
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rh(III)-catalyzed regioselective intermolecular N-methylene Csp3–H bond carbenoid insertion†
Haisheng
Xie
 a,
Zongren
Ye
b,
Zhuofeng
Ke
*b,
Jianyong
Lan
a,
Huanfeng
Jiang
*a and
Wei
Zeng
*a
a,
Zongren
Ye
b,
Zhuofeng
Ke
*b,
Jianyong
Lan
a,
Huanfeng
Jiang
*a and
Wei
Zeng
*a
aSchool of Chemistry and Chemical Engineering, South China University of Technology, No. 381 Wushan Road, Guangzhou, 510641, P. R. China. E-mail: jianghf@scut.edu.cn; zengwei@scut.edu.cn
bSchool of Materials Science & Engineering, PCFM Lab, Sun Yat-sen University, Guangzhou, 510275, P. R. China. E-mail: kezhf3@mail.sysu.edu.cn
First published on 27th November 2017
Abstract
A Rh(III)-catalyzed regioselective intermolecular carbenoid insertion into the N-methylene Csp3–H bond of acyclic aliphatic amides has been achieved, taking advantage of bidentate–chelation assistance. This methodology has been successfully applied to a broad range of linear and branched-chain N-alkylamides, thus providing a practical method for the assembly of diverse beta-amino esters. Mechanism studies and density functional theory (DFT) calculations revealed that a singlet Fischer type carbene insertion via an outer-sphere pathway was involved in this N-methylene Csp3–H bond carbenoid insertion.
Introduction
Alkyl C–H bond carbenoid functionalization is one of the most challenging topics for atom-economical C–C bond formations.1 Over the past few decades, transition-metal catalyzed intra- and intermolecular heteroatom-adjacent Csp3–H carbenoid insertions have been well-established for assembling structurally complex molecules, but substrate-specific problems have not yet been overcome.2 In terms of the intermolecular version of N-adjacent Csp3–H bond carbenoid insertion to acyclic amines, the existing transition metal catalytic systems only tolerate N-methyl Csp3–H bonds instead of N-methylene Csp3–H bonds (Scheme 1a).2a The intermolecular carbenoid insertion into the N-methylene Csp3–H bonds of acyclic aliphatic amines is very difficult to achieve because of the delicate balance between steric and electronic factors.3 | ||
| Scheme 1 Approaches to versatile Csp3–H carbenoid insertion. | ||
Recently, chelation-assisted intermolecular Csp2–H bond carbenoid functionalization has obtained a breakthrough via the “Inner-Sphere Pathway4 (ISP)”; this strategy provides a powerful approach for site-selective aryl C–H carbenoid insertion. In this regard, Yu,5 Glorious,6 Rovis,7 Li8 and others9 successively reported that Rh(III) and Co(III)-catalyzed ortho aryl Csp2–H cross-coupling reactions with diazo compounds could conveniently install C–C bonds into arenes by employing oximes, hydroxamic acids, pyridines or quaternary ammoniums as directing groups. In sharp contrast, ligand-directed alkyl Csp3–H carbenoid functionalization remains almost undeveloped, owing to such bonds possessing a relatively smaller s-orbital contribution and larger bond dissociation energy.10 To date, only Martin and Zhou have ever reported that transition metal-catalyzed intermolecular Csp3–H bond alkylation with diazo compounds could occur through the ISP using aryl bromides, arylamine N-oxides and quinoline as reaction platforms, but these tactics were limited to only primary methyl Csp3–H bonds (Scheme 1b).11 Therefore, developing various types of methylene Csp3–H carbenoid insertion remains challenging yet highly desirable. Very recently, we have accomplished a novel Ir(III)-catalyzed bidentate-assisted regioselective methylene Csp3–H nitrene insertion,12 in which the “Outer-Sphere Pathway (OSP)”4,12 is involved in the transformation. Undoubtedly, the chelation-assisted OSP has already brought us a rising innovative concept, thus providing a promising approach to achieve versatile methylene Csp3–H functionalizations. Herein, we report an unprecedented Rh(III)-catalyzed bidentate-assisted regioselective intermolecular carbenoid insertion of N-methylene Csp3–H bonds via the OSP (Scheme 1c). This protocol constitutes a unique tool to rapidly build up complex linear beta-amino acid derivatives, which are among the most important precursors of beta-peptides and beta-lactams13 and feature in a large number of naturally occurring and unnatural compounds.14
Results and discussion
We initiated our study by investigating the cross-coupling reaction of N-butyl-pyridine-2-carboxylic acid amide (1a) and α-diazo-β-ketoester (2a) in the presence of metal catalysts including [Cp*IrCl2]2, Cp*Co(CO)I2, Cp*Co(MeCN)3SbF6, RhCl3, Rh2(OAc)4 and [Cp*RhCl2]2 (5 mol%) and Ag2CO3 (20 mol%) in CH3CN at 100 °C for 24 h (see Table S-1 in ESI†). To our delight, screening of the catalysts quickly revealed that [Cp*RhCl2]2 could provide the coupling product 3a with a promising 47% yield (Table 1, entry 2), in which the C–H carbenoid insertion occurred highly regioselectively at the N-methylene C–H bond. Unfortunately, other transition metal salts such as [Cp*IrCl2]2, Cp*Co(CO)I2, Rh2(OAc)4, etc. were not efficient at all. Then, various types of silver additive were evaluated (entries 3–7) and it was found that employing AgOAc as an additive could moderately improve the yield of 3a from 47% to 65% (compare entry 2 with 7, also see Table S-2 in ESI†). The reaction conversion could be further promoted when the transformation was conducted in TFE, which delivered an 89% yield of 3a (entry 8); however, switching to other solvents such as 1,4-dioxane or DMSO led to a significantly lower coupling efficiency (see Table S-3 in ESI†). Also, lowering or increasing the reaction temperature resulted in worse results (entries 9 and 10).|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Additive | Solvent | Yieldb (%) |
| a Unless otherwise noted, all of the reactions were carried out using N-butyl-pyridine-2-carboxylic acid amide (1a) (0.10 mmol) and diazo compound (2a) (0.20 mmol) with a metal catalyst (5.0 mol%) in the presence of a silver salt (20 mol%) in solvent (1.0 mL) at 100 °C for 24 h under Ar in a sealed reaction tube, followed by flash chromatography on SiO2. b Isolated yield. c TFE refers to 2,2,2-trifluoroethanol. d The reaction temperature is 80 °C. e The reaction temperature is 110 °C. | ||||
| 1 | Rh2(OAc)4 | Ag2CO3 | CH3CN | 0 |
| 2 | [Cp*RhCl2]2 | Ag2CO3 | CH3CN | 47 |
| 3 | [Cp*RhCl2]2 | AgClO4 | CH3CN | — |
| 4 | [Cp*RhCl2]2 | AgSbF6 | CH3CN | 28 |
| 5 | [Cp*RhCl2]2 | AgBF4 | CH3CN | 15 |
| 6 | [Cp*RhCl2]2 | AgNTf2 | CH3CN | 63 |
| 7 | [Cp*RhCl2]2 | AgOAc | CH3CN | 65 |
| 8 | [Cp*RhCl2]2 | AgOAc | TFEc | 89 |
| 9 | [Cp*RhCl2]2 | AgOAc | TFE | 69d |
| 10 | [Cp*RhCl2]2 | AgOAc | TFE | 71e |
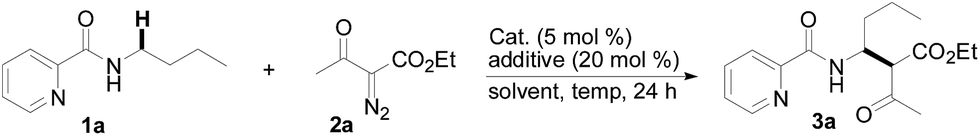
With this protocol in hand, the scope of the Rh(III)-catalyzed N-methylene C–H carbenoid insertion of N-butyl-pyridine-2-carboxylic acid amide (1a) was first investigated with a range of diazo compounds 2 (Table 2). As illustrated for 3a–3i, diacceptor- and donor/acceptor-substituted diazo compounds underwent smooth cross-coupling reactions with N-methylene C–H bonds to furnish beta-amino esters (3a–3i, 43–91% yields). Among them, alpha-diazo-beta-ketoesters participated in the transformation to produce alpha-acyl-beta-amino esters (3a and 3b, 89% and 43% yield, respectively). Moreover, various alpha-diazo-beta-arylesters are also applicable to the present transformation, leading to the formation of the corresponding alpha-aryl and beta-amino esters, in which the substituent on the aryl ring had an important effect on the yield of the reaction. These alpha-diazo-beta-arylesters with electron-deficient phenyl rings gave the products in moderate to excellent yields (3c–3h, 50–91% yields). On the contrary, compared with diazo-phenyl-acetic acid methyl ester (3i, 51% yield), when an electron-richer aryl group-containing diazo ester such as diazo-(4-methoxy-phenyl)-acetic acid methyl ester was used, an unexpected alkene 3k (Z/E = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was formed in 67% yield, and no desired beta-amino ester 3j was observed.
