol. Thus, the sequential ETPT mechanism dominates the entire pH range and there is no concerted EPT process with either water/added base functions as the proton acceptor in a bimolecular MSEPT process. In the case of ADDDP, the PCET reaction kinetics decrease with the decrease in pH which indicates the concerted transfer of electron and proton in the completely intramolecular MSEPT process. Thus, in the highly oxidized environment, a simple phenol always prefers to undergo an electron first ETPT pathway under bimolecular conditions when water or even base is the proton acceptor. Whereas under unimolecular conditions when proton acceptors are intramolecularly hydrogen bonded to the phenolic group it prefers to exhibit a CPET pathway facilitated by a direct or water mediated intramolecular hydrogen bond even in a highly oxidizing environment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SKT and RS thank CSIR (New Delhi) for the award of Senior Research Fellowship. The authors thank Dr C. Selvaraju, National Centre for Ultrafast Processes, University of Madras for his help with the femtosecond upconversion studies. The authors thank Dr Karunakaran Venugopal, Senior Scientist at NIIST in the Photosciences and Photonics department for his help wotj the femtosecond transient absorption studies. The authors also thank Dr V. Subramanian, Sr Principal Scientist CSIR-CLRI, Chennai for his help with the theoretical studies.References
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- J. M. Savéant, Annu. Rev. Anal. Chem., 2014, 7, 537–560 CrossRef PubMed.
- J. L. Dempsey, J. R. Winkler and H. B. Gray, Chem. Rev., 2010, 110, 7024–7039 CrossRef CAS PubMed.
- C. J. Gagliardi, B. C. Westlake, C. A. Kent, J. J. Paul, J. M. Papanikolas and T. J. Meyer, Chem. Soc. Rev., 2010, 254, 2459–2471 CAS.
- M. N. Jackson and Y. Surendranath, J. Am. Chem. Soc., 2016, 138, 3228–3234 CrossRef CAS PubMed.
- A. Magnuson, M. Anderlund, O. Johansson, P. Lindblad, R. Lomoth, T. Polivka, S. Ott, K. Stensjö, S. Styring, V. Sundström and L. Hammarström, Acc. Chem. Res., 2009, 42, 1899–1909 CrossRef CAS PubMed.
- J. M. Mayer and I. J. Rhile, Biochim. Biophys. Acta, 2004, 1655, 51–58 CrossRef CAS PubMed.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- M. Sjödin, S. Styring, B. Åkermark, L. Sun and L. Hammarström, J. Am. Chem. Soc., 2000, 122, 3932–3936 CrossRef.
- C. Carra, N. Iordanova and S. H. Schiffer, J. Am. Chem. Soc., 2003, 125, 10429–10436 CrossRef CAS PubMed.
- M. Sjödin, S. Styring, H. Wolpher, Y. Xu, L. Sun and L. Hammarström, J. Am. Chem. Soc., 2005, 127, 3855–3863 CrossRef PubMed.
- M. Sjödin, T. Irebo, J. E. Utas, J. Lind, G. Merényi, B. Åkermark and L. Hammarström, J. Am. Chem. Soc., 2006, 128, 13076–13083 CrossRef PubMed.
- S. J. Edwards, A. V. Soudackov and S. H. Schiffer, J. Phys. Chem. A, 2009, 113, 2117–2126 CrossRef CAS PubMed.
- L. MacAleese, S. Hermelin, K. El Hage, P. Chouzenoux, A. Kulesza, R. Antoine, L. Bonacina, M. Meuwly, J. P. Wolf and P. Dugourd, J. Am. Chem. Soc., 2016, 138, 4401–4407 CrossRef CAS PubMed.
- P. Goyal and S. H. Schiffer, ACS Energy Lett., 2017, 2, 512–519 CrossRef CAS.
- C. Costentin, M. Robert and J. M. Savéant, Phys. Chem. Chem. Phys., 2010, 12, 11179–11190 RSC.
- J. Nomrowski and O. S. Wenger, Inorg. Chem., 2015, 54, 3680–3687 CrossRef CAS PubMed.
- G. F. Manbeck, E. Fujita and J. J. Concepcion, J. Am. Chem. Soc., 2016, 138, 11536–11549 CrossRef CAS PubMed.
- T. Irebo, M. T. Zhang, T. F. Markle, A. M. Scott and L. Hammarström, J. Am. Chem. Soc., 2012, 134, 16247–16254 CrossRef CAS PubMed.
- S. H. Schiffer, Energy Environ. Sci., 2012, 5, 7696–7703 Search PubMed.
- J. Soetbeer, P. Dongare and L. Hammarström, Chem. Sci., 2016, 7, 4607–4612 RSC.
- G. T. Babcock, B. A. Barry, R. J. Debus, C. W. Hoganson, M. Atamian, L. McIntosh, I. Sithole and C. F. Yocum, Biochemistry, 1989, 28, 9557–9565 CrossRef CAS PubMed.
- P. Gilli, L. Pretto, V. Bertolasi and G. Gilli, Acc. Chem. Res., 2009, 42, 33–44 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- S. H. Schiffer and A. A. Stuchebrukhov, Chem. Rev., 2010, 110, 6939–6960 CrossRef PubMed.
- S. H. Schiffer, J. Am. Chem. Soc., 2015, 137, 8860–8871 CrossRef PubMed.
- T. Maki, Y. Araki, Y. Ishida, O. Onomura and Y. Matsumura, J. Am. Chem. Soc., 2001, 123, 3371–3372 CrossRef CAS PubMed.
- I. J. Rhile and J. M. Mayer, J. Am. Chem. Soc., 2004, 126, 12718–12719 CrossRef CAS PubMed.
- I. J. Rhile, T. F. Markle, H. Nagao, A. G. DiPasquale, O. P. Lam, M. A. Lockwood, K. Rotter and J. M. Mayer, J. Am. Chem. Soc., 2006, 128, 6075–6088 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2007, 129, 9953–9963 CrossRef CAS PubMed.
- T. F. Markle and J. M. Mayer, Angew. Chem., Int. Ed., 2008, 47, 738–740 CrossRef CAS PubMed.
- M. T. Zhang, T. Irebo, O. Johansson and L. Hammarström, J. Am. Chem. Soc., 2011, 133, 13224–13227 CrossRef CAS PubMed.
- J. N. Schrauben, M. Cattaneo, T. C. Day, A. L. Tenderholt and J. M. Mayer, J. Am. Chem. Soc., 2012, 134, 16635–16645 CrossRef CAS PubMed.
- L. Sun, M. Burkitt, M. Tamm, M. K. Raymond, M. Abraham-sson, D. LeGourriérec, Y. Frapart, A. Magnuson, P. H. Kenéz, P. Brandt, A. Tran, L. Hammarström, S. Styring and B. Åkermark, J. Am. Chem. Soc., 1999, 121, 6834–6842 CrossRef CAS.
- L. Sun, L. Hammarström, B. Åkermark and S. Styring, Chem. Soc. Rev., 2001, 30, 36–49 RSC.
- L. Hammarström and S. Styring, Energy Environ. Sci., 2011, 4, 2379–2388 Search PubMed.
- C. J. Fecenko, H. H. Thorp and T. J. Meyer, J. Am. Chem. Soc., 2007, 129, 15098–15099 CrossRef CAS PubMed.
- T. Irebo, S. Y. Reece, M. Sjödin, D. G. Nocera and L. Hammarström, J. Am. Chem. Soc., 2007, 129, 15462–15464 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2007, 129, 5870–5879 CrossRef CAS PubMed.
- M. T. Zhang and L. Hammarström, J. Am. Chem. Soc., 2011, 133, 8806–8809 CrossRef CAS PubMed.
- J. Bonin, C. Costentin, M. Robert, M. Routier and J. M. Savéant, J. Am. Chem. Soc., 2013, 135, 14359–14366 CrossRef CAS PubMed.
- P. Dongare, S. Maji and L. Hammarström, J. Am. Chem. Soc., 2016, 138, 2194–2199 CrossRef CAS PubMed.
- L. Biczók and H. Linschitz, J. Phys. Chem., 1995, 99, 1843–1845 CrossRef.
- L. Biczók, T. Bérces and H. Linschitz, J. Am. Chem. Soc., 1997, 119, 11071–11077 CrossRef.
- S. Prashanthi and P. R. Bangal, Chem. Commun., 2009, 1757–1759 RSC.
- P. Hemant Kumar, Y. Venkatesh, S. Prashanthi, D. Siva, B. Ramakrishna and P. R. Bangal, Phys. Chem. Chem. Phys., 2014, 16, 23173–23181 RSC.
- C. C. Hsieh, C. M. Jiang and P. T. Chou, Acc. Chem. Res., 2010, 43, 1364–1374 CrossRef CAS PubMed.
- A. P. Demchenko, K. C. Tangb and P. T. Chou, Chem. Soc. Rev., 2013, 42, 1379–1408 RSC.
- M. Fischer and P. Wan, J. Am. Chem. Soc., 1998, 120, 2680–2681 CrossRef CAS.
- M. Fischer and P. Wan, J. Am. Chem. Soc., 1999, 121, 4555–4562 CrossRef CAS.
- M. Flegel, M. Lukeman, L. Huck and P. Wan, J. Am. Chem. Soc., 2004, 126, 7890–7897 CrossRef CAS PubMed.
- M. K. Nayak and P. Wan, Photochem. Photobiol. Sci., 2008, 7, 1544–1554 CAS.
- M. Rini, B. Z. Magnes, E. Pines and E. T. J. Nibbering, Science, 2003, 301, 349–351 CrossRef CAS PubMed.
- O. F. Mohammed, D. Pines, J. Dreyer, E. Pines and E. T. J. Nibbering, Science, 2005, 310, 83–85 CrossRef CAS PubMed.
- J. E. Utas, M. Kritikos, D. Sandström and B. Åkermark, Biochim. Biophys. Acta, 2006, 1757, 1592–1596 CrossRef CAS PubMed.
- M. Högbom, Y. Huque, B. M. Sjöberg and P. Nordlund, Biochemistry, 2002, 41, 1381–1389 CrossRef.
- H. J. Timpe, S. Ulrich and J. P. Fouassier, J. Photochem. Photobiol., A, 1993, 73, 139–150 CrossRef CAS.
- B. Venkatachalapathy and P. Ramamurthy, Phys. Chem. Chem. Phys., 1999, 1, 2223–2230 RSC.
- V. Thiagarajan, C. Selvaraju, E. J. Padma Malar and P. Ramamurthy, ChemPhysChem, 2004, 5, 1200–1209 CrossRef PubMed.
- P. Ashokkumar, V. T. Ramakrishnan and P. Ramamurthy, Eur. J. Org. Chem., 2009, 5941–5947 CrossRef CAS.
- G. J. Zhao and K. L. Han, Acc. Chem. Res., 2012, 45, 404–413 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer Science, New York, USA, 3rd edn, 2006 Search PubMed.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS.
- M. E. Tessensohn, H. Hirao and R. D. Webster, J. Phys. Chem. C, 2013, 117, 1081–1090 CAS.
- L. P. McMahon, W. J. Colucci, M. L. McLaughlin and M. D. BarMey, J. Am. Chem. Soc., 1992, 114, 8442–8448 CrossRef CAS.
- L. R. Khundkar, J. W. Perry, J. E. Hanson and P. B. Dervan, J. Am. Chem. Soc., 1994, 116, 9700–9709 CrossRef CAS.
- Y. Kaizu, Inorg. Chem., 1998, 37, 3371–3375 CrossRef.
- B. Venkatachalapathy, P. Ramamurthy and V. T. Ramaklishnan, J. Photochem. Photobiol., A, 1997, 163–169 CrossRef CAS.
- A. Harriman, J. Phys. Chem., 1987, 91, 6102–6104 CrossRef CAS.
- P. Ashokkumar, V. T. Ramakrishnan and P. Ramamurthy, ChemPhysChem, 2011, 12, 389–396 CrossRef CAS PubMed.
- M. Kuss-Petermann, H. Wolf, D. Stalke and O. S. Wenger, J. Am. Chem. Soc., 2012, 134, 12844–12854 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, IR, NMR, HRMS and necessary supplementing photophysical spectra along with corresponding data are provided in the form of a table. See DOI: 10.1039/c7sc03161k |
| This journal is © The Royal Society of Chemistry 2018 |