
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganocatalytic controlled/living ring-opening polymerization of cyclotrisiloxanes initiated by water with strong organic base catalysts†
Keita
Fuchise
*,
Masayasu
Igarashi
,
Kazuhiko
Sato
* and
Shigeru
Shimada
 *
*
Interdisciplinary Research Center for Catalytic Chemistry, National Institute of Advanced Industrial Science and Technology (AIST), 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan. E-mail: k-fuchise@aist.go.jp; k.sato@aist.go.jp; s-shimada@aist.go.jp
First published on 19th February 2018
Abstract
Organocatalytic controlled/living ring-opening polymerization of cyclotrisiloxanes, such as hexamethylcyclotrisiloxane, 1,3,5-trimethyl-1,3,5-triphenylcyclotrisiloxane, 1,3,5-trimethyl-1,3,5-trivinylcyclotrisiloxane, and 1,3,5-trimethyl-1,3,5-tris(3,3,3-trifluoropropyl)cyclotrisiloxane, using water as an initiator and strong organic bases, such as amidines, guanidines, phosphazene bases, and proazaphosphatrane, as catalysts produced a variety of polysiloxanes with controlled number-average molecular weights (Mn = 2.64–102.3 kg mol−1), narrow polydispersity (Đ = 1.03–1.16), and well-defined symmetric structures. Controlled syntheses of statistical copolymers and triblock copolymers were achieved by copolymerizations of two cyclotrisiloxanes. Various terminal functionalities were successfully introduced by the end-capping reaction of propagating polysiloxanes using functional chlorosilanes. Kinetic investigations demonstrated that the polymerization proceeded through the initiator/chain-end activation mechanism, namely activations of water in the initiation reaction and of terminal silanols in propagating polysiloxanes in the propagation reaction. Catalytic activities of strong organic bases were revealed to depend on their Brønsted basicity and efficiency of the proton transfer in the initiation and propagation reactions. Guanidines possessing an R–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(N)–NH–R′ unit, in particular 1,3-trimethylene-2-propylguanidine, showed excellent performance as a catalyst. In this system, even non-dehydrated solvents are usable for the polymerization.
C(N)–NH–R′ unit, in particular 1,3-trimethylene-2-propylguanidine, showed excellent performance as a catalyst. In this system, even non-dehydrated solvents are usable for the polymerization.
Introduction
Polysiloxanes are major components of organosilicon materials that have a wide range of applications including lubricants, insulating and coating materials, sealants, adhesives in the form of oil, rubber, gel, and resin due to their excellent chemical and physical properties.1 Precise control of the linear, branched, and cross-linked structure of polysiloxanes is difficult, but would be essential for preparing the next generation of organosilicon materials with much superior properties. The linear polysiloxanes are industrially synthesized by (1) hydrolysis and simultaneous polycondensation of dichlorosilanes or dialkoxysilanes or (2) ring-opening polymerization (ROP) of cyclooligosiloxanes, in particular cyclotetrasiloxanes, using acidic species as catalysts or hydroxide salts as initiators, although the molecular weight distribution of the products becomes broad and the formation of cyclic oligomers as by-products are unavoidable.2 Anionic ROP of cyclotrisiloxanes initiated by lithium compounds, such as organolithiums and lithium silanolates, has been the sole method to synthesize polysiloxanes and their copolymers with narrow molecular weight distributions and well-defined structures.3–17 However, the biggest drawback of this method is that highly purified and dried starting materials must be used to avoid undesired side reactions, such as termination, backbiting, and chain-transfer reactions. The synthetic difficulty of polysiloxanes with a controlled structure has been a hurdle to actively employ them for developing organosilicon materials.In the field of polymer science, there has been continuous motivation to develop controlled polymerization that can be conducted with simple starting materials and procedures. Organocatalytic polymerization has been studied over the last decades to realize it.18–21 In particular, organocatalytic ROP of cyclic monomers, such as lactones, cyclic carbonates, epoxides, and cyclic phosphoesters, has been studied using organic acids, such as sulfonic acids, bis(sulfonyl)imides, and phosphoric acids, as well as organic bases, such as amines, amidines, guanidines, phosphazene bases, proazaphosphatranes, cyclopropenimines, and N-heterocyclic carbenes (NHC), as catalysts.18–21 Many of them proceed in a controlled/living fashion and have been extensively applied to controlled synthesis of various well-defined polymers, including end-functionalized polymers, block copolymers, and star-shaped polymers. The success of the organocatalytic ROP inspired us to apply it to cyclooligosiloxanes and develop a convenient method to synthesize various polysiloxanes with well-defined structures.
ROP of cyclooligosiloxanes12,22–32 and cyclic carbosiloxanes33,34 have been attempted with organic acids and bases as catalysts. Regarding acidic catalysts, HOSO2CF3,22,23 HN(SO2CF3)2,24 HB(C6F5)4,25 Ph3CB(C6F5)4,26 and B(C6F5)3 (ref. 27,28) have been used for ROP of hexamethylcyclotrisiloxane (D(Me2)3). However, none of them produced poly(dimethylsiloxane) (PDMS) with a narrow molecular weight distribution. For the basic catalysts, only those with quite high Brønsted basicity, such as phosphazene bases, NHCs, and bicyclic guanidines, have been employed. Möller and coworkers were the first to employ an electronically neutral strong organic base. They reported a polymerization of octamethylcyclotetrasiloxane (D(Me2)4) using 1-tert-butyl-4,4,4-tris(dimethylamino)-2,2-bis[tris(dimethylamino)phosphoranylidenamino]-2λ5,4λ5-catenadi(phosphazene) (tBu-P4) as a catalyst and methanol as an initiator.12,29 The polymerization was very rapid and the resulting PDMS had a very high number-average molecular weight (Mn) up to 440 kg mol−1, although polydispersity (Đ) was broad, 1.7–1.9. Hupfeld and Taylor also reported that a polymerization of D(Me2)4 using tBu-P4 as a catalyst almost immediately produced PDMS with Mn of up to 4 × 103 kg mol−1 with Đ of 1.5–1.9.30 Clarson's group reported that a polymerization of 1,3,5,7-tetramethyl-1,3,5,7-tetraphenylcyclotetrasiloxane (D(Me,Ph)4) using tBu-P4 as a catalyst and methanol as an initiator at ambient temperature produced poly[methyl(phenyl)siloxane] (PMPS) with Mn of 122–249 kg mol−1 and Đ of 1.5.31 Baceiredo and colleagues reported ROP of D(Me2)4 using NHCs, such as 1,3-dicyclohexylimidazol-2-ylidene and 1,3-di-tert-butyl-4,5-dimethylimidazol-2-ylidene, as catalysts and alcohols as initiators to give PDMS with high Đ of 1.5–1.7.32 Waymouth and Hedrick and coworkers reported the only example that succeeded in obtaining PDMS with narrow Đ (<1.2) by organocatalytic ROP of D(Me2)3. They used 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) as a catalyst and 4-pyrenebutan-1-ol as an initiator, although spectral and chromatographic data of the products were not provided.33 Thus, controlled synthesis of polysiloxanes by organocatalytic polymerization has not been well established. We considered that an appropriate choice of a catalyst and an initiator is the key to develop a controlled/living ROP of cyclooligosiloxanes. The acidity/basicity of the catalyst should not be too high to avoid side reactions, such as main chain scission and condensation of propagating polysiloxanes, that affect Mn, Đ, and terminal structures of resulting polysiloxanes. Furthermore, an initiation reaction between a cyclooligosiloxane and an initiator should generate propagating polysiloxanes with a sufficiently stable terminal structure at a sufficiently fast rate in comparison with a propagation reaction.
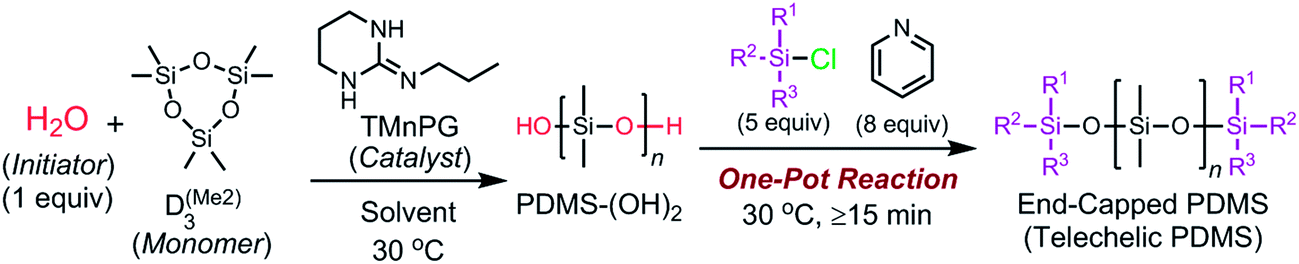
We herein report a controlled/living ROP of cyclotrisiloxanes using water as an initiator, strong organic bases as catalysts, and organochlorosilanes as end-capping agents. As shown in Scheme 1, the developed system is capable of polymerizing various cyclotrisiloxane in a controlled/living fashion and producing a variety of (telechelic) polysiloxanes with controlled Mn, narrow molecular weight distributions, and well-defined symmetric structure, which are difficult to obtain by the conventional anionic ROP using lithium compounds as initiators.10,35–38 This new polymerization method has a further advantage that non-dehydrated solvents can be used to give well-defined polysiloxanes.
 | ||
| Scheme 1 Ring-opening polymerization (ROP) of cyclotrisiloxanes using water as an initiator and strong organic bases as catalysts. | ||
Results and discussion
Optimization of catalyst for the polymerization of D(Me2)3
We found that water initiates a polymerization of D(Me2)3 in the presence of strong organic bases acting as catalysts to give PDMS with narrow molecular weight distributions. We first optimized the catalysts for the polymerization by testing the catalytic activity of various strong organic bases with different Brønsted basicity and structural classes. The strong organic bases tested in this study were classified into the following four categories. Amidines/guanidines A are amidines and guanidines with no hydrogen atoms on their amino groups, such as 1,1,3,3-tetramethylguanidine (TMGa),39 1,5-diazabicyclo[4.3.0]non-5-ene (DBN),39 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU),39 and 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (MTBD).39,40 Guanidines B are guanidines with an R–NC(N)–NH–R′ unit, such as 1,5,7-triazabicyclo[4.3.0]non-6-ene (TBN)39 and TBD.39,40 Phosphazene bases include 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP),39tert-butylimino-tri(pyrrolidino)phosphorane (tBu-P1(pyrr)),39 and 1-ethyl-2,2,4,4,4-pentakis(dimethylamino)-2λ5,4λ5-catenadi(phosphazene) (Et-P2).39 As proazaphosphatranes, only 2,8,9-triisobutyl-2,5,8,9-tetraaza-1-phosphabicyclo[3.3.3]undecane (TiBP)39 was employed. The Brønsted basicity of the bases in MeCN (MeCNpKBH) are shown in Scheme 1 and Table 1.
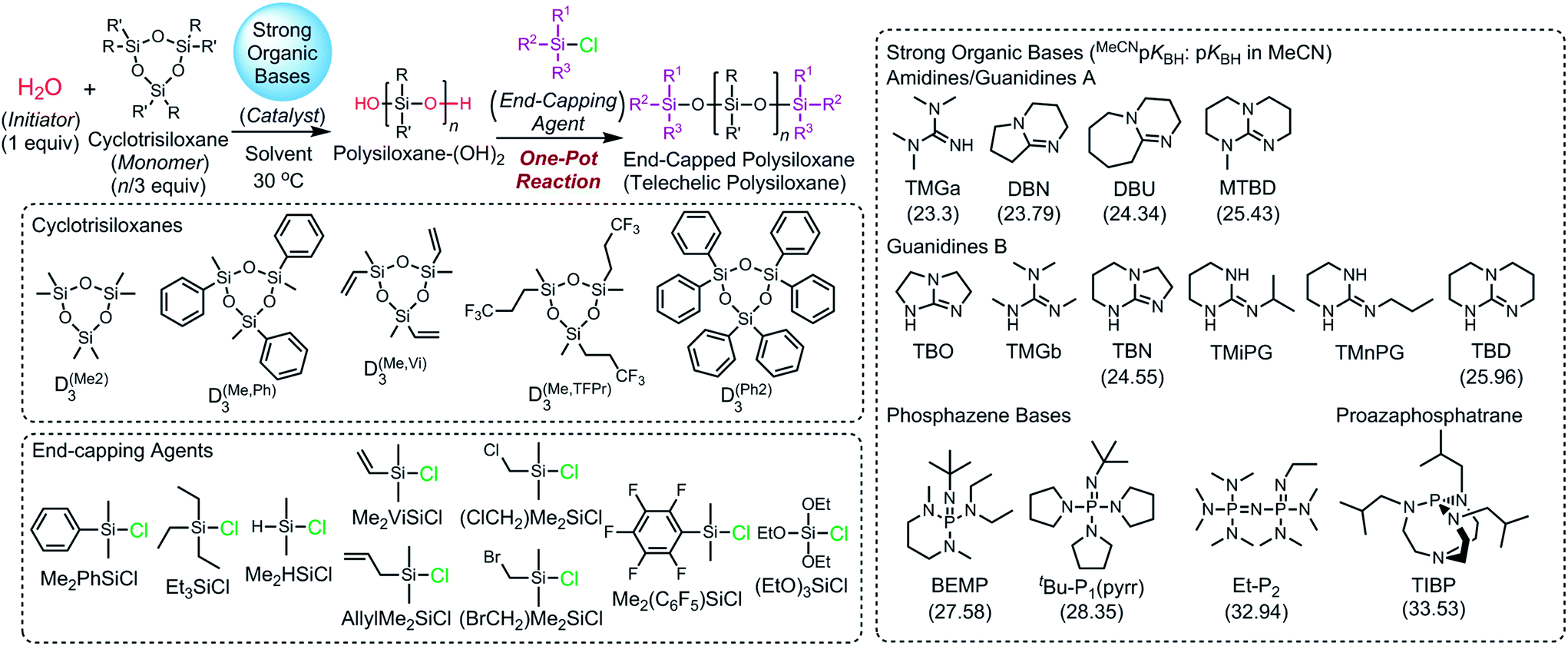
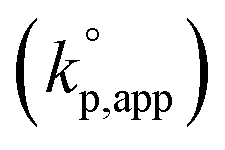 for various strong organic base catalysts observed in the polymerizations of D(Me2)3 using water as an initiator in THF at 30 °C a
for various strong organic base catalysts observed in the polymerizations of D(Me2)3 using water as an initiator in THF at 30 °C a
| Entry | Catalyst | MeCNpKBH |
|
Đ of the productc (Conv. (%)) |
|---|---|---|---|---|
a The polymerizations were carried out under the conditions of [D(Me2)3]0 = 1.80 mol L−1 and [D(Me2)3]0/[H2O]0/[C]0 = 10/1/0.005 for Et-P2 and TiBP, 10/1/0.007 for TBD, and 10/1/0.10 for the other bases.
b Calculated with 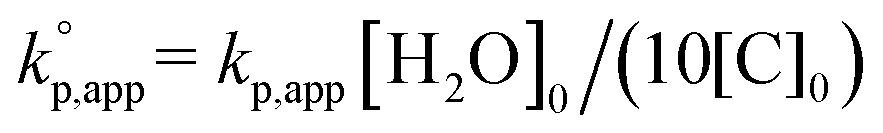 .
c
Đ of the obtained PDMS at the indicated conversion of monomer.
d Not reported.
e Not determined. .
c
Đ of the obtained PDMS at the indicated conversion of monomer.
d Not reported.
e Not determined.
|
||||
| 1 | TMGa | 23.3 | 0.0026 | 1.08 (50.7) |
| 2 | DBN | 23.79 | 0.019 | 1.10 (98.5) |
| 3 | DBU | 24.34 | 0.039 | 1.11 (99.1) |
| 4 | MTBD | 25.43 | 0.29 | 1.11 (99.6) |
| 5 | TBO | 0.0036 | n.d.e | |
| 6 | TMGb | 0.093 | 1.13 (99.3) | |
| 7 | TBN | 24.55 | 0.16 | 1.11 (99.9) |
| 8 | TMiPG | 1.7 | 1.11 (99.5) | |
| 9 | TMnPG | 2.6 | 1.11 (98.0) | |
| 10 | TBD | 25.96 | 6.3 | 1.12 (99.1) |
| 11 | BEMP | 27.58 | 0.14 | 1.14 (99.2) |
| 12 | tBu-P1(pyrr) | 28.35 | 0.60 | 1.23 (99.5) |
| 13 | Et-P2 | 32.94 | 1.1 × 103 | 1.22 (99.8) |
| 14 | TiBP | 33.53 | 5.2 × 102 | 1.13 (99.9) |
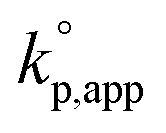
The catalytic activity of the bases was determined from the apparent rate coefficients of propagation (kp,app, h−1) observed in the polymerizations in tetrahydrofuran (THF) at 30 °C under the conditions of [D(Me2)3]0 = 1.80 mol L−1 and [D(Me2)3]0/[H2O]0/[C (catalyst)]0 = 10/1/0.005 for Et-P2 and TiBP, 10/1/0.007 for TBD, and 10/1/0.10 for other bases. The polymerization was initiated by adding a stock solution of water in THF and that of the catalysts in dry THF to a solution of D(Me2)3 in dry THF in this order. A linear relationship between the polymerization time, t (h), and −ln(1 − c), where c is monomer conversion, was observed in the first-order kinetic plot of each polymerization. According to the eqn (1), the kp,app values were determined from the slope observed in the plots.
| −ln(1 − c) = kp,appt | (1) |
The observed kp,app values were normalized to 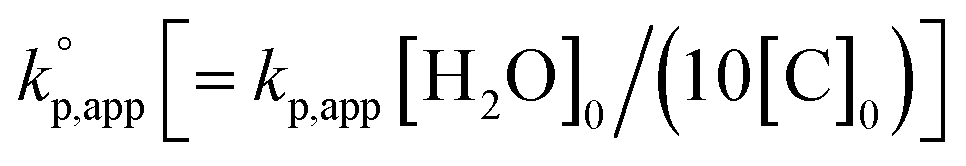 , as listed in Table 1 to directly compare kp,app values observed in the polymerizations using different amounts of the catalysts. This conversion should be rational, since we confirmed an almost linear relationship of kp,app with the initial molar ratio of the catalyst and water, [C]0/[H2O]0, as discussed in the next section (see Fig. 5).
, as listed in Table 1 to directly compare kp,app values observed in the polymerizations using different amounts of the catalysts. This conversion should be rational, since we confirmed an almost linear relationship of kp,app with the initial molar ratio of the catalyst and water, [C]0/[H2O]0, as discussed in the next section (see Fig. 5).
Fig. 1 shows the dependence of 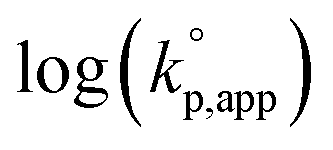 values on MeCNpKBH of strong organic bases. MeCNpKBH instead of those in THF can be used for the discussion, since Leito's group has reported that pKBH values in MeCN and THF have a good linear correlation.41,42 The catalytic activity of an organic base per its Brønsted basicity,
values on MeCNpKBH of strong organic bases. MeCNpKBH instead of those in THF can be used for the discussion, since Leito's group has reported that pKBH values in MeCN and THF have a good linear correlation.41,42 The catalytic activity of an organic base per its Brønsted basicity, 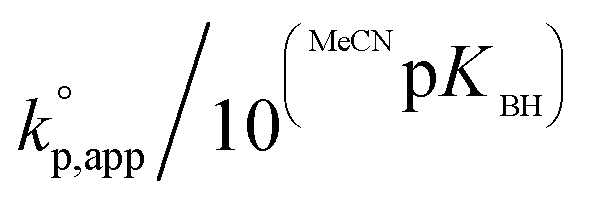 , increased in the following order: TiBP < phosphazene bases < guanidines/amidines A < guanidines B, which indicated that MeCNpKBH values of the catalysts were not the only single factor that affected the rates of polymerization. Interestingly, the amidines/guanidines A and the phosphazene bases independently showed linear relationships between
, increased in the following order: TiBP < phosphazene bases < guanidines/amidines A < guanidines B, which indicated that MeCNpKBH values of the catalysts were not the only single factor that affected the rates of polymerization. Interestingly, the amidines/guanidines A and the phosphazene bases independently showed linear relationships between 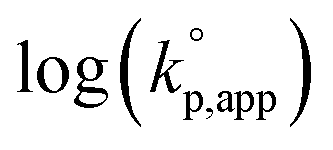 and their MeCNpKBH.
and their MeCNpKBH.
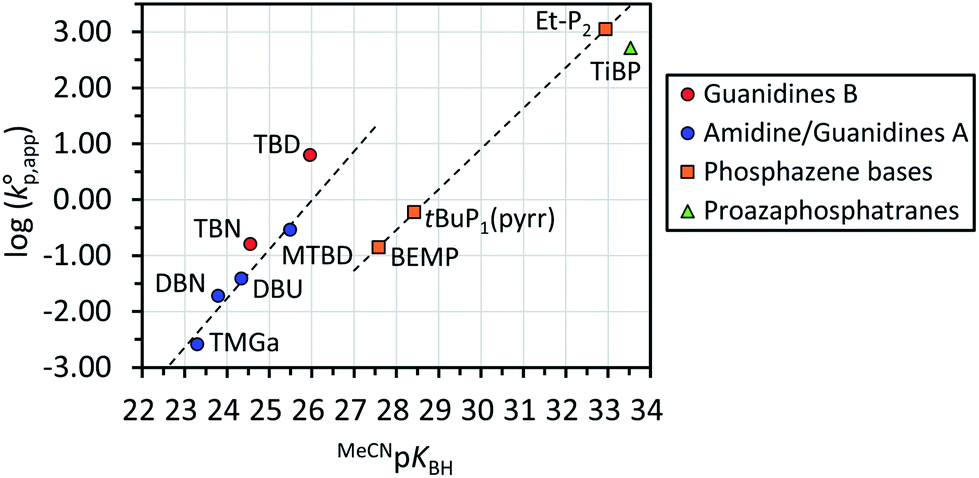 | ||
Fig. 1 A dependence of the logarithm of the normalized apparent rate coefficients of propagation 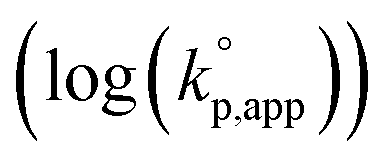 on MeCNpKBH of strong organic bases observed in the polymerizations of D(Me2)3 using water as an initiator and strong organic bases as a catalyst in THF at 30 °C under the conditions of [D(Me2)3]0/[H2O]0 = 10 and [D(Me2)3]0 = 1.80 mol L−1. on MeCNpKBH of strong organic bases observed in the polymerizations of D(Me2)3 using water as an initiator and strong organic bases as a catalyst in THF at 30 °C under the conditions of [D(Me2)3]0/[H2O]0 = 10 and [D(Me2)3]0 = 1.80 mol L−1. | ||
Among the phosphazene bases and TiBP with MeCNpKBH values of 27.5–33.5 (Table 1, entries 11–14), Et-P2 (1.1 × 103 h−1) and TiBP (5.2 × 102 h−1) showed significantly higher catalytic activity than the other bases due to their very high Brønsted basicity. On the other hand, BEMP (0.14 h−1) and tBu-P1(pyrr) (0.60 h−1) showed catalytic activities that were only comparable to MTBD (0.29 h−1) and TBN (0.16 h−1), which have more than 100 times weaker Brønsted basicity. The very high Brønsted basicity of the phosphazene bases and TiBP caused the frequent occurrence of undesired side reactions (vide infra), in particular, in the polymerizations catalyzed by Et-P2 and tBu-P1(pyrr), which made these bases unfavorable for controlling the polymerization as can be seen from the Đ of the products in each polymerization.
Regarding the amidines/guanidines A with MeCNpKBH values of 23–25.5 (Table 1, entries 1–4), TMGa (0.0026 h−1), DBN (0.019 h−1), DBU (0.039 h−1), and MTBD (0.29 h−1), showed only low catalytic activity, although undesired side reactions almost never occurred within the observed time range. In contrast, the guanidines B with MeCNpKBH values of 24.5–26 (Table 1, entries 7 and 10), such as TBD (6.3 h−1) and TBN (0.16 h−1), showed much higher 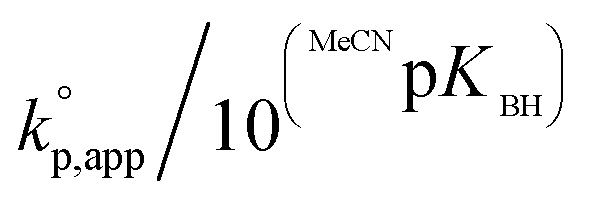 than the amidines/guanidines A as shown in Fig. 1, even though their structures and Brønsted basicity were not very different. For example, TBD showed 22 times higher catalytic activity than MTBD, although its Brønsted basicity was only around 3.4 times stronger than MTBD. This high catalytic activity of the guanidines B may have originated from the R–NC(N)–NH–R′ unit that enables high efficiency of the initiation and propagation reactions as described in the section regarding the mechanism (see Scheme 3).
than the amidines/guanidines A as shown in Fig. 1, even though their structures and Brønsted basicity were not very different. For example, TBD showed 22 times higher catalytic activity than MTBD, although its Brønsted basicity was only around 3.4 times stronger than MTBD. This high catalytic activity of the guanidines B may have originated from the R–NC(N)–NH–R′ unit that enables high efficiency of the initiation and propagation reactions as described in the section regarding the mechanism (see Scheme 3).
We hence evaluated the catalytic activity of four more guanidines B of which MeCNpKBH values have not been reported, such as 1,4,6-triazabicyclo[3.3.0]oct-4-ene (TBO), 1,1,2,3-tetramethylguanidine (TMGb), 1,3-trimethylene-2-isopropylguanidine (TMiPG), and 1,3-trimethylene-2-propylguanidine (TMnPG), since undesired side reactions rather frequently occurred in the polymerizations catalyzed by TBD and TBN. TMGb and TMiPG were newly synthesized in this study. TBO (0.0036 h−1), a bicyclic guanidine with two five-membered rings, showed much lower catalytic activity than TBD and TBN, presumably due to its much lower Brønsted basicity than TBD and TBN.43 TMGb (0.093 h−1), an acyclic tetramethylguanidine with an R–NC(N)–NH–R′ unit, showed 36 times higher catalytic activity than TMGa. The replacement of an H–NC(N)–NMe2 unit of TMGa with an Me–NC(N)–NH–Me unit certainly contributed to the increase in the catalytic activity. TMiPG (1.7 h−1) and TMnPG (2.6 h−1), monocyclic guanidines with a six-membered ring and the same number of carbon atoms as TBD, showed high catalytic activity next to TBD. The Brønsted basicity of TMnPG has been reported to be at least higher than TBN in CD3OD/D2O = 8/2 (w/w).43 TMiPG showed a slightly lower catalytic activity than TMnPG presumably because of the higher steric hindrance of the isopropyl group than that of the propyl group. It is noteworthy that the undesired side reactions were much less frequent in the polymerization catalyzed by TMiPG and TMnPG in comparison with that catalyzed by other bases with comparable catalytic activity. Hence, we identified TMnPG as the most suitable catalyst for the polymerization of cyclotrisiloxanes initiated by water.
Characteristics of the polymerization of D(Me2)3 using water and TMnPG
TMnPG catalyzed a polymerization of D(Me2)3 in THF at 30 °C under the conditions of [D(Me2)3]0/[H2O]0/[TMnPG]0 = 10/1/0.10 and [D(Me2)3]0 = 1.80 mol L−1 to give α,ω-dihydroxy-terminated PDMS (PDMS-(OH)2) with Mn estimated by 1H NMR measurement (Mn,NMR) of 2.64 kg mol−1 and Đ of 1.14 (Table 2, entry 1). The observed Mn,NMR corresponded well to the Mn calculated from the polymerization conditions (Mn,calcd) of 2.20 kg mol−1. The conversion of monomer reached 98.0% after 90 min of polymerization. The purification of the product was easily achieved by neutralizing TMnPG with an excess amount of benzoic acid and by washing the concentrated oily crude product with acetonitrile (MeCN).44 Removal of the MeCN layer with a pipette and drying the residue under vacuum gave pure PDMS-(OH)2 as a colorless viscous liquid. End-capping of the propagating PDMS-(OH)2 was achieved by directly adding pyridine (8 equiv.) and chlorodimethyl(phenyl)silane (Me2PhSiCl, 5 equiv.) to the reaction mixture at 30 °C and continuing the reaction for 15 min. The purification of the end-capped product (PDMS-(OSiMe2Ph)2), was also easily achieved by washing the concentrated crude product with methanol or MeCN.44Mn,NMR and Đ of the obtained PDMS-(OSiMe2Ph)2 were determined to be 3.17 kg mol−1 and 1.11, respectively. The end-capping reaction hence did not affect the Đ of the original product. Fig. 2 shows the molecular weight distribution of the products measured by the size-exclusion chromatography (SEC) using THF as an eluent and polystyrene as calibration standards.|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | [D(Me2)3]0/[H2O]0/[C]0 | Solventb | End-capping agent | Time (h) | Conv (%)c | M n (kg mol−1) | Đ | k p,app (h−1) | ||
| Calcdd | SECe | NMRc | ||||||||
| a [D(Me2)3]0 = 1.80 mol L−1. b Volume ratios of two solvents are shown for the mixed solvents. c Determined by 1H NMR. d Calculated from Mn,calcd = [D(Me2)3]0/[H2O]0 × Conv. × (MW. of D(Me2)3 = 222.46) + (MW. of terminal structures). e Determined by SEC measurements in THF using polystyrene standards. | ||||||||||
| 1 | 10/1/0.10 | THF | None | 1.5 | 98.0 | 2.20 | 1.66 | 2.64 | 1.14 | 2.6 |
| Me2PhSiCl | 2.47 | 1.90 | 3.17 | 1.11 | ||||||
| 2 | 10/1/0.05 | THF | Me2PhSiCl | 3 | 98.1 | 2.47 | 1.49 | 2.85 | 1.13 | 1.3 |
| 3 | 10/1/0.01 | DMAc/THF = 57/43 | None | 0.50 | 98.6 | 2.21 | 0.83 | 2.81 | 1.33 | 8.2 |
| 4 | 10/1/0.10 | CH2Cl2/THF = 65/35 | None | 1 | 98.6 | 2.21 | 1.80 | 2.70 | 1.12 | 4.6 |
| 5 | 25/1/0.25 | CH2Cl2/THF = 84/16 | None | 2 | 99.5 | 5.54 | 4.09 | 5.70 | 1.06 | 2.4 |
| 6 | 50/1/0.50 | CH2Cl2/THF = 91/9 | Me2PhSiCl | 1.57 | 93.8 | 10.7 | 9.75 | 12.2 | 1.04 | 1.7 |
| 7 | 100/1/1.0 | CH2Cl2/THF = 94/6 | Me2PhSiCl | 3 | 97.3 | 21.9 | 15.6 | 18.9 | 1.04 | 1.2 |
| 8 | 200/1/2.0 | CH2Cl2/THF = 96/4 | Me2PhSiCl | 5.5 | 95.0 | 42.5 | 31.8 | 39.9 | 1.06 | 0.47 |
| 9 | 550/1/5.5 | CH2Cl2/THF = 97/3 | None | 18 | 93.0 | 113.8 | 80.0 | 102.3 | 1.03 | 0.15 |
| 10 | 10/1/0.10 | CH2Cl2/THF = 57/43 | Me2HSiCl | 1 | 99.5 | 2.35 | 1.60 | 2.95 | 1.15 | |
| 11 | 10/1/0.10 | CH2Cl2/THF = 57/43 | Me2ViSiCl | 1 | 99.1 | 2.39 | 1.71 | 3.00 | 1.14 | |
| 12 | 10/1/0.10 | CH2Cl2/THF = 57/43 | AllylMe2SiCl | 1 | 99.3 | 2.42 | 1.67 | 2.77 | 1.13 | |
| 13 | 10/1/0.10 | CH2Cl2/THF = 57/43 | (ClCH2)Me2SiCl | 1 | 99.5 | 2.44 | 1.84 | 3.14 | 1.10 | |
| 14 | 10/1/0.10 | CH2Cl2/THF = 57/43 | (BrCH2)Me2SiCl | 1 | 98.8 | 2.52 | 1.71 | 3.08 | 1.13 | |
| 15 | 10/1/0.10 | CH2Cl2/THF = 57/43 | Me2(C6F5)SiCl | 1 | 99.3 | 2.68 | 1.72 | 3.38 | 1.13 | |
| 16 | 10/1/0.10 | CH2Cl2/THF = 57/43 | (EtO)3SiCl | 1 | 99.5 | 2.57 | 1.84 | 3.25 | 1.11 | |
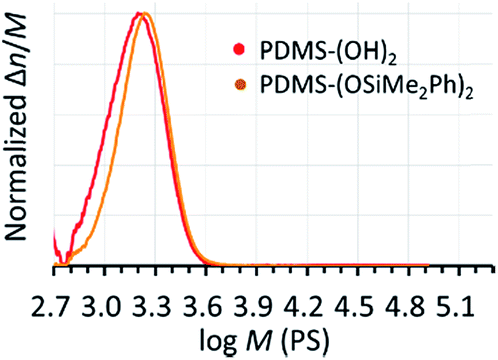 | ||
| Fig. 2 SEC chromatograms of the PDMS-(OH)2 (Mn,NMR = 2.64 kg mol−1, Đ = 1.14) and the PDMS-(OSiMe2Ph)2 (Mn,NMR = 3.17 kg mol−1, Đ = 1.11) synthesized in THF at 30 °C under the conditions of [D(Me2)3]0/[H2O]0/[TMnPG]0 = 10/1/0.10 and [D(Me2)3]0 = 1.80 mol L−1 (Table 2, entry 1). | ||
The well-defined structures of the obtained PDMS-(OH)2 and PDMS-(OSiMe2Ph)2 were proven by 1H and 29Si{1H} NMR analysis as shown in Fig. 3. In each spectrum, signals due to the terminal groups and several monomeric units from the termini were separately observed from that due to the inner repeating units. In both 1H and 29Si{1H} NMR spectra of the PDMS-(OSiMe2Ph)2, the signals due to the PDMS-(OH)2 were not observed, which evidenced the quantitative end-capping of the PDMS-(OH)2.
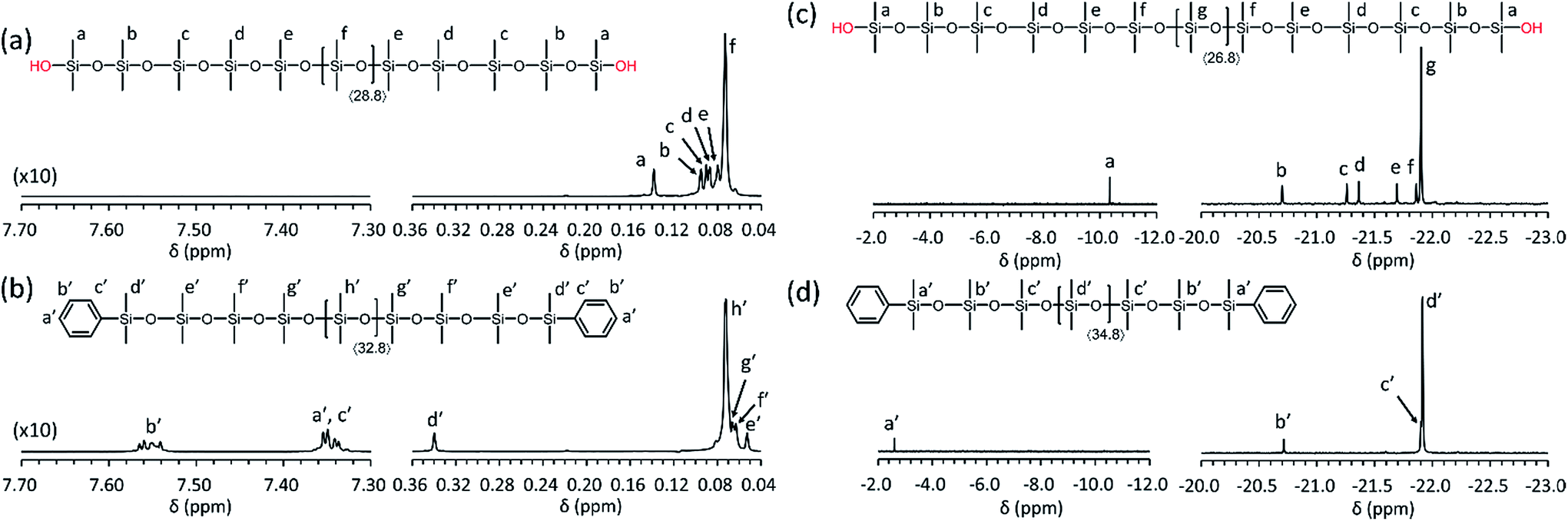 | ||
| Fig. 3 1H and 29Si{1H} NMR spectra of the synthesized PDMSs (Table 2, entry 1) in CDCl3: (a) 1H NMR spectrum of PDMS-(OH)2, (b) 1H NMR spectrum of PDMS-(OSiMe2Ph)2, (c) 29Si{1H} NMR spectrum of PDMS-(OH)2, (d) 29Si{1H} NMR spectrum of PDMS-(OSiMe2Ph)2. | ||
Structures of the obtained PDMS-(OH)2 and PDMS-(OSiMe2Ph)2 were further analyzed by positive ion matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) using trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) as a matrix and sodium trifluoroacetate as a cationization agent. Only one series of peaks was observed in both of the spectra shown in Fig. 4, which indicated that the obtained PDMS-(OH)2 and PDMS-(OSiMe2Ph)2 consisted of only linear PDMS with two terminal hydroxy groups and two terminal dimethyl(phenyl)siloxy groups, respectively. The m/z values of the observed molecular ion peaks corresponded to the calculated molar mass of PDMS-(OH)2 and PDMS-(OSiMe2Ph)2 cationized by a sodium cation. Interestingly, the obtained PDMSs contained PDMSs of which the degree of polymerization was not a multiple of 3, although those PDMSs should not be produced if only a simple ring-opening reaction of D(Me2)3 occurred in the polymerization. This point will be discussed in a later section regarding the mechanism of the polymerization.
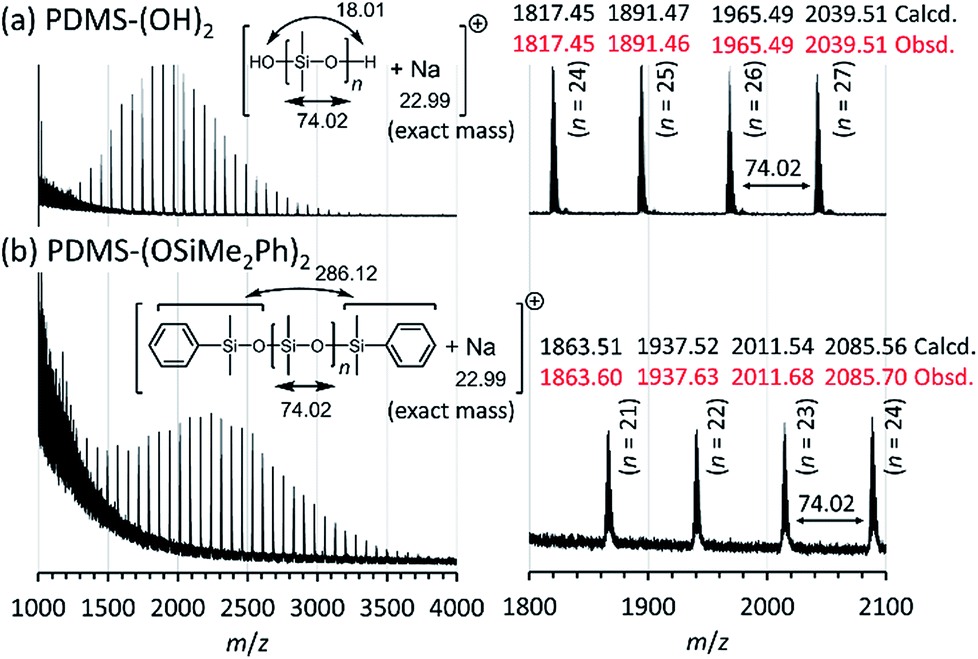 | ||
| Fig. 4 Positive ion matrix-assisted laser desorption/ionization time-of-flight mass (MALDI-TOF MS) spectra of the synthesized PDMSs (Table 2, entry 1) measured in the reflector mode using DCTB as a matrix and sodium trifluoroacetate as a cationization agent. (a) PDMS-(OH)2 (Mn,NMR = 2.64 kg mol−1, Đ = 1.14) and (b) PDMS-(OSiMe2Ph)2 (Mn,NMR = 3.17 kg mol−1, Đ = 1.11). | ||
As already mentioned in the previous section, a linear relationship was observed in the first-order kinetic plot of the polymerizations carried out with [D(Me2)3]0/[H2O]0/[TMnPG]0 = 10/1/0.10 and 10/1/0.05 (Table 2, entries 1 and 2) as shown in Fig. 5a. The kp,app for the former and the latter polymerizations were determined to be 2.6 and 1.3, which suggested that the kp,app varies linearly with the initial ratio of the catalyst and water, i.e., [C]0/[H2O]0.
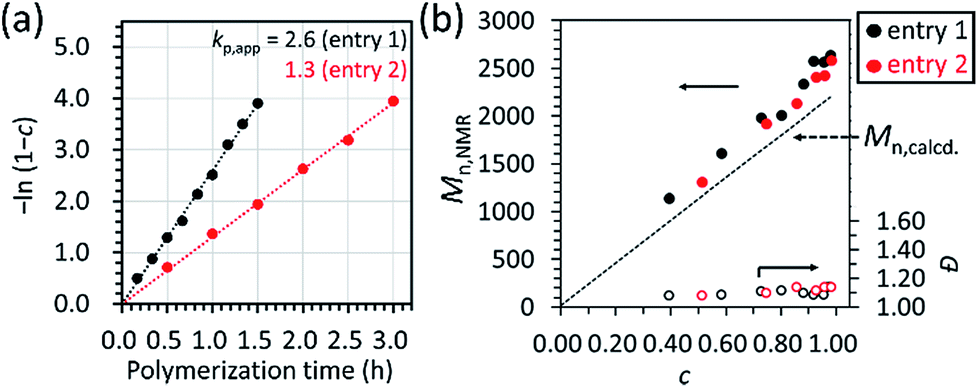 | ||
| Fig. 5 (a) First-order kinetic plot and (b) the dependence of number-average molecular weight (Mn,NMR) and polydispersity index (Đ) of the resulting PDMS on the monomer conversion, c, for the polymerization of D(Me2)3 catalyzed by TMnPG in THF at 30 °C under the conditions of [D(Me2)3]0 = 1.80 mol L−1 and [D(Me2)3]0/[H2O]0/[TMnPG]0 = 10/1/0.10 (Table 2, entry 1) and 10/1/0.05 (Table 2, entry 2). | ||
Fig. 5b shows the dependence of Mn,NMR and Đ on monomer conversion (c). In both of the polymerizations, Mn,NMR increased linearly as c increased, while Đ remained in a range of 1.08–1.14 even until the late stage of polymerization. It was hence found that the propagation reaction was the dominant process in the polymerizations and undesired side reactions were not frequent.
Other than THF, dichloromethane (CH2Cl2) was usable as a solvent for the polymerization in homogeneous solutions. N,N-Dimethylacetamide (DMAc), an aprotic polar solvent, could also be applied for the polymerization (Table 2, entry 3), although reactions in this medium proceeded as an inhomogeneous mixture due to the low solubility of PDMS in DMAc. The contribution of each solvent to the increase in the rate of polymerization varied in the following order: DMAc (entry 3) ≫ CH2Cl2 (entry 4) > THF (entry 1). The occurrence of undesired side reactions (vide infra) increased depending on the content of each solvent in the following order: DMAc ≫ THF > CH2Cl2. Hence, CH2Cl2 was found to be the most suitable solvent for the polymerization.
The Mn,NMR of PDMS was linearly controllable by changing the initial feed ratio of D(Me2)3 and water for the polymerization performed in a mixed solvent of CH2Cl2 and THF at 30 °C for 1–18 h under the conditions of [D(Me2)3]0 = 1.80 mol L−1, [D(Me2)3]0/[H2O]0 = 10–550, and [D(Me2)3]0/[TMnPG]0 = 100 (Table 2, entries 4–9). The Mn,NMR values of the products linearly increased from 2.70 kg mol−1 to 102.3 kg mol−1, which agreed well with the values of Mn,calcd for each polymerization. Furthermore, all the products had low Đ ranging from 1.03 to 1.12 as determined by the SEC analysis as shown in Fig. 6. A condensation of propagating polymers gradually occurred when the conversion of monomers increased above 80–90%, in particular in the polymerizations carried out with high [D(Me2)3]0/[H2O]0 ratios (vide infra). The occurrence of the condensation was indicated by the appearance of an additional peak with an almost double molecular weight of the main peak in the SEC chromatogram.
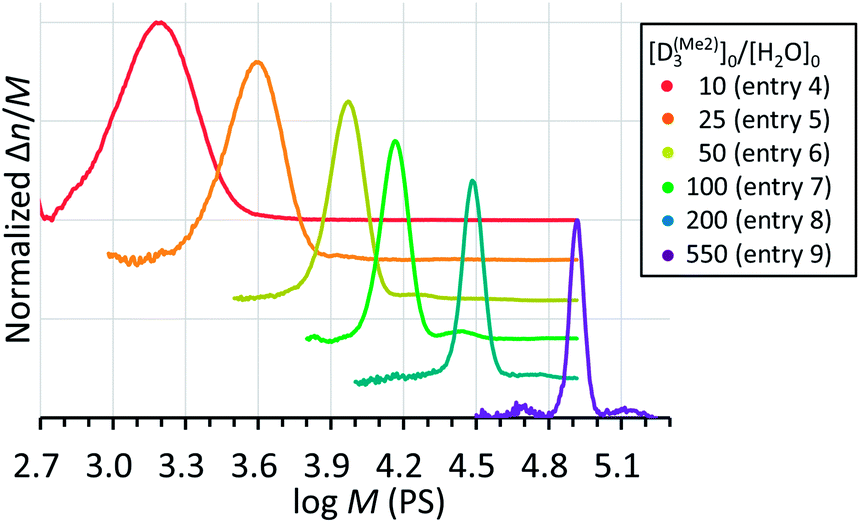 | ||
| Fig. 6 SEC chromatograms of PDMS synthesized by the polymerizations of D(Me2)3 initiated by water and catalyzed by TMnPG in a mixed solvent of CH2Cl2 and THF at 30 °C under different initial feed ratio of D(Me2)3 and water (Table 2, entries 4–9). | ||
Controlled synthesis of various telechelic polysiloxanes was easily achieved by an end-capping reaction using functional chlorosilanes. This was so far achieved solely with conventional anionic ROP using specially synthesized lithium compounds as an initiator.10,35–38 PDMS end-functionalized with dimethylhydrosilyl, dimethyl(vinyl)silyl, allyldimethylsilyl, (chloromethyl)dimethylsilyl, (bromomethyl)dimethylsilyl, dimethyl(2,3,4,5,6-pentafluorophenyl)silyl, and triethoxysilyl groups were obtained by the end-capping with the corresponding chlorosilanes at 30 °C for 15 min after the polymerization (Table 2, entries 10–16). 1H NMR, 29Si{1H} NMR, and MALDI-TOF MS spectra of the products proved the quantitative introduction of the functional groups as shown in Fig. S9–S15.† The products exhibited narrow Đ ranging from 1.10 to 1.15. Notably, telechelic polysiloxanes with halogenomethyl, 2,3,4,5,6-pentafluorophenyl, and triethoxysilyl groups with narrow Đ values were also obtained by our method, although these functional groups are potentially sensitive to the reaction conditions of the conventional anionic ROP of cyclotrisiloxanes using lithium compounds.
It is worth noting that, in contrast to the conventional anionic ROP initiated by lithium compounds, even non-dehydrated solvents were usable for the polymerization. D(Me2)3 using non-dehydrated THF (purity: >99.5%, stabilizer-free) as a solvent produced PDMS-(OSiMe2Ph)2 with Mn,NMR of 2.89 kg mol−1 and Đ of 1.11 under the same conditions as the polymerization shown in entry 1 of Table 2. Non-dehydrated CH2Cl2 (purity: >99.5%, stabilized with 2-methylbut-2-ene) was also usable (see ESI†). The maximum Mns reachable with non-dehydrated reagents and solvents depend on their water contents.45
Scope of monomers
Controlled/living polymerizations of cyclotrisiloxanes other than D(Me2)3 were also achieved using water as an initiator and TMnPG as a catalyst. 1,3,5-Trimethyl-1,3,5-triphenylcyclotrisiloxane (D(Me,Ph)3, cis/trans = 24/76), 1,3,5-trimethyl-1,3,5-trivinylcyclotrisiloxane (D(Me,Vi)3, cis/trans = 23/77), and 1,3,5-trimethyl-1,3,5-tris(3,3,3-trifluoropropyl)cyclotrisiloxane (D(Me,TFPr)3, cis/trans = 20/80) were polymerized in a mixed solvent of CH2Cl2 and THF at 30 °C under the conditions of [M]0/[H2O]0 = 10/1 and [M]0 = 1.80 mol L−1. Chlorotriethylsilane (Et3SiCl) was used as an end-capping agent for the polymerization of D(Me,Ph)3, while Me2PhSiCl was used for the polymerizations of D(Me,Vi)3 and D(Me,TFPr)3 to give well-defined PMPS, poly[methyl(vinyl)siloxane] (PMVS), and poly[methyl(3,3,3-trifluoropropyl)siloxane] (PMTFPS) with Mn,NMR (Đ) of 5.60 kg mol−1 (1.16), 3.64 kg mol−1 (1.11), and 6.00 kg mol−1 (1.12), respectively (Table 3, entries 1, 3, and 5). The stereoregularity of the resulting polymers was found to be atactic since the methyl groups in the inner repeating units of the PMPS and the PMVS showed three different signals in the 1H NMR spectra and the silicon atoms in the inner repeating units of the three polymers also showed three different signals in the 29Si{1H} NMR spectra (Fig. S16–18†).46 MALDI-TOF MS spectra shown in Fig. S16–18† evidenced that the obtained PMPS, PMVS, and PMTFPS consisted of only linear PMPS with two terminal triethylsiloxy groups as well as PMVS and PMTFPS with two terminal dimethyl(phenyl)siloxy groups. The polymerizations needed to be terminated when the monomer conversion reached 80–90%, since the condensation of two propagating polymers and the backbiting (vide infra) became noticeable.47 Indeed, the formation of D(Me,Ph)4 and 1,3,5,7-tetramethyl-1,3,5,7-tetravinylcyclotetrasiloxane (D(Me,Vi)4) was observed in the polymerizations of D(Me,Ph)3 and D(Me,Vi)3 (Table 3, entries 1–4 and Fig. S2 and S3†). Mn values of PMPS, PMVS, and PMTFPS were also controllable by changing [M]0/[H2O]0 (Table 3, entries 1 vs. 2, entries 3 vs. 4, and entries 5 vs. 6) as already demonstrated for PDMS.48 The values of the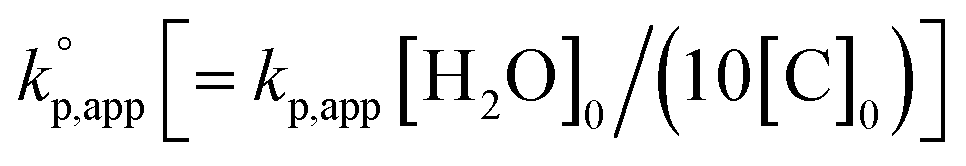 observed in the polymerizations of D(Me,Ph)3 (17 h−1, Table 3, entry 1), D(Me,Vi)3 (35 h−1, Table 3, entry 3), and D(Me,TFPr)3 (99 h−1, Table 3, entry 5) carried out with [M]0/[H2O]0 = 10 were all higher than that observed for D(Me2)3 (4.6 h−1, Table 2, entry 4), presumably due to the effect of electron-withdrawing phenyl, vinyl, and 3,3,3-trifluoropropyl groups, which increased the electrophilicity of the silicon atoms in the three monomers. Interestingly, the polymerizations of D(Me,Ph)3 showed almost 1 h of an induction period at the initial stage (Table 3, entries 1 and 2), although both of them produced PMPS with narrow Đ. Considering from the mechanism of the polymerization discussed in a later section, the reaction of water and D(Me,Ph)3 would proceed slowly while the activation of water by TMnPG was much more dominant than that of α,ω-dihydroxy-terminated oligo[methyl(phenyl)siloxane]s (vide infra).
observed in the polymerizations of D(Me,Ph)3 (17 h−1, Table 3, entry 1), D(Me,Vi)3 (35 h−1, Table 3, entry 3), and D(Me,TFPr)3 (99 h−1, Table 3, entry 5) carried out with [M]0/[H2O]0 = 10 were all higher than that observed for D(Me2)3 (4.6 h−1, Table 2, entry 4), presumably due to the effect of electron-withdrawing phenyl, vinyl, and 3,3,3-trifluoropropyl groups, which increased the electrophilicity of the silicon atoms in the three monomers. Interestingly, the polymerizations of D(Me,Ph)3 showed almost 1 h of an induction period at the initial stage (Table 3, entries 1 and 2), although both of them produced PMPS with narrow Đ. Considering from the mechanism of the polymerization discussed in a later section, the reaction of water and D(Me,Ph)3 would proceed slowly while the activation of water by TMnPG was much more dominant than that of α,ω-dihydroxy-terminated oligo[methyl(phenyl)siloxane]s (vide infra).
| Entry | Monomer | [M]0/[H2O]0/[C]0 | CH2Cl2/THF (v/v) | End-capping agent | Time (min) | Conv.a,b (%) | M n (kg mol−1) | Đ | k p,app (h−1) |
|
|||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| To polym. | To D4 | Calcd c | SEC d | NMR a | |||||||||
| a Determined by 1H NMR. b Mol% of the monomer converted to the corresponding polysiloxanes and cyclotetrasiloxanes. c Calculated from Mn,calcd = [M]0/[H2O]0 × (Conv. to polymer) × (MW. of monomer) + (MW. of terminal structures). d Determined by SEC measurements in THF using polystyrene standards. e Not determined. f Around 1 h of an induction period was observed. | |||||||||||||
| 1 | D(Me,Ph)3 | 10/1/0.01 | 57/43 | Et3SiCl | 219 | 92.4 | 5.8 | 4.02 | 4.39 | 5.60 | 1.16 | 1.7f | 17 |
| 2 | D(Me,Ph)3 | 30/1/0.03 | 79/21 | Et3SiCl | 240 | 83.3 | 10.6 | 10.5 | 5.04 | 9.92 | 1.15 | 1.0f | |
| 3 | D(Me,Vi)3 | 10/1/0.01 | 57/43 | Me2PhSiCl | 72 | 95.5 | 3.1 | 2.76 | 2.73 | 3.64 | 1.11 | 3.5 | 35 |
| 4 | D(Me,Vi)3 | 30/1/0.03 | 79/21 | Me2PhSiCl | 51 | 80.6 | 5.0 | 6.65 | 5.28 | 5.68 | 1.15 | 2.4 | |
| 5 | D(Me,TFPr)3 | 10/1/0.004 | 70/30 | Me2PhSiCl | 40 | 92.8 | n.d.e | 4.63 | 2.94 | 6.00 | 1.12 | 3.9 | 99 |
| 6 | D(Me,TFPr)3 | 30/1/0.012 | 88/12 | Me2PhSiCl | 100 | 94.2 | n.d.e | 13.5 | 6.38 | 13.3 | 1.13 | 1.7 | |
| 7 | D(Me2)4 | 7.5/1/0.10 | 65/35 | None | 1500 | 6.2 | n.d.e | 0.16 | n.d.e | n.d.e | n.d.e | ||
| 8 | D(Me,Vi)4 | 7.5/1/0.01 | 57/43 | None | 1500 | 10.6 | n.d.e | 0.29 | n.d.e | n.d.e | n.d.e | ||
| 9 | D(Me2)5 | 6/1/0.10 | 65/35 | None | 1500 | 1.5 | n.d.e | 0.051 | n.d.e | n.d.e | n.d.e | ||
| 10 | D(Me2)3 + D(Me,Vi)3 | 25 + 8/1/0.05 | 70/30 | Me2PhSiCl | 540 | 64.9, >99.9 | n.d.e | 6.02 | 4.68 | 6.34 | 1.13 | ||
| 11 | 1st: D(Me2)3 | 25/1/0.05 | 67/33 | Me2PhSiCl | 540 | 84.9 | n.d.e | 4.74 | 4.16 | 5.11 | 1.09 | ||
| 2nd: D(Me,Vi)3 | 25/1/0.05 | 28 | 80.8 | 2.9 | 10.2 | 9.56 | 11.2 | 1.07 | |||||
| 12 | 1st: D(Me2)3 | 25/1/0.25 | 84/16 | Et3SiCl | 60 | 92.7 | n.d.e | 5.17 | 4.48 | 5.89 | 1.08 | ||
| 2nd: D(Ph2)3 | 7.5/1/0.25 | 44 | 87.4 | n.d.e | 9.30 | 8.71 | 11.7 | 1.06 | |||||
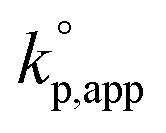
In contrast to cyclotrisiloxanes, cyclotetrasiloxanes and cyclopentasiloxanes, such as D(Me2)4, D(Me,Vi)4 and decamethylcyclopentasiloxane (D(Me2)5), were almost non-reactive under similar reaction conditions for D(Me2)3 and D(Me,Vi)3 (Table 3, entries 7–9), even though tBu-P4 (MeCNpKBH = 42.7)39 and NHCs, such as 1,3-dialkylimidazol-2-ylidene and 1,3-dialkyl-4,5-dimethylimidazol-2-ylidene (estimated MeCNpKBH = 32–34 and 34–36, respectively),49 with much higher Brønsted basicity than TMnPG are known to catalyze the polymerization of cyclotetrasiloxanes.29–32
Characteristics of copolymerization
Both statistical copolymerization and block copolymerization of two cyclotrisiloxanes were achieved with the polymerization using water as an initiator and TMnPG as a catalyst. Statistical copolymerization of D(Me2)3 and D(Me,Vi)3 was achieved by periodically adding 1 equiv. (to the initial amount of water) of D(Me,Vi)3 8 times into a reaction mixture of the polymerization of D(Me2)3 in CH2Cl2/THF (70/30, v/v) at 30 °C under the conditions of [D(Me2)3]0/[H2O]0/[TMnPG]0 = 25/1/0.05 (Table 3, entry 10) in a manner similar to the ‘semibatch method’ performed by Cypryk's group for a copolymerization of D(Me2)3 and D(Me,TFPr)3 using n-butyllithium as an initiator.50 D(Me,Vi)3 was added at 28, 60, 95, 135, 182, 238, 308, and 401 min from the initiation so that a constant amount of D(Me2)3 was consumed in the time interval between the two additions. The number-average content of the dimethylsiloxane and methyl(vinyl)siloxane units (n(D(Me2))/n(D(Me,Vi))) in the resulting poly[dimethylsiloxane-co-methyl(vinyl)siloxane] was 52/26 as determined by 1H NMR analysis (Fig. S19†). Mn,NMR and Đ of the product were determined to be 6.34 kg mol−1 and 1.13, respectively. 29Si{1H} NMR analysis revealed that the molar ratios of the triad monomeric sequences in the obtained copolymer were D(Me2)D(Me2)D(Me2)/D(Me2)D(Me2)D(Me,Vi)/D(Me,Vi)D(Me2)D(Me,Vi) = 71/29/0 and D(Me,Vi)D(Me,Vi)D(Me,Vi)/D(Me,Vi)D(Me,Vi)D(Me2)/D(Me2)D(Me,Vi)D(Me2) = 43/44/13, and the terminal monomeric unit of the product was D(Me2) as shown in Fig. S20.† The monomeric sequence of the obtained copolymer was visualized based on the population of the triad monomeric sequences, n(D(Me2))/n(D(Me,Vi)), and Đ as shown in Fig. S21,† which indicated the almost segregated presence of D(Me2)D(Me2) and D(Me,Vi)D(Me,Vi) units. In contrast, copolymerization of D(Me2)3 and D(Me,Vi)3 by adding TMnPG to a premixed solution of the two monomers resulted in a rapid and preferential polymerization of D(Me,Vi)3 followed by a slow polymerization of D(Me2)3 as previously observed in a copolymerization of D(Me2)3 and D(Me,Vi)3 using n-butyllithium as an initiator.51 The difference of each monomer consumption and the time-dependent SEC chromatogram changes are shown in Fig. S4 and S5,† respectively. The product was almost segmented copolymers of PMVS and PDMS with a broad molecular weight distribution as determined from Fig. S19 and S20.† A terminal methyl(vinyl)silanol unit, Si(Me,Vi)-OH, generated from D(Me,Vi)3 preferentially reacted with D(Me,Vi)3 rather than D(Me2)3 presumably due to higher electrophilicity of the former than the latter. Besides, side reactions involved by the Si(Me,Vi)-OH were more frequent than those of a dimethylsilanol unit, Si(Me2)-OH, and caused the broadening of the molecular weight distribution.Consecutive copolymerizations of D(Me2)3 and D(Me,Vi)3 (ref. 52,53) as well as D(Me2)3 and hexaphenylcyclotrisiloxane (D(Ph2)3)54 gave triblock copolymers of PDMS and PMVS (PMVS-b-PDMS-b-PMVS, Table 3, entry 11) as well as PDMS and poly(diphenylsiloxane) (PDPS) (PDPS-b-PDMS-b-PDPS, Table 3, entry 12). The syntheses were successful even though homopolymerization of D(Ph2)3 using water and TMnPG was hard to control because of very low solubility of PDPS in common organic solvents.54–56 The polymerizations of D(Me2)3 were first carried out in CH2Cl2/THF at 30 °C under the conditions of [D(Me2)]0/[H2O]0/[TMnPG]0 = 25/1/0.05 for the former and 25/1/0.25 for the latter. The polymerizations were further continued by adding 25 equiv. (with respect to the initial amount of water) of D(Me,Vi)3 or 7.5 equiv. Of D(Ph2)3 after 540 min or 75 min from the initiation of the first polymerization. SEC chromatograms of the obtained PDMS-(OH)2, PMVS-b-PDMS-b-PMVS, and PDPS-b-PDMS-b-PDPS indicated that PDMS-(OH)2 generated in the first polymerization quantitatively initiated the second polymerization as shown in Fig. 7. The products did not have a gradient structure between the segments of PDMS and PMVS as well as PDMS and PDPS as observed in the 29Si{1H} NMR spectra shown in Fig. S20 (ref. 57) and S22,†22,54,58 although the conversions of D(Me2)3 in the first polymerization were not quantitative in both of the syntheses. This result indicated that D(Me2)3 almost did not react in the second stage of the polymerization due to very low reactivity of the propagating end of PMVS and PDPS against D(Me2)3. The obtained PDPS-b-PDMS-b-PDPS had a much narrower molecular weight distribution (Đ = 1.06) than those synthesized by the conventional anionic ROP using dilithium diphenylsilanediolate as an initiator (Đ = 1.4–1.8).54 These successful block copolymerizations demonstrated the controlled/living nature of the ROP of cyclotrisiloxanes using water and strong organic bases.
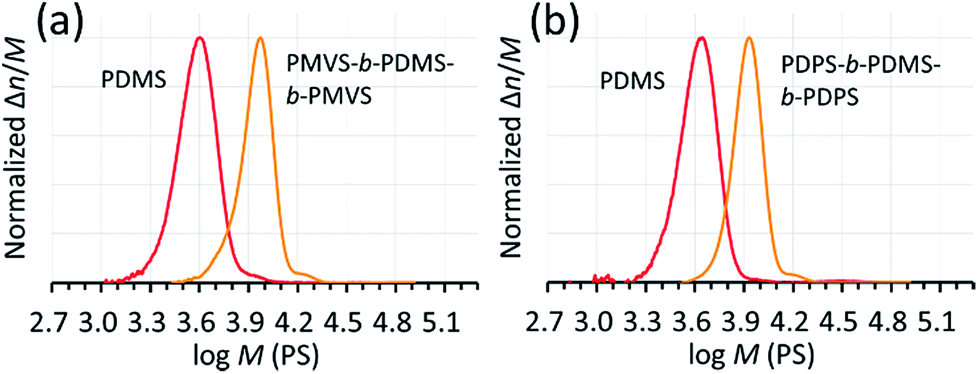 | ||
| Fig. 7 SEC chromatograms of the products obtained in the consecutive polymerizations of (a) D(Me2)3 and D(Me,Vi)3 (Table 3, entry 11) and (b) D(Me2)3 and D(Ph2)3 (Table 3, entry 12). | ||
Mechanism of the polymerization
It is worth discussing the mechanism of the polymerization catalyzed by the strong organic bases, since it has different characteristics compared to that of the conventional anionic ROP of cyclooligosiloxanes using hydroxide salts, organolithiums, and lithium silanolates as initiators. The structure of the propagating end in the former is a silanol, while that of the latter is a silanolate. Scheme 2 shows the possible elementary reactions in the ROP of cyclotrisiloxanes using water as an initiator and strong organic bases as catalysts. The polymerization is considered to proceed through the initiator/chain-end activation mechanism. (a) In the initiation reaction (Scheme 2a), a strong organic base first activates a water molecule. The ring-opening reaction of a cyclotrisiloxane occurs through the nucleophilic attack of the activated water at a silicon atom of a cyclotrisiloxane, followed by a proton transfer from the water to the cyclotrisiloxane to form a 1,5-dihydroxytrisiloxane. (b) The propagation reaction (Scheme 2b) proceeds through the activation of a terminal silanol group of a propagating polysiloxane by the strong organic base. The degree of polymerization of a propagating polysiloxane, n, increases by three after one propagation reaction. Kw and Ks can be defined as the equilibrium constants for the activation of water and a terminal silanol, and ki and kp can be defined as the rate coefficients for the ring-opening reaction of a monomer with the activated water and terminal silanol. The high catalytic activity of the guanidines B may have originated from the high efficiency of the proton transfer in the initiation and propagation reactions. The R–NC(N)–NH–R′ unit in the guanidines B would simultaneously activate water or a silanol on a propagating polymer by the imino group and transfer the hydrogen atom on the amino group to an incoming monomer based on the ‘proton shuttling mechanism’ as depicted in Scheme 3a.43 In contrast, the bases in the other categories are incapable of ‘proton shuttling’ and need to directly transfer the hydrogen atom as depicted in Scheme 3b. Ks would increase as the acidity of the terminal silanol increases and the steric hindrance of the silanol decreases. kp would increase as nucleophilicity of the activated silanol increases, electrophilicity of the monomer increases, and the steric hindrance of the silanol and the monomer decrease. (c) ‘Intermolecular transfer’ of a terminal hydroxysilyl group from one propagating polymer to another one (disproportionation) can be considered to occur by a nucleophilic attack of the activated terminal silanol to another terminal silanol (Scheme 2c). This reaction increases and decreases the degree of polymerization of the two propagating polymers by one and causes the formation of polysiloxanes, of which degree of polymerization is not a multiple of 3, as observed in Fig. 4. A model reaction using 1,5-dihydroxy-1,1,3,3,5,5-hexamethyltrisiloxane (D(Me2)3-(OH)2) and TMnPG produced a mixture of α,ω-dihydroxy-terminated permethyloligosiloxanes with different numbers of siloxy units in 6 min in THF at 30 °C under the conditions of [D(Me2)3-(OH)2]0/[TMnPG]0 = 1/0.1 (Fig. S6†). Similar reactions, ‘intermolecular transfer’ of terminal lithium silanolates on PDMS14,59 as well as ‘disproportionation’ of hydroxy-terminated oligodimethylsiloxanes,60–62 have also been reported to occur in the conventional anionic ROP of D(Me2)3 as well as in a transformation of oligosiloxanol in the presence of a strong base. The intermolecular transfer is an allowable side reaction since it does not largely affect Mn and Đ of the resulting polysiloxanes. (d) Condensation (dimerization, Scheme 2d) also occurs during the polymerization by a similar mechanism to (c) as an undesired side reaction. As already mentioned, the condensation of two propagating polysiloxanes becomes pronounced, in particular in the late stage of polymerization, and causes increasing of Đ of the resulting polysiloxanes. It occurs much less frequently than (c). The similar tendency was observed in the disproportionation of oligosiloxanol using phosphazenium hydroxide.62 (e) Backbiting (depolymerization, Scheme 2e) occurs by intramolecular nucleophilic attack of the activated terminal silanol of a propagating polymer to a siloxy unit in its inner repeating units. The backbiting produces cyclooligosiloxanes, such as cyclotetrasiloxane and cyclopentasiloxane, and reduces the degree of polymerization and yield of propagating polymers. On the other hand, (f) the intermolecular chain-transfer reaction (Scheme 2f), which is known to occur in a conventional anionic ROP of cyclotrisiloxane,14 did not occur at least in the polymerization of D(Me2)3 catalyzed by TMnPG in THF at 30 °C, although it is a possible undesired side reaction. Trimethylsiloxy-terminated PDMS was not produced in a model polymerization of D(Me2)3 in the presence of dodecamethylpentasiloxane (see ESI, Fig. S7†). The difference in the chances of side reactions (e) and (f) would be originated from the fact that the silanol and the silicon atom to be attacked always closely present for the former. The terminal silicon atom bearing a hydroxy group would be sterically and electronically more susceptible to a nucleophilic attack than the silicon atoms in the inner repeating units, since a hydroxy group is less bulky and a stronger electron withdrawing group than a dialkylsiloxy group.63,64 The frequency of the side reactions increased in the following order: (e) < (d) < (c).
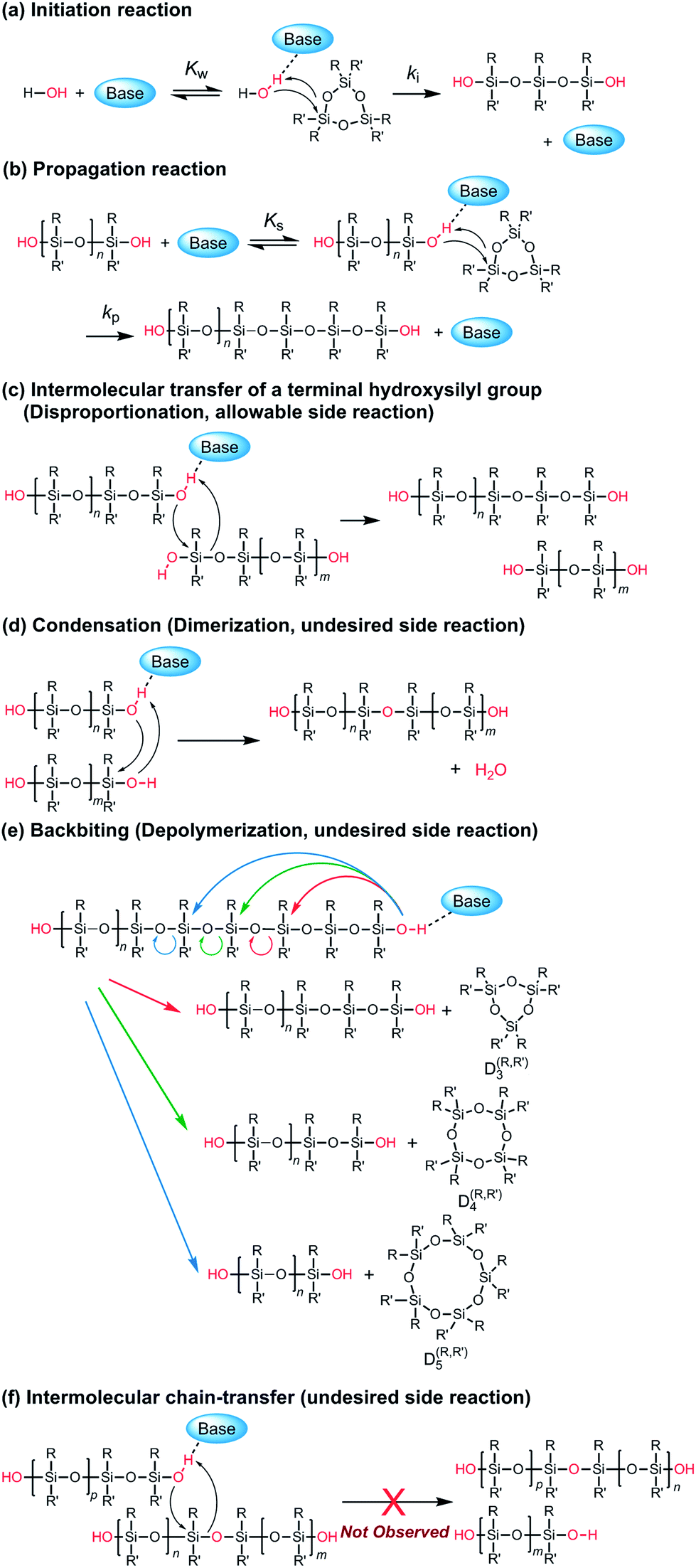 | ||
| Scheme 2 Possible elementary reactions in the polymerization of cyclotrisiloxane using water as an initiator and strong organic bases as catalysts. | ||
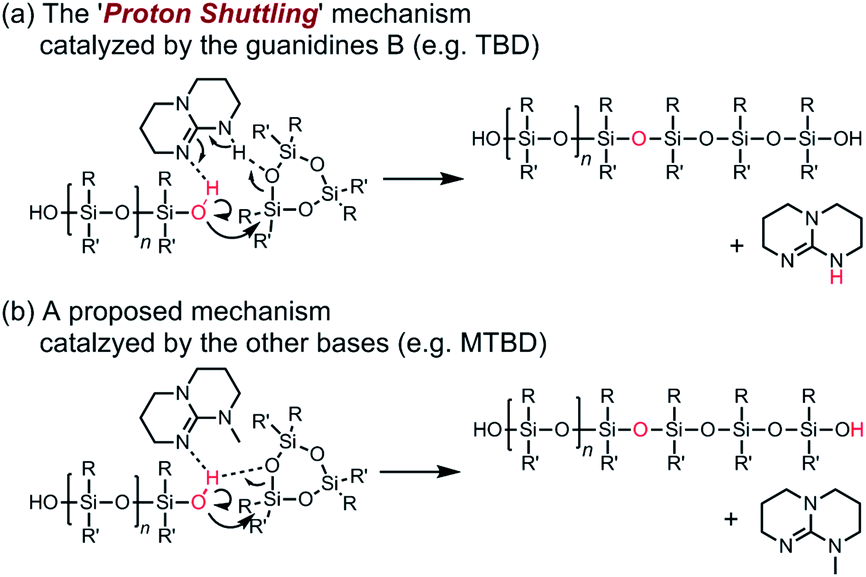 | ||
| Scheme 3 Proposed mechanism for the ring-opening reaction of cyclotrisiloxanes catalyzed by strong organic bases. | ||
Although only the intramolecular condensation of a propagating polysiloxane can be considered as a possible termination reaction, we did not observe this process in any polymerizations that we conducted. Hence, the polymerization is characterized by the absence of termination reactions, which ensures the controlled/living nature of the polymerization. The total numbers of reactive hydroxy groups in water, which has ‘two’ hydroxyl groups, and propagating polysiloxanes do not change throughout the polymerization even when side reactions (c), (d), and (e) occur. The polymerization can be terminated only by neutralization of the catalyst or end-capping of propagating polymers.
Another possible polymerization mechanism is the ‘nucleophilic monomer activation mechanism’ that is based on the activation of the monomer by the nucleophilic attack of the catalyst.19 However, if so, (1) catalytic activity of the strong organic bases would not be linear to their Brønsted basicity as shown in Fig. 1, since the nucleophilicity (‘silicophilicity’) of the bases and their basicity are independent as it was demonstrated in a condensation of a slanol and a organosilanes with a leaving group catalyzed by an organic base.65–67 Besides, (2) the first-order kinetic plot would show a convex curve when the interaction of a catalyst and a monomer is high.68
Effects of reaction conditions on kinetics of the polymerization
The plausibility of the proposed polymerization mechanism was confirmed by comparing a rate equation derived from the kinetics of the elementary reactions, eqn (2), with the experimentally obtained rate equation, eqn (1). The rate of propagation, r, can be expressed by the product of the rate coefficient of propagation, kp (L mol−1 h−1), the concentration of hydroxy groups activated by catalyst, [P*] (mol L−1), and the concentration of unreacted monomer, [M] (mol L−1), when both Kw and ki are comparable to or greater than Ks and kp.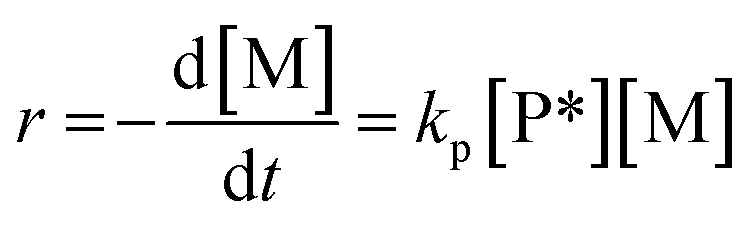 | (2) |
[P*] is expressed by the product of the concentration of hydroxy groups not being activated, [P] (mol L−1), the concentration of catalyst not interacting with hydroxy groups, [C] (mol L−1), and the equilibrium constant for the activation of silanol groups by the catalyst, Ks (L mol−1), i.e., [P*] = Ks[P][C], since Ks is defined as Ks = [P*]/[P][C]. [P*] is hence expressed by eqn (3) using the initial concentration of hydroxy groups, [P]0 (mol L−1), the initial concentration of the catalyst, [C]0 (mol L−1), since [P] = [P]0 − [P*] and [C] = [C]0 − [P*].
| [P*] = Ks([P]0 − [P*])([C]0 − [P*]) | (3) |
From the quadratic formula, [P*] is expressed by eqn (4) as the function of three constants, i.e., [P]0, [C]0, and Ks that varies depending on the catalyst, the temperature, and the solvent employed for a polymerization:
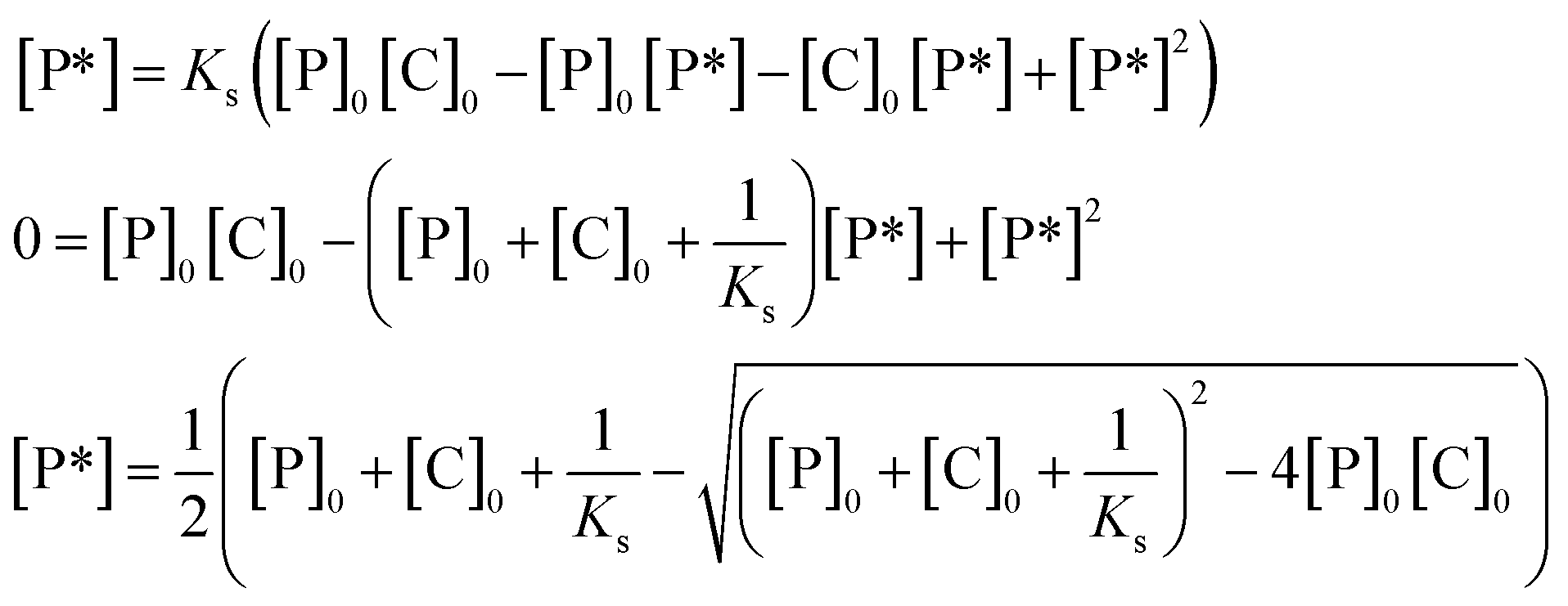 | (4) |
[P*] is hence constant, regardless of monomer conversion, c, and frequency of the side reactions (c), (d), and (e). The integration of eqn (2) gives eqn (5):
| −ln(1 − c) = kp[P*]t | (5) |
| kp,app = kp[P*] | (6) |
Hence, the proposed mechanism for the polymerization shown in Scheme 2 were proven to be reasonable. The polymerization is considered to proceed through the initiator/chain-end activation mechanism.
Effects of reaction conditions on the kinetics of polymerization can be expected with eqn (4)–(6). Fig. 8a shows the expected dependence of kp,app on Ks and [C]0/[P]0 under the conditions of [M]0 = 1.80 mol L−1 and [M]0/[P]0 = 5. The calculated kp,app was normalized by dividing it by the kp,app on [M]0/[P]0 = 1. It was found that kp,app is almost proportional to [C]0/[P]0 when [C]0/[P]0 ≤ 1 regardless of Ks, which corresponded to the results shown in Fig. 5. In contrast, the increase in kp,app with increasing [C]0/[P]0 depends on Ks when [C]0/[P]0 > 1 and its rate of change decreases with increasing Ks. Fig. 8b shows the expected dependence of kp,app on Ks and [M]0/[P]0 under the conditions of [M]0 = 1.80 mol L−1 and [M]0/[C]0 = 100. The calculated 1/kp,app was normalized by multiplying it by the kp,app on [M]0/[P]0 = 5. It was found that kp,app is almost inversely proportional to [M]0/[P]0 when Ks is small, while kp,app only gradually decreases with increasing [M]0/[P]0 when Ks is large. Considering the values of kp,app observed in the polymerizations of D(Me2)3, D(Me,Ph)3, D(Me,Vi)3, and D(Me,TFPr)3 with different [M]0/[H2O]0 (entries 4, 5 of Table 2 and entries 1–6 of Table 3), the magnitude of Ks would increase in the following order: D(Me,TFPr)3 ≈ D(Me2)3 < D(Me,Ph)3 < D(Me,Vi)3.
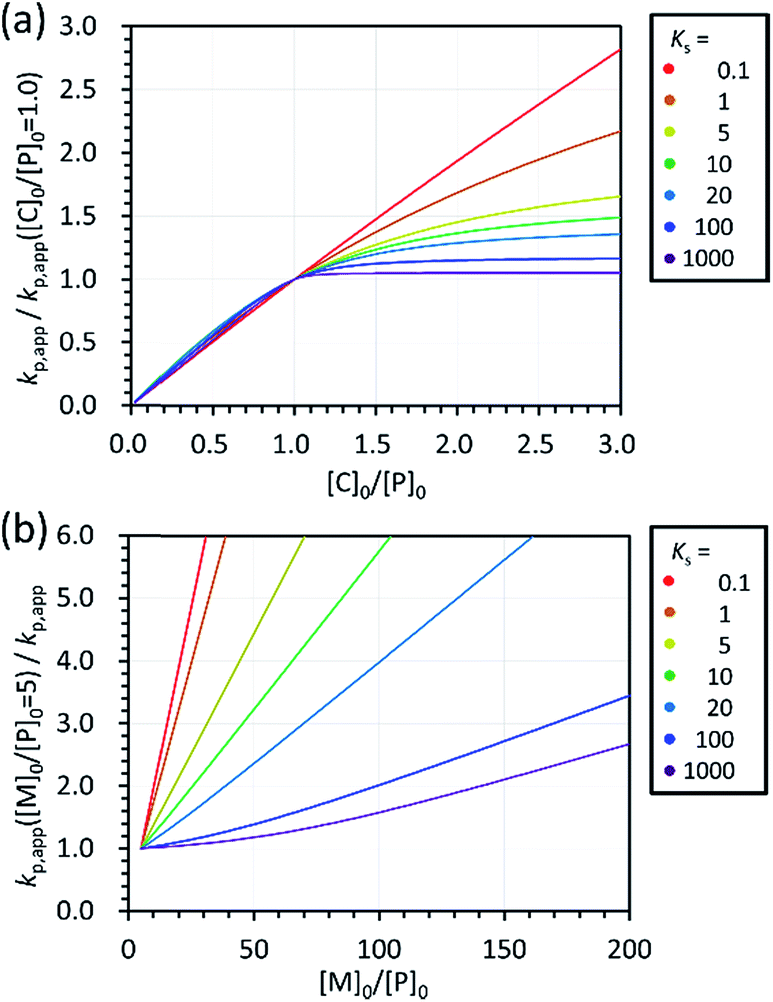 | ||
| Fig. 8 Calculated dependence of (a) kp,app/kp,app([C]0/[P]0 = 1.0) on Ks and [C]0/[P]0 ([M]0 = 1.80 mol L−1, [M]0/[P]0 = 5) and that of (b) kp,app([M]0/[P]0 = 5)/kp,app on Ks and [M]0/[P]0 ([M]0 = 1.80 mol L−1, [M]0/[C]0 = 100). | ||
Conclusions
We have developed an organocatalytic controlled/living ROP of cyclotrisiloxanes using water as an initiator, strong organic bases as catalysts, and organochlorosilanes as end-capping agents as a convenient and efficient method to synthesize (telechelic) linear polysiloxanes and their copolymers with controlled Mn, narrow molecular weight distributions, and desired terminal structures. The catalytic activity of each strong organic base was found to depend on its Brønsted basicity, MeCNpKBH, and the efficiency of the proton transfer from water or terminal silanols on propagating polysiloxanes toward an incoming monomer. The guanidines B, namely guanidines possessing an R–NC(N)–NH–R′ unit, showed the highest catalytic activity per their Brønsted basicity among the tested organic bases, probably due to their high efficiency of the proton transfer through the ‘proton shuttling mechanism’. In particular, the performance of TMnPG, a monocyclic guanidine B, was the best in terms of its high catalytic activity and low frequency of side reactions. The results of the kinetic investigations and the model reactions suggested that the polymerization proceeds through the initiator/chain-end activation mechanism. One of the most remarkable features of the polymerization is that the polymerization proceeds in a controlled/living fashion even in a non-dehydrated solvent as long as the moisture content of the solvent is taken into account, since it has been considered ‘common sense’ that intensive dehydration and purification of reagents and solvents is essential for conventional anionic ROP initiated by lithium compounds. This new convenient method to synthesize well-defined polysiloxanes will enable the synthesis of organosilicon and organic/inorganic hybrid materials with diverse architectures and may lead to the development of new advanced materials with improved properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the “Development of Innovative Catalytic Processes for Organosilicon Functional Materials” project (PL: K. S.) from the New Energy and Industrial Technology Development Organization (NEDO).Notes and references
- Silicon-Based Polymer Science, ed. J. M. Zeigler and F. W. G. Fearon, ACS Publication, Washington, DC, 1990 Search PubMed.
- M. A. Brook, Silicon in Organic, Organometallic, and Polymer Chemistry, Wiley-Interscience, New York, 1999 Search PubMed.
- B. Jiang, X. Zhan, L. Yi, X. Zhang, M. Xu and L. Sun, Prog. Chem., 2010, 22, 1169–1176 CAS.
- S. Carlotti and F. Peruch, Cyclic Monomers: Epoxides, Lactide, Lactones, Lactams, Cyclic Silicon-Containing Monomers, Cyclic Carbonates, and Others, in Anionic Polymerization: Principles, Practice, Strength, Consequences and Applications, ed. N. Hadjichristidis and A. Hirao, Springer Japan KK, Tokyo, 2015, pp. 191–305 Search PubMed.
- J. C. Saam, D. J. Gordon and S. Lindsay, Macromolecules, 1970, 3, 1–4 CrossRef CAS.
- H. J. Hölle and B. R. Lehnen, Eur. Polym. J., 1975, 11, 663–667 CrossRef.
- J. G. Zilliox, J. E. L. Roovers and S. Bywater, Macromolecules, 1975, 8, 573–578 CrossRef CAS.
- S. Boileau, Use of Cryptates in Anionic Polymerization of Heterocyclic Compounds, in Anionic Polymerization, Kinetics, Mechanisms and Synthesis, ed. J. E. McGrath, American Chemical Society, Washington, DC, 1981, pp. 283–306 Search PubMed.
- S. Boileau, Anionic Polymerization of Cyclosiloxanes with Cryptates as Counterions, in Ring-Opening Polymerization Kinetics, Mechanisms, and Synthesis, ed. J. E. McGrath, American Chemical Society, Washington, DC, 1985, pp. 23–35 Search PubMed.
- Y. Gnanou and P. Rempp, Makromol. Chem., 1988, 189, 1997–2005 CrossRef CAS.
- U. Maschke, T. Wagne and X. Coqueret, Makromol. Chem., 1992, 193, 2453–2466 CrossRef CAS.
- B. Eßwein, A. Molenberg and M. Möller, Macromol. Symp., 1996, 107, 331–340 CrossRef.
- A. Molenberg and M. Möller, Macromol. Chem. Phys., 1997, 198, 717–726 CrossRef CAS.
- D. M. Haddleton, S. A. Bon, K. L. Robinson, N. J. Emery and I. Moss, Macromol. Chem. Phys., 2000, 201, 694–698 CrossRef CAS.
- V. Bellas, H. Iatrou and N. Hadjchristidis, Macromolecules, 2000, 33, 6993–6997 CrossRef CAS.
- P. Boehm, M. Mondeshki and H. Frey, Macromol. Rapid Commun., 2012, 33, 1861–1867 CrossRef CAS PubMed.
- H.-F. Fei, X. Han, B. Liu, X. Gao, Q. Wang, Z. Zhang and Z. Xie, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 835–843 CrossRef CAS.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- C. Thomasa and B. Bibal, Green Chem., 2014, 16, 1687–1699 RSC.
- W. N. Ottou, H. Sardon, D. Mecerreyes, J. Vignolle and D. Taton, Prog. Polym. Sci., 2016, 56, 64–115 CrossRef CAS.
- J. Chojnowski, M. Cypryk and K. Kaźmierski, Macromolecules, 2002, 35, 9904–9912 CrossRef CAS.
- G. Toskas, M. Moreau and P. Sigwalt, Macromol. Symp., 2006, 240, 68–77 CrossRef CAS.
- J.-R. Desmurs, L. Ghosez, J. Martins, T. Deforth and G. Mignani, J. Organomet. Chem., 2002, 646, 171–178 CrossRef CAS.
- A. Grzelka, J. Chojnowski, W. Fortuniak, R. Taylor and P. H. Hupfield, J. Inorg. Organomet. Polym., 2004, 14, 101–116 CrossRef CAS.
- Q. Wang, H. Zhang, G. K. S. Prakash, T. E. Hogen-Esch and G. A. Olah, Macromolecules, 1996, 29, 6691–6694 CrossRef CAS.
- G. Toskas, M. Moreau, M. Masure and P. Sigwalt, Macromolecules, 2001, 34, 4730–4736 CrossRef CAS.
- J. Chojnowski, S. Rubinsztajn, W. Fortuniak and J. Kurjata, J. Inorg. Organomet. Polym. Mater., 2007, 17, 173–187 CrossRef CAS.
- A. Molenberg and M. Möller, Macromol. Rapid Commun., 1995, 16, 449–453 CrossRef CAS.
- P. C. Hupfeld and R. G. Taylor, J. Inorg. Organomet. Polym., 1999, 9, 17–34 CrossRef.
- M. E. Van Dyke and S. J. Clarson, J. Inorg. Organomet. Polym., 1998, 8, 111–117 CrossRef CAS.
- M. Rodriguez, S. Marrot, T. Kato, S. Stérin, E. Fleury and A. Baceiredo, J. Organomet. Chem., 2007, 692, 705–708 CrossRef CAS.
- B. G. G. Lohmeijer, G. Dubois, L. Leibfarth, R. C. Pratt, F. Nederberg, A. Nelson, R. M. Waymouth, C. Wade and J. L. Hedrick, Org. Lett., 2006, 8, 4683–4686 CrossRef CAS PubMed.
- H. A. Brown, Y. A. Chang and R. M. Waymouth, J. Am. Chem. Soc., 2013, 135, 18738–18741 CrossRef CAS PubMed.
- S. L. Bontems, J. Stein and M. A. Zumbrum, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2697–2710 CrossRef CAS.
- K. P. Battjes, C.-M. Kuo, R. L. Miller and J. C. Saam, Macromolecules, 1995, 28, 790–792 CrossRef CAS.
- W. Peng and Z. Xie, Macromol. Chem. Phys., 2000, 201, 1292–1294 CrossRef CAS.
- A. Saxena, S. Rajaraman and M. Leatherman, Macromolecules, 2007, 40, 752–755 CrossRef CAS.
- D. Margetić, Physico-Chemical Properties of Organosuperbases, in Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalysts, ed. T. Ishikawa, Wiley, New York, 2009, pp. 9–48 Search PubMed.
- B. Kovačević and Z. B. Maksić, Org. Lett., 2001, 3, 1523–1526 CrossRef.
- I. Kaljurand, A. Kütt, L. Sooväli, T. Rodima, V. Mäemets, I. Leito and I. Koppel, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS PubMed.
- K. Vazdar, R. Kunetskiy, J. Saame, K. Kaupmees, I. Leito and U. Jahn, Angew. Chem., Int. Ed., 2014, 53, 1435–1438 CrossRef CAS PubMed.
- pKBHs of TBO, TBN, TMGa, TMnPG, MTBD, and TBD in CD3OD/D2O = 8/2 (w/w) were reported to be 11.09, 12.9, 13.2, >14, >14, and >14. C. Ghobril, P. Hammar, S. Kodepelly, B. Spiess, A. Wagner, F. Himo and R. M. Baati, ChemCatChem, 2010, 2, 1573–1581 CrossRef CAS.
- The low-molecular-weight parts of the products were removed during the purification procedures when Mns of the products were low as shown in Fig. S1.†.
- According to Williams et al., the water content in non-dehydrated THF and CH2Cl2 (wsolv.) are 107.8 ppm and 22.4 ppm, respectively. Mn,calcd can be calculated by Mn,calcd = 103 (MWmonomer)(MWH2O)[M]0/(dsolv. × 10−6wsolv.), when only water from the solvents is considered (dTHF = 0.852, dCH2Cl2 = 1.327 g mL−1). The maximum Mn of PDMS was calculated to be 78.4 kg mol−1 and 243 kg mol−1 for polymerizations of D(Me2)3 in THF and CH2Cl2 under the conditions of [M]0 = 1.8 mol L−1. D. B. G. Williams and M. Lawton, J. Org. Chem., 2010, 75, 8351–8354 CrossRef CAS PubMed.
- T. N. Baratova, V. P. Mileshkevich and V. I. Gurari, Polym. Sci. U.S.S.R., 1982, 24, 27–33 CrossRef.
- Kelling's group has reported that electron-withdrawing substituents on a silanol increase the rate of condensation under basic conditions. S. Bilda, G. Röhr, D. Lange, E. Popowski and H. Kelling, Z. Anorg. Allg. Chem., 1988, 564, 155–160 CrossRef CAS.
- We assume that suppression of backbiting (depolymerization) will be necessary for obtaining PMPS, PMVS, and PMTFPS with high Mn.
- A. M. Magill, K. J. Cavell and B. F. Yates, J. Am. Chem. Soc., 2004, 126, 8717–8724 CrossRef CAS PubMed.
- M. Cypryk, B. Delczyk, A. Juhari and K. Koynov, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1204–1216 CrossRef CAS.
- J. Chojnowski, M. Cypryk, W. Fortuniak, M. Ścibiorek and K. Rózga-Wijas, Macromolecules, 2003, 36, 3890–3897 CrossRef CAS.
- M. Ścibiorek, N. K. Gladkova and J. Chojnowski, Polym. Bull., 2000, 44, 377–384 CrossRef.
- J. Bauer, N. Hüsing and G. Kickelbick, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3975–3985 CrossRef CAS.
- T. M. Gädda and W. P. Weber, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3629–3639 CrossRef.
- B. R. Harkness, M. Tachikawa, H. Yue and I. Mita, Chem. Mater., 1998, 10, 1700–1705 CrossRef CAS.
- V. S. Papkov, M. I. Buzin, M. V. Gerasimov and E. S. Obolonkova, Macromolecules, 2002, 35, 1079–1090 CrossRef CAS.
- 29Si{1H} NMR spectrum of poly[dimethylsiloxane-co-methyl(vinyl)siloxane] with different monomer sequences has been measured in CDCl3. C. Ivanenko, C. Maitre, F. Ganachaud and P. Hémery, E Polymer, 2003, 010 Search PubMed.
- M. Cypryk, K. Kaźmierski, W. Fortuniak and J. Chojnowski, Macromolecules, 2000, 33, 1536–1545 CrossRef CAS.
- T. Suzuki, Polymer, 1989, 30, 333–337 CrossRef CAS.
- J. Chojnowski, S. Rubinsztajn, W. Stańczyk and M. Ścibiorek, Makromol. Chem., Rapid Commun., 1983, 4, 703–706 CrossRef CAS.
- J. Chojnowski, K. Kaźmierski, S. Rubinsztajn and W. Stańczyk, Makromol. Chem., 1986, 187, 2039–2052 CrossRef CAS.
- A. Grzelka, J. Chojnowski, M. Cypryk, W. Fortuniak, P. C. Hupfield and R. G. Taylor, J. Organomet. Chem., 2002, 660, 14–26 CrossRef CAS.
- A hydroxy group should be a stronger electron-withdrawing group than a dimethylsiloxy group, since the relative acidity of the hydroxy group in Me2Si(OH)2 was reported to be higher than that in Me3Si–O–SiMe2–O–SiMe2OH. R50 H. J. Holdt, E. Popowski and H. Kelling, Z. Anorg. Allg. Chem., 1984, 519, 233–240 CrossRef CAS.
- W. Rutz, D. Lange, E. Popowski and H. Kelling, Z. Anorg. Allg. Chem., 1986, 542, 217–222 CrossRef CAS.
- S. Rubinsztajn, M. Cypryk and J. Chojnowski, J. Organomet. Chem., 1989, 367, 27–37 CrossRef CAS.
- M. Cypryk, S. Rubinsztajn and J. Chojnowski, J. Organomet. Chem., 1989, 377, 197–204 CrossRef CAS.
- J. Pikies, K. Przyjemska and W. Wojnowski, Z. Anorg. Allg. Chem., 1987, 551, 209–214 CrossRef CAS.
- K. Fuchise, Y.-G. Chen, K. Takada, T. Satoh and T. Kakuchi, Macromol. Chem. Phys., 2012, 213, 1604–1611 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and 1H NMR, 29Si{1H} NMR, and MALDI-TOF MS spectra of the synthesized polysiloxanes. See DOI: 10.1039/c7sc04234e |
| This journal is © The Royal Society of Chemistry 2018 |