:1) was formed in 67% yield, and no desired beta-amino ester 3j was observed.
|
|
|---|
| a All of the reactions were carried out using amides (1) (0.10 mmol) and diazo compounds (2) (0.20 mmol) with [Cp*RhCl2]2 (5.0 mol%) in the presence of AgOAc (20 mol%) in 2,2,2-trifluoroethanol (1.0 mL) at 100 °C for 24 h under Ar in a sealed reaction tube, followed by flash chromatography on SiO2. b Isolated yield. c d.r. values were determined by 1H NMR spectroscopy, please see ESI. |

|

|

|
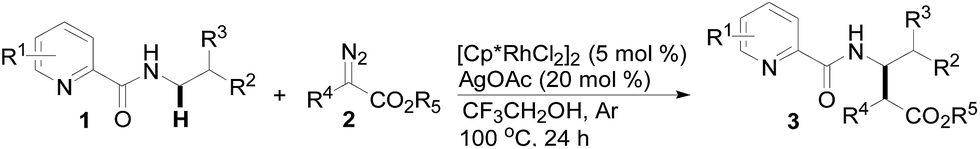
Subsequently, we prepared the 3- or 4-substituted pyridine-2-carboxylic acid butylamides, and investigated the substitution effect of pyridine moieties on N-methylene Csp3–H bond carbenoid insertion with 2-diazo-3-oxo-butyric acid methyl ester 2a. It was found that introducing a methyl group or bromo group into the 3- or 4-position of the pyridine ring could lead to moderate yields of 3l and 3n (48%), and a pyridine ring with a strong electron-withdrawing group (–NO2) was not tolerated for this transformation (3m).
The scope of the present procedure with regard to different types of N-amido alkane has been further evaluated. Compared with the N-n-butyl-substituted amide (1a), the shorter or longer straight-chain alkylamine-based amides could be smoothly regioselectively installed a Csp3–Csp3 bond into the alpha-position of the alkylamine moiety in 78–86% yields (3o–3r). Branched-chain 3-methyl-butylamine-based amides and phenylpropylamine- or phenylethylamine-based amides could also tolerate this reaction system and afforded structurally complex beta-amino acid derivatives 3s (76%), 3t (83%) and 3u (73%). Meanwhile, we also observed that the intermolecular N-methylene Csp3–H bond carbenoid insertion of 2-thiophen-3-yl-ethylamine-based amides also proceeded well to give a 66% yield of 3v, and thiophenyl C–H carbenoid insertion did not occur.15 However, N-benzyl-substituted amides made the transformation a little sluggish, possibly due to steric hindrance from the phenyl ring suppressing the N-methylene Csp3–H bond insertion (3w–3zand 3-1a–3-1c). To our surprise, the present protocol was also applicable to an alkenyl functional group-containing amidoalkane, in which the carbon–carbon double bond could be kept intact (3-1d, 40% yield).16 More importantly, in addition to the N-allylamide, the N-cyclopropylmethyleneamine-based amide was also amenable to the reaction, furnishing the desired beta-cyclopropyl-beta-amino ester 3-1e in a 49% yield. This transformation was not only limited to the N-methylene Csp3–H bond;17N-cyclopropylamide could also couple with diazoester 2a through an N-methyne Csp3–H bond carbenoid insertion to provide the target product 3-1f (46%). Unfortunately, pyridine-2-carboxylic acid (2-acetylamino-ethyl)-amide was not tolerated for this transformation, possibly due to the coordination between Rh(III) and 1,2-bisamide “N” inhibiting the Csp3–H bond carbenoid insertion (3-1g, 0%). Finally, the post-synthetic utility of this transformation revealed that 2-pyridyl carboxyamide 3e could be smoothly converted into a N–H free beta-amino acid (4a) in a 67% yield via a one-pot process (see ESI† for more details).
