
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBinding of anions in triply interlocked coordination catenanes and dynamic allostery for dehalogenation reactions†
Linlin
Yang‡
,
Xu
Jing‡
,
Bowen
An
,
Cheng
He
 *,
Yang
Yang
and
Chunying
Duan
*
*,
Yang
Yang
and
Chunying
Duan
*
State Key Laboratory of Fine Chemicals, College of Zhang Dayu, Dalian University of Technology, Dalian, 116024, P. R. China. E-mail: cyduan@dlut.edu.cn
First published on 30th November 2017
Abstract
By synergistic combination of multicomponent self-assembly and template-directed approaches, triply interlocked metal organic catenanes that consist of two isolated chirally identical tetrahedrons were constructed and stabilized as thermodynamic minima. In the presence of suitable template anions, the structural conversion from the isolated tetrahedral conformers into locked catenanes occurred via the cleavage of an intrinsically reversible coordination bond in each of the tetrahedrons, followed by the reengineering and interlocking of two fragments with the regeneration of the broken coordination bonds. The presence of several kinds of individual pocket that were attributed to the triply interlocked patterns enabled the possibility of encapsulating different anions, allowing the dynamic allostery between the unlocked/locked conformers to promote the dehalogenation reaction of 3-bromo-cyclohexene efficiently, as with the use of dehalogenase enzymes. The interlocked structures could be unlocked into two individual tetrahedrons through removal of the well-matched anion templates. The stability and reversibility of the locked/unlocked structures were further confirmed by the catching/releasing process that accompanied emission switching, providing opportunities for the system to be a dynamic molecular logic system.
Introduction
Biomolecular machines are fascinating systems in which integrated functions are controlled in a synergistic manner. The constitution of the individual components within the inter-connected networks governs their capacity to perform specific functions and to interact with other relevant species in the recognition mediated systems.1–5 Inspired by sophisticated structures and mechanisms in biological systems, molecular recognition and self-assembly have been exploited as efficient driving forces for the fabrication of artificial molecular systems that operate far from equilibrium in a preorganized manner through well-controlled molecular design.6,7 Of these reported artificial systems, molecules containing mechanical bonds, such as catenanes,8–10 knots11 and rotaxanes,12–14 have captured the attention of the scientific community because of their intriguing architectures and topologies as well as the ability of their components to undergo controllable intra-molecular movements.A template-directed approach that preorganizes building blocks into entwined or threaded systems by non-covalent interactions was developed to precisely control the architectures and topologies of the mechanically interlocked molecules.15–18 For more complex molecules with non-trivial topologies that rely on dynamic bonds, new strategies are required to ensure that the systems undergo ‘error checking’ reactions to obtain their thermodynamic minima.19–22 In these cases, their intrinsic natures, including well-defined geometries and fast exchange kinetics, allow metal–ligand interactions to act as promising dynamic bonds for the creation of interlocked structures that reproduce key structural or functional aspects of natural systems and their artificial analogues.23,24 However, compared to the clear understanding of template-directed approaches for the creation of mechanically interlocked systems, the requirements for integrating the self-replication of machines with recognition mediated processes relevant to the revisable locked/unlocked architectures and dynamic control of their optical, electronic and transport properties remain largely unexplored.25–27
On the other hand, enzymes accelerate biochemical reactions by providing scaffolds to recognize and activate substrates and stabilize transition states between reactants and products.28,29 It is postulated that strongly halophilic molecular knots can promote anion abstraction reactions, echoing the properties of dehalogenase enzymes which contain halide-binding sites that facilitate the cleavage of carbon–halogen bonds.30 The pathway involving substrate capture surmounts the kinetic barriers of the reaction through a “mild” equilibrium control method. Multiply interlocked catenanes exhibit high stabilities and have the potential to form different kinds of pocket that encapsulate several kinds of guest with different sizes and shapes.31–36 In these cases, positively charged, multiply coordinated interlocked structures are promising promoters to selectively and simultaneously bind a large number of guest anions.37–39 Inspired by these systems, a new approach based on interlocked catenane was developed to realize the dehalogenation process, simulating the mechanism in dehalogenation enzymes.
Herein, through the synergistic combination of multi-component self-assembly with a template-directed approach, we report a new approach to construct triply interlocked coordination catenanes from 40 components. The one-pot assembly reaction was well controlled from several types of commercially available fragment40–43 including a tris(4-amino phenyl)amine (TPA) group extended by a 2,2′-bipyridine-5-carbaldehyde chelator and the dynamics active zinc(II) ions. These triply interlocked catenanes could also be formed and unlocked into individual tetrahedrons upon the addition or removal of an anion template with a well-matched size and conformational structure. Additionally, the large inner pocket and the multiply interlocked fashion lead to a cavity that is divided into several individual pockets, enabling the encapsulation of different kinds of anion and promoting the dehalogenation reaction of 3-bromo-cyclohexene (Scheme 1).
