
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLayered zinc hydroxide monolayers by hydrolysis of organozincs†
Alice H. M.
Leung
a,
Sebastian D.
Pike
 a,
Adam J.
Clancy
b,
Hin Chun
Yau
b,
Won Jun
Lee
b,
Katherine L.
Orchard
b,
Milo S. P.
Shaffer
*bc and
Charlotte K.
Williams
*ab
a,
Adam J.
Clancy
b,
Hin Chun
Yau
b,
Won Jun
Lee
b,
Katherine L.
Orchard
b,
Milo S. P.
Shaffer
*bc and
Charlotte K.
Williams
*ab
aChemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, UK OX1 3TA. E-mail: charlotte.williams@chem.ox.ac.uk
bDepartment of Chemistry, Imperial College London, London, UK SW7 2AZ. E-mail: m.shaffer@imperial.ac.uk
cDepartment of Materials, Imperial College London, London, UK SW7 2AZ
First published on 25th January 2018
Abstract
2D inorganic materials and their exfoliated counterparts are both of fundamental interest and relevant for applications including catalysis, electronics and sensing. Here, a new bottom-up synthesis route is used to prepare functionalised nanoplatelets, in apolar organic solvents, via the hydrolysis of organometallic reagents; the products can be prepared in high yield, at room temperature. In particular, a series of layered zinc hydroxides, coordinated by aliphatic carboxylate ligands, were produced by the hydrolysis of diethyl zinc and zinc carboxylate mixtures, optimally at a molar ratio of [COOR]/[Zn] = 0.6. Layered zinc hydroxides coordinated by oleate ligands form high concentration solutions of isolated monolayers (3 nm thick x ∼ 26 nm) in apolar organic solvents (up to 23 mg mL−1 in toluene), as confirmed by both atomic force and transmission electron microscopies of deposited species. The high solubility of the product allows the synthetic pathway to be monitored directly in situ through 1H NMR spectroscopy. The high solubility also provides a route to solution deposition of active functional materials, as illustrated by the formation of nanoporous films of optically transparent porous zinc oxide (1 μm thickness) after annealing at 500 °C. This new organometallic route to 2D materials obviates common complications of top-down exfoliation syntheses, including sonochemical-degradation and low yields of aggregated polydispersed layers, and may potentially be extended to a wide range of systems.
Introduction
Inorganic nanoplatelets combine an atomic layer thickness with lateral, 2D crystalline lattices, usually of nano- or micrometer size.1 Bulk quantities, particularly of liquid phase dispersions, are commonly prepared by the exfoliation or separation of (mono)layers, from a layered crystalline feedstock. Inorganic clays have been well known for at least half a century,2–4 and more recently, the isolation of monolayer graphene5 and other 2D nanoplatelets, including transition metal dichalcogenides, black phosphorous and metal hydroxides/oxides, have been explored.6–14 The enormous current interest in 2D nanoplatelets stems from an appreciation of their unique and diverse (opto)electronic properties, arising from their physical size and versatile composition.6 On the other hand, nanoplatelets also show very high specific surface areas, with potentially highly controllable and defined surface chemistry, leading to a high density of exposed active sites. Additionally, their aspect ratio allows them to be assembled into hierarchical structures with good transport characteristics, short critical diffusion lengths and, in many cases, good conductivity for ions and/or electrons.15–17 These properties have resulted in promising performances as catalysts, supports, photo-redox materials, ion sensors and as the active layer in electronics or energy storage devices.6,17–20 Nonetheless, it remains challenging to synthesise inorganic nanoplatelets efficiently and controllably, and in particular to produce high yields of exfoliated monolayers.Nanoplatelets of inorganic layered metal hydroxides have been reported for most transition metals, main group elements and lanthanides, but are most widely applied with Zn(II), Mg(II) or Al(III) metal centres, often as layered double hydroxides (LDH).1,8,9,21–23 Layered zinc hydroxides (LZHs) have the general formula [Zn5(OH)8(A)2·nH2O], where A is an intercalated anion such as a carbonate, nitrate, carboxylate or sulphate group.22,24–27 Their structures are related to the natural mineral Brucite and feature octahedral, edge-sharing sheets of zinc-hydroxide units, where two tetrahedral zinc hydroxide units are situated above and below a vacant octahedral site (Fig. 1). The intercalated ligand, A, coordinates to the tetrahedral zinc atom, at the apex of the tetrahedron, and thereby directs the structure to form a sequence of alternating inorganic hydroxide and intercalated anionic layers. The preparation of LZH nanoplatelets is generally accomplished in two stages: (1) inorganic salts undergo a base-assisted decomposition to form a bulk material and (2) the layers of the agglomerated material are exfoliated to yield the nanoplatelets.9 The base-assisted decomposition of zinc salts/oxides forms agglomerated layered zinc hydroxides, typically under harsh conditions (usually at temperatures >80 °C, for 1–5 days), which limit precise control of the layer thickness.21,24,26,28 Similar methods have been reported at lower temperature (60 °C), using amino acids and dipeptides to stablise LZH over ZnO, but relatively long synthesis times are still required.29 Syntheses at room temperatures have been reported, though in some cases requiring a higher temperature (>100 °C) for precursor dissolution.24,27 Urea is commonly used as the base, however, its use releases carbonate ions which contaminate the nanoplatelets (also a common problem with LZH syntheses conducted under air) and complicate exfoliation into individual layers.30–32 The second step involves a physically aggressive treatment of the agglomerated bulk in solvent to exfoliate the nanoplatelets; for example, ultrasonication in dimethylformamide and/or butanol is common, often with the use of additional surfactants.27,33,34 By using these methods, it is possible to form kinetically-stabilised mixtures of dispersed monolayers and stacks,6,34 but there can be significant sonochemical degradation of both the LZHs and solvents.35 Exfoliation is also possible in polar solvents using conventional mechanical shear, but the process can be slow and is also poorly controlled.27,34 These physical processes tend to limit reproducibility and monolayer yield, while generating only metastable products. LZH nanoplatelets prepared via these routes are nonetheless broadly useful and have been applied in biomedical technologies (such as encapsulation/intercalation and release of drug molecules),29,36,37 photocatalysis38 and in electronic devices.39–43 For example, a LZH-derived ZnO film formed a microstructure particularly suited to the preparation of dye-sensitised solar cells (DSSCs) with good performance.41 It is likely that there are many other potential applications based on the known properties of the related layered double hydroxide structures, such as sorbents and fire retardants.9,44
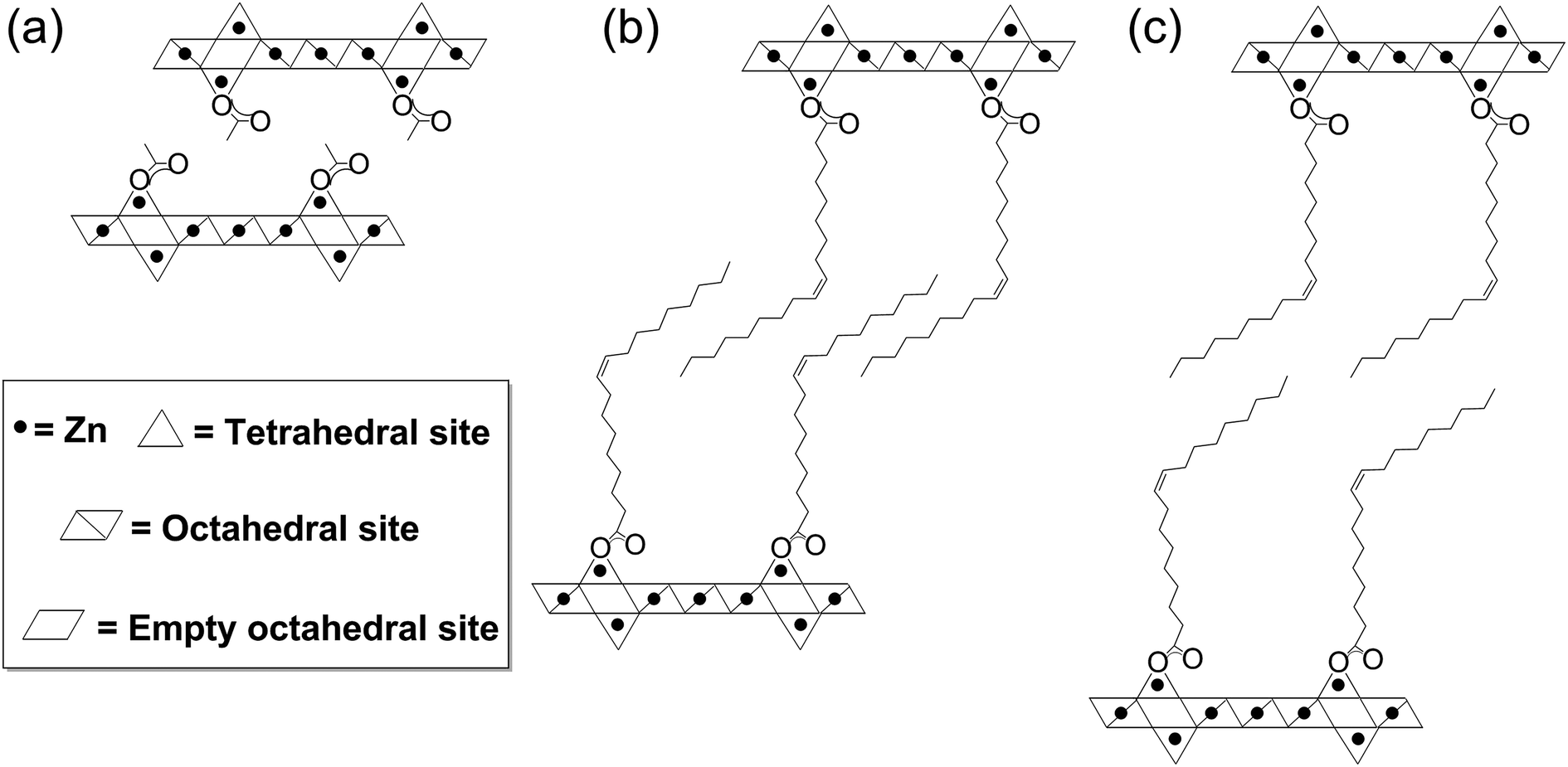 | ||
| Fig. 1 Schematic structure of (a) layered zinc hydroxide acetate (LZH-OAc), (b) layered zinc hydroxide oleate (LZH-Ole) with partial interdigitation and (c) LZH-Ole with no interdigitation occurring between the oleate chains. | ||
Given the range of applications and intrinsic interest in layered nanoplatelets, there is a need to explore low temperature and more specific routes to prepare and exfoliate this class of inorganic material. Our approach, which uses the controlled hydrolysis of organo-zinc reagents to prepare nanoplatelets directly and with minimal contamination, is quite distinct from conventional syntheses of LZHs. We were inspired by discoveries that the hydrolysis of organo-zinc species, such as diethyl zinc, can be used to prepare crystalline Wurtzite ZnO nanoparticles, in a range of organic solvents, at room temperature.45–55 Although the detailed mechanisms of the hydrolysis reactions remain under-explored, it is proposed that the Zn–C bonds rapidly react with water to generate transient Zn–OH species,56 which subsequently undergo a series of condensation and rearrangement reactions to ultimately deliver crystalline ZnO. It is also known that the addition of a second zinc source, in particular a non-hydrolysable zinc carboxylate complex, results in the formation of colloidal dispersions of 3–4 nm ZnO nanoparticles, surface coordinated by carboxylate ligands.46,57–59 In the presence of sufficient carboxylate ligands, it may be feasible to synthesise nanoplatelets of layered zinc hydroxides directly, and hence avoid the challenges of top-down exfoliation. This type of organometallic hydrolysis route has not previously been reported, for 2D materials, yet it offers a number of attractions, including the high efficiency of Zn–OH bond formation under mild conditions and its potential to deliver a one-pot route to exfoliated nanoplatelets.
Results & discussion
ZnO nanoparticles were previously prepared via the hydrolysis of mixtures of diethyl zinc and sub-stoichiometric quantities of zinc carboxylate complexes or carboxylic acids, in various organic solvents, at room temperature.46 The zinc carboxylate complex or carboxylic acid reagent provides carboxylate ligands which are located on the ZnO nanoparticle surfaces and help control the size at 3–4 nm. These syntheses were conducted using molar ratios [COOR]/[Zn] = 0.20 to produce fully surface ligated particles; at lower loadings (<0.2), the nanoparticle size remained similar but the carboxylate-nanoparticle surface coverage was reduced.46 Thus far, there are no reports of hydrolyses conducted using higher carboxylate loadings (0.2 < [COOR]/[Zn] < 1). Since the formula unit of layered zinc hydroxides, [Zn5(OH)8(A)2·nH2O], is consistent with a ratio of [COOR]/[Zn] = 0.40, we investigated carboxylate loadings at or above this value.The synthesis reactions were conducted by the reaction of commercially available diethyl zinc with the appropriate amount of a zinc bis(carboxylate) complex ([COOR]/[Zn] = 0.40–0.60). A series of zinc bis(carboxylate) complexes were tested including acetate, hexanoate and oleate, so as to determine the influence of the anion (A) over the exfoliation process. The zinc carboxylate complexes are either commercially available or easily prepared using standard syntheses.60,61 The precursor complexes were first dissolved in toluene, then pure (de-ionized) water was added to the solution, and white powders containing LZH were subsequently isolated as products. Initially, the influence of the carboxylate/zinc molar ratio was explored, using oleate ligands (Fig. 2a–c).
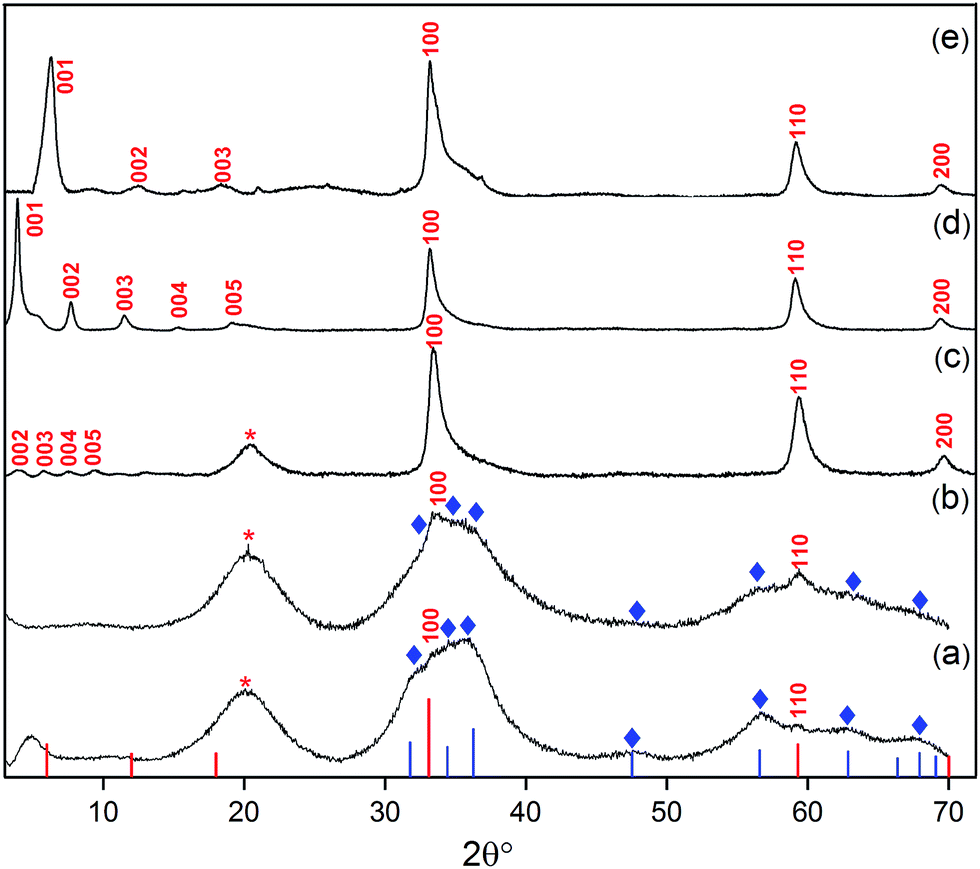 | ||
| Fig. 2 XRD patterns of products from different loadings of oleate within the synthesis mixture: (a) [Ole]/[Zn] = 0.40, (b) [Ole]/[Zn] = 0.50 and (c) [Ole]/[Zn] = 0.6, i.e. LZH-Ole. A loading of [COOR]/[Zn] = 0.60 was used to synthesise (d) LZH-OHex & (e) LZH-OAc. Reference data: ZnO (blue lines and ◊, JCPDS card no.: 00-001-1136), LZH (the red lines and labels), LZH-OAc was reported by Fiévet et al.24 No literature diffraction data have been reported for LZH-OHex and LZH-Ole. Peak marked with ‘*’ is due to hydrocarbon chain of the longer carboxylates.46,65 | ||
In all cases, X-ray diffraction (XRD) provided good evidence for the presence of LZH, in particular the appearance of X-ray diffraction peaks at low angles is highly characteristic of layered inorganic sheets (see further discussion below). However, at the nominally stoichiometric ratio of 0.40, a significant proportion of ZnO was also observed in the XRD patterns, as seen by the signals at 31°, 34°, 36°, 47°, 56°, 62° and 67° 2θ, which are indexed against ZnO (Fig. 2a–c). When the carboxylate ratio was increased to 0.50, the formation of LZH becomes more favoured and at a ratio of 0.60, LZH becomes the only obvious crystalline product. Compared to the expected stoichiometry, excess carboxylate ligands are needed to suppress unwanted over-hydrolysis to the oxide. It is tentatively proposed that the (excess) carboxylate anions are located in the layers between the nanoplatelets, existing as zinc bis(carboxylates), as shown by a comparison of IR spectra of LZH-Ole and Zn(Ole)2 (Fig. S1†); this suggestion is also consistent with the proposed localization of excess carbonate anions during hydrothermal syntheses. It is also supported by elemental analysis which confirms the original reagent stoichiometry is maintained in the products (Table S3†); in other words, none of the volatile diethyl zinc is lost to evaporation, and none of the ligand is washed away.
The purity of the LZH materials was also analysed using UV-vis spectroscopy (Fig. S4†) which confirmed that only a very small proportion of ZnO nanoparticles was present. By calibrating the extinction coefficient of these nanoparticles, the quantity of ZnO was estimated to represent 20 mol% Zn or 6.4 wt% of the final product (see ESI,† page 17); such a quantity, in the form of small nanoparticles, is too low to appear in the powder XRD analysis. In previous reports, using conventional syntheses, ZnO contamination is apparent in significantly greater quantities by XRD.27,34 Assuming that the only other product is excess zinc carboxylate, the LZH-Ole synthesis yields ∼63 mol% Zn in the form of LZH (see ESI,† page 16). Specific yields are scarce in the literature, presumably due to the difficulty of identifying byproducts, but the value compares reasonably to ∼90 mol% Zn and 60–89 mol% Zn reported for LZH–dodecyl sulphate62 and LZH–benzoate,63,64 respectively.
In order to explore the generality of the new route to LZH materials, the syntheses were performed using three different zinc carboxylates (CnH2n−1O2): acetate (n = 2), hexanoate (n = 6) and oleate (n = 18, with cis-alkene at C9 position). In each case, the ratio [COOR]/[Zn] = 0.60 was applied and the XRD patterns of all three products are consistent with the formation of LZH materials (Fig. 2c–e, S2 & S3†). One important difference compared to hydrothermal/base assisted synthesis routes, is that there are no contaminating signals due to hydrozincite22 (which is the product of carbonate contamination and is common when urea is applied as the base). The higher reactivity of the organometallic zinc reagent also ensures excellent conversion efficiency under ambient conditions. Analysis of all three powder XRD patterns reveals characteristic peaks at 33°, 59° and 70° 2θ which correspond to the in-plane spacings of zinc hydroxide layers. The peak marked with ‘*’ corresponds to the lateral packing of alkyl chains.46,65 There is some asymmetry to these peaks as is characteristic for a turbostratic layered material.24,66 The peaks below 2θ = 10° correspond to the interlayer distances between the zinc hydroxide planes and vary systematically with different ligand chain lengths. Increasing the number of carbon atoms in the carboxylate ligand leads to larger interlayer spacing.64 In the acetate product, the two signals below 10° 2θ are assigned to (001) and (002), in agreement with previous data for the same material, synthesised using base-assisted decomposition or hydrolysis in polyol media.24,67 As the alkyl chain length increases, i.e. for hexanoate and oleate, a greater number of higher order layer spacings are observed (Table S2†).64,68,69 The (001) peak of LZH-OHex indicates a basal spacing, d, of 22.7 Å (°2θ = 3.9). For LZH-Ole, features at 22.1 (°2θ = 4.0°), 15.2 (5.8°), 11.6 (7.6°) and 9.5 Å (9.3°) correspond to (002), (003), (004) and (005) planes, indicating layer spacing of around 4.6 ± 0.1 nm. The peaks corresponding to the 00l planes of LZH-Ole are of low intensities due to the disorder in the oleate chain packing and analogous systems, such as LDH-Ole, which also exhibit similar low intensity peaks for these higher order Bragg reflections.13,68,70,71 The value of 4.6 nm suggests no interdigitation of oleate chains and is larger than the distances estimated for LZH structures showing either significantly interdigitated68 (3.3 nm) or non-interdigitated (4.0 nm) oleate chains (Fig. 1b and c). This longer (001) plane spacing in LZH-Ole was investigated by SAXS (small-angle X-ray scattering; Fig. S5 & S6†) to give a broadly consistent average spacing of 3.8 ± 0.6 nm, given the experimental uncertainties. It should also be noted that the error in the data from the SAXS measurement is rather high due to the low resolution of the measurement.
The IR spectra of the three LZHs (Fig. S7†) showed intense signals at ∼1600 cm−1 and ∼1400 cm−1, which are assigned to the asymmetric and symmetric stretches of the carboxylate ligands, respectively.72 All three materials also showed broad signals at 3100–3600 cm−1 which are assigned to hydroxyl groups and intercalated water molecules coordinated to zinc hydroxide layers.63 The peak breadth is likely due to hydrogen bonding between the hydroxyl groups and the lattice.24,63,64,69,73 Indeed, the peak width of the hydroxyl resonance decreases as the carboxylate alkyl chain length increases. In the cases of LZH-Ole and LZH-OHex, some sharp features are observed in this region; for example, LZH-Ole shows two sharp peaks at 3512 cm−1 and 3395 cm−1. It is proposed that longer alkyl chains increase hydrophobicity and help to create more ordered head group coordination.74
The LZHs were also analysed using SEM (Fig. 3): LZH-Ole (Fig. 3c–d) displayed particulates which were hundreds of micrometres wide (∼350 μm) with smooth surfaces. In contrast, LZH-OAc and LZH-OHex showed platelet structures only at high magnification (Fig. 3a–b), as previously reported for other syntheses used to prepare LZH-OAc.67,75 The smooth surface of LZH-Ole may be a direct consequence of its higher solubility (see below) which facilitates sample deposition and preparation. The thermal analyses of the LZHs, by TGA and DTA, showed the first major mass loss at ∼100 °C which is attributed to the removal of intercalated water and the dehydroxylation of the zinc hydroxide layers (endothermic process), followed by the subsequent decomposition of organic carboxylate groups (exothermic process) at temperatures above 300 °C (Fig. S8–S10†).24,64,67 As the chain length of the carboxylate group increases, the weight loss at 300 °C increases; the absolute values are somewhat higher than predicted by the nominal stoichiometry (Zn5(OH)8(A)2) but consistent with the excess carboxylate sequestered in the layers during synthesis, as discussed already for the elemental analysis data (Table S3†).
 | ||
| Fig. 3 SEM images showing the surface morphology of (a) LZH-OAc, (b) LZH-OHex, (c) LZH-Ole and (d) the macrostructure of LZH-Ole. | ||
This new route to prepare organically-modified LZH nanomaterials can be useful in a wide range of contexts; however, one striking finding was that, in certain cases, nanoplatelets could be isolated directly, as individual solvated monolayers. The LZH–OAc and LZH-OHex materials were rather insoluble in most common solvents (i.e. tetrahydrofuran, dichloromethane, toluene, hexane, ethanol, water) which is in line with previous reports.9 However, increasing the length of carboxylate alkyl chain to oleate dramatically improved the solubility of the LZH in solvents such as toluene and hexane. The product remains in solution through the synthesis and isolated, dried product redissolves in such apolar organic solvents (Table S4†). The cis-alkene in the middle of the oleate chain is known, in general, to lower the degree of interdigitation (as demonstrated for LDHs intercalated with oleate and elaidate68); presumably, the lower degree of ordering assists the swelling of the layers in apolar organic solvents for exfoliation through redissolution of the original LZH monolayers. LZH-Ole dissolves in toluene to form a transparent colloidal solution at a concentration of 23 mg mL−1. The dissolution occurs spontaneously, over a period of days, as evidenced by increased Rayleigh scattering from the clear solution over time (Fig. S11†). With gentle mechanical stirring to remove concentration gradients, a saturated solution can be produced in under 2 h. It is important to emphasise that the exfoliation was obtained quantitatively, simply by stirring the LZH in toluene using normal magnetic stirrer plate; the result is in contrast to previous routes to LZH dispersion that required ultrasonication and filtration to reach 22 mg mL−1 for LZH-DS in formamide.27,33,34 As a control, ultrasound was applied to the exfoliation of LZH-Ole in toluene, but found to be less effective; significant degrees of aggregation were observed, potentially due to sonochemical degradation.
The toluene solution of LZH-Ole was deposited onto silicon wafers and gold grids for analysis by atomic force microscopy (AFM) and transmission electron microscopy (TEM), respectively. The TEM analysis showed large dried agglomerations of small, sheet-like structures (Fig. S12†) which likely form when the exfoliated LZH-Ole solution dries on the grid. In comparison, when dried LZH-Ole is simply shaken briefly in toluene, without allowing time for dissolution, bulk unexfoliated LZH is observed (Fig. S12g & h†), consistent with the SEM images of the powdered products (Fig. 3c & d). Within the thickest section of agglomerates of the exfoliated sample, small stacks of ∼5–8 layers of LZH-Ole nanoplatelets are present (Fig. 4a). For the stacks orientated parallel to the electron beam, clear diffraction fringes from the (001) layer planes are visible, marked by line profile [1] (Fig. 4b) and [2] (Fig. 4c). The intensity of line profiles [1] and [2] show an interlayer spacing of 5.3 ± 1.3 nm (N ∼ 30); the distance is consistent, within error, with that obtained by XRD (4.6 ± 0.1 nm)/SAXS (solid sample: 3.8 ± 0.6 nm) data for the (001) plane. A similar side-on morphology was reported in TEM measurements of LZH-DS exfoliated in formamide.34 The edges of the agglomerates are thinner and do not contain any obvious stacks. The FFT (Fig. 4e) of HR-TEM image (Fig. 4d) shows the presence of three diffraction planes, which correspond to the three in-plane peaks seen in XRD at 33° ((100), d = 2.7 Å), 59° ((110), d = 1.5 Å) and 70° ((200), d = 1.3 Å) 2θ (Fig. 4e). The gaps between the sheets can be attributed to the presence of amorphous oleate chains. The thickness of the nanoplatelets cannot be easily measured using TEM, but was determined by AFM.
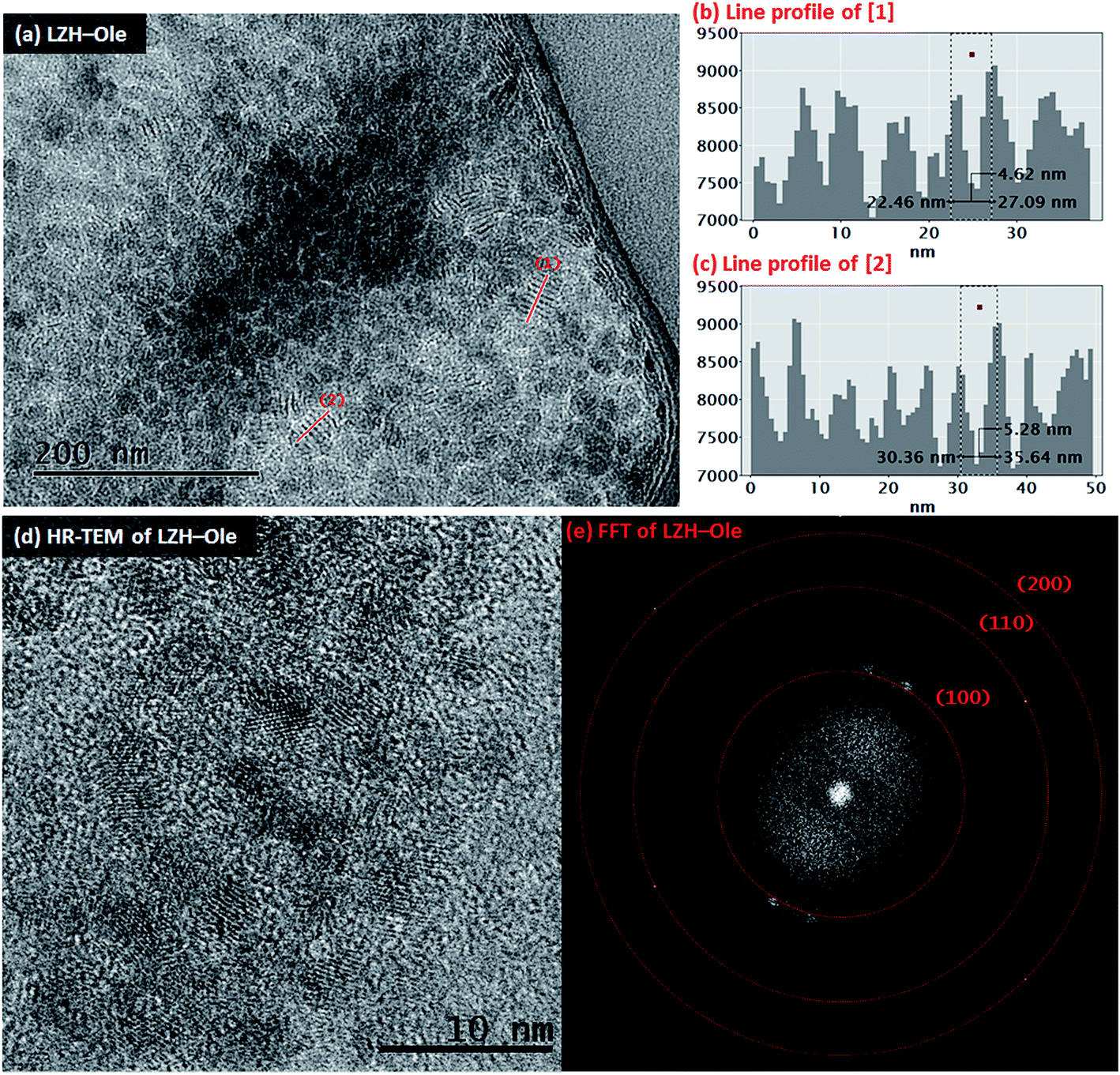 | ||
| Fig. 4 TEM image of (a) LZH-Ole after exfoliation by mechanical stirring for 2 h in toluene. Line profiles (b) & (c) correspond to lines [1] & [2] in (a), respectively. Within the line profiles, the interlayer distances are marked by the dashed line box. (d) HR-TEM image of the edge of agglomerate of LZH-Ole after exfoliation treatment (mechanical stirring, 2 h, toluene) and (e) the FFT of image (d). | ||
Individual LZH-Ole nanoplatelets were observed in AFM (Fig. 5), with height profiles typical for 2D layered materials. The average thickness of individual nanoplatelets is 3.62 nm ± 0.07 nm (σ = 0.75 nm, N = 127, Fig. S12†), in good agreement with solid-state SAXS monolayer thickness (3.8 ± 0.6 nm). The average radius of the monolayers was 13.25 nm (σ = 3.38 nm, N = 127), but this value is expected to be a significant overestimate due to tip convolution. Occasional large agglomerates (Fig. 5c) similar to the ones found in TEM were also observed; the adjacent flat regions had a height around 3 nm (Fig. 5d), consistent with a monolayer of particles separated by gaps too small to resolve with AFM. Lower magnification images (Fig. S13†) show similar structures over a larger area. The microscopic analysis of deposited sample LZH-Ole solution strongly suggests that LZH-Ole exists as solvated monolayers in toluene; the occasional short stacks observed in both AFM and TEM are likely an artefact of drying during sample preparation for microscopic analyses.
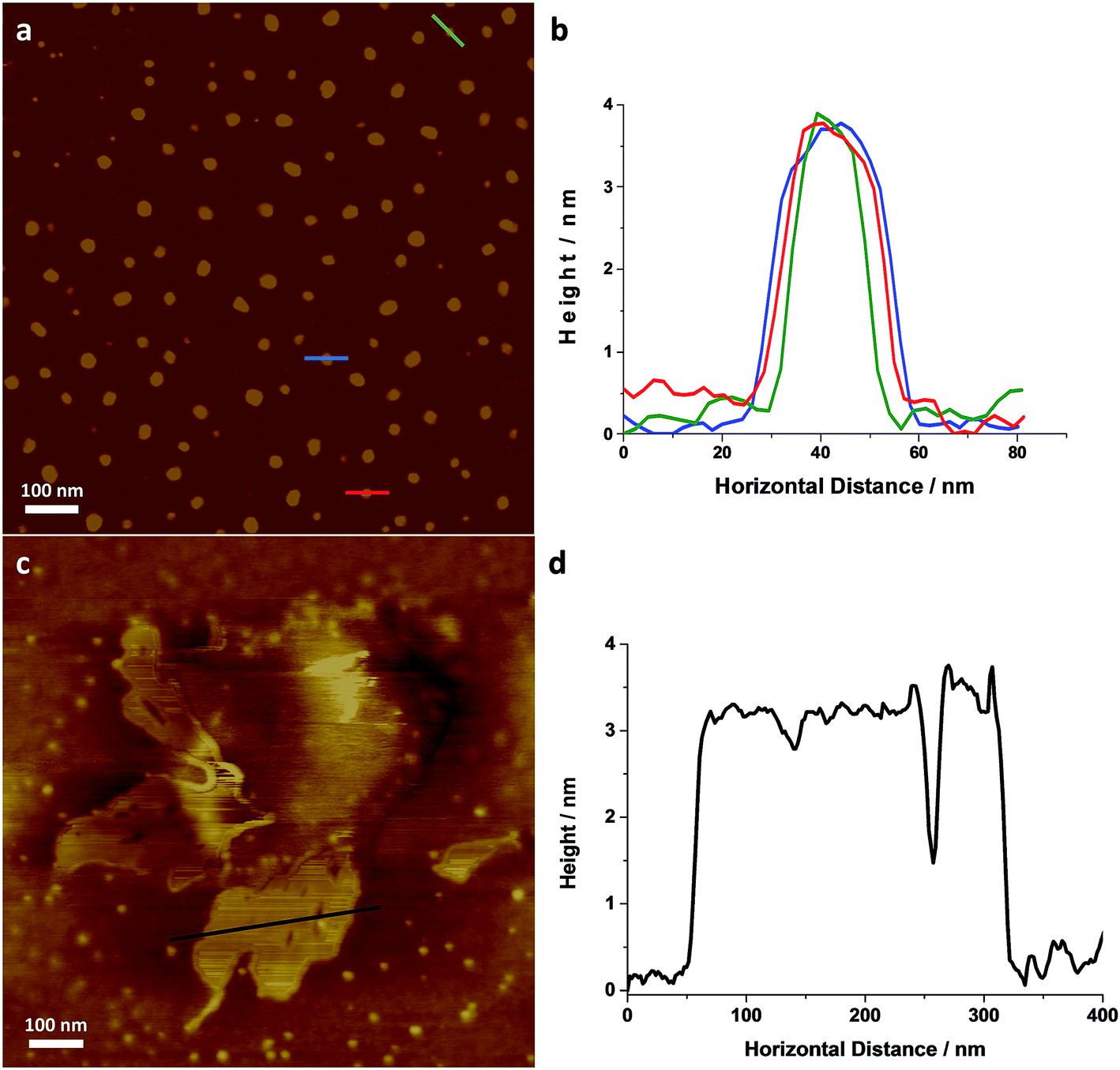 | ||
| Fig. 5 (a) AFM image of individual LZH-Ole and (b) height profiles of the individual platelets highlighted in (a) [full histogram of platelet thickness available in ESI, S12†]. (c) AFM image of self-assembled LZH-Ole and (d) height profile of the self-assembled platelets highlighted in (d). All scale bars are 100 nm. | ||
The high solubility of the oleate nanoplatelets provides an exciting opportunity to monitor the organometallic synthesis of LZH-Ole, in situ, using 1H NMR spectroscopy (Fig. S14†). The 1H NMR spectrum of the LZH-Ole product showed quite broad peaks where the characteristic oleate signals are shifted compared to the zinc bis(oleate) precursor complex.76,77 The low intensity broad peak at δ 0.53 ppm is tentatively assigned to intercalated water molecules which are hydrogen-bonded to the zinc hydroxide layers; the chemical shift of H2O dissolved in C6D6 is δ 0.40 ppm.78
The first step of the LZH synthesis (at [Ole]/[Zn] = 0.60), before the addition of water, involves the reaction of ZnEt2 and Zn(Ole)2 to form the pentanuclear cluster, [Zn5(Et)4(Ole)6] (Fig. 6a, signals marked ‘C’, S15), alongside excess unreacted ZnEt2 (Fig. 6a signals marked X, S15). The formation of such clusters is known, for other carboxylates in apolar solvents such as toluene, and sequesters all carboxylate even in the presence of excess diethyl zinc.58 In the oleate case, the cluster is clearly identified by oleate signals in the range δ 5.60–0.90 ppm, along with signals at δ 1.70 ppm (t, 21H), which consist of overlapping signals of oleate and the ethyl groups of the pentanuclear cluster, and δ 0.78 ppm (q, 8H) belonging to ethyls of the same cluster. The relative integrals of signals at δ 5.52 and δ 0.78 ppm are roughly 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, in line with the values expected for the pentanuclear cluster. The signals at δ 1.12 (t, 18H) and δ 0.10 ppm (q, 12H) correspond to residual diethyl zinc. The relative integrals of the oleate signal at δ 5.52 ppm, the ethyl signal at δ 0.78 ppm and the diethyl zinc signal at δ 0.12 ppm indicate 1 mole of cluster per 1.5 moles of residual diethyl zinc. The mixture of cluster and diethyl zinc was hydrolysed progressively, by sequential addition of known quantities of water, added under a flow of N2. The solutions remained homogeneous throughout and therefore, the 1H NMR spectra (Fig. 6b–d) are representative of all proton-containing species present. The addition of ∼0.5 equiv. of water (vs. total Zn) results in an immediate decrease in intensity of the ZnEt2 signals (Fig. 6a and b, marked ‘X’, δ 0.10 & δ 1.12 ppm) since it is more reactive to hydrolysis than the cluster. Further addition of water to a total of ∼1 equiv., results in the hydrolysis of the ethyl ligands in the pentanuclear cluster complex, [Zn5(Et)4(Ole)6], as evidenced by the disappearance of the sharp signals at δ 0.78 and δ 1.70 ppm (Fig. 6b and c). The putative zinc hydroxide product(s) also shows broadened carboxylate resonances consistent with coordination to zinc (Fig. 6c). The evolution of ethane was also clearly observed, as expected, via the increasing signal intensity at δ 0.80 ppm (Fig. 6b–d, marked ‘#’). The changes in chemical shifts and broadening of the oleate signals (L: δ 5.52, 2.30, 2.11, 1.58, 1.42, 1.40, 1.30, 1.17, 0.92 ppm) are consistent with the formation of LZH-Ole. Indeed, clear signals assigned to LZH-Ole are observed after the addition of ∼1.6 equiv. of water; the 1H NMR spectrum at this stage of the reaction is closely comparable to that of independently synthesized LZH-Ole (Fig. 6d–e). There are two other very low intensity signals (δ 3.30 ppm, δ 5.61 ppm) which are not currently assigned. It is notable that the low intensity signals are distinct from LZH-Ole and they are not present in samples of zinc oxide coordinated by oleate (Fig. S16†); it may be that these signals arise due to intercalated water/carboxylate ligands.
:4, in line with the values expected for the pentanuclear cluster. The signals at δ 1.12 (t, 18H) and δ 0.10 ppm (q, 12H) correspond to residual diethyl zinc. The relative integrals of the oleate signal at δ 5.52 ppm, the ethyl signal at δ 0.78 ppm and the diethyl zinc signal at δ 0.12 ppm indicate 1 mole of cluster per 1.5 moles of residual diethyl zinc. The mixture of cluster and diethyl zinc was hydrolysed progressively, by sequential addition of known quantities of water, added under a flow of N2. The solutions remained homogeneous throughout and therefore, the 1H NMR spectra (Fig. 6b–d) are representative of all proton-containing species present. The addition of ∼0.5 equiv. of water (vs. total Zn) results in an immediate decrease in intensity of the ZnEt2 signals (Fig. 6a and b, marked ‘X’, δ 0.10 & δ 1.12 ppm) since it is more reactive to hydrolysis than the cluster. Further addition of water to a total of ∼1 equiv., results in the hydrolysis of the ethyl ligands in the pentanuclear cluster complex, [Zn5(Et)4(Ole)6], as evidenced by the disappearance of the sharp signals at δ 0.78 and δ 1.70 ppm (Fig. 6b and c). The putative zinc hydroxide product(s) also shows broadened carboxylate resonances consistent with coordination to zinc (Fig. 6c). The evolution of ethane was also clearly observed, as expected, via the increasing signal intensity at δ 0.80 ppm (Fig. 6b–d, marked ‘#’). The changes in chemical shifts and broadening of the oleate signals (L: δ 5.52, 2.30, 2.11, 1.58, 1.42, 1.40, 1.30, 1.17, 0.92 ppm) are consistent with the formation of LZH-Ole. Indeed, clear signals assigned to LZH-Ole are observed after the addition of ∼1.6 equiv. of water; the 1H NMR spectrum at this stage of the reaction is closely comparable to that of independently synthesized LZH-Ole (Fig. 6d–e). There are two other very low intensity signals (δ 3.30 ppm, δ 5.61 ppm) which are not currently assigned. It is notable that the low intensity signals are distinct from LZH-Ole and they are not present in samples of zinc oxide coordinated by oleate (Fig. S16†); it may be that these signals arise due to intercalated water/carboxylate ligands.
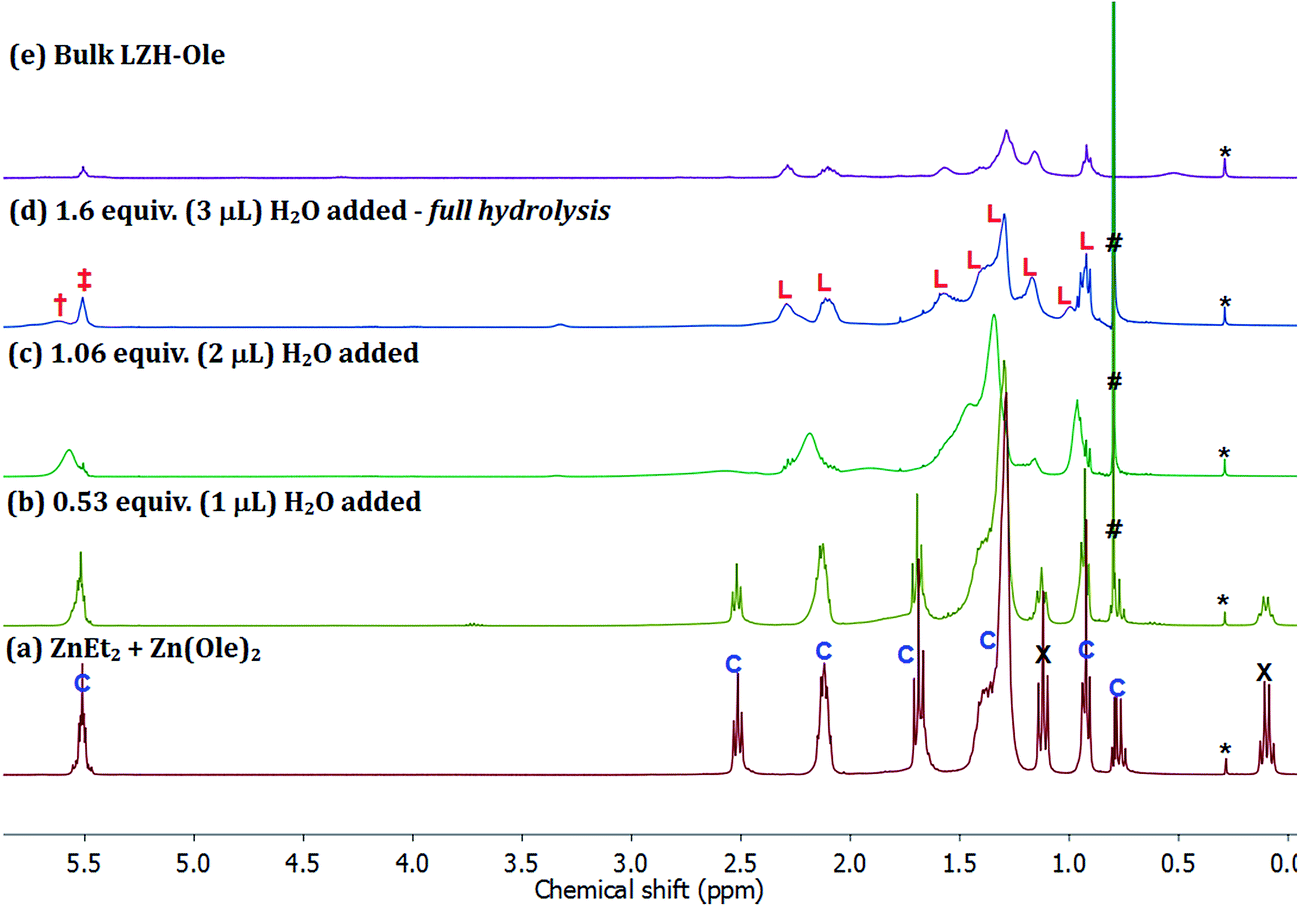 | ||
| Fig. 6 1H NMR spectra of (a) ZnEt2 and [Zn5(Et)4(Ole)6], the hydrolysed products observed at (b) 0.53 equiv. H2O added, (c) 1.06 equiv. H2O added, (d) 1.60 equiv. H2O added and (e) the bulk LZH-Ole. *: silicon grease, #: ethane (δ 0.80 ppm), C: [Zn5(Et)4(Ole)6], L: LZH-Ole, X: ZnEt2 (δ 0.10 & δ 1.12 ppm), ‡ & †: alkenyl protons of oleate (δ 5.51 & δ 5.61 ppm). | ||
The hydrolysis pathway, therefore, proceeds via a number of clear stages: (1) the organometallic reagents react to form a pentanuclear zinc cluster compound [Zn5(Et)4(COOR)6] and excess diethyl zinc; (2) the hydrolysis progresses by initial hydrolysis of the excess diethyl zinc; (3) then, the hydrolysis of the ethyl ligands on the cluster occurs, along with the formation of the LZH. The organometallic hydrolysis reaction is controlled, by reagent stoichiometry, to yield either the selective formation of carboxylate-coordinated ZnO nanoparticles ([COOR]/[Zn] < 0.2)46,56 or to form 2D layered zinc hydroxide materials ([COOR]/[Zn] > 0.4 and selective formation at 0.6).
This new method to prepare LZH could be useful in a wide range of contexts, including the deposition of functional materials. A proof of concept study was undertaken to prepare zinc oxide thin films using the LZH-Ole as a precursor. Thermal decomposition of LZH-coated substrates is already reported to produce ZnO films relevant for the fabrication of photovoltaics, although they are relatively thick (typically 17–33 μm),41,42,67,75,79 and more suited to DSSCs, whereas sub-micrometer ZnO films have been proposed for higher efficiency hybrid solar cells.80 The LZH-Ole solutions offer the opportunity to deposit thin, uniform precursor films, as illustrated by immersing a glass slide vertically into a toluene solution of LZH and evaporating the solvent. LZH-Ole was coated homogeneously onto the substrate with a uniform film thickness of ∼1 μm, as measured from SEM (Fig. 7a). TGA of LZH-Ole (Fig. S10†) shows complete calcination at temperatures above 490 °C. Hence, the LZH thin film was calcined at 500 °C, for 15 min in air, which led to the formation of a transparent ZnO film (Fig. S17†). The UV-vis spectrum of the ZnO film showed a peak at 374 nm corresponding to the band edge of ZnO (Fig. S18†). SEM images showed the formation of a homogeneous, ∼1 μm thick film of ZnO nanoparticles with maximum diameter of 25 nm (Fig. 7b, d, f and S19†). The surface morphology clearly shows the particulate, porous structure of the ZnO which is quite distinct from the denser packing observed for the LZH (Fig. 7c and e). It is, nonetheless, promising that both the thickness and homogeneity of the films were retained upon conversion of the LZH to ZnO. Such thin ZnO films may be useful for hybrid photovoltaic devices or as transistor films, where thicknesses ranging from 100 nm to 1 μm are optimal.81–83
 | ||
| Fig. 7 SEM images LZH-Ole film and ZnO film: (a) cross section of LZH film, (b) cross section of ZnO film, (c) & (e) LZH film surface and (d) & (f) ZnO film surface with inset in (f) displaying the shape of ZnO nanoparticles formed to be close to spherical. The ZnO films were formed by annealing LZH films at 500 °C for 15 min under air. | ||
Conclusions
Layered zinc hydroxides were successfully synthesised through the reaction of diethyl zinc with the appropriate zinc carboxylate and water. The reaction occurred at room temperature and in apolar organic solvents. These conditions are markedly different to those normally used in the high temperature, base-catalysed decomposition of zinc salts. The organometallic hydrolysis route, applying the optimum ratio of [COOR]/[Zn] = 0.60, led to the most selective formation of LZH nanoplatelets. In contrast to conventional routes, there was no contamination of the LZH product with the carbonate salt, hydrozincite, and modest contamination by ZnO and zinc bis(carboxylates). The layered zinc hydroxide products featuring acetate, hexanoate and oleate anions were characterised by IR spectroscopy, XRD, TGA, SEM and solid state SAXS measurements. The oleate layered zinc hydroxide showed high solubility in apolar solvents (up to 23 mg mL−1 in toluene) which is similar to common layered zinc hydroxides that have previously been exfoliated by sonication in formamide. However, the LZH-Ole product was exfoliated by spontaneous dissolution in toluene, completed with simple stirring in under 2 h. The exfoliated monolayers were deposited onto silicon wafers and TEM grids, enabling detailed analysis by atomic force microscopy and transmission electron microscopy. Both techniques indicate the formation of nanoplatelets with an average thickness of 3.6 nm and lateral size of ∼10 nm. The synthesis reaction was studied using solution NMR spectroscopy which revealed that the organometallic reagents react together to form a zinc cluster species and that the hydrolysis proceeds directly to form LZH. The facility to direct the product formation is rather useful, particularly given that the reactions occur readily at room temperature and in organic solvents. In principle, the nanoplatelets could be formed in situ with a range of resins or other reaction media, since the byproduct is only volatile ethane gas.84 The production of exfoliated layered zinc hydroxides allows the deposition of thin films on glass substrates which were subsequently thermally annealed to produce films of nanoporous ZnO around 1 μm thick that are relevant for optoelectronic applications. In general, the new route to LZH takes advantage of the high reactivity of organo-zinc reagents to efficiently deliver nanoplatelets of 2D layered zinc hydroxides. Future investigations may extend the new method to other compositions, including doped85,86 or layered double hydroxide nanoplatelets and monolayers.Experimental section
Materials and methods
The syntheses of LZHs were carried out in a dry nitrogen-filled glovebox and Schlenk line techniques were used for hydrolysis of the organometallic precursor to form LZHs. All solvents were purchased from VWR, UK; chemicals from Sigma Aldrich and used as supplied unless otherwise stated. Toluene used in the syntheses of LZHs was dried and distilled by refluxing over metallic Na and then degassed by freeze–thaw cycles (x3) to remove oxygen. Diethyl zinc is a highly pyrophoric and volatile liquid where extreme caution must be taken when using the chemical. HPLC-grade water was used for the controlled hydrolysis of LZH synthesis mixture. Anhydrous zinc bis(acetate) was purchased from Sigma Aldrich and used as received. The syntheses of zinc bis(hexanoate) and zinc bis(oleate) salts were performed according to reported routes.60,61The carboxylate loading ([COOR]/[Zn]) was varied (the molar ratios and moles of reagents for each loading are listed in Table S1†), whilst maintaining the total concentration of zinc at 0.15 M. For the synthetic methods reported below for LZH-OAc, LZH-OHex and LZH-Ole, a loading of [COOR]/[Zn] = 0.60 was used and the total Zn content was fixed to 1.37 mmol for all carboxylate loadings.
1H NMR spectra were recorded at 400 MHz using Bruker Av400 spectrometer operating at 9.4 T in C6D6 unless otherwise stated. The recycle delays (D1) for all spectra were 1 s, with 16 scans per spectrum. Chemical shifts are expressed in parts per million (ppm) and referenced to residual solvent resonance. For the 1H NMR study of the synthesis of LZH-Ole, the addition of water for the hydrolysis process was performed under a dynamic flow of N2 and a 1H NMR spectrum was taken 15 min after each addition, at which point no visible water droplets are present. The full hydrolysis of the reaction mixture requires 3 μL of water (1.6 equiv. to total moles of zinc atoms present in the reaction) and the stepwise addition of water was conducted by adding 3 × 1 μL to reach full hydrolysis. Fourier transform infrared (FT-IR) spectra were recorded using a Perkin-Elmer Spectrum 100 spectrometer fitted with an ATR attachment and each spectrum was collected with 4 or 32 scans with a resolution of 4 cm−1. Powder X-ray diffraction (XRD) experiments were performed using a PANalytical Xpert PRO diffractometer at an operating voltage of 40 kV with a step size of 0.033° 2θ, scan step time of 70 s and scan range of 3–75° 2θ. Small angle X-ray scattering (SAXS) measurements were performed using a PANalytical Empyrean diffractometer equipped with ScatterX78 module, from 0–5° 2θ, using a step size of 0.039° 2θ and scan step time of 195 s. Data analysis was performed using PANalytic EasySAXS software and error given is derived from instrumental resolution. Thermogravimetric analysis (TGA) and differential thermal analysis (DTA) were carried out using a Mettler Toledo TGA/DSC 1, under a flow of dry air with flow rate of 60 mL min−1, from 50–600 °C, at a heating rate of 5 °C min−1. UV-vis absorption spectra of ZnO nanoparticles were recorded using Perkin-Elmer Lambda 950 spectrometer in transmission mode from 300–500 nm with a scan speed of 1 nm s−1. The ZnO nanoparticle diameters were estimated from the UV-vis spectra, using the empirical method developed by Meulenkamp.87
Scanning electron microscopy (SEM) samples were prepared by adhering samples to stubs with silver paint and sputtering with a 10 nm layer of chromium. Micrographs were taken with a LEO Gemini 1525 FEGSEM (field emission gun SEM) with an accelerating voltage of 5 keV and a 30 μm aperture, using an in-lens secondary electron detector. Transmission electron microscopy (TEM) samples were drop-cast (toluene solution, 0.05 mg mL−1) onto 300 mesh Au grids, with a 3 nm ultra-thin carbon film and holey carbon support (Agar Scientific). TEM images were obtained on JEOL JEM 2100F scanning transmission electron microscope, equipped with an Oxford X-Max 80 SDD EDX detector, at an operating voltage of 200 kV in bright field imaging mode. Atomic force microscopy (AFM) samples were prepared on silicon wafer pre-cleaned in 3:1 mixture of H2SO4 (98%) and H2O2 (30%). Samples were drop-cast (0.05 mg mL−1 in toluene) onto the Si wafer and dried under vacuum for 16 h at room temperature. AFM measurements were performed with tapping mode on a Bruker Digital Instruments Multimode VIII AFM with Nanoscope IV Digital Instruments AFM controller. All AFM micrographs were recorded with a resolution of 512 lines and with a typical scanning speed of 1 Hz, and processed using Bruker NanoScope Analysis v1.40 (R2Sr). Height profiles were recorded from individual particle maximum heights (N = 127).
FT-ATR-IR (ν cm−1): 3367 (O–H), 1550 (COO)asymm, 1406 (COO)symm. Elemental analysis: C6H17O14Zn5 C, 11.26; H, 2.68; found C, 12.72; H, 2.27.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(CH2)7CH3)2 (0.260 g, 0.414 mmol) in toluene (9.1 mL). The mixture was stirred for 16 h at room temperature, which yielded a clear solution. Under a flow of N2, HPLC-grade water (39 μL, 2.18 mmol, 1.6 equiv. to the total moles of Zn of 1.37 mmol) was directly added to the reaction and the mixture was stirred for 3 h. A clear solution remained after stirring and all volatiles were removed in vacuo, which yielded LZH-Ole as a white, glassy solid. FT-ATR-IR (ν cm−1): 3389 (O–H), 3003 (C–H)asymm, 2920 (C–H)asymm, 2851 (C–H)symm, 1570 (COO)asymm, 1465 (C–H)symm, 1411 (COO)symm. 1H NMR (400 MHz, C6D6, 298 K, D1 = 1 s, see ESI Fig. S13† for proton assignment): δ 5.52 (cis-alkene H, He), 2.30 (Ha), 2.11 (Hd), 1.58 (Hb), 1.42 (Hc), 1.40 (Hc), 1.30 (Hc), 1.17 (Hc), 0.92 (terminal CH3 of oleate, Hf). Elemental analysis: C54H107O14Zn5 C, 49.61; H, 8.25; found C, 46.74; H, 8.34.
CH(CH2)7CH3)2 (0.260 g, 0.414 mmol) in toluene (9.1 mL). The mixture was stirred for 16 h at room temperature, which yielded a clear solution. Under a flow of N2, HPLC-grade water (39 μL, 2.18 mmol, 1.6 equiv. to the total moles of Zn of 1.37 mmol) was directly added to the reaction and the mixture was stirred for 3 h. A clear solution remained after stirring and all volatiles were removed in vacuo, which yielded LZH-Ole as a white, glassy solid. FT-ATR-IR (ν cm−1): 3389 (O–H), 3003 (C–H)asymm, 2920 (C–H)asymm, 2851 (C–H)symm, 1570 (COO)asymm, 1465 (C–H)symm, 1411 (COO)symm. 1H NMR (400 MHz, C6D6, 298 K, D1 = 1 s, see ESI Fig. S13† for proton assignment): δ 5.52 (cis-alkene H, He), 2.30 (Ha), 2.11 (Hd), 1.58 (Hb), 1.42 (Hc), 1.40 (Hc), 1.30 (Hc), 1.17 (Hc), 0.92 (terminal CH3 of oleate, Hf). Elemental analysis: C54H107O14Zn5 C, 49.61; H, 8.25; found C, 46.74; H, 8.34.
Exfoliation of LZH-Ole
The solubility of LZH-Ole was assessed by gradual addition to 1 mL of aliquots toluene, hexane, chloroform, dichloromethane, ethanol and water. The point of saturation was determined to be when insoluble solid was seen in solution. A second method of solubility measurement of LZH-Ole in toluene was achieved by obtaining an over-saturated solution of LZH-Ole (50 mg) in toluene (2 mL), undissolved solids was allowed to settle and an aliquot of solution (1 mL) was taken and solvent was removed under a flow of N2. The weight of the sample from the aliquot was measured and the solubility of LZH-Ole in toluene was determined to be 20 mg mL−1. LZH-Ole (10 mg) was added to toluene (10 mL) and the mixture was mechanically stirred for 2 h at room temperature. A clear colloidal solution of LZH-Ole in toluene was obtained. The solution was then diluted to concentration of 0.05 mg mL−1 and then drop-cast onto TEM grids and Si wafers for TEM and AFM.Fabrication of ZnO thin films
Glass microscope slides were used as substrates for deposition of thin films of LZH-Ole. Slides were placed vertically into a solution of LZH-Ole in toluene (1 mg mL−1), the solution was then evaporated under a stream of nitrogen overnight and a uniform film of LZH-Ole was deposited on substrate. For the production of ZnO thin films on glass substrates, slides coated with LZH-Ole were annealed in a three-zone tube furnace (PTF 12/38/500, Lenton Ltd, UK) at 500 °C, with a heating ramp rate of 50 °C min−1, for 15 min, under a flow of air. All fabricated films were characterised by SEM and UV-vis spectroscopy in transmission mode.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The Engineering and Physical Sciences Research Council are acknowledged for research funding (EP/H046380, EP/K035274/1; DTA studentships to KO, AL).References
- R. Ma and T. Sasaki, Adv. Mater., 2010, 22, 5082–5104 CrossRef CAS PubMed.
- R. Allman, Acta Crystallogr., 1968, 24, 972–977 CrossRef.
- H. F. W. Taylor, Mineral. Mag., 1973, 39, 377–389 CAS.
- G. W. Brindley and S. Kikkawa, Am. Mineral., 1979, 64, 86–843 Search PubMed.
- K. S. Novoselov, A. K. Geim, S. Morozov, D. Jiang, Y. Zhang, S. A. Dubonos, I. Grigorieva and A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De and R. J. Smith, Science, 2011, 331, 568–571 CrossRef CAS PubMed.
- F. Wang, J.-H. Seo, G. Luo, M. B. Starr, Z. Li, D. Geng, X. Yin, S. Wang, D. G. Fraser and D. Morgan, Nat. Commun., 2016, 7, 10444 CrossRef CAS PubMed.
- H. Yin and Z. Tang, Chem. Soc. Rev., 2016, 45, 4873–4891 RSC.
- Q. Wang and D. O'Hare, Chem. Rev., 2012, 112, 4124–4155 CrossRef CAS PubMed.
- H. Yin, S. Zhao, K. Zhao, A. Muqsit, H. Tang, L. Chang, H. Zhao, Y. Gao and Z. Tang, Nat. Commun., 2015, 6, 6430 CrossRef CAS PubMed.
- H. Tang, J. Wang, H. Yin, H. Zhao, D. Wang and Z. Tang, Adv. Mater., 2015, 27, 1117–1123 CrossRef CAS PubMed.
- S. Zhao, Y. Wang, J. Dong, C.-T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- Z. Liu, R. Ma, M. Osada, N. Iyi, Y. Ebina, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2006, 128, 4872–4880 CrossRef CAS PubMed.
- R. Ma, Z. Liu, L. Li, N. Iyi and T. Sasaki, J. Mater. Chem., 2006, 16, 3809 RSC.
- L. Qiu, W. Chen and B. Qu, Colloid Polym. Sci., 2005, 283, 1241–1245 CAS.
- W. Chen and B. Qu, J. Mater. Chem., 2004, 14, 1705–1710 RSC.
- W. Shi, Y. Lin, X. Kong, S. Zhang, Y. Jia, M. Wei, D. G. Evans and X. Duan, J. Mater. Chem., 2011, 21, 6088 RSC.
- J. Lee, D. C. Sorescu and X. Deng, J. Phys. Chem. Lett., 2016, 7, 1335–1340 CrossRef CAS PubMed.
- Y. Zhao, G. Chen, T. Bian, C. Zhou, G. I. Waterhouse, L. Z. Wu, C. H. Tung, L. J. Smith, D. O'Hare and T. Zhang, Adv. Mater., 2015, 27, 7824–7831 CrossRef CAS PubMed.
- J. Wang, L. Zhao, H. Shi and J. He, Angew. Chem., Int. Ed. Engl., 2011, 50, 9171–9176 CrossRef CAS PubMed.
- G. G. C. Arizaga, K. G. Satyanarayana and F. Wypych, Solid State Ionics, 2007, 178, 1143–1162 CrossRef CAS.
- S. Ghose, Acta Crystallogr., 1964, 17, 1051–1057 CrossRef CAS.
- G. Rogez, C. Massobrio, P. Rabu and M. Drillon, Chem. Soc. Rev., 2011, 40, 1031–1058 RSC.
- L. Poul, N. Jouini and F. Fiévet, Chem. Mater., 2000, 12, 3123–3132 CrossRef CAS.
- Z. P. Xu and P. S. Braterman, J. Mater. Chem., 2003, 13, 268–273 RSC.
- F. Wypych, G. G. Arizaga and J. E. da Costa Gardolinski, J. Colloid Interface Sci., 2005, 283, 130–138 CrossRef CAS PubMed.
- J. Miao, M. Xue, H. Itoh and Q. Feng, J. Mater. Chem., 2006, 16, 474–480 RSC.
- V. Briois, C. Giorgetti, F. Baudelet, S. Blanchandin, M. Tokumoto, S. H. Pulcinelli and C. V. Santilli, J. Phys. Chem. C, 2007, 111, 3253–3258 CAS.
- P. Gerstel, P. Lipowsky, O. Durupthy, R. C. Hoffmann, P. Bellina, J. Bill and F. Aldinger, J. Ceram. Soc. Jpn., 2006, 114, 911–917 CrossRef CAS.
- O. Altuntasoglu, Y. Matsuda, S. Ida and Y. Matsumoto, Chem. Mater., 2010, 22, 3158–3164 CrossRef CAS.
- Z. Liu, R. Ma, Y. Ebina, N. Iyi, K. Takada and T. Sasaki, Langmuir, 2007, 23, 861–867 CrossRef CAS PubMed.
- T. Hibino, Chem. Mater., 2004, 16, 5482–5488 CrossRef CAS.
- L. Zhao, J. Miao, H. Wang, Y. Ishikawa and Q. Feng, J. Ceram. Soc. Jpn., 2009, 117, 1115–1119 CrossRef CAS.
- J. Demel, J. Plestil, P. Bezdicka, P. Janda, M. Klementova and K. Lang, J. Colloid Interface Sci., 2011, 360, 532–539 CrossRef CAS PubMed.
- H. C. Yau, M. K. Bayazit, J. H. Steinke and M. S. Shaffer, Chem. Commun., 2015, 51, 16621–16624 RSC.
- H. Nabipour and M. H. Sadr, Bull. Mater. Sci., 2015, 38, 1561–1567 CrossRef CAS.
- H. Nabipour, M. Hosaini Sadr and N. Thomas, J. Exp. Nanosci., 2015, 10, 1269–1284 CrossRef CAS.
- J. Hynek, V. Kalousek, R. Zouzelka, P. Bezdicka, P. Dzik, J. Rathousky, J. Demel and K. Lang, Langmuir, 2014, 30, 380–386 CrossRef CAS PubMed.
- E. Hosono, S. Fujihara, T. Kimura and H. Imai, J. Colloid Interface Sci., 2004, 272, 391–398 CrossRef CAS PubMed.
- H. Wang and C. Xie, J. Cryst. Growth, 2006, 291, 187–195 CrossRef CAS.
- Y.-H. Lai, C.-Y. Lin, H.-W. Chen, J.-G. Chen, C.-W. Kung, R. Vittal and K.-C. Ho, J. Mater. Chem., 2010, 20, 9379 RSC.
- T. Yuki, S. Ueno, M. Hagiwara and S. Fujihara, Journal of Asian Ceramic Societies, 2015, 3, 144–150 CrossRef.
- M.-K. Liang, M. J. Limo, A. Sola-Rabada, M. J. Roe and C. C. Perry, Chem. Mater., 2014, 26, 4119–4129 CrossRef CAS.
- G. Fan, F. Li, D. G. Evans and X. Duan, Chem. Soc. Rev., 2014, 43, 7040–7066 RSC.
- M. Monge, M. L. Kahn, A. Maisonnat and B. Chaudret, Angew. Chem., Int. Ed. Engl., 2003, 42, 5321–5324 CrossRef CAS PubMed.
- K. L. Orchard, M. S. P. Shaffer and C. K. Williams, Chem. Mater., 2012, 24, 2443–2448 CrossRef CAS.
- A. Gonzalez-Campo, K. L. Orchard, N. Sato, M. S. P. Shaffer and C. K. Williams, Chem. Commun., 2009, 4034–4036, 10.1039/b905353k.
- C. Lizandara-Pueyo, S. Siroky, M. R. Wagner, A. Hoffmann, J. S. Reparaz, M. Lehmann and S. Polarz, Adv. Funct. Mater., 2011, 21, 295–304 CrossRef CAS.
- M. L. Kahn, M. Monge, V. Collière, F. Senocq, A. Maisonnat and B. Chaudret, Adv. Funct. Mater., 2005, 15, 458–468 CrossRef CAS.
- Y. Coppel, G. Spataro, C. Pages, B. Chaudret, A. Maisonnat and M. L. Kahn, Chem.–Eur. J., 2012, 18, 5384–5393 CrossRef CAS PubMed.
- C. Pagès, Y. Coppel, M. L. Kahn, A. Maisonnat and B. Chaudret, ChemPhysChem, 2009, 10, 2334–2344 CrossRef PubMed.
- M. L. Kahn, A. Glaria, C. Pages, M. Monge, L. Saint Macary, A. Maisonnat and B. Chaudret, J. Mater. Chem., 2009, 19, 4044 RSC.
- S. Saliba, Y. Coppel, P. Davidson, C. Mingotaud, B. Chaudret, M. L. Kahn and J.-D. Marty, J. Mater. Chem., 2011, 21, 6821 RSC.
- C. Amiens, B. Chaudret, D. Ciuculescu-Pradines, V. Collière, K. Fajerwerg, P. Fau, M. Kahn, A. Maisonnat, K. Soulantica and K. Philippot, New J. Chem., 2013, 37, 3374 RSC.
- A. Ryzhikov, J. Jońca, M. Kahn, K. Fajerwerg, B. Chaudret, A. Chapelle, P. Ménini, C. H. Shim, A. Gaudon and P. Fau, J. Nanopart. Res., 2015, 17, 280 CrossRef.
- S. D. Pike, E. R. White, M. S. P. Shaffer and C. K. Williams, Nat. Commun., 2016, 7, 13008 CrossRef CAS PubMed.
- N. J. Brown, J. Weiner, K. Hellgardt, M. S. Shaffer and C. K. Williams, Chem. Commun., 2013, 49, 11074–11076 RSC.
- K. L. Orchard, A. J. P. White, M. S. P. Shaffer and C. K. Williams, Organometallics, 2009, 28, 5828–5832 CrossRef CAS.
- N. J. Brown, A. García-Trenco, J. Weiner, E. R. White, M. Allinson, Y. Chen, P. P. Wells, E. K. Gibson, K. Hellgardt, M. S. P. Shaffer and C. K. Williams, ACS Catal., 2015, 5, 2895–2902 CrossRef CAS.
- S. Barman and S. Vasudevan, J. Phys. Chem. B, 2007, 111, 5212–5217 CrossRef CAS PubMed.
- O. Berkesi, I. Dreveni, J. Andor and J. Mink, Inorg. Chim. Acta, 1992, 195, 169–173 CrossRef CAS.
- D. Zhang, X. Liu, H. Wan, N. Zhang, S. Liang, R. Ma and G. Qiu, ACS Sustainable Chem. Eng., 2017, 5, 5869–5879 CrossRef CAS.
- S. Inoue and S. Fujihara, Langmuir, 2010, 26, 15938–15944 CrossRef CAS PubMed.
- S. Inoue and S. Fujihara, Inorg. Chem., 2011, 50, 3605–3612 CrossRef CAS PubMed.
- R. A. Taylor, H. A. Ellis, P. T. Maragh and N. A. S. White, J. Mol. Struct., 2006, 787, 113–120 CrossRef CAS.
- D. Yang and R. Frindt, J. Mater. Res., 1996, 11, 1733–1738 CrossRef CAS.
- A. Moezzi, A. McDonagh, A. Dowd and M. Cortie, Inorg. Chem., 2013, 52, 95–102 CrossRef CAS PubMed.
- Z. P. Xu, P. S. Braterman, K. Yu, H. Xu, Y. Wang and C. J. Brinker, Chem. Mater., 2004, 16, 2750–2756 CrossRef CAS.
- J. W. Lee, W. C. Choi and J.-D. Kim, CrystEngComm, 2010, 12, 3249 RSC.
- C. Manzi-Nshuti, P. Songtipya, E. Manias, M. M. Jimenez-Gasco, J. M. Hossenlopp and C. A. Wilkie, Polymer, 2009, 50, 3564–3574 CrossRef CAS.
- Y. Kameshima, H. Yoshizaki, A. Nakajima and K. Okada, J. Colloid Interface Sci., 2006, 298, 624–628 CrossRef CAS PubMed.
- J. Oomens and J. D. Steill, J. Phys. Chem. A, 2008, 112, 3281–3283 CrossRef CAS PubMed.
- K. Nakamoto, Infrared and Raman spectra of inorganic and coordination compounds, Wiley Online Library, 1986 Search PubMed.
- S. Musić, S. Popović, M. Maljković and Đ. Dragčević, J. Alloys Compd., 2002, 347, 324–332 CrossRef.
- H. Morioka, H. Tagaya, M. Karasu, J.-i. Kadokawa and K. Chiba, Inorg. Chem., 1999, 38, 4211–4216 CrossRef CAS.
- Z. Hens and J. C. Martins, Chem. Mater., 2013, 25, 1211–1221 CrossRef CAS.
- M. Schönhoff, Curr. Opin. Colloid Interface Sci., 2013, 18, 201–213 CrossRef.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- H. Kajihara, M. Hagiwara and S. Fujihara, J. Ceram. Soc. Jpn., 2016, 124, 673–677 CrossRef CAS.
- S. Ueno and S. Fujihara, J. Electrochem. Soc., 2011, 158, K1 CrossRef CAS.
- T. Gershon, Mater. Sci. Technol., 2013, 27, 1357–1371 CrossRef.
- J. S. Park, W.-J. Maeng, H.-S. Kim and J.-S. Park, Thin Solid Films, 2012, 520, 1679–1693 CrossRef CAS.
- E. Fortunato, P. Barquinha, A. Pimentel, A. Gonçalves, A. Marques, L. Pereira and R. Martins, Thin Solid Films, 2005, 487, 205–211 CrossRef CAS.
- A. Gonzalez-Campo, K. L. Orchard, N. Sato, M. S. Shaffer and C. K. Williams, Chem. Commun., 2009, 4034–4036 RSC.
- M. Jobbágy, G. J. d. A. Soler-Illia, A. E. Regazzoni and M. A. Blesa, Chem. Mater., 1998, 10, 1632–1637 CrossRef.
- H. Nishizawa and K. Yuasa, J. Solid State Chem., 1998, 141, 229–234 CrossRef CAS.
- E. A. Meulenkamp, J. Phys. Chem. B, 1998, 102, 5566–5572 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: LZH-OAc and LZH-OHex characterisation data (powder XRD, IR, SEM, TGA, DTA), bulk LZH-Ole solid-state SAXS data, exfoliated LZH-Ole TEM data, AFM data with histogram and line profiles of samples (N ∼ 100) taken from image, UV-vis spectra of ZnO@Ole and ZnO thin films, 1H NMR of LZH-Ole, ZnO@Ole and pre-hydrolysis mixture of synthesis of LZH-Ole, photographs of thin films of LZH-Ole and ZnO on glass substrate, method used for determining the content of ZnO nanoparticles in synthesis mixture, calculation of yields of LZHs based on Zn. See DOI: 10.1039/c7sc04256f |
| This journal is © The Royal Society of Chemistry 2018 |