Designed control experiments, as well as DFT studies (see ESI† for more details), were performed to elucidate the plausible reaction mechanism (Scheme 2). Treatment of N-butyl-benzamide (1y) or N,N-dibutyl-benzamide (1z) with alpha-diazo ester (2a) under our standard conditions did not provide the corresponding target products 5a or 5b (Scheme 2a and b),18 and thus demonstrated that the pyridyl group and amide “N” played a significant bichelate-directing role in enabling the N-methylene C–H carbenoid insertion. Meanwhile, when 1a was subjected to 1.0 equiv. of AcOD in the presence of diazo compound 2a, no H/D exchange was detected at the alpha- or beta-position of the beta-aminoester 3a (Scheme 2c). Although this experiment implied that an irreversible concerted metalation–deprotonation (CMD) process followed by metal protonation was possibly involved in this transformation,19 our DFT study further excluded an inner-sphere mechanism via bidentate-assisted N-methylene Csp3–H bond activation, which is required to overcome an activation free energy of 49.1 kcal mol−1 (TS3, Fig. S-5, ESI†) due to the three-membered ring strain. Moreover, treatment of d-1m (78% D) with 2a afforded the deuterated product d-3w, in which 81% D was inserted at both the alpha- and beta-positions of the beta-amino ester d-3w (Scheme 2d); this result clearly indicated that a two-electron carbenoid insertion into the Csp3–H bond occurred in the presence of bidentate–chelation assistance. DFT calculations (Fig. 1, detailed pathways are shown in Fig. S-5, ESI†) were carried out to further confirm the carbenoid insertion process. In the Rh-carbenoid formation stage, the Rh-carbenoid is formed via transition state TS1, with a calculated activation free energy of 33.8 kcal mol−1 (Cat → TS1). Subsequently, we further evaluated both the singlet and the triplet carbenoid insertion pathways. The corresponding DFT results suggest that the carbenoid insertion proceeds in a singlet Fischer type carbene manner (21.9 kcal mol−1, TS2S-a; −2.5 kcal mol−1, TS2S-b), and the triplet pathway through radical recombination is less feasible due to the high activation free energy (43.4 kcal mol−1, TS2T-a). The singlet carbene pathway was further confirmed by the control experiment (Scheme 2g), in which using TEMPO (1.0 equiv.) did not significantly decrease the reaction yield (91% of 3a).
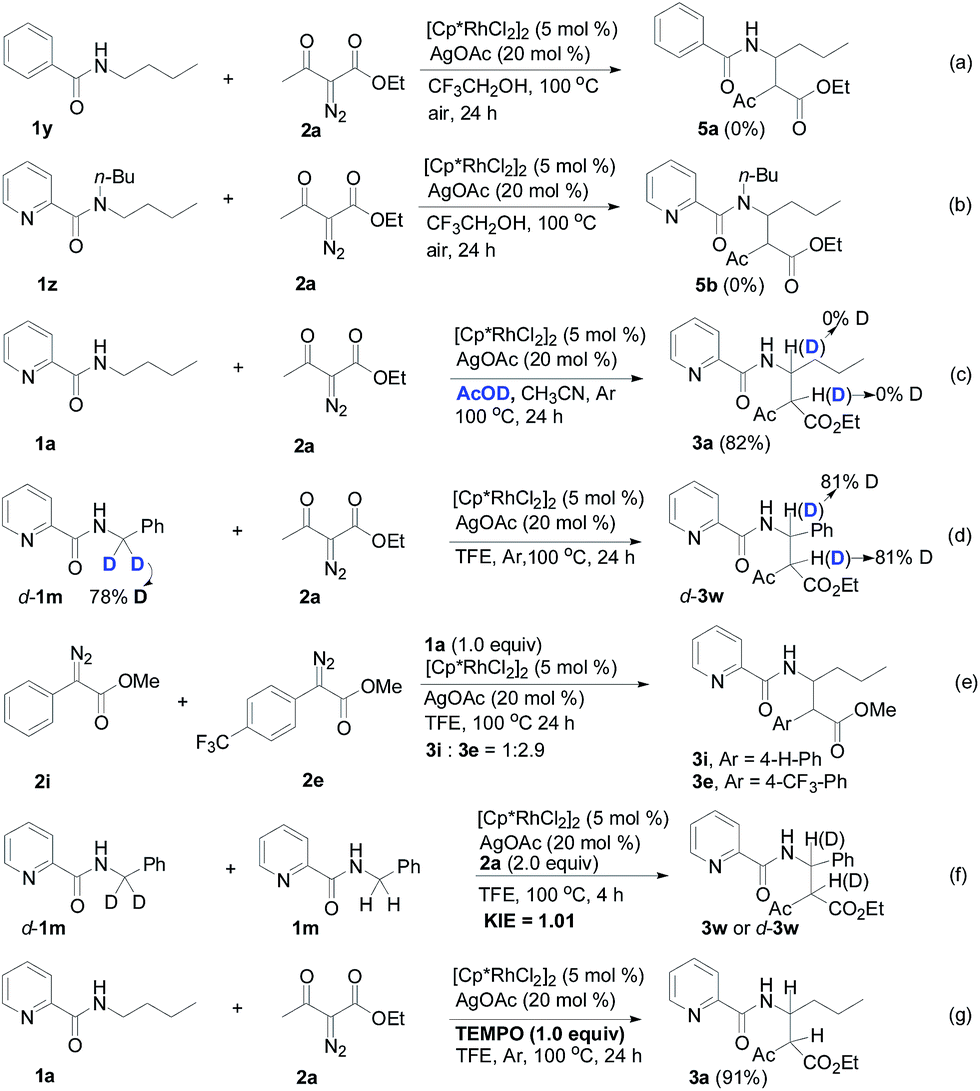 | ||
| Scheme 2 Preliminary mechanism studies. | ||
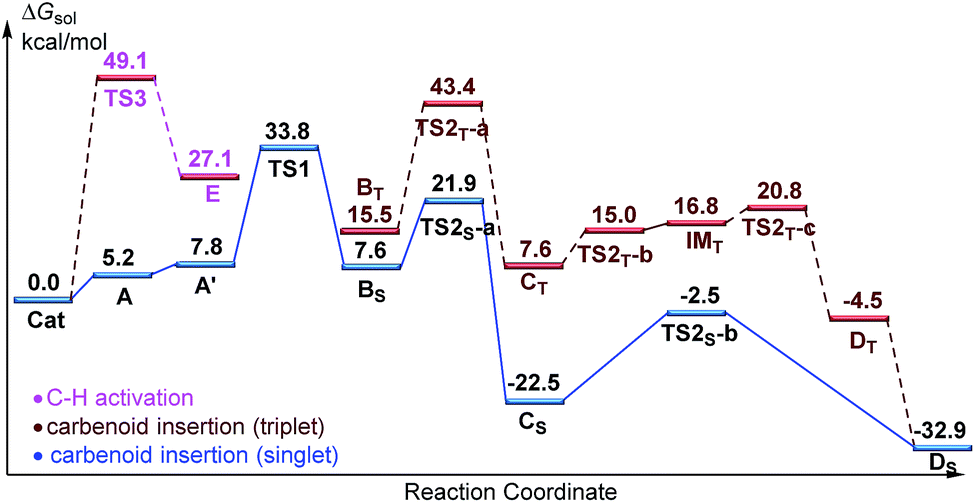 | ||
| Fig. 1 The free energy profiles for the Rh(III)-catalyzed regioselective N-methylene Csp3–H bond carbenoid functionalization. The free energies are reported in kcal mol−1 at the M06-L/BSII/SMD(2,2,2-trifluoroethanol)//M06-L/BSI level of theory. | ||
The N-methylene C–H carbenoid insertion between alpha-aryl-alpha-diazo esters differing in electronic effects indicates that an electron-deficient diazo compound tended to form a rhodium carbene at a relatively higher rate (Scheme 2e). Moreover, a competitive cross-coupling of an equimolar mixture of d-1m and 1m with alpha-diazo ester 2a also gave a kH/kD value of 1.01 on the basis of the 1H NMR spectrum (Scheme 2f), suggesting that C–H bond carbenoid insertion did not involve the rate-limiting step of this transformation. These results are further supported by our DFT data, which demonstrated that the formation of Rh-carbenoid viaTS1 (ΔG‡ = 33.8 kcal mol−1) should be the rate-determining step.
A mechanism rationale coherent with these results and the DFT studies is proposed in Scheme 3. The initial coordination of the pyridyl nitrogen and amide nitrogen of substrate 1f to an active Rh(III) catalyst affords complex A. Subsequent interaction of complex A with diazo compound 2a is followed by denitrogenation to generate Rh carbene species B. The rhodium complex B undergoes a bichelate-assisted singlet Fischer type carbenoid insertion into the N-methylene Csp3–H bond via an outer-sphere pathway, successively producing the corresponding imine intermediate C and Rh(III) complex D. Further protonization of complex D furnishes the desired beta-amino ester 3p with the regeneration of the Rh(III) catalyst.
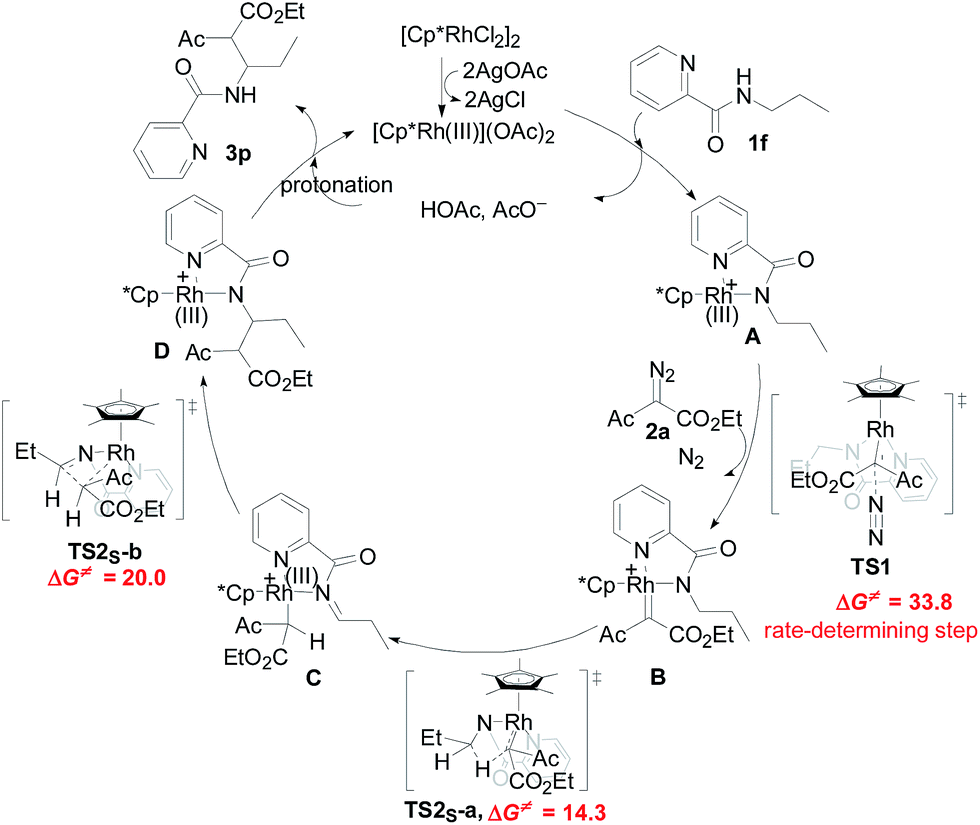 | ||
| Scheme 3 Proposed reaction mechanism. | ||
Conclusions
In summary, we have developed the first Rh(III)-catalyzed intermolecular N-methylene Csp3–H bond carbenoid insertion of acyclic aliphatic amides with high regioselectivity. In these systems, bidentate–chelation acts as a unique platform to enable the cross-coupling of N-methylene Csp3–H bonds with diazo compounds through the “Outer-Sphere Pathway”. This strategy could have broad implications on future research directions on selective Csp3–H functionalization. Moreover, this reaction tolerates a broad scope of substrates and provides an effective approach to diverse beta-amino esters. Further efforts to achieving an asymmetric version of this transformation are underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Key Research Program of China (No. 2016YFA0602900), NSFC (No. 21372085, 21473261, and 21673301) and Guangdong Natural Science Funds for Distinguished Young Scholars (No. 2015A030306027) for financial support.Notes and references
- For selected reviews, see: (a) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861 CrossRef CAS PubMed; (b) H. M. L. Davies and Y. Lian, Acc. Chem. Res., 2012, 45, 923 CrossRef CAS PubMed.
- For the cross-coupling reaction of diazo compounds with N-methyl Csp3–H bonds of linear aliphatic amines, see: (a) H. M. L. Davies and C. Venkataramani, Angew. Chem., Int. Ed., 2002, 41, 2197 CrossRef CAS; (b) H. M. L. Davies and Q. Jin, Org. Lett., 2004, 6, 1769 CrossRef CAS PubMed; (c) H. M. L. Davies and A. Ni, Chem. Commun., 2006, 3110 RSC. For the cross-coupling reaction of diazo compounds with N-methylene Csp3–H bonds of cyclic amines, see: (d) H. M. L. Davies, C. Venkataramani, T. Hansen and D. W. Hopper, J. Am. Chem. Soc., 2003, 125, 6462 CrossRef CAS PubMed; (e) H. M. L. Davies, T. Hansen, D. W. Hopper and S. A. Panaro, J. Am. Chem. Soc., 1999, 121, 6509 CrossRef CAS; (f) J. M. Axten, R. Ivy, L. Krim and J. D. Winkler, J. Am. Chem. Soc., 1999, 121, 6511 CrossRef CAS. For the cross-coupling reaction of diazo compounds with O-adjacent Csp3–H bonds of ethers, see: (g) D. M. Guptill and H. M. L. Davies, J. Am. Chem. Soc., 2014, 136, 17718 CrossRef CAS PubMed; (h) H. M. L. Davies and T. Hansen, J. Am. Chem. Soc., 1997, 119, 9075 CrossRef CAS.
- (a) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed; (b) S. M. Nicolle, W. Lewis, C. J. Hayes and C. J. Moody, Angew. Chem., Int. Ed., 2016, 55, 3749 CrossRef CAS PubMed; (c) K. Liao, S. Negretti, D. G. Musaev, J. Bacsa and H. M. L. Davies, Nature, 2016, 533, 230 CrossRef CAS PubMed.
- (a) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (b) K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040 CrossRef CAS PubMed; (c) S. H. Park, J. Kwak, K. Shin, J. Ryu, Y. Park and S. Chang, J. Am. Chem. Soc., 2014, 136, 2492 CrossRef CAS PubMed.
- W.-W. Chan, S.-F. Lo, Z. Zhou and W.-Y. Yu, J. Am. Chem. Soc., 2012, 134, 13565 CrossRef CAS PubMed.
- D. Zhao, J. H. Kim, L. Stegemann, C. A. Strassert and F. Glorious, Angew. Chem., Int. Ed., 2015, 54, 4508 CrossRef CAS PubMed.
- T. K. Hyster, K. E. Ruhl and T. Rovis, J. Am. Chem. Soc., 2013, 135, 5364 CrossRef CAS PubMed.
- S. Yu, S. Liu, Y. Lan, B. Wan and X. Li, J. Am. Chem. Soc., 2015, 137, 1623 CrossRef CAS PubMed.
- (a) J. Jeong, P. Patel, H. Hwang and S. Chang, Org. Lett., 2014, 16, 4598 CrossRef CAS PubMed; (b) J. Wang, M. Wang, K. Chen, S. Zha, C. Song and J. Zhu, Org. Lett., 2016, 18, 1178 CrossRef CAS PubMed; (c) Z. Liu and J. Wang, J. Org. Chem., 2013, 78, 10024 CrossRef CAS PubMed.
- (a) D. G. Musaev and S. B. Blakey, Organometallics, 2012, 31, 4950 CrossRef CAS; (b) J. Xie, C. Pan, A. Abdukader and C. Zhu, Chem. Soc. Rev., 2014, 43, 5245 RSC.
- (a) Á. Gutiérrez-Bonet, F. Juliá-Hernández, B. de Luis and R. Martin, J. Am. Chem. Soc., 2016, 138, 6384 CrossRef PubMed; (b) B. Zhou, Z. Chen, Y. Yang, W. Ai, H. Tang, Y. Wu, W. Zhu and Y. Li, Angew. Chem., Int. Ed., 2015, 54, 12121 CrossRef CAS PubMed; (c) W. Hou, Y. Yang, Y. Wu, H. Feng, Y. Li and B. Zhou, Chem. Commun., 2016, 52, 9672 RSC.
- X. Xiao, C. Hou, Z. Zhang, Z. Ke, J. Lan, H. Jiang and W. Zeng, Angew. Chem., Int. Ed., 2016, 55, 11897 CrossRef CAS PubMed.
- For selected examples, see: (a) T. N. Salzmann, R. W. Ratcliffe, B. G. Christensen and F. A. Bouffard, J. Am. Chem. Soc., 1980, 102, 6161 CrossRef CAS; (b) D. Seebach, S. Abele, K. Gademann and B. Jaun, Angew. Chem., Int. Ed., 1999, 38, 1595 CrossRef CAS; (c) Z. Juaristi, D. Quintania and J. Escalante, Aldrichimica Acta, 1994, 27, 3 Search PubMed.
- For selected reviews, see: (a) R. M. Williams and J. A. Hendrix, Chem. Rev., 1992, 92, 889 CrossRef CAS; (b) S. France, A. Weatherwax, A. E. Taggi and T. Lectka, Acc. Chem. Res., 2004, 37, 592 CrossRef CAS PubMed; (c) G. P. Pollini, S. Benetti, C. De Risi and V. Zanirato, Chem. Rev., 2006, 106, 2434 CrossRef CAS PubMed; (d) Y.-F. Wang, Q.-W. Shi, M. Dong, H. Kiyota, Y.-C. Gu and B. Cong, Chem. Rev., 2011, 111, 7652 CrossRef CAS PubMed.
- For selected examples, see: (a) R. J. Gillespie, J. Murray-Rust, P. Murray-Rust and A. E. A. Porter, Tetrahedron, 1981, 37, 743 CrossRef CAS; (b) T. Bowles, R. J. Gillespie, A. E. A. Porter, J. A. Rechka and H. S. Rzepa, J. Chem. Soc., Perkin Trans. 1, 1988, 4, 803 RSC.
- For selected examples, see: (a) R. Sambasivan and Z. T. Ball, Angew. Chem., Int. Ed., 2012, 51, 8568 CrossRef CAS PubMed; (b) L. Gao, G.-S. Hwang and D. H. Ryu, J. Am. Chem. Soc., 2011, 133, 20708 CrossRef CAS PubMed.
- Rh(III)-catalyzed Csp3–H bond carbenoid insertion is only selective for the N-methylene Csp3–H bond. N-methyl pyridine-2-carboxylic acid amide (1x) could not tolerate the reaction conditions.
- It should be noted that starting materials 1y, 1z and 2a were almost completely recovered, and also no dimers derived from diazo compound 2a were detected.
- We also ran the H/D exchange experiment in TF3CD2OD (1.0 mL) instead of the solvent system (AcOD/CH3CN, Scheme 2c), and we still did not observe H/D exchange at the alpha- or beta-position of the beta-aminoester 3a, please see the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc03802j |
| This journal is © The Royal Society of Chemistry 2018 |