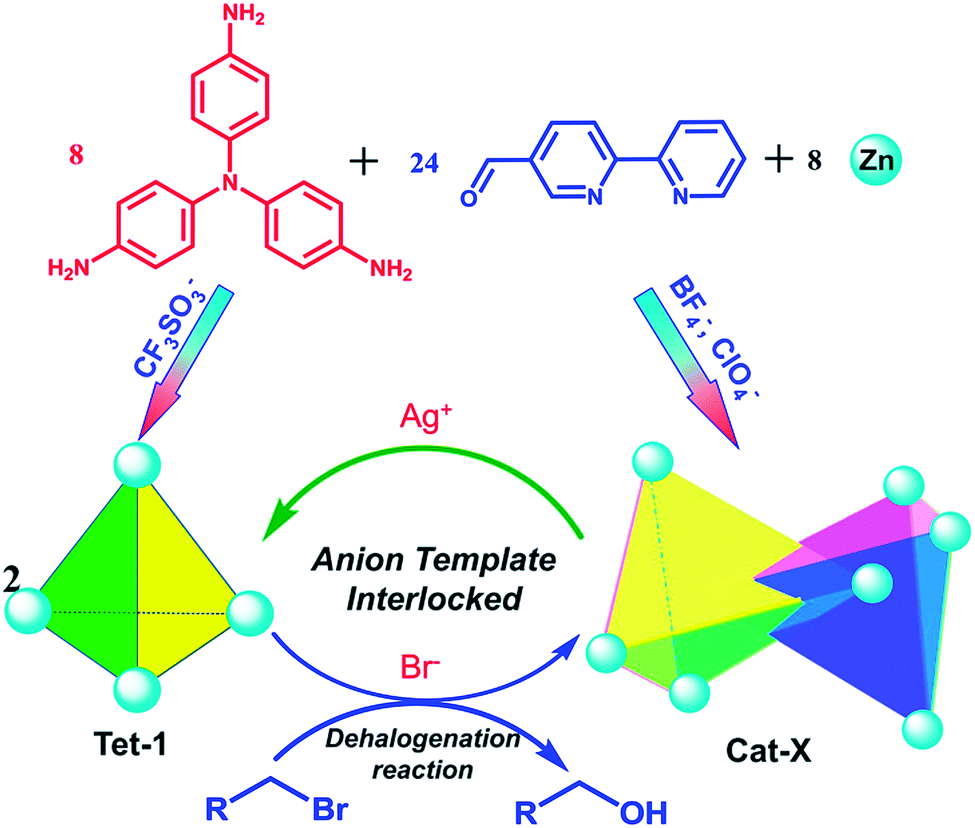 | ||
| Scheme 1 The illustration of the reversible assembly and dehalogenation reaction of triply interlocked catenanes via anion template and subcomponent self-assembly. | ||
Results and discussion
The reaction of tris(4-aminophenyl)amine (TPA, 8 equiv.) with 2,2′-bipyridine-5-carbaldehyde (24 equiv.) and Zn(BF4)2 (8 equiv.) provided the complex Cat-BF4 with a 60% yield. The ESI-MS spectrum revealed a series of m/z species corresponding to [Zn8L8][BF4]n(16−n)+ (where L is the postulated Schiff-base ligand derived from the condensation of TPA and 2,2′-bipyridine-5-carbaldehyde, Scheme S1; †n = 7–10, Fig. S25†). The splitting of the signals in the 19F-NMR spectrum of the BF4− anions revealed the encapsulation of BF4− anions within the molecular architecture (Fig. S3†).44 The fact that the one-pot self-assembly from 40 individual pieces led to the metal–organic catenane suggested the high thermodynamic stability of Cat-BF4 in the solution.X-ray crystallographic analysis of Cat-BF4 confirmed the formation of the metal–organic catenane and revealed that the triply locked identical cages feature seven pockets, each filled with a BF4− anion (Fig. 1). In the asymmetrical unit, four zinc centers with identical Δ or Λ stereochemistries of one tetrahedron are positioned at the terminal apical sites and are bridged by four tripodal ligands that cap the four faces. Within one tetrahedron, the average Zn⋯Zn separation is 19.1 Å, and a tetrahedron has an approximate volume of 820 Å3. Two chirally identical tetrahedrons interlocked in C2 symmetry with one Zn(bipy)3 node situated at the center of the internal cavity of the counterpart. Three bidentate chelating arms coordinated to one catenane-coordinated zinc center inter-penetrate across three of the four openings of the counterpart to form a triply interlocked cage.45,46
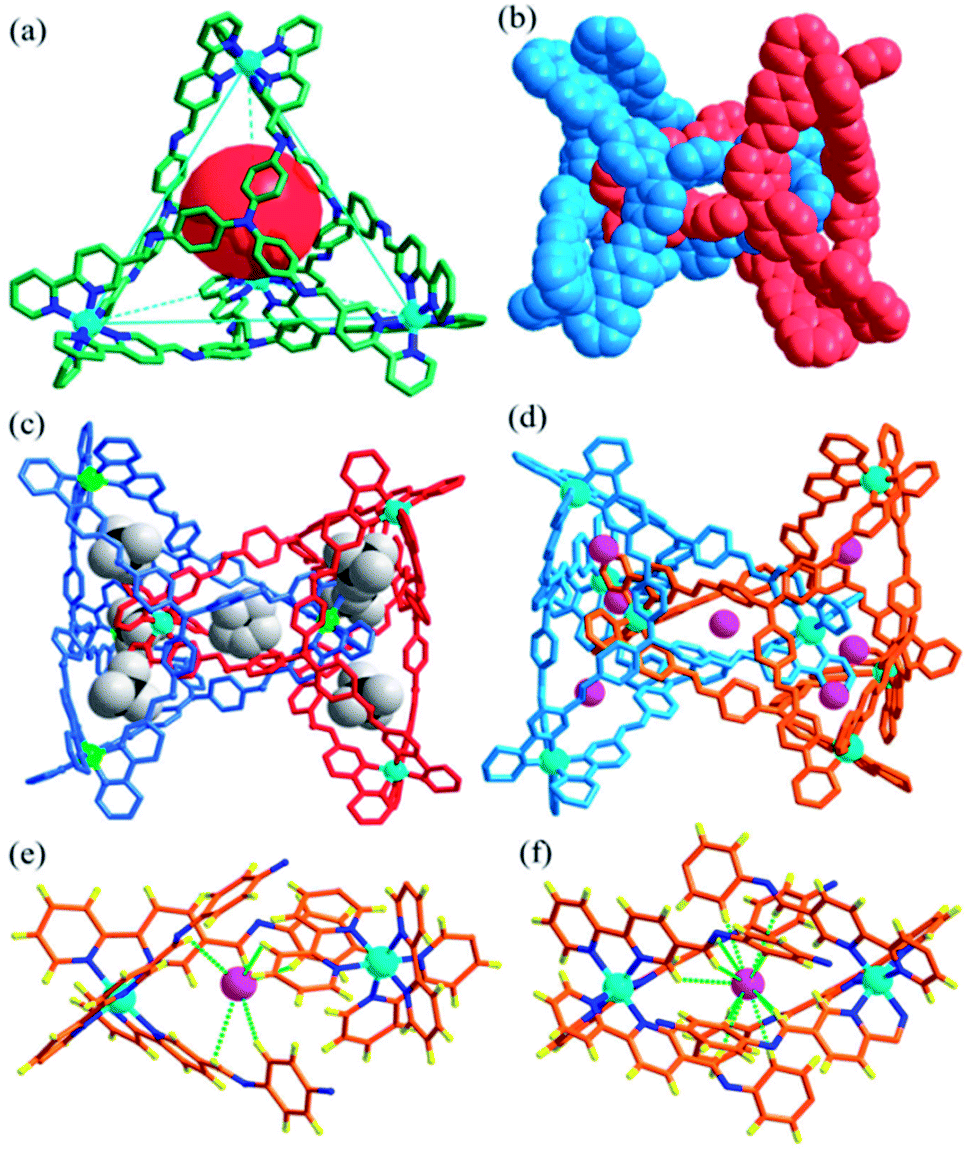 | ||
| Fig. 1 The structure of the triply interlocked catenane Cat-BF4, showing the discrete polyhedrons (a) (the interlocked partner was omitted for clarity) and the space-filling model of the interlocked structure (b). Structures of the interlocked catenanes Cat-BF4 (c) and Cat-I/BF4 (d) filled with BF4− and I− anions in the pockets, respectively; and a view of the hydrogen bonding interactions with I− anions in the outer (e) and central (f) pocket. | ||
The six locked arms face one another to create an inner core – a molecular bowl that is similar to an ‘egg cup’.47 One of the BF4− anions occupies the center of the molecular bowl. Abundant C–H⋯F interactions, which stabilize the interlocked structure, are found between the CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N groups and BF4− anions (Fig. S21†). It should also be noted that the large inner pocket and the multiply interlocked fashion lead to a cavity that is divided into 7 individual pockets. Beside the center pocket (inner pocket) that is filled by a BF4− anion, the other six pockets (outer pockets) formed each encapsulate a BF4− anion. These anions occupy the middle point of the seven shortest separated zinc pairs of the catenane zinc(II) nodes to adhere the charged zinc(II) nodes together through electrostatic interactions and to partly balance the positive charges of the catenane tetrahedrons. These cooperative weak interactions allow the subcomponents of Cat-BF4 to recognize each other well and to finely compact in an interlocked nature, benefiting the construction of complicated systems using the same constituent fragments.48
N groups and BF4− anions (Fig. S21†). It should also be noted that the large inner pocket and the multiply interlocked fashion lead to a cavity that is divided into 7 individual pockets. Beside the center pocket (inner pocket) that is filled by a BF4− anion, the other six pockets (outer pockets) formed each encapsulate a BF4− anion. These anions occupy the middle point of the seven shortest separated zinc pairs of the catenane zinc(II) nodes to adhere the charged zinc(II) nodes together through electrostatic interactions and to partly balance the positive charges of the catenane tetrahedrons. These cooperative weak interactions allow the subcomponents of Cat-BF4 to recognize each other well and to finely compact in an interlocked nature, benefiting the construction of complicated systems using the same constituent fragments.48
Replacing Zn(BF4)2 with Zn(CF3SO3)2 under the same assembly conditions gave an isolated tetrahedron Tet-1. ESI-MS analysis of the resulting solution of Tet-1 revealed a family of prominent signals at m/z = 619.15, 772.77, 1003.2 and 1387.25 that are assigned to [Zn4L4(CF3SO3)n](8−n)+ (n = 2, 3, 4 and 5, Fig. S27†) species based on a comparison with simulated and natural isotopic abundances (Fig. 4a). The presence of one set of ligand signals in the 1H NMR (Fig. 2b) and 19F-NMR (Fig. S10†) spectra suggests that the four ligands and eight CF3SO3− anions in Tet-1 are identical. Important evidence for the interlocked structure of Cat-BF4 is provided in the NOESY spectrum (Fig. S5†), where H–H interactions between the bipyridyl protons (Hj) and the TPA protons were found. However, no relevant signals were observed for Tet-1 because of the long distance of these interactions (Fig. S9†). The different self-assembly products should be impacted by the different sizes and shapes of the template anions.
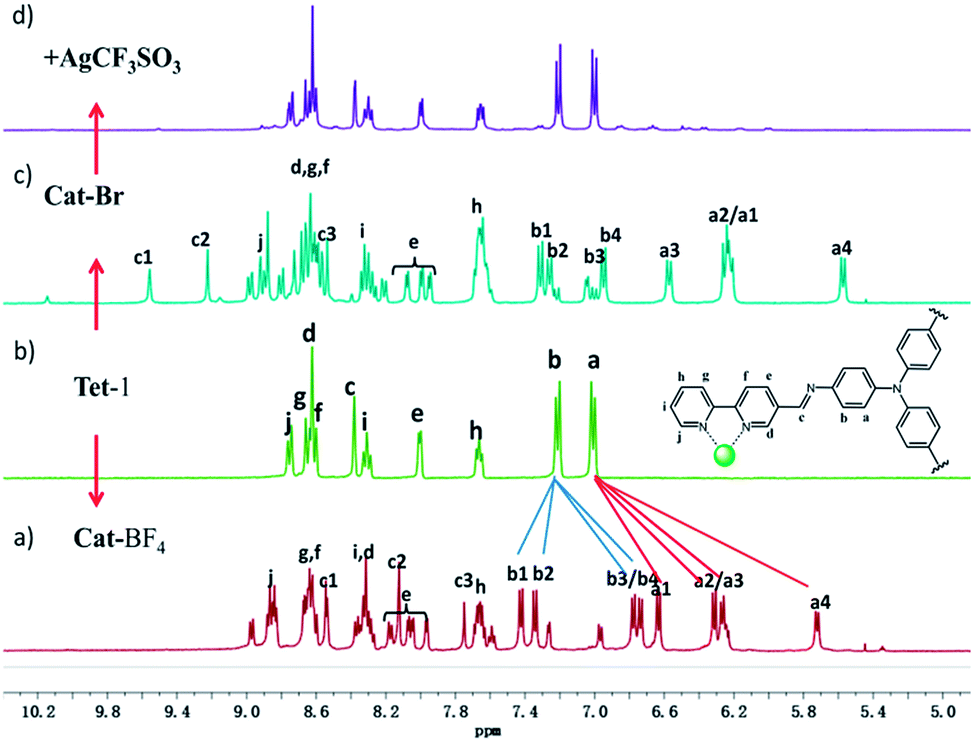 | ||
| Fig. 2 1H-NMR spectra of (a) the interlocked cage Cat-BF4 and (b) the monomeric tetrahedron Tet-1. 1H-NMR spectra of the monomeric tetrahedron after addition of 7.0 equiv. Br− ions (c); followed by subsequent addition of excess (11 equiv.) AgCF3SO3 (d) which unlocks the assembly and restores the structure to a discrete tetrahedron. | ||
Generally, the intrinsic reversibility of the coordinated bonds allows the dynamic self-assembly to occur through an ‘error checking’ mode, allowing the system to reach its thermodynamic minimum. Thus, this reversibility allows for the interlocked structure to be assembled from isolated fragments of the components with extra stimulating template anions.49,50 Treating the solution containing the isolated tetrahedron Tet-1 with ClO4− at room temperature gave direct evidence for the formation of the inter-locked architecture. As shown in Fig. S49,† the simple and sharp peaks split into several parts corresponding to the loss of symmetry in the interlocked architecture.51 The yield of the interlocked catenane increased directly with increasing concentration of ClO4− (Fig. 3a). This result indicates that the interlocked catenane sites are at the thermodynamic minimum, and could be rapidly transformed if a suitable template anion is present. The moderate and clear formation of Cat-ClO4 allowed the conversion process to be monitored in the 1H-NMR along with the changes of ClO4− anions. For Cat-ClO4, the inner binding core contains one rigidly binding perchlorate anion sited in its central pocket. A simple thermodynamics model indicated that two individual tetrahedrons and one ClO4− anion are involved, and the mechanism of the conversion process was assumed (see the ESI, Section 5.2†). The well-matched fitting of the 1H-NMR titration profile generated from the addition of Bu4NClO4 at different concentrations to the solution of Tet-1 (2 mM) with an assumed plot gave an equilibrium constant Kass (ClO4) of 7.0 × 104 M−2 (ΔG = −27.63 kJ mol−1) at room temperature (Fig. 3a). Meanwhile, with BF4− as the template anion (Fig. S51†), both the rate and conversion of Tet-1 to catenane were quite low compared to those in the presence of ClO4−, whereas the addition of the PF6− anion could not lead to the formation of the interlocked structure (Fig. S47†), further confirming that the size and shape of the template anions were essential to the interlocked system.
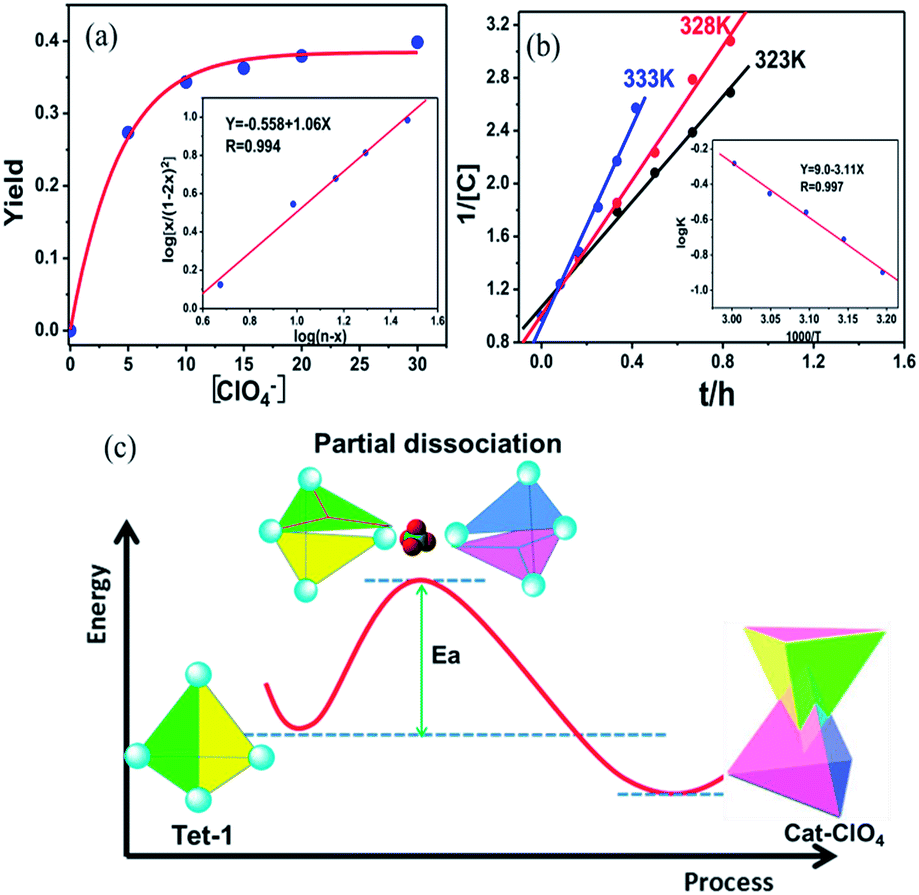 | ||
| Fig. 3 (a) The fitting curve of the conversion of Cat-ClO4 with the addition of different equiv. of ClO4−. (b) The kinetic plot fitting for the dimerization process at different temperatures in the presence of 20 equiv. of ClO4−. (c) The schematic representation of a putative partial dissociation mechanism for the formation of the interlocked Zn8L8 catenane. | ||
The formation of interlocked Cat-ClO4 was dynamically active, and the well-fitted linear relationship of 1/[Tet-1] with the reaction time at a fixed temperature suggested a second-order kinetic behaviour (Fig. 3b). The efficient collision between the two tetrahedrons was assumed to be the rate-determining step, which agreed well with the aforementioned thermodynamics assumption for the interlocked transformation. From the determination of kinetic constants at different temperatures, the activation energy of the catenane construction reaction from two isolated tetrahedrons was calculated to be 59.5 kJ mol−1, according to the Arrhenius equation (Table S3†). Generally, the interlocked transformation involves the breaking of at least two of the interacting bonds followed by the recovery of these bonds to interlock together. The fact that the activation energy is lower than the cleavage energy of one set of the chelating Zn–N (380 kJ mol−1) coordination bonds52 suggests that the structural conversion likely occurs through the random cleavage of one high-probability coordination bond out of the twelve active positions of each tetrahedron, followed by the reengineering of the two colloidal activated tetrahedrons with the formation of new chelating coordination bonds (Fig. 3c).53 Control experiments involving the use of more inert metal ions, with Fe(II) or Co(II) replacing Zn(II) in the subcomponent assembly, gave only the isolated tetrahedron (Fig. S31–S34†). The inactivity of the interlocked transformation with the kinetically inert metal ions confirmed that the structural formation was controlled by the partial dissociation dynamics of the Werner-type interactions and the suitable metal–ligand interactions.54–56
The kinetic study suggested that the addition of anions with a similar size which could weakly coordinate to the Zn centers would lower the reaction energy barrier and accelerate the conversion. In this case, the addition of halide anions as NBu4+ salts to the solution of Tet-1 resulted in the quickly exclusive detection of signals corresponding to the interlocked species [7X ⊂ Zn8L8] (X = I− and Br−) and a number of CF3SO3− anions as counter anions in the ESI-MS spectra (Fig. 4). This result demonstrates that the halide anions I− and Br− have suitable sizes and trigger hydrogen bonding similarly to BF4−. In addition, the driving forces for structure allostery were strong enough to direct the equilibrium controlled transformation to the thermodynamic minimum of the system. When excess Bu4NBF4 salt was added to the solution of the interlocked structure [7I− ⊂ Zn8L8], crystals of Cat-I/BF4 were obtained and characterized using X-ray diffraction. As shown in Fig. 1d, the cage morphology of Cat-I/BF4 is slightly altered in the presence of I−. Clearly, one I− anion occupies the ‘egg cup’-like inner core, and six I− or BF4− anions statistically occupy the middle points of the shortest separated zinc pairs to adhere the charged nodes together via electrostatic interactions.57,58 In this structure, the I− anion in the central cavity was strongly fixed, while the outer six I− anions could be randomly exchanged with the added BF4− anions. The results showed that there are two kinds of micro-environment in this catenane. The central cavity can bind anions more powerfully than the other six “outer” cavities, which is likely due to the presence of more hydrogen bonding interaction sites (Fig. 1f).
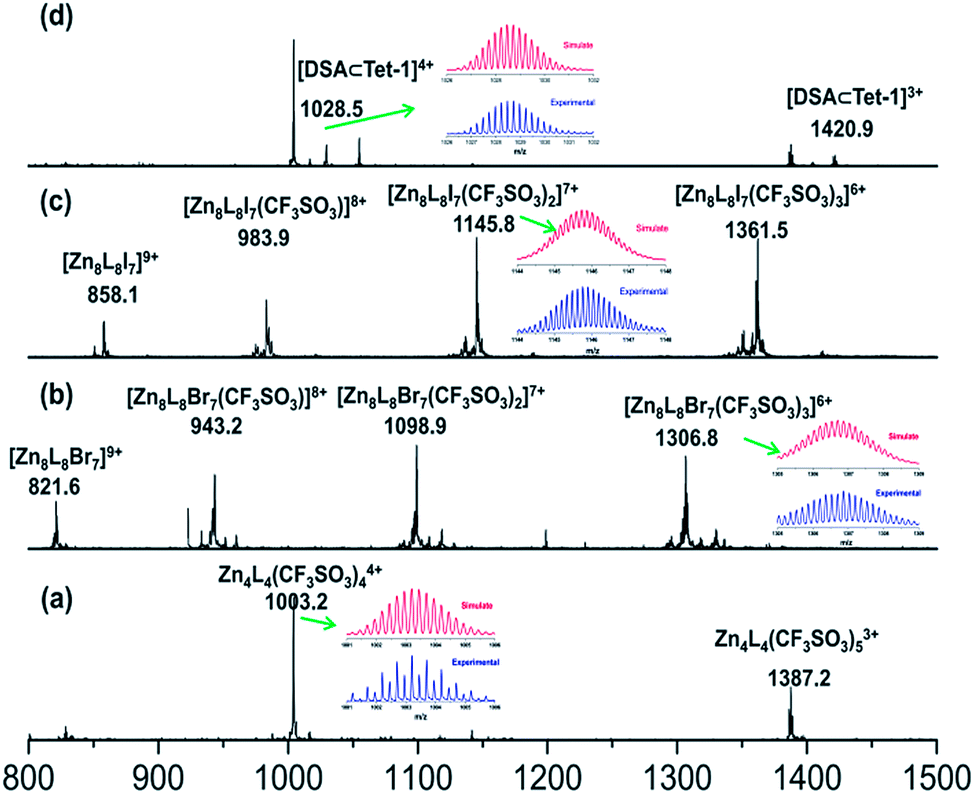 | ||
| Fig. 4 ESI-MS spectra of the solution containing the isolated tetrahedron Tet-1 (a). The aforementioned solution upon the addition of (b) Bu4NBr, (c) Bu4NI and (d) Bu4NDSA, showing the formation of interlocked structures with a 7Br− ⊂ Zn8L8 core, 7I− ⊂ Zn8L8 core and host–guest DSA ⊂ Tet-1 species, respectively. | ||
To elucidate the driving force for dimerization and the origin of the thermodynamic stability of the interlocked tetrahedrons, we evaluated the thermodynamic parameters using isothermal titration calorimetry (ITC).59 A typical titration curve is shown in Fig. 5a. The observed inclusion number between Tet-1 and Br− anions was 3.5, which is in good agreement with the seven Br− anions in the ESI-MS spectra of Cat-Br. Curve fitting by the computer simulation using an “independent” model reveals the activation enthalpy ΔH = 8.1 kJ mol−1, the activation entropy ΔS = 123.8 J mol−1, and the Gibbs free energy ΔG = −28.77 kJ mol−1 (Fig. S69†).60,61 These results demonstrate that the dimerization of Tet-1 is an entropy-driven endothermic reaction, which is attributed to the increase of molecular disorder and release of solvents.62 Similarly, the ΔG between the dimerization of Tet-1 with I− anions is −30.15 kJ mol−1 (Fig. S70†). The sequence of the Gibbs free energy in the dimerization process seems to be ranked as BF4− < ClO4− < Br− < I−.
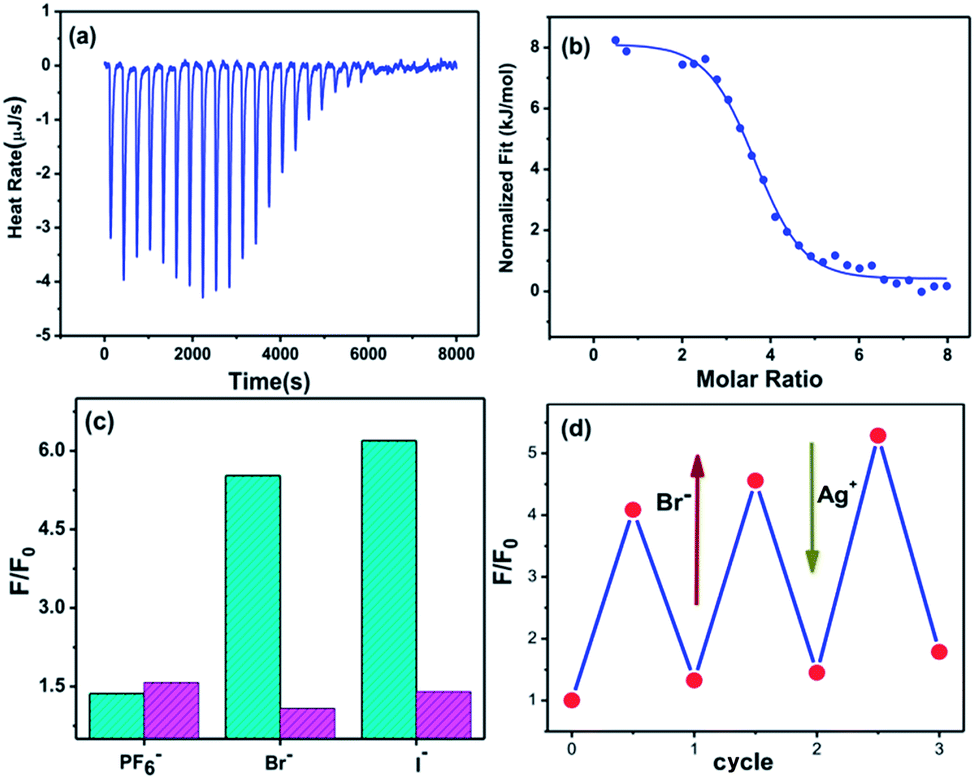 | ||
| Fig. 5 (a) The isothermal titration calorimetry (ITC) data for sequential injections (10 μL per injection) of Br− solution (5 mM) into Tet-1 solution (0.125 mM). (b) The apparent reaction heat obtained from the integration of calorimetric traces. (c) The fluorescence of DSA ⊂ Tet-1 species with the addition of 10 equiv. of different template ions (PF6−, Br− and I−, cyan column) followed by titration of 20 equiv. of AgCF3SO3 (red column) to the aforementioned solution (F0 corresponds to the fluorescence of DSA ⊂ Tet-1). (d) The reversible Br−/Ag+ controlled uptake and release of DSA monitored by fluorescence. | ||
Since silver salts can remove halide anions from solution via the formation of an insoluble AgX precipitate, we identified that the bound halide anions in Cat-X could also be precipitated by the addition of AgCF3SO3 with the catenane unlocked. As shown in Fig. 2d, the 1H-NMR titration of AgCF3SO3 into a solution containing Cat-Br resulted in the deterioration of the splitting of the signals of the interlocked structure. The simple NMR pattern is attributed to the tetrahedron Tet-1, and the ESI-MS spectrum (Fig. S42†) confirmed the formation of the isolated tetrahedron from the locked/unlocked equilibrium. The conformational flexibility and dynamic reversibility of the interlocked catenane from the isolated tetrahedron with a large inner cavity provided the possibility of introducing photo or electro-active elements to functionalize the transformation processes.63,64 Since emission is a sensitive method to recognize responsive behavior, Bu4NDSA (where DSA is dansyl acid), a widely used luminescent indicator, was chosen as a guest to visually show the reversibility of the allostery of the structure. As shown in Fig. 4d, upon the addition of NBu4DSA, the ESI-MS spectrum of a Tet-1 solution exhibited new peaks at m/z = 1028.5 and 1420.9 that were assigned to [DSA ⊂ Tet-1]4+ and [(DSA ⊂ Tet-1) (CF3SO3)]3+ species, respectively. The 1H NMR spectrum of Tet-1 was largely affected upon the addition of 5 equiv. of DSA, demonstrating the formation of the complexes of DSA in solution (Fig. S81†). Preliminary results also indicated that the formation of the host–guest species DSA ⊂ Tet-1 led to a strong decrease in luminescence.65
Furthermore, the addition of Br− and I− to a solution containing both NBu4DSA and Tet-1 resulted in the direct enhancement of the luminescence (Fig. 5c). The recovered fluorescence was quenched and returned to its initial value when Ag+ ions were added to the solution containing the interlocked structure, and the same cycle could be repeated several times. These results showed that, during the locked/unlocked process, the individual cage as well as the interlocked structure in solution are quite stable and exhibit excellent structure reversibility. The halide-communicated catching/releasing conformational switching resembles a key feature of dehalogenase enzymes in which the halide anion binding pathway efficiently surmounts the kinetic barriers associated with the carbon–halide bonds through a “mild” equilibrium control method. The locked/unlocked behavior allowed the system to become a dynamic molecular logic system, enabling it to adopt what are otherwise thermodynamically inaccessible functional conformations.
We wondered if such a strong halide allostery process might be applied to the dehalogenation reaction, which has been traditionally performed by halophilic silver salts. We envisaged that the seven pockets in the catenane might be able to promote the formation of a carbocation intermediate by abstracting the Br− anions from the C–Br bond cleavage reactions.30 In a typical procedure, Tet-1 was mixed with 3-bromocyclohexene (3.6 μM) in CD3CN in the presence of 2,4,6-collidine and MeOTf at room temperature, resulting in conversion to 2-cyclohexene-1-ol after stirring for 3 hours. A loading of 10% mole ratio of the Tet-1 tetrahedron resulted in 65% conversion to 2-cyclohexene-1-ol. The bromide ions bound within the cavities of Cat-Br react with methyl triflate to form bromomethane gas. Clearly, the presence of MeOTf restores the formed Cat-Br to the unlocked Tet-1 as shown in Fig. S73.† However, Cat-BF4 only reacts with a stoichiometric amount of 3-bromo-cyclohexene, and peaks of Cat-Br were detected in the dehalogenation of 3-bromo-cyclohexene in the absence of MeOTf (Fig. S90†). Control experiments showed that neither the ligand components (TPA + 2,2′-bipyridine-5-carbaldehyde), the zinc(II) salt, nor Zn(bpy)3 could promote this reaction in a decent yield (Table S5†).
As shown in Fig. 6, the transformation exhibited a first order kinetic behavior. When the concentration of the 3-bromo-cyclohexene was fixed, the initial reaction rate of the dehalogenation reaction exhibited a linear relationship with the concentration of Tet-1. When the concentration of Tet-1 was fixed, the initial turnover frequency of the reaction hardly changed with the concentration of the substrate. It seems that the rate of the reaction only depends on the concentration of the bromide encapsulated interlocked complex. The formation of the bromide encapsulated interlocked complex was possibly the rate-limited step and the dehalogenation reaction took place through the direct SN1 cleavage of the C–Br bond.
