
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-selective 18F fluorination of unactivated C–H bonds mediated by a manganese porphyrin†
Wei
Liu
a,
Xiongyi
Huang
a,
Michael S.
Placzek
bc,
Shane W.
Krska
d,
Paul
McQuade
e,
Jacob M.
Hooker
*bc and
John T.
Groves
 *a
*a
aDepartment of Chemistry, Princeton University, Princeton, New Jersey 08544, USA. E-mail: jtgroves@princeton.edu
bAthinoula A. Martinos Center for Biomedical Imaging, Massachusetts General Hospital, Harvard Medical School, Charlestown, Massachusetts 02129, USA. E-mail: hooker@nmr.mgh.harvard.edu
cDivision of Nuclear Medicine and Molecular Imaging, Department of Radiology, Massachusetts General Hospital, Boston, Massachusetts 02114, USA
dDepartment of Process Chemistry, Merck Research Laboratories, Rahway, New Jersey 07065, USA
eImaging Research, Merck Research Laboratories, West Point, Pennsylvania 19486, USA
First published on 5th December 2017
Abstract
The first direct C–H 18F fluorination reaction of unactivated aliphatic sites using no-carrier-added [18F]fluoride is reported. Under the influence of a manganese porphyrin/iodosylbenzene system, a variety of unactivated aliphatic C–H bonds can be selectively converted to C–18F bonds. The mild conditions, broad substrate scope and generally inaccessible regiochemistry make this radio-fluorination a powerful alternate to established nucleophilic substitution for the preparation of 18F labeled radio tracers.
Introduction
Positron emission tomography (PET) is a noninvasive nuclear medicine imaging technique with clinical and research applications in diverse areas of medicine, notably in oncology, cardiology, neuroscience and in drug development.1 Following administration of a radiolabeled bioactive molecule into a patient or research subject, PET can provide a quantitative time course of three-dimensional images stemming from molecular interactions in vivo. Among the commonly used PET radioisotopes, 18F is the preferred radionuclide largely due to its suitable half-life (110 min) for synthesis and imaging.2 Because of the importance of 18F-containing radiotracers in clinical and research domains, much effort has been devoted to developing new strategies to introduce 18F substituents into complex bioactive molecules.3Despite the recent advances, current methods for incorporating an 18F atom at C sp3 positions are still dominated by traditional nucleophilic substitution of no-carrier-added 18F fluoride with a precursor molecule, typically bearing a suitable leaving group such as an alkyl halide or sulfonate at the labeling site (Fig. 1a).1b One major drawback of this displacement approach is that the preparation of the precursor molecules usually requires multistep synthesis and the highly activated precursor are prone to degradation, for example through elimination of the leaving group. Accessibility of precursors, in fact, often hinders the development of a radiotracer in PET imaging for research applications.
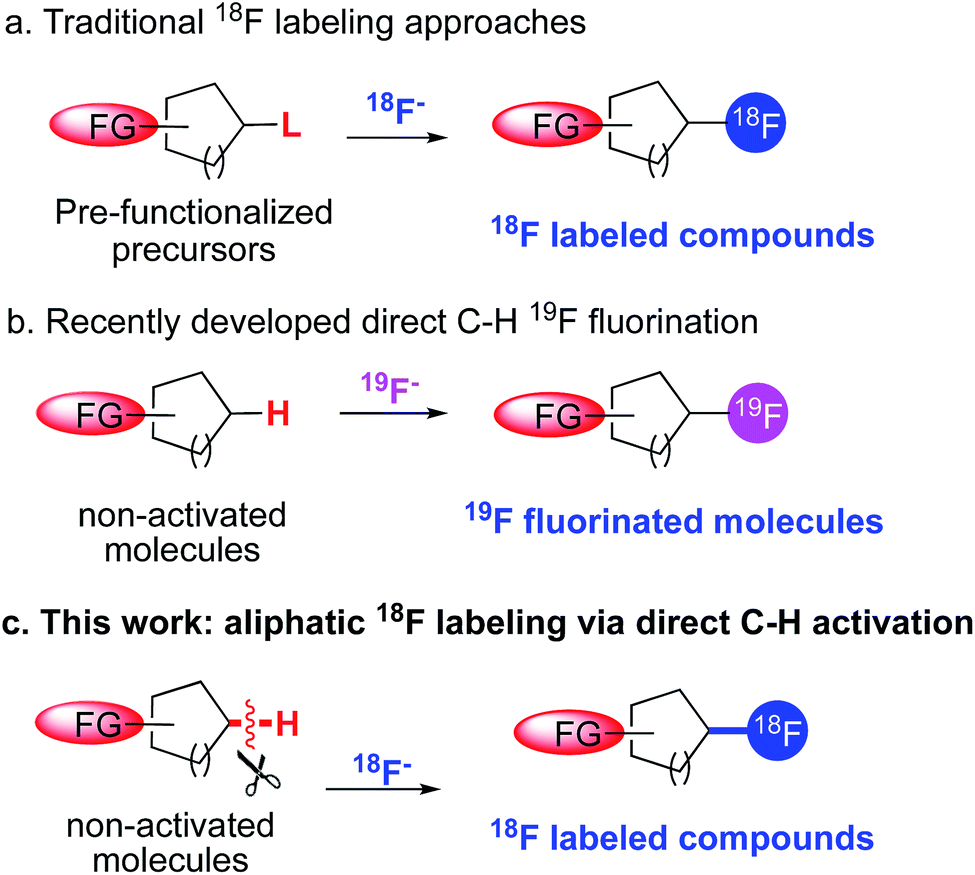 | ||
| Fig. 1 (a) Traditional 18F labeling methods rely on nucleophilic substitution of pre-functionalized precursors with 18F fluorides. (b) Recent studies allow direct conversion of aliphatic C–H bonds to C–F with nucleophilic fluoride sources. (c) Current work enables 18F labeling of aliphatic sites by direct C–H activation. | ||
A potential solution to issues related to pre-functionalization of labeling precursors would be to develop methods to directly and selectively convert C–H bonds to C–18F bonds, thus obviating the complex precