
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExciton coupling in diketopyrrolopyrrole–helicene derivatives leads to red and near-infrared circularly polarized luminescence†
Kais
Dhbaibi
ag,
Ludovic
Favereau
 *a,
Monika
Srebro-Hooper
b,
Marion
Jean
c,
Nicolas
Vanthuyne
c,
Francesco
Zinna‡
d,
Bassem
Jamoussi
e,
Lorenzo
Di Bari
*d,
Jochen
Autschbach
*f and
Jeanne
Crassous
*a
*a,
Monika
Srebro-Hooper
b,
Marion
Jean
c,
Nicolas
Vanthuyne
c,
Francesco
Zinna‡
d,
Bassem
Jamoussi
e,
Lorenzo
Di Bari
*d,
Jochen
Autschbach
*f and
Jeanne
Crassous
*a
aInstitut des Sciences Chimiques de Rennes UMR 6226, Campus Beaulieu, 35042 Rennes Cedex, France. E-mail: ludovic.favereau@univ-rennes1.fr; jeanne.crassous@univ-rennes1.fr
bFaculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30-387 Krakow, Poland
cAix Marseille University, CNRS, Centrale Marseille, iSm2, Marseille, France
dDipartimento di Chimica e Chimica Industriale (University of Pisa), via Moruzzi 13, 56124, Pisa, Italy. E-mail: lorenzo.dibari@unipi.it
eLaboratoire de Chimie Organique et Analytique, Institut Supérieur de l’Education et de la Formation Continue, 2000 Bardo, Tunisia
fDepartment of Chemistry, University at Buffalo, State University of New York, Buffalo, NY 14260, USA. E-mail: jochena@buffalo.edu
gUniversity of Gabés, Faculty of Science of Gabès, Zrig, 6072 Gabès, Tunisia
First published on 15th November 2017
Abstract
Molecular and macromolecular chiral π-conjugated diketopyrrolopyrrole (DPP)–helicene derivatives were prepared and their chiroptical properties examined experimentally and theoretically. Exciton coupling leads to red and near-infrared circularly polarized absorption and luminescence arising from the achiral DPP units in the helical environment, highlighting an interesting synergy between the chiral helicene and the organic dye.
Introduction
Circularly polarized luminescence (CPL) emitters have received renewed attention due to their potential in several (chiro)-optoelectronic applications (stereoscopic displays, light-emitting diodes, optical information processing and bio-imaging).1 At the molecular level, lanthanide complexes are known to show CPL with luminescence dissymmetry factors (glum = 2(IL − IR)/(IL + IR)) that can reach more than 1 thanks to their formally f → f Laporte forbidden transitions.2 On the other hand, chiral organic molecules display lower glum values of 10−4–10−2, due to electric dipole-allowed transitions (i.e. IL − IR is not necessarily smaller than for lanthanide complexes, but IL + IR is much larger).3 However, their tuneable photophysical properties, associated with easy processing and integration into optoelectronic devices, have made chiral organic molecules valuable candidates for CPL applications.4 Helicenes and their derivatives have been the archetypal class of chiral molecules displaying intense chiroptical properties, with glum up to 10−3–10−2.5 Up to now however, their spectral responses limited to the blue domain and modest emission quantum yields ϕ have restricted their examination in chiral optoelectronic devices or bio-imaging, and only few examples have displayed chiroptical properties above 600 nm either through metalation or via functionalization by electron push–pull groups.6 Developing chirally perturbed π-extended achiral chromophores has emerged as an alternative strategy to design efficient CPL emitters, mainly based on C2-symmetric chiral moieties (chiral binaphthyl or 1,2-diamino-cyclohexane derivatives) linked to bodipy or perylene organic dyes.7 Accordingly, to further expand the chiroptical properties of helicenes to the visible and near-infrared (near-IR) region spectrum, we decided to investigate this approach by functionalizing the chiral 3-dimensional helical π-conjugated core with diketopyrrolopyrrole (DPP) chromophore, which possesses interesting and tuneable photophysical properties across the whole visible spectrum (ϕ up to 70% at 620 nm).8 Moreover, while achiral DPP derivatives have been widely investigated in numerous areas of material science like OLEDs, photovoltaic devices, organic transistors, and fluorescent probes,8b,9 to our knowledge no CPL emitter based on DPP has been reported so far.Herein, we thus report the synthesis and chiroptical properties of π-conjugated diketopyrrolopyrrole–helicene derivatives ranging from discrete dyads and triads, i.e. P- and M-H6DPP and -H6(DPP)2, to helical small oligomers (P,P)- and (M,M)-DPP(H6DPP)2 (Fig. 1), with the aim of developing new CPL emitters. This new family of chiral organic dyes shows intense electronic circular dichroism (ECD) in the visible region and near-IR CPL with ϕ up to 41% and glum of ca. 10−3. Moreover, experimental and theoretical investigations show that the chiral [6]helicene backbone allows for efficient exciton coupling between achiral organic DPP dyes, thus highlighting a new aspect in helicenic-type chiral molecular materials.
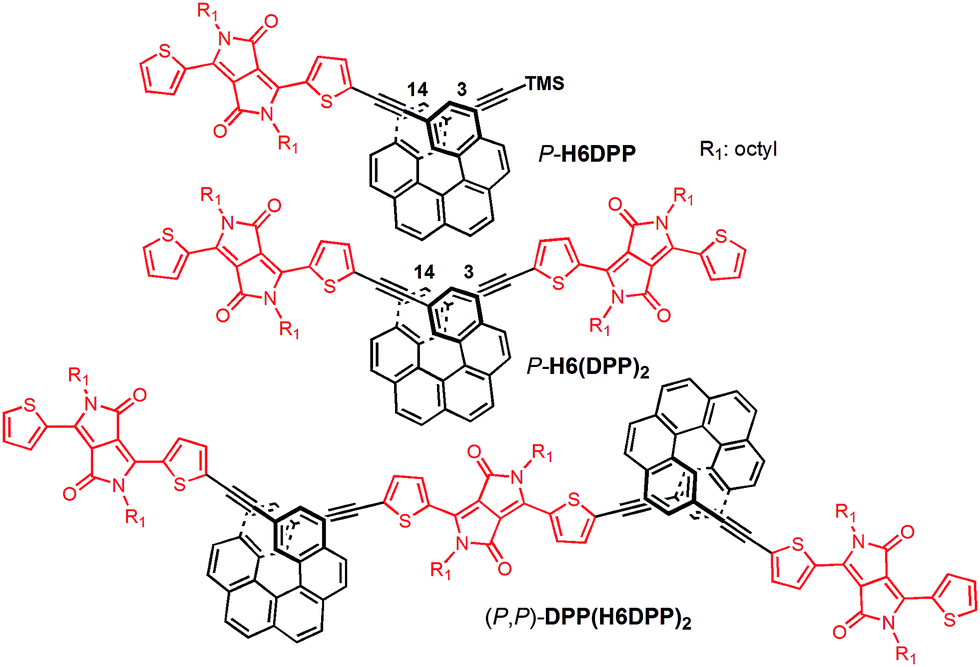 | ||
| Fig. 1 Chemical structures of dyad H6DPP, triad H6(DPP)2, and oligomer DPP(H6DPP)2 synthesized and investigated during this study. For clarity, only P stereochemistry is presented. | ||
Results and discussion
Synthesis of monohelicenic dyads H6DPP and triads H6(DPP)2
The chiral emitters described here are based on the classic carbo[6]helicene (H6) and bis-2-thienyl-diketopyrrolopyrrole (DPP) units, connected through ethynyl bridges: H6DPP, H6(DPP)2, and DPP(H6DPP)2 with either P or M stereochemistry (Fig. 1). Initially, we anticipated that ensuring a strong electronic coupling between the chiral helicene and the DPP unit may lead to an induced chiroptical response at the achiral DPP chromophores.3b,7a–c,10 Therefore we first focused on H6DPP and H6(DPP)2 and investigated their synthesis and their chiroptical properties.The convergent synthesis of H6DPP and H6(DPP)2 is depicted in Scheme 1 for the P enantiomers. See also ESI† for detailed experimental conditions and full characterization. It firstly involves a deprotection of P-2,15-bis-(trimethylsilyl-ethynyl)[6]helicene (P-H6(TMS)2), either partially (P-H6a) or fully (P-H6b).11 The resulting statistical mixture was directly engaged in the following step, i.e. a Sonogashira coupling with an excess of 2-bromothienyl-2-thienyldiketopyrrolopyrrole,12DPPBr, to obtain P-H6DPP and P-H6(DPP)2 within the same reaction, in 70–75% yield for both compounds. These chiral dyes were then separated by column chromatography and characterized by NMR and mass spectrometry, displaying typical signatures of both the [6]helicene and DPP units. For example, the C1-symmetric H6DPP shows two characteristic 1H NMR signals (doublets of doublets) at 7.30 and 7.40 ppm corresponding to H3 and H14 (see Fig. 1) of the unsymmetrical H6 unit, while the C2-symmetric H6(DPP)2 displays only one 1H NMR signal at 7.39 ppm for these protons (ESI†). The inversion barriers of H6(TMS)2 and H6(DPP)2 were measured experimentally (see ESI†) and found to be 180.7 kJ mol−1 (207 °C, 1,2-dimethoxybenzene) and 163.6 kJ mol−1 (182 °C, 1,2-dichlorobenzene), respectively. These barriers correspond to half-life times higher than 200 million years at 50 °C and confirm that the DPP–helicene systems are configurationally stable, but nevertheless there is an influence of the substitution on their stability.
 | ||
| Scheme 1 Synthesis of enantiopure DPP–helicene derivatives P-H6DPP and P-H6(DPP)2. TMS: trimethylsilyl. Reaction conditions: (i) TBAF, CHCl3; (ii) Pd(PPh3)4, CuI, Et3N/toluene, 50 °C, DPPBr, 70% (H6DPP) and 75% (H6(DPP)2). | ||
UV-vis spectroscopy
UV-vis absorption spectra of the novel helicene–ethynyl-diketopyrrolopyrrole derivatives H6DPP and H6(DPP)2 were recorded in dichloromethane (DCM) solutions and compared with their corresponding precursors H6(TMS)2 and DPPBr (Fig. 2). The UV-vis spectrum of H6DPP shows strong absorptions between 300 and 400 nm (ε = 1.5 to 5.5 × 104 M−1 cm−1) and between 550 and 600 nm (ε ∼ 3 × 104 M−1 cm−1) that visibly correspond to absorption maxima of H6(TMS)2 and DPPBr, respectively. The H6DPP UV-vis bands are however red-shifted in comparison with both precursors (by about 20 nm relative to DPPBr), which clearly reflects an extension of the π-conjugation between the DPP dye and the helicene via the alkynyl bridge. Interestingly, going from one DPP substituent in H6DPP to two in H6(DPP)2 does not further red-shift the absorption, which indicates lack of electronic conjugation through the whole helix. Accordingly, the high molar extinction coefficients in the visible part of the spectrum are approximately proportional to the number of DPP units within the molecule, for example ε = 3.5 × 104 M−1 cm−1 and ε = 8.5 × 104 M−1 cm−1 at 580 nm for H6DPP and H6(DPP)2, respectively.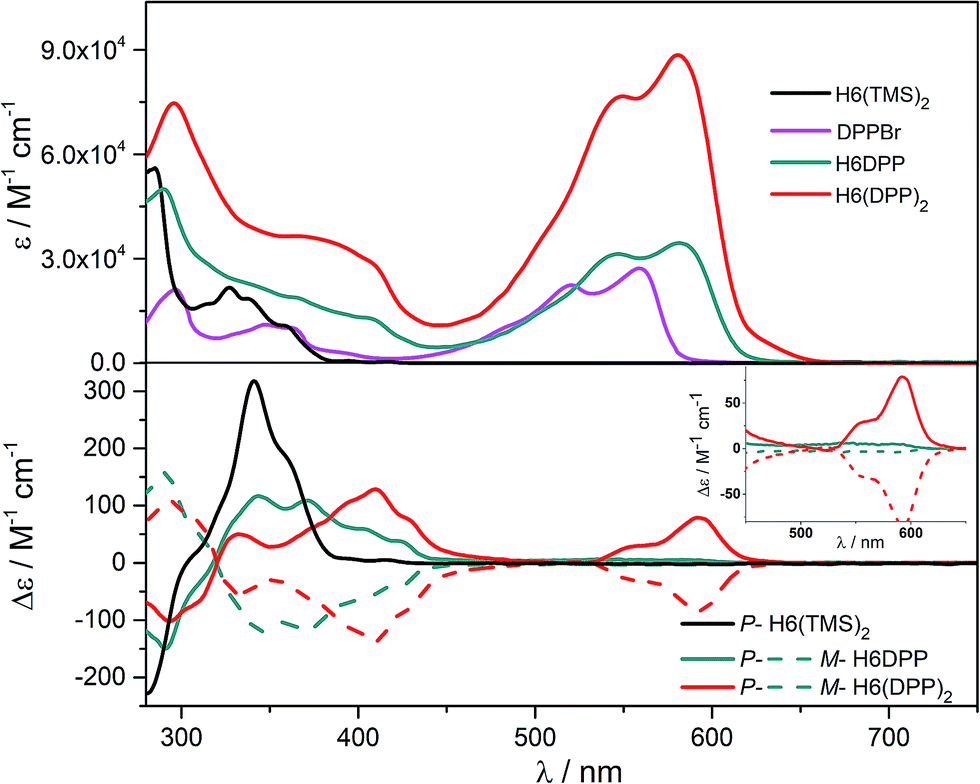 | ||
| Fig. 2 UV-vis (top) and ECD (bottom) spectra of H6(TMS)2 (black), DPPBr (purple), H6DPP (green), and H6(DPP)2 (red) in dichloromethane at 298 K (∼10−5 M). Inset: enlargement of 450–650 nm region of the ECD spectra. | ||
Computational analyses (with (time-dependent) Kohn–Sham, (TD-)KS, DCM continuum solvent model)13 on truncated systems (with n-octyl groups replaced by methyls) support these interpretations. The full set of theoretical results and all computational details are provided in the ESI.† Structural optimizations (BP/SV(P)) of the H6DPP and H6(DPP)2 systems considered different rotamers for the relative orientations of the helicene and the DPP substituents. Fig. 3 shows the lowest-energy nearly isoenergetic structures (with the thiophene rings cis or trans with respect to the helicene) found for H6DPP and H6(DPP)2. Within the numerical accuracy of the KS calculations, the different conformers can be assumed to be present in equal amounts. As seen in Fig. 4a, the calculated (BHLYP/SV(P)) UV-vis absorption spectra are in good agreement with the experimental ones, correctly reproducing a red-shift of the H6DPP/H6(DPP)2 absorption bands compared to H6(TMS)2 and DPPBr precursors and a significant increase in the absorption intensity when going from H6DPP to H6(DPP)2. The computations assign the lowest-energy (ca. 580 nm) strong band for H6DPP to an almost pure (95%) HOMO-to-LUMO π-to-π* transition within DPP (excitation no. 1, calculated at 552 nm, vide infra). Indeed, the frontier molecular orbitals (FMOs) in H6DPP are essentially DPP-centered but also π-conjugated with the adjacent phenyl ring through the alkynyl bridge which goes along with an increase/decrease in the energy of HOMO/LUMO and consequently a reduction of the HOMO–LUMO gap as compared to DPPBr (Fig. 4b). This accounts for a strong red-shift and increase in the oscillator strength of the excitation compared to DPPBr (Fig. 4a and ESI†). The lowest-energy UV-vis band of H6(DPP)2 has the same DPP-centered π-to-π* assignment, involving predominantly contributions from nearly degenerate HOMO − 1, HOMO and LUMO, LUMO + 1 (Fig. 4 and ESI†). Noteworthy, an intense pair of excitations (no. 1 and 2, calculated at respectively 561 and 544 nm) contributes to this band in the case of H6(DPP)2 which may indicate exciton coupling (vide infra).
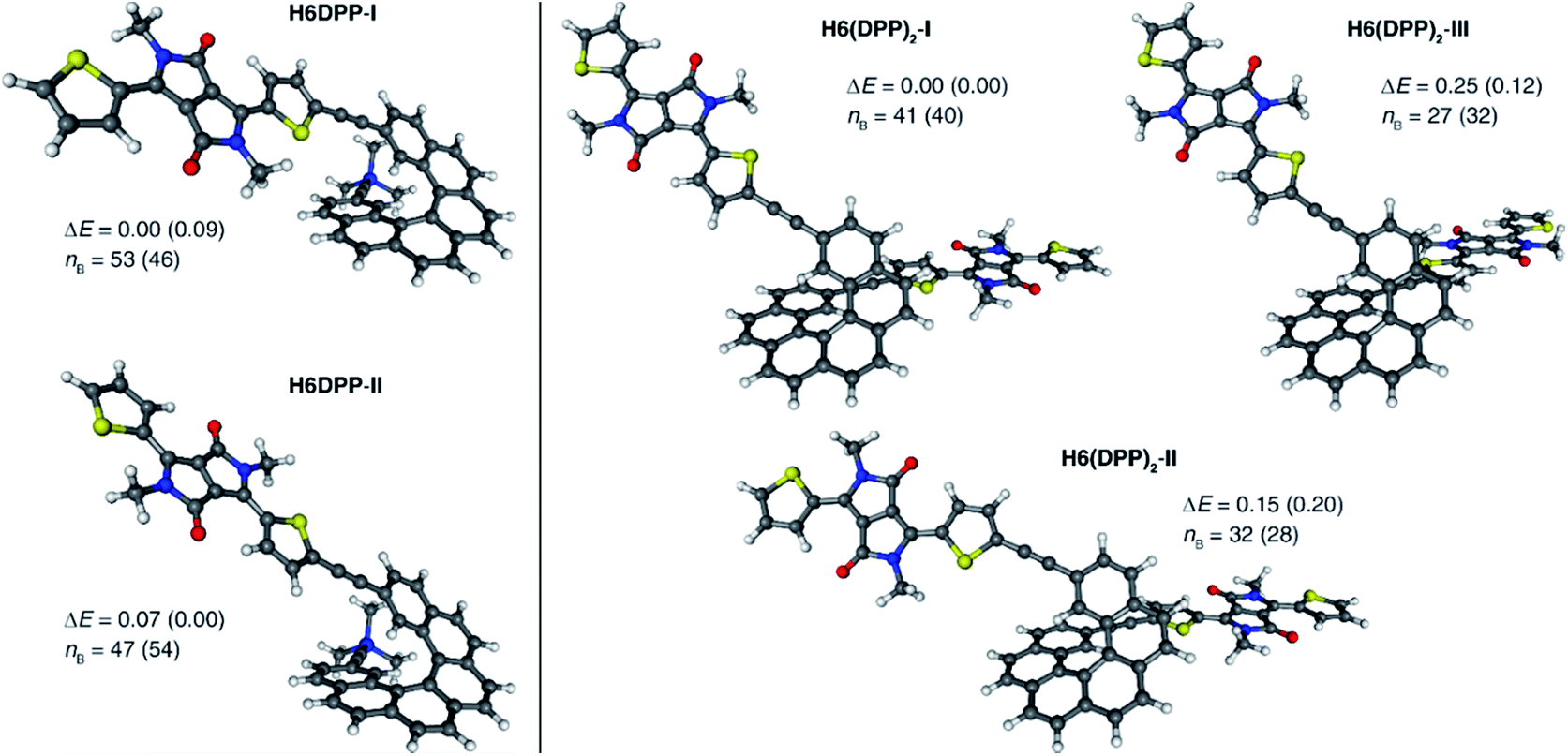 | ||
| Fig. 3 Selected BP/SV(P) (continuum solvent model for DCM) optimized low-energy structures of H6DPP (left) and H6(DPP)2 (right). ΔE and nB values are, respectively, relative energies (in kcal mol−1) and the corresponding Boltzmann populations (in %, at 25 °C); in parentheses are data for calculations without the solvent model. | ||
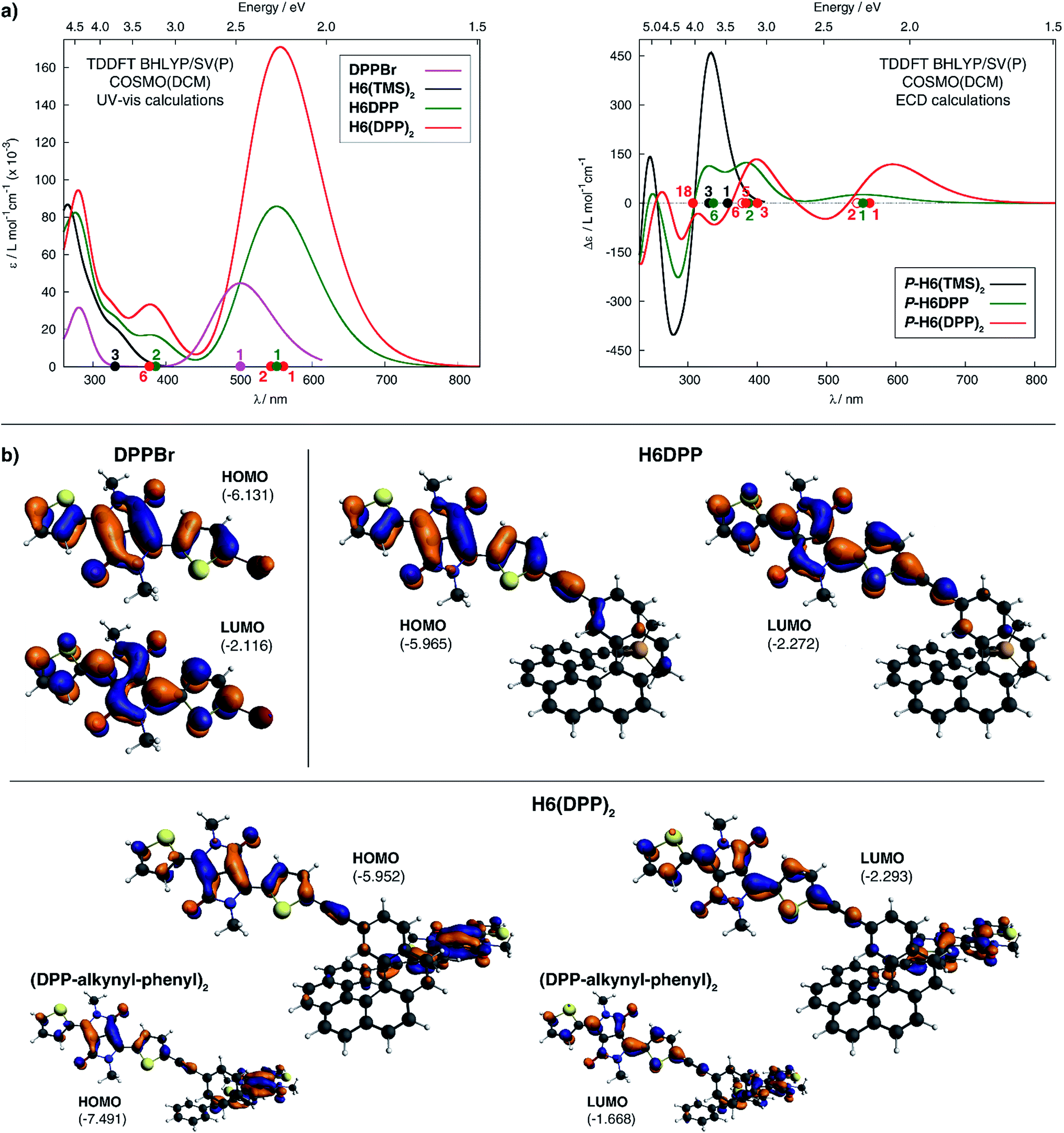 | ||
| Fig. 4 Panel (a): comparison of the simulated UV-vis (left) and ECD (right) spectra of H6DPP and H6DPP2 with H6(TMS)2 and DPPBr. H6DPP and H6DPP2 spectra shown have been Boltzmann-averaged (25 °C) for conformers presented in Fig. 3. No spectral shift has been applied. Selected excitation energies have been indicated by dots on the abscissa. Filled/unfilled dots in the ECD spectra indicate positive/negative rotatory strength value. Panel (b): isosurfaces (±0.04 au) of frontier MOs for DPPBr, H6DPP (conformer I) and H6(DPP)2 (conformer I). The corresponding MOs for (DPP–CC–Ph)2 model for the exciton coupling have been inserted for comparison. Values listed in the parentheses are orbital energies, in eV. | ||
The calculated HOMO and LUMO in H6DPP and H6(DPP)2 are DPP-centered, with nearly identical energies for both species (Fig. 4b). Accordingly, cyclic voltammetry revealed almost identical redox potentials of ca. +0.9 V and −1.1 V vs. SCE for both compounds (Fig. 5 and ESI†). These reversible redox processes were assigned to the oxidation and reduction of the DPP units by comparison with the corresponding DPPBr precursor, which show nearly identical redox properties (EOx = +0.93 V and ERed = −1.25 V vs. SCE, Fig. 5), reflecting the minor electronic contribution of the helicene in the HOMO and LUMO levels of the new compounds. Moreover, the fact that only one oxidation or reduction process is observed on the voltammogram of H6(DPP)2 confirms that the DPP units interact only weakly through the π-conjugated helix.
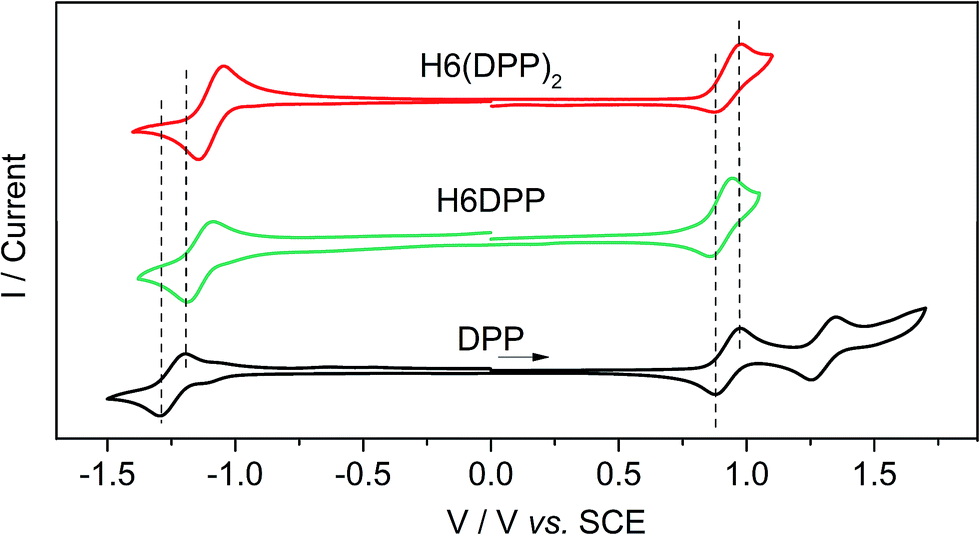 | ||
| Fig. 5 Cyclic voltammograms of DPPBr, H6DPP and H6DPP2versus saturated calomel electrode (SCE) as the reference and 0.1 M Bu4NPF6 in dichloromethane as the electrolyte. | ||
ECD spectroscopy and exciton coupling
The ECD of P- and M-H6(TMS)2 are strongly modified upon functionalization by one or two DPP (Fig. 2). For instance, P-H6DPP gives a strong negative ECD band (Δε = −150 M−1 cm−1) at 291 nm which is 16 nm red-shifted compared to P-H6(TMS)2, a large positive band between 320 and 440 nm (Δε = +88M−1 cm−1 at 400 nm) displaying some vibronic progression, and a very weak ECD signal (Δε = +5 M−1 cm−1 at 580 nm, Fig. 2 inset). P-H6(DPP)2 displays a slightly red-shifted but otherwise similar ECD below 500 nm as P-H6DPP, such as a negative band at 294 nm (Δε = −100 M−1 cm−1) and a broad positive band between 340 and 450 nm (Δε = +130 M−1 cm−1 at 410 nm). Interestingly, the ECD at 592 nm originating from the DPP–alkynyl transitions (+79 M−1 cm−1, vide supra) is much more intense than for P-H6DPP, with an associated dissymmetry factor gabs = Δε/ε = +9.0 × 10−4, i.e. six times higher than for P-H6DPP (gabs = +1.5 × 10−4). This significant ECD increase for P-H6(DPP)2 and the presence of a weak negative band at 540 nm, Δε = −2 M−1 cm−1 (Fig. 2 inset), suggests the presence of exciton coupling between the two DPP–alkynyl π-to-π* transitions (vide infra), with a substantial cancellation of the higher energy (540 nm) couplet component owing to the nearby intense positive bands.In Fig. 4a, simulated ECD spectra of the DPP-substituted systems are compared to spectra of H6(TMS)2. The calculations reproduce the experimental features well. An analysis of the dominant excitations in the low- and medium-energy parts of the simulated spectra of P-H6DPP conformers (Fig. 3) assigns the intense positive ECD band below 400 nm to a combination of helicene-to-DPP and DPP-to-helicene charge transfers (CT), and helicene-centered π-to-π* transitions (excitations no. 2 and 6 calculated at respectively ca. 385 nm and 335 nm, Fig. 4, ESI†). The weak ECD band at 580 corresponds to the HOMO-to-LUMO π-to-π* transition within the DPP–alkynyl group (excitation no. 1). This excitation has additional small contributions from helicene π-orbitals that are also delocalized over the alkynyl bridge, and therefore electronically coupled to the DPP π-orbitals. Accordingly, the excitation acquires a sizable calculated rotatory strength (see ESI†). Vibrational bending of the alkynyl–C bonds is expected to break the conjugation between DPP and H6 moieties and lead to the overall weaker intensity observed experimentally at wavelengths > 500 nm (vide infra).
The assignment of most of the H6(DPP)2 ECD spectrum is qualitatively similar to that of H6DPP, involving DPP-alkynyl- and helicene-centered π-to-π* transitions along with helicene-to-DPP and DPP-to-helicene CT excitations (Fig. 4, ESI†). In the following we focus on the most striking new spectral feature of H6(DPP)2, i.e. the exciton couplet signature in the visible range of the spectrum, with peaks around 600 nm (Fig. 2 inset). The intensity of the low-energy positive band at ca. 600 nm for P-H6(DPP)2 is only slightly overestimated by the calculation. However, the first positive ECD band in the calculation is followed by a weaker negative band around 500 nm, while the corresponding experimental ECD only slightly dips below zero in this energy range. The calculated H6(DPP)2 ECD spectrum at low energy is caused by an intense pair of excitations with opposite rotatory strengths (R) at ca. 560 (+) and 545 nm (−), respectively (excitations no. 1 and 2, Fig. 4a and ESI†), followed by two intense excitations with positive rotatory strengths at ca. 400 and 380 nm (excitations no. 3 and 5). The negative second band in the simulated spectrum is thus due to the second excitation with negative R, which is not completely suppressed by the positive ECD intensity of surrounding excitations. However, the intensity of all three bands is seen to be sensitive to the rotamer structures (with the second (higher-energy) positive band being affected the most, see ESI†). Since the experimental spectra show vibronic fine structure, it is conceivable that vibronic effects lead to some suppression of the negative ECD band in the experiment (vide infra).
Compared to H6DPP, the rotatory strengths of the first two excitations of H6(DPP)2 are staggeringly large, with R between +1708 and +2657 for the first excitation, and between −1332 and −2320 for the second, depending on the rotamer (ESI†). The MO pair contributions to the first excitation are 51% HOMO-to-LUMO and 44% HOMO − 1-to-LUMO + 1 for all rotamers. For the second excitation, the contributions are 48% each from HOMO − 1-to-LUMO and from HOMO-to-LUMO + 1, for all rotamers. The HOMO − 1, HOMO, LUMO, and LUMO + 1 of the rotamers of H6(DPP)2 are essentially ±linear combinations of the DPP substituents' FMOs (Fig. 4b, ESI†). As for H6DPP, these orbitals are DPP-centered but also delocalized over the alkynyl bridge and conjugated with the adjacent rings of the helicene. The DPP-centered π-orbitals of H6(DPP)2 are weakly interacting with each other through the helicene, as indicated by the small (0.03 to 0.04 eV) energetic splittings between HOMO − 1 and HOMO, and between LUMO and LUMO + 1, respectively. The large rotatory strengths and the opposite signs of the first two excitations have the same characteristics as an exciton CD couplet, whereby the two electric transition dipoles of the π-to-π* excitations of a DPP dimer form a chiral arrangement, couple with each other, and create strong magnetic transition dipoles that are not perpendicular to the coupled electric transition dipoles.
In order to confirm the presence of an exciton coupling mechanism, calculations were performed on a (DPPBr)2 dimer in the same arrangement as the substituents in H6(DPP)2, as well as a (DPP–CC–H)2 dimer including the alkynyl group and a (DPP–CC–Ph)2 model including the alkynyl and the first phenyl group of the helicene (see ESI† for the full set of data and an analysis). The corresponding calculated ECD spectra of the dimer models indeed show very similar spectral shapes as H6(DPP)2 below 450 nm. For the extended coupled chromophores, as in the (DPP–CC–Ph)2 model, the exciton couplet increases dramatically in intensity, which goes along with a delocalization of the DPP frontier orbitals through the alkynyl into the phenyl groups (Fig. 4b, ESI†) and an energetic splitting of the coupled excitations (ca. 0.06 eV) that is almost as large as in H6(DPP)2 (0.07 eV). The rotatory strengths calculated for the lowest-energy exciton couplet of (DPP–CC–Ph)2 come close to those of H6(DPP)2. However, the simulated ECD intensities remain lower for the model, showing that the full helicene bridge in H6(DPP)2 enhances the intensity of the couplet even further. The resulting exciton CD couplet may appear conservative or not, in the full system as well as in the dimer models, depending on the relative orientations of the coupled chromophores to each other.
The assignment of the two lowest energy excitations for all three conformers of the (DPP–CC–Ph)2 is an approximately 50–50 mix of HOMO − 1-to-LUMO and HOMO-to-LUMO + 1, respectively, with orbitals appearing as ±linear combinations of the DPP orbitals, similar to those in the full H6(DPP)2 system. The small energetic splitting between HOMO − 1 and HOMO, and LUMO and LUMO + 1, respectively, indicates weak through-space electrostatic interactions in the dimer model ground state. The models for P-H6(DPP)2 therefore show unambiguously that the longest-wavelength ECD band is caused mainly by exciton coupling between the electric transition dipoles of the DPP–alkynyl–phenyl fragments at the extremities of P-H6(DPP)2, with minor contributions of the central part of the helicene. Thus, the sign of the resulting exciton CD couplet is determined by the sense of the helical arrangement of the coupled electric transition dipole moments which is opposite for the P and M structures (Fig. 6).14 To our knowledge, such an exciton coupling CD of substituents of a helicene is a new aspect in helicene chemistry, from which we can take benefit to perform efficient engineering to obtain chirally arranged π-conjugated derivatives with strong chiral absorption response in the near-IR region and as a direct consequence a strong near-IR chiral emission (vide infra).7
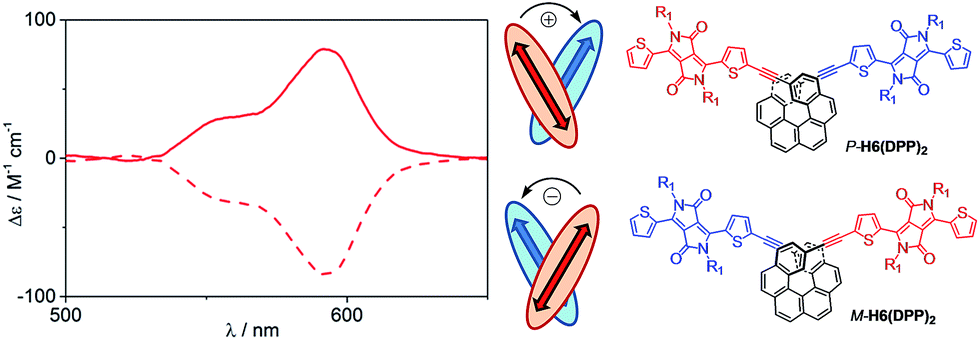 | ||
| Fig. 6 Relation between the helical arrangement of the DPP units around the P- or M-helicenic core and the respective positive or negative exciton coupling signature. | ||
Synthesis and (chir)optical properties of enantiopure oligomer DPP(H6DPP)2
The efficient synthesis of H6DPP and H6(DPP)2 and the intense ECD band obtained for the visible DPP π–π* transitions in P- and M-H6(DPP)2 prompted us to further explore the exciton coupling in longer diketopyrrolopyrrole-helicene derivatives. We thus synthesized enantiopure oligomers (P,P)- and (M,M)-DPP(H6DPP)2, as described in Scheme 2 for the (P,P) enantiomer. Deprotection of the remaining TMS group in P-H6(DPP), followed by a Sonogashira coupling with bis-(2-bromo-2-thienyl-)diketopyrrolopyrrole, DPPBr2, afforded (P,P)-DPP(H6DPP)2 in 65% yield. DPP(H6DPP)2 was characterized by NMR and mass spectroscopy (see ESI†) and can be viewed as a dimer of H6(DPP)2 sharing one DPP unit. | ||
| Scheme 2 Synthesis of enantiopure (P,P)-DPP(H6DPP)2. TMS = trimethylsilyl. Reaction conditions: (i) TBAF, CH2Cl2, rt; (ii) Pd(PPh3)4, CuI, Et3N/toluene, 50 °C, DPPBr2, 65%. | ||
Optical and chiroptical properties of oligomer DPP(H6DPP)2 are similar to those of H6(DPP)2 (Fig. 7). The main difference in the UV-vis spectrum of DPP(H6DPP)2 arises from the shoulder at 622 nm, which may be attributed to the more π-conjugated bis(ethynyl)-DPP unit. This extended conjugation may also account for a red-shift of ECD spectrum of DPP(H6DPP)2 as compared to H6(DPP)2. Thanks to the further separation of the lowest-energy DPP-centered transition from the rest of the absorption spectrum, the excitonic signature (bisignate signal) is even clearer for P-DPP(H6DPP)2 than for P-H6(DPP)2, with positive ECD response obtained at 606 nm (Δε = +94 M−1 cm−1) and negative at 534 nm (Δε = −15 M−1 cm−1). Interestingly, gabs of the DPP π–π* transitions increases again when going from P-H6(DPP)2 to P-DPP(H6DPP)2 (+1.3 × 10−3 and +9.0 × 10−4 for (P,P)-DPP(H6DPP)2 and P-H6(DPP)2, respectively), which means that it is possible to make chiral absorbers (and emitters, vide infra) deeper into the near-infrared by increasing the π-conjugation of the compounds without affecting the excitonic coupling.
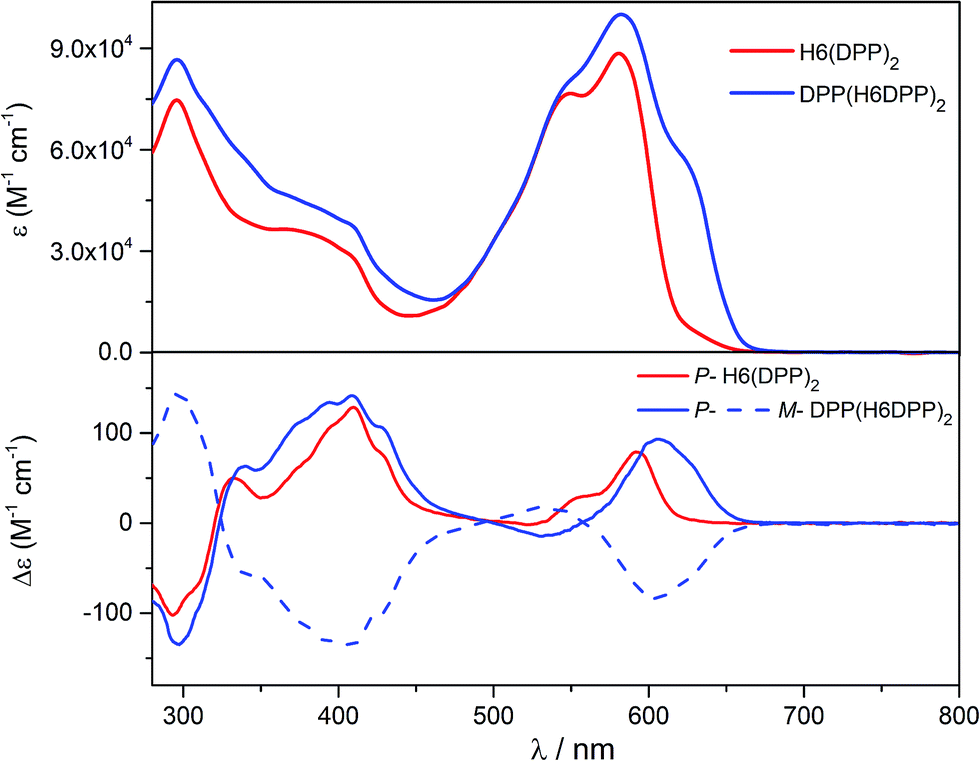 | ||
| Fig. 7 UV-vis (top) and ECD (bottom) spectra of H6(DPP)2 (red) and DPP(H6DPP)2 (blue) in dichloromethane solution at 298 K (∼10−5 M). | ||
Unpolarized and circularly polarized luminescence
Emission properties of the DPP–helicene derivatives were recorded in DCM solution and corresponding spectra are displayed in Fig. 8.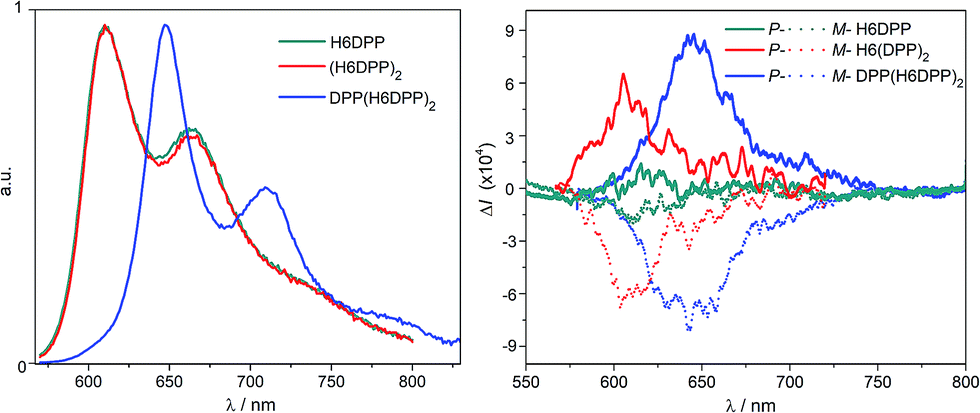 | ||
| Fig. 8 Normalized fluorescence (left) and CPL (right) spectra of H6DPP (green), H6(DPP)2 (red) and DPP(H6DPP)2 (blue) in dichloromethane at 298 K. CPL spectra for P and M enantiomers are shown respectively in solid and dotted lines. | ||
All the compounds display intense nonpolarized and circularly polarized emission properties above 600 nm, arising from the DPP-ethynyl unit with a characteristic structured signal.8aH6DPP and H6(DPP)2 exhibit exactly the same emission maximum at 610 nm, which indicate a weak electronic coupling between the DPP-ethynyl units also in the excited state. As in the case of the UV-vis absorption, the emission spectra of DPP(H6DPP)2 is significantly red-shifted by about 50 nm due to the extension of the π-conjugation on the central DPP unit, as discussed above. In terms of emission efficiency of these novel near-IR emitters, high quantum yields of 41% for both H6DPP and H6(DPP)2 and 35% for DPP(H6DPP)2 were measured. For comparison, H6(TMS)2 precursor displays a much lower quantum yield of ca. 7% at 450 nm (Table S1.2†).15 Interestingly, the reported chiral derivatives keep the characteristic photophysical properties of the DPP attached dyes, which makes them appealing targets for chiral optoelectronic applications.3–7 Indeed, circularly polarized luminescence (CPL) spectra for each enantiomer of H6DPP, H6(DPP)2 and DPP(H6DPP)2 were recorded in dichloromethane solutions and all products gave mirror-image signals corresponding to the same wavelength as the unpolarised fluorescence (Fig. 8). The obtained reliable signals allowed us to determine glum factors ranging from 1.0 × 10−4 (P-H6DPP) to 9.0 × 10−4 (P-DPP(H6DPP)2), which follow the same trend observed in the ECD spectra with gabs. This similarity strongly suggests that both ground and emitting excited states have a similar chiral geometry. The measured glum values are in the same range as previously reported chiral organic dye ones (10−4–10−2) but the obtained CPL signatures are among the most red-shifted reported to date for (small) organic molecules.3c,4e,5i,7e,g,16 Especially, extension of the exciton coupling in oligomer (P,P)- (or (M,M-)) DPP(H6DPP)2 appears to be a highly promising approach for reaching intense emission and CPL in the red and near-infrared region.
Conclusions
We have synthesized new chiral organic near-infrared CPL emitters based on an unprecedented association between enantiopure [6]helicene and diketopyrrolopyrrole which provides molecular and macromolecular helical π-conjugated molecules with strong ECD signal in the visible region (∼600 nm), intense red and near-infrared luminescence (ϕ ∼ 0.4) and corresponding CPL activity up to 650 nm with glum higher than 10−3. Our results show that chiral helicenes symmetrically functionalized by DPP units display an unprecedented exciton coupling effect, thus resulting in a strong synergy between the chiral helical π-conjugated core and the photophysical properties of the dye. This constitutes a novel strategy of chemical engineering of a π-helical platform to further improve its chiroptical responses especially in the near-IR and red regions. With potential offered by the DPP and the helicene units in numerous optoelectronic applications (OLEDs, organic transistors, bio-imaging), this work may open new opportunities for chiral organic molecules.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Ministère de l’Education Nationale, de la Recherche et de la Technologie, the Centre National de la Recherche Scientifique (CNRS). K. D. thanks the University of Gabès, the University of Rennes 1 and Campus France for financial support. J. A. thanks the National Science Foundation (CHE 1560881) for financial support and the Center for Computational Research (CCR) at the University at Buffalo for computational resources. M. S. H. acknowledges a young researchers' T-subsidy from the Ministry of Science and Higher Education in Poland. The PRISM core facility (Biogenouest©, UMS Biosit, Université de Rennes 1 – Campus de Villejean – 35043 RENNES Cedex, FRANCE) is thanked for the NMR characterizations and ECD spectroscopy.References
- (a) F. S. Richardson and J. P. Riehl, Chem. Rev., 1977, 77, 773–792 CrossRef CAS; (b) J. P. Riehl and F. S. Richardson, Chem. Rev., 1986, 86, 1–16 CrossRef CAS; (c) Chirality at the Nanoscale, Nanoparticles, Surfaces, Materials and more, ed. D. Amabilino, Wiley-VCH, 2009 Search PubMed.
- (a) F. Zinna and L. Di Bari, Chirality, 2015, 27, 1–13 CrossRef CAS PubMed; (b) F. Zinna, U. Giovanella and L. Di Bari, Adv. Mater., 2015, 27, 1791–1795 CrossRef CAS PubMed; (c) F. Zinna, M. Pasini, F. Galeotti, C. Botta, L. Di Bari and U. Giovanella, Adv. Funct. Mater., 2017, 27, 1603719 CrossRef.
- (a) H. Maeda and Y. Bando, Pure Appl. Chem., 2013, 85, 1967 CrossRef CAS; (b) J. Kumar, T. Nakashima and T. Kawai, J. Phys. Chem. Lett., 2015, 6, 3445–3452 CrossRef CAS PubMed; (c) E. M. Sánchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem.–Eur. J., 2015, 21, 13488–13500 CrossRef PubMed.
- (a) J. Gilot, R. Abbel, G. Lakhwani, E. W. Meijer, A. P. H. J. Schenning and S. C. J. Meskers, Adv. Mater., 2010, 22, E131–E134 CrossRef CAS PubMed; (b) Y. Yang, R. C. da Costa, M. J. Fuchter and A. J. Campbell, Nat. Photonics, 2013, 7, 634–638 CrossRef CAS; (c) Y. Yang, R. C. da Costa, D.-M. Smilgies, A. J. Campbell and M. J. Fuchter, Adv. Mater., 2013, 25, 2624–2628 CrossRef CAS PubMed; (d) J. R. Brandt, X. Wang, Y. Yang, A. J. Campbell and M. J. Fuchter, J. Am. Chem. Soc., 2016, 138, 9743–9746 CrossRef CAS PubMed; (e) S. Feuillastre, M. Pauton, L. Gao, A. Desmarchelier, A. J. Riives, D. Prim, D. Tondelier, B. Geffroy, G. Muller, G. Clavier and G. Pieters, J. Am. Chem. Soc., 2016, 138, 3990–3993 CrossRef CAS PubMed; (f) M. Schulz, M. Mack, O. Kolloge, A. Lutzen and M. Schiek, Phys. Chem. Chem. Phys., 2017, 19, 6996–7008 RSC; (g) P. Josse, L. Favereau, C. Shen, S. Dabos-Seignon, P. Blanchard, C. Cabanetos and J. Crassous, Chem.–Eur. J., 2017, 23, 6277–6281 CrossRef CAS PubMed; (h) J. R. Brandt, F. Salerno and M. J. Fuchter, Nat. Rev. Chem., 2017, 1, 0045 CrossRef.
- (a) Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed; (b) M. Gingras, Chem. Soc. Rev., 2013, 42, 1051–1095 RSC; (c) Y. Shen and C.-F. Chen, Helicenes Chemistry: From Synthesis to Applications, Springer, Berlin, 2017 Search PubMed; (d) M. Li, H.-Y. Lu, C. Zhang, L. Shi, Z. Tang and C.-F. Chen, Chem. Commun., 2016, 52, 9921–9924 RSC; (e) H. Oyama, M. Akiyama, K. Nakano, M. Naito, K. Nobusawa and K. Nozaki, Org. Lett., 2016, 18, 3654–3657 CrossRef CAS PubMed; (f) H. Sakai, T. Kubota, J. Yuasa, Y. Araki, T. Sakanoue, T. Takenobu, T. Wada, T. Kawai and T. Hasobe, J. Phys. Chem. C, 2016, 120, 7860–7869 CrossRef CAS; (g) Y. Yamamoto, H. Sakai, J. Yuasa, Y. Araki, T. Wada, T. Sakanoue, T. Takenobu, T. Kawai and T. Hasobe, J. Phys. Chem. C, 2016, 120, 7421–7427 CrossRef CAS; (h) H. Oyama, K. Nakano, T. Harada, R. Kuroda, M. Naito, K. Nobusawa and K. Nozaki, Org. Lett., 2013, 15, 2104–2107 CrossRef CAS PubMed; (i) A. Ushiyama, S. Hiroto, J. Yuasa, T. Kawai and H. Shinokubo, Org. Chem. Front., 2017, 4, 664–667 RSC.
- (a) N. Saleh, C. Shen and J. Crassous, Chem. Sci., 2014, 5, 3680–3694 RSC; (b) C. Shen, E. Anger, M. Srebro, N. Vanthuyne, K. K. Deol, T. D. Jefferson, G. Muller, J. A. G. Williams, L. Toupet, C. Roussel, J. Autschbach, R. Reau and J. Crassous, Chem. Sci., 2014, 5, 1915–1927 RSC; (c) N. Saleh, B. Moore, M. Srebro, N. Vanthuyne, L. Toupet, J. A. G. Williams, C. Roussel, K. K. Deol, G. Muller, J. Autschbach and J. Crassous, Chem.–Eur. J., 2015, 21, 1673–1681 CrossRef CAS PubMed; (d) I. H. Delgado, S. Pascal, A. Wallabregue, R. Duwald, C. Besnard, L. Guenee, C. Nancoz, E. Vauthey, R. C. Tovar, J. L. Lunkley, G. Muller and J. Lacour, Chem. Sci., 2016, 7, 4685–4693 RSC; (e) J. Bosson, G. M. Labrador, S. Pascal, F. A. Miannay, O. Yushchenko, H. Li, L. Bouffier, N. Sojic, R. C. Tovar, G. Muller, D. Jacquemin, A. D. Laurent, B. Le Guennic, E. Vauthey and J. Lacour, Chem.–Eur. J., 2016, 22, 18394–18403 CrossRef CAS PubMed; (f) H. Sakai, T. Kubota, J. Yuasa, Y. Araki, T. Sakanoue, T. Takenobu, T. Wada, T. Kawai and T. Hasobe, Org. Biomol. Chem., 2016, 14, 6738–6743 RSC; (g) T. Otani, A. Tsuyuki, T. Iwachi, S. Someya, K. Tateno, H. Kawai, T. Saito, K. S. Kanyiva and T. Shibata, Angew. Chem., Int. Ed., 2017, 56, 3906–3910 CrossRef CAS PubMed; (h) C. Shen, M. Srebro-Hooper, M. Jean, N. Vanthuyne, L. Toupet, J. A. Williams, A. R. Torres, A. J. Riives, G. Muller, J. Autschbach and J. Crassous, Chem.–Eur. J., 2017, 23, 407–418 CrossRef CAS PubMed; (i) D. Q. He, H. Y. Lu, M. Li and C. F. Chen, Chem. Commun., 2017, 53, 6093–6096 RSC.
- (a) T. Kawai, K. Kawamura, H. Tsumatori, M. Ishikawa, M. Naito, M. Fujiki and T. Nakashima, ChemPhysChem, 2007, 8, 1465–1468 CrossRef CAS PubMed; (b) E. M. Sánchez-Carnerero, F. Moreno, B. L. Maroto, A. R. Agarrabeitia, M. J. Ortiz, B. G. Vo, G. Muller and S. d. l. Moya, J. Am. Chem. Soc., 2014, 136, 3346–3349 CrossRef PubMed; (c) X. Ma, E. A. Azeem, X. Liu, Y. Cheng and C. Zhu, J. Mater. Chem. C, 2014, 2, 1076–1084 RSC; (d) F. Li, Y. Li, G. Wei, Y. Wang, S. Li and Y. Cheng, Chem.–Eur. J., 2016, 22, 12910–12915 CrossRef CAS PubMed; (e) M. Saikawa, T. Nakamura, J. Uchida, M. Yamamura and T. Nabeshima, Chem. Commun., 2016, 52, 10727–10730 RSC; (f) Y. Wu, S. Wang, Z. Li, Z. Shen and H. Lu, J. Mater. Chem. C, 2016, 4, 4668–4674 RSC; (g) F. Zinna, T. Bruhn, C. A. Guido, J. Ahrens, M. Broring, L. Di Bari and G. Pescitelli, Chem.–Eur. J., 2016, 22, 16089–16098 CrossRef CAS PubMed.
- (a) M. Grzybowski and D. T. Gryko, Adv. Opt. Mater., 2015, 3, 280–320 CrossRef CAS; (b) M. Kaur and D. H. Choi, Chem. Soc. Rev., 2015, 44, 58–77 RSC.
- (a) P. Data, A. Kurowska, S. Pluczyk, P. Zassowski, P. Pander, R. Jedrysiak, M. Czwartosz, L. Otulakowski, J. Suwinski, M. Lapkowski and A. P. Monkman, J. Phys. Chem. C, 2016, 120, 2070–2078 CrossRef CAS; (b) E. Heyer, P. Lory, J. Leprince, M. Moreau, A. Romieu, M. Guardigli, A. Roda and R. Ziessel, Angew. Chem., Int. Ed., 2015, 54, 2995–2999 CrossRef CAS PubMed.
- (a) F. Li, Y. Wang, Z. Wang, Y. Cheng and C. Zhu, Polym. Chem., 2015, 6, 6802–6805 RSC; (b) Y. Wang, Y. Li, S. Liu, F. Li, C. Zhu, S. Li and Y. Cheng, Macromolecules, 2016, 49, 5444–5451 CrossRef CAS; (c) T. Goto, Y. Okazaki, M. Ueki, Y. Kuwahara, M. Takafuji, R. Oda and H. Ihara, Angew. Chem., Int. Ed., 2017, 56, 2989–2993 CrossRef CAS PubMed.
- E. Anger, M. Srebro, N. Vanthuyne, L. Toupet, S. Rigaut, C. Roussel, J. Autschbach, J. Crassous and R. Réau, J. Am. Chem. Soc., 2012, 134, 15628–15631 CrossRef CAS PubMed.
- X.-F. Wu, W.-F. Fu, Z. Xu, M. Shi, F. Liu, H.-Z. Chen, J.-H. Wan and T. P. Russell, Adv. Funct. Mater., 2015, 25, 5954–5966 CrossRef CAS.
- (a) J. Autschbach, Chirality, 2009, 21, E116–E152 CrossRef CAS PubMed; (b) M. Srebro-Hooper and J. Autschbach, Annu. Rev. Phys. Chem., 2017, 68, 399–420 CrossRef CAS PubMed.
- (a) N. Harada, K. Nakanishi and N. Berova, in Comprehensive Chiroptical Spectroscopy, John Wiley & Sons, Inc., 2012, pp. 115–166 Search PubMed; (b) N. Berova, L. Di Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914–931 RSC.
- (a) M. Sapir and E. V. Donckt, Chem. Phys. Lett., 1975, 36, 108–110 CrossRef CAS; (b) K. Nagarajan, A. R. Mallia, K. Muraleedharan and M. Hariharan, Chem. Sci., 2017, 8, 1776–1782 RSC.
- (a) S. P. Morcillo, D. Miguel, L. Alvarez de Cienfuegos, J. Justicia, S. Abbate, E. Castiglioni, C. Bour, M. Ribagorda, D. J. Cardenas, J. M. Paredes, L. Crovetto, D. Choquesillo-Lazarte, A. J. Mota, M. C. Carreno, G. Longhi and J. M. Cuerva, Chem. Sci., 2016, 7, 5663–5670 RSC; (b) M. Gon, Y. Morisaki and Y. Chujo, Chem.–Eur. J., 2017, 23, 6323–6329 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04312k |
| ‡ Current address: Université de Genève, Département de Chimie Organique, Quai Ernest-Ansermet 30, 1211 Genève 4, Switzerland. |
| This journal is © The Royal Society of Chemistry 2018 |