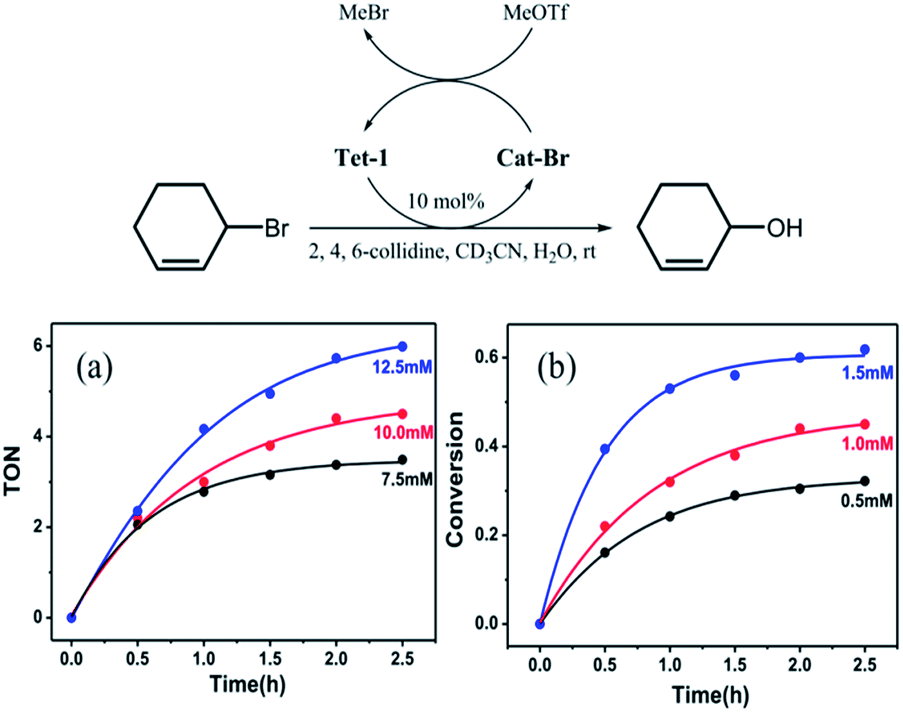 | ||
| Fig. 6 The kinetics study of the dehalogenation reaction of 3-bromo-cyclohexene. (a) The variation in turnover number (per mole of the catalyst Tet-1) with the concentration of the substrate 3-bromo-cyclohexene varies in the system containing 2,4,6-collidine (20 mM), MeOTf (20 mM), and Tet-1 (1 mM), and (b) the conversion of the substrate 3-bromo-cyclohexene (10.0 mM) with the concentration of Tet-1 varies in the system containing 2,4,6-collidine (20 mM) and MeOTf (20 mM). | ||
Conclusions
In summary, we report a new approach to construct triply interlocked coordination catenanes that consist of two chirally identical tetrahedrons. The new approach included the synergic combination of multi-component self-assembly and molecular recognition, and at the same time also involved seeking the thermodynamic minima of the interlocked/unlocked equilibrium based on the intrinsic dynamic active nature of the coordination assembly. The presence of several kinds of individual pocket that were attributed to the triply interlocked patterns provided the possibility to encapsulate different kinds of anion, allowing the dynamic allostery between the locked/unlocked conformers to efficiently promote the elimination of 3-bromo-cyclohexene.Experimental
Preparation of the interlocked tetrahedron Cat-BF4 [Zn8L8][BF4]16
TPA (29 mg, 0.1 mmol, 4 equiv.), 2,2′-bipyridine-5-carbaldehyde (56 mg, 0.3 mmol, 12 equiv.), Zn(BF4)2 (34 mg, 0.1 mmol, 4 equiv.), and dry acetonitrile (30 mL) were added to a Schlenk tube. The solution was degassed for three evacuation/nitrogen fill cycles. The tube was kept at 343 K for 24 h. Diethyl ether was diffused into an acetonitrile solution and the desired product Cat-BF4 was isolated by filtration as red square crystals in 60% yield. 1H NMR (500 MHz, 298 K, CD3CN): δ = 9.00(6H, d), 8.86(24H, m), 8.59–8.70(36H, m), 8.56(12H, s), 8.27–8.42(36H, m), 8.15(12H, s), 7.9–8.1(18H, d), 7.77(6H, s), 7.6–7.7(24H, m), 7.45(12H, d), 7.37(12H, d), 7.29(6H, d), 7.0(6H, d), 6.65–6.79(24H, dd), 6.66(12H, d), 6.25–6.36(24H, dd), 5.7(12H, d). ESI-MS: m/z [Zn8L8(BF4)7]9+ 826.8, [Zn8L8(BF4)8]8+ 941.0, [Zn8L8(BF4)9]7+ 1087.9, [Zn8L8(BF4)10]6+ 1283.7, [Zn8L8(BF4)11]5+ 1557.8. Elemental analysis (Zn8C408H288N80B16F64·14H2O) calcd (%): C 57.82, H 3.76, N 13.22; found (%): C 57.91, H 3.35, N 13.48. IR (KBr, cm−1), ν 3547, 3074, 1605, 1498, 1474, 1440, 1318, 1286, 1060, 835, 796, 750, 736.Preparation of the discrete tetrahedron Tet-1
TPA (29 mg, 0.1 mmol, 4 equiv.), 2,2′-bipyridine-5-carbaldehyde (56 mg, 0.3 mmol, 12 equiv.), Zn(CF3SO3)2 (36 mg, 0.1 mmol, 4 equiv.), and dry acetonitrile (30 mL) were added to a Schlenk tube. The solution was degassed by three evacuation/nitrogen fill cycles. The tube was kept at 343 K for 24 h. Diethyl ether was diffused into the acetonitrile solution and the desired product Tet-1 was isolated by centrifugation as a red solid. The isolated yield was 75% (based on the product dried under vacuum). 1H NMR (500 MHz, 298 K, CD3CN): δ = 8.75(12H, d), 8.58–8.67(36H, m), 8.38(12H, s, J = 1.3 Hz, imine), 8.31(12H, m), 8.0(12H, d), 7.6(12H, m), 7.22(24H, d, J = 8.8 Hz), 7.01(24H, d, J = 8.8 Hz). 13C NMR (500 MHz, 298 K, CD3CN): δ = 153.3, 149.9, 148.6, 148.5, 148.1, 146.1, 144.6, 141.7, 138.9, 134.9, 127.5, 124.2, 123.7, 123.3, 122.6. ESI-MS: m/z, [Zn4L4(CF3SO3)2]6+ 619.15, [Zn4L4(CF3SO3)3]5+ 772.77, [Zn4L4(CF3SO3)4]4+ 1003.2 and [Zn4L4(CF3SO3)5]3+ 1387.25. Elemental analysis (Zn4C212H144N40F24S8O24·2CH3CN·3H2O) calcd (%): C 54.66, H 3.31, N 12.39; found (%): C 54.41, H 3.25, N 12.48. IR (KBr, cm−1), ν 3378, 3063, 1605, 1499, 1474, 1440, 1317, 1280, 1164, 1030, 833, 796, 751, 638.Preparation of the interlocked tetrahedron Cat-ClO4 [Zn8L8][ClO4]16
TPA (29 mg, 0.1 mmol, 4 equiv.), 2,2′-bipyridine-5-carbaldehyde (56 mg, 0.3 mmol, 12 equiv.), Zn(ClO4)2 (37 mg, 0.1 mmol, 4 equiv.), and dry acetonitrile (30 mL) were added to a Schlenk tube. The solution was degassed by three evacuation/nitrogen fill cycles. The tube was kept at 343 K for 24 h. Diethyl ether was diffused into an acetonitrile solution and the desired product Cat-ClO4 was isolated by filtration as red square crystals in 65% yield (based on the product dried under vacuum). 1H NMR (500 MHz, 298 K, CD3CN): δ = 8.88(24H, m), 8.60–8.64(64H, m), 8.52(6H, s), 8.37(6H, s), 8.28(36H, m), 8.19(12H, m), 8.06–7.97(24H, d), 7.62(36H, m), 7.47(12H, d), 7.38(12H, d), 6.8(12H, d), 6.67(12H, d), 6.6(12H, d), 6.31–6.36(24H, dd), 5.9(12H, d). ESI-MS: m/z [Zn8L8(ClO4)7]9+ 836.8, [Zn8L8(ClO4)8]8+ 953.9, [Zn8L8(ClO4)9]7+ 1104.3, [Zn8L8(ClO4)10]6+ 1304.9, [Zn8L8(ClO4)11]5+ 1585.2. Elemental analysis: (Zn8C408H288N80Cl16O64·2CH3CN·7H2O) calcd (%): C 57.3, H 3.59, N 13.3; found (%): C 56.8, H 3.65, N 13.42. IR (KBr, cm−1): ν 3520, 3065, 1604, 1498, 1474, 1439, 1317, 1280, 1088, 834, 795, 750, 624.Preparation of the interlocked tetrahedron Cat-X (X = Br or I)
TPA (29 mg, 0.1 mmol, 4 equiv.), 2,2′-bipyridine-5-carbaldehyde (56 mg, 0.3 mmol, 12 equiv.), Zn(CF3SO3)2 (36 mg, 0.1 mmol, 4 equiv.), and dry acetonitrile (30 mL) were added to a Schlenk tube. The solution was degassed by three evacuation/nitrogen fill cycles. The tube was kept at 343 K for 24 h. Then Bu4NX (X = Br or I) (7 equiv.) was added to give a solution containing the interlocked cage [Zn8L8] with X− inclusion.Preparation of Cat-I/BF4 [Zn8L8I9(BF4)7]
TPA (29 mg, 0.1 mmol, 4 equiv.), 2,2′-bipyridine-5-carbaldehyde (56 mg, 0.3 mmol, 12 equiv.), Zn(CF3SO3)2 (36 mg, 0.1 mmol, 4 equiv.), and dry acetonitrile (30 mL) were added to a Schlenk tube. The solution was degassed by three evacuation/nitrogen fill cycles. The tube was kept at 343 K for 24 h. Bu4NI (7 equiv.) was added to give a solution containing the interlocked cage, and subsequently the salt Bu4NBF4 (10 equiv.) was added to replace the CF3SO3− anions. Then diethyl ether was diffused into an acetonitrile solution and the desired product Cat-I/BF4 was isolated by filtration as red square crystals in 55% yield. Elemental analysis (Zn8C408H288N80I9B7F28·2CH3CN·16H2O) calcd (%): C 54.26, H 3.67, N 12.82; found (%): C 54.31, H 3.45, N 12.98. IR (KBr, cm−1), ν 3381, 3073, 1605, 1503, 1474, 1440, 1318, 1281, 1060, 835, 796, 750, 736.X-ray crystallography
Crystals of the interlocked Cat-BF4 suitable for X-ray diffraction were obtained by slow diffusion of diethyl ether into an acetonitrile solution of the complex over a few days. Crystals of the interlocked Cat-I/BF4 suitable for X-ray diffraction were obtained by slow diffusion of diethyl ether into an acetonitrile solution of complex Cat-I in the presence of 10 equivalents of BF4− over a few days. The crystals were very susceptible to loss of solvent. Despite rapid handling times and a low temperature collection, the quality of data was less than ideal.X-ray intensity data were measured on a Bruker SMART APEX CCD-based diffractometer (Mo-Kα radiation, λ = 0.71073 Å) using the SMART and SAINT programs.66,67 The crystal data was solved by direct methods and further refined by full-matrix least-squares refinements on F2 using the SHELXL-97 software.68 Non-H atoms were refined with anisotropic displacement parameters. The hydrogen atoms within the ligand backbones and the solvent CH3CN molecules were fixed geometrically at calculated distances and allowed to ride on the parent non-hydrogen atoms.
Crystal data for Cat-BF4: Zn4C204H158N40O7B8F32, M = 4237.66, monoclinic, space group C2/c, red block, a = 47.616(2) Å, b = 31.748(1) Å, c = 56.289(2) Å, β = 111.304(2)°, V = 79![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 279(5) Å3, Z = 8, ρcalc = 0.712 g cm−3, μ(Mo-Kα) = 0.289 mm−1, T = 100(2) K [Rint = 0.1629]. Final R1 [with I > 2σ(I)] = 0.0985, wR2 (all data) = 0.2896. CCDC number 1515722.
279(5) Å3, Z = 8, ρcalc = 0.712 g cm−3, μ(Mo-Kα) = 0.289 mm−1, T = 100(2) K [Rint = 0.1629]. Final R1 [with I > 2σ(I)] = 0.0985, wR2 (all data) = 0.2896. CCDC number 1515722.
Crystal data for Cat-I/BF4: Zn4C206H163N41O8B3.5I4.5F14, M = 4477.14, monoclinic, space group C2/c, red block, a = 47.061(2) Å, b = 32.042(1) Å, c = 55.568(3) Å, β = 110.4(1)°, V = 78535(7) Å3, Z = 8, ρcalc = 0.757 g cm−3, μ(Mo-Kα) = 0.637 mm−1, T = 150(2) K [Rint = 0.1617]. Final R1 [with I > 2σ(I)] = 0.0885, wR2 (all data) = 0.2659. CCDC number 1515723.
For the refinement of Cat-BF4, several atoms on some pyridine rings of the ligands were disordered into two parts, with the site occupancy factors (s.o.f.) of each part being fixed at 0.5. Several fluorine atoms in the BF4− anions were disordered into two parts with the s.o.f. of each part being refined with a fixed value. Except for the disordered parts, the partially occupied solvent molecules and the partially occupied anions, the other non-hydrogen atoms were refined anisotropically. The hydrogen atoms within the ligand backbones were fixed geometrically at calculated distances and allowed to ride on the parent non-hydrogen atoms. The bond distance in some of the pyridine and benzene rings, as well as in several BF4− anions, was confined as idealized values. The thermal parameters of adjacent atoms of the ligand backbone and the BF4− groups were confined to be similar. The SQUEEZE subroutine in PLATON was used.
For the refinement of Cat-I/BF4, except for the half occupied iodide ion sited at the center of the catenane, the other iodide ions share the site with the boron atoms of the BF4− anions, with the s.o.f. of each part being fixed at a suitable value. Some of the pyridine rings of the ligands were disordered into two parts, with the site occupancy factors (s.o.f.) of each part being fixed as a certain value. Several of the fluoride atoms in a partially occupied BF4− anion were disordered into two parts with the s.o.f. of each part being refined with a fixed value. Except for the disordered parts, the partially occupied solvent molecules and the partially occupied anions, the other non-hydrogen atoms were refined anisotropically. The hydrogen atoms within the ligand backbones were fixed geometrically at calculated distances and allowed to ride on the parent non-hydrogen atoms. The bond distance in some of the pyridine and benzene rings, as well as in several BF4− anions, was confined as idealized values. The thermal parameters of adjacent atoms of the disordered parts of the ligand backbone and the particularly occupied BF4− groups were confined to be similar. The SQUEEZE subroutine in PLATON was used.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This study was supported by the National Natural Science Foundation of China (21231003, 21531001 and 21501041).References
- M. Yoshizawa, K. Kumazawa and M. Fujita, J. Am. Chem. Soc., 2005, 127, 13456 CrossRef CAS PubMed.
- G. H. Clever and M. Shionoya, Chem.–Eur. J., 2010, 16, 11792 CrossRef CAS PubMed.
- H. Dube, D. Ajami and J. Rebek Jr, Angew. Chem., 2010, 122, 3260 CrossRef.
- G. H. Clever, S. Tashiro and M. Shionoya, J. Am. Chem. Soc., 2010, 132, 9973 CrossRef CAS PubMed.
- J. D. Barrio, S. T. J. Ryan, P. G. Jambrina, E. Rosta and O. A. Scherman, J. Am. Chem. Soc., 2016, 138, 5745 CrossRef PubMed.
- R. Vilar, Angew. Chem., Int. Ed., 2003, 42, 1460 CrossRef CAS PubMed.
- G. T. Spence and P. D. Beer, Acc. Chem. Res., 2013, 46, 571 CrossRef CAS PubMed.
- T. J. Hubin and D. H. Busch, Coord. Chem. Rev., 2000, 200, 5 CrossRef.
- G. Zhang, O. Presly, F. White, I. M. Oppel and M. Mastalerz, Angew. Chem., Int. Ed., 2014, 126, 5226 CrossRef.
- G. Gil-Ramirez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 127, 6110 CrossRef PubMed.
- J. F. Ayme, J. E. Beves, C. J. Campbella and D. A. Leigh, Chem. Soc. Rev., 2013, 42, 1700 RSC.
- C. J. Bruns and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2186 CrossRef CAS PubMed.
- M. J. Langton and P. D. Beer, Acc. Chem. Res., 2014, 47, 1935 CrossRef CAS PubMed.
- A. Joosten, Y. Trolez, J. P. Collin, V. Heitz and J. P. Sauvage, J. Am. Chem. Soc., 2012, 134, 1802 CrossRef CAS PubMed.
- K. S. Chichak, S. J. Cantrill, A. R. Pease, S.-H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308 CrossRef CAS PubMed.
- M. S. Vickers and P. D. Beer, Chem. Soc. Rev., 2007, 36, 211 RSC.
- F. L. Thorp-Greenwood, A. N. Kulak and M. J. Hardie, Nat. Chem., 2015, 7, 526 CrossRef CAS PubMed.
- S. L. Huang, T. S. A. Hor and G. X. Jin, Coord. Chem. Rev., 2017, 333, 1 CrossRef CAS.
- S. J. Loeb, Chem. Soc. Rev., 2007, 36, 226 RSC.
- S. M. Goldup, D. A. Leigh, P. J. Lusby, R. T. McBurnery and A. M. Z. Slawin, Angew. Chem., Int. Ed., 2008, 47, 6999 CrossRef CAS PubMed.
- S. Yi, V. Brega, B. Captaina and A. E. Kaifer, Chem. Commun., 2012, 48, 10295 RSC.
- E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778 RSC.
- C. S. Wood, T. K. Ronson, A. M. Belenguer, J. J. Holstein and J. R. Nitschke, Nat. Chem., 2015, 7, 354 CrossRef CAS PubMed.
- J. E. M. Lewis, P. D. Beer, S. J. Loeb and S. M. Goldup, Chem. Soc. Rev., 2017, 46, 2577 RSC.
- L. Fang, M. A. Olson, D. Benítez, E. Tkatchouk, W. A. Goddard III and J. F. Stoddart, Chem. Soc. Rev., 2010, 39, 17 RSC.
- V. Balzani, A. Credi, S. Silvi and M. Venturi, Chem. Soc. Rev., 2006, 35, 1135 RSC.
- A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19 RSC.
- M. D. Levin, D. M. Kaphan, C. M. Hong, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2016, 138, 9682 CrossRef CAS PubMed.
- T. H. Kim, P. Mehrabi, Z. Ren, A. Sljoka, C. Ing, A. Bezginov, L. Ye, R. Pomès, R. S. Prosser and E. F. Pai, Science, 2017, 355, eaag2355 CrossRef PubMed.
- V. Marcos, A. J. Stephens, J. Jaramillo-Garcia, A. L. Nussbaumer, S. L. Woltering, A. Valero, J. F. Lemonnier, I. J. Vitorica-Yrezabal and D. A. Leigh, Science, 2016, 352, 1555 CrossRef CAS PubMed.
- M. Frank, M. D. Johnstone and G. H. Clever, Chem.–Eur. J., 2016, 22, 14104 CrossRef CAS PubMed.
- S. Löffler, J. Lübben, L. Krause, D. Stalke, B. Dittrich and G. H. Clever, J. Am. Chem. Soc., 2015, 137, 1060 CrossRef PubMed.
- J. F. Ayme, J. E. Beves, C. J. Campbell, G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, J. Am. Chem. Soc., 2015, 137, 9812 CrossRef CAS PubMed.
- D. Samanta and P. S. Mukherjee, J. Am. Chem. Soc., 2014, 136, 17006 CrossRef CAS PubMed.
- A. K. Bar, S. Raghothama, D. Moon and P. S. Mukherjee, Chem.–Eur. J., 2012, 18, 3199 CrossRef CAS PubMed.
- S. Loffler, J. Lubben, A. Wuttke, R. A. Mata, M. John, B. Dittrich and G. H. Clever, Chem. Sci., 2016, 7, 4676 RSC.
- S. Freye, R. Michel, D. Stalke, M. Pawliczek, H. Frauendorf and G. H. Clever, J. Am. Chem. Soc., 2013, 135, 8476 CrossRef CAS PubMed.
- D. Preston, J. E. M. Lewis and J. D. Crowley, J. Am. Chem. Soc., 2017, 139, 2379 CrossRef CAS PubMed.
- D. Preston, A. Fox-Charles, W. K. C. Lo and J. D. Crowley, Chem. Commun., 2015, 51, 9042 RSC.
- T. K. Ronson, S. Zarra, S. P. Black and J. R. Nitschke, Chem. Commun., 2013, 49, 2476 RSC.
- M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728 RSC.
- D. A. Leigh, R. G. Pritchard and A. J. Stephens, Nat. Chem., 2014, 6, 978 CrossRef CAS PubMed.
- L. Yang, X. Jing, C. He, Z. Chang and C. Y. Duan, Chem.–Eur. J., 2016, 22, 18107 CrossRef CAS PubMed.
- S. Freye, D. M. Engelhard, M. John and G. H. Clever, Chem.–Eur. J., 2013, 19, 2114 CrossRef CAS PubMed.
- M. Fujita, N. Fujita, K. Ogura and K. Yamaguchi, Nature, 1999, 400, 52 CrossRef CAS.
- A. Westcott, J. Fisher, L. P. Harding, P. Rizkallah and M. J. Hardie, J. Am. Chem. Soc., 2008, 130, 2950 CrossRef CAS PubMed.
- T. Hasell, X. Wu, T. A. Jones, J. Basca, A. Steiner, T. Mitra, A. Trewin, D. J. Adams and A. I. Cooper, Nat. Chem., 2010, 2, 750 CrossRef CAS PubMed.
- J. Mosquera, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2016, 138, 1812 CrossRef CAS PubMed.
- R. Sekiya, M. Fukuda and R. Kuroda, J. Am. Chem. Soc., 2012, 134, 10987 CrossRef CAS PubMed.
- H. Lee, P. Elumalai, N. Singh, H. Kim, S. U. Lee and K.-W. Chi, J. Am. Chem. Soc., 2015, 137, 4674 CrossRef CAS PubMed.
- Y. H. Li, J. J. Jiang, Y. Z. Fan, Z. W. Wei, C. X. Chen, H. J. Yu, S. P. Zheng, D. Fenske, C. Y. Su and M. Barboiu, Chem. Commun., 2016, 52, 8745 RSC.
- Y. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, 2007 Search PubMed.
- M. D. Pluth and K. N. Raymond, Chem. Soc. Rev., 2007, 36, 161 RSC.
- W. Wang, Y. X. Wang and H. B. Yang, Chem. Soc. Rev., 2016, 45, 2656 RSC.
- A. V. Davis, D. Fiedler, G. Seeber, A. Zahl, R. van Eldik and K. N. Raymond, J. Am. Chem. Soc., 2006, 128, 1324 CrossRef CAS PubMed.
- A. V. Davis and K. N. Raymond, J. Am. Chem. Soc., 2005, 127, 7912 CrossRef CAS PubMed.
- S. Freye, J. Hey, A. Torras-Galan, D. Stalke, R. Herbsy-Irmer, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 2191 CrossRef CAS PubMed.
- R. Zhu, J. Lubben, B. Dittrich and G. H. Clever, Angew. Chem., Int. Ed., 2015, 54, 2796 CrossRef CAS PubMed.
- G. B. Huang, S. H. Wang, H. Ke, L. P. Yang and W. Jiang, J. Am. Chem. Soc., 2016, 138, 14550 CrossRef CAS PubMed.
- J.-P. Demers and A. Mittermaier, J. Am. Chem. Soc., 2009, 131, 4355 CrossRef CAS PubMed.
- C.-F. Zhang, S.-H. Li, C.-C. Zhang and Y. Liu, Org. Biomol. Chem., 2015, 13, 10808 CAS.
- Y. M. Zhang, X. J. Zhang, X. Xu, X. N. Fu, H. B. Hou and Y. Liu, J. Phys. Chem. B, 2016, 120, 3932 CrossRef CAS PubMed.
- H. Dube, M. R. Ams Jr and J. Rebek, J. Am. Chem. Soc., 2010, 132, 9984 CrossRef CAS PubMed.
- D. H. Qu, Q. C. Wang, Q. W. Zhang, X. Ma and H. Tian, Chem. Rev., 2015, 115, 7543 CrossRef CAS PubMed.
- X. Jing, C. He, Y. Yang and C. Y. Duan, J. Am. Chem. Soc., 2015, 137, 3967 CrossRef CAS PubMed.
- SMART Data collection software, version 5.629, Bruker AXS Inc., Madison, WI, 2003 Search PubMed.
- SAINT Data reduction software, version 6.45, Bruker AXS Inc., Madison, WI, 2003 Search PubMed.
- G. M. Sheldrick, SHELX-97: Program for crystal structure analysis, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization data and additional tables and figures. CCDC 1515722 and 1515723. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04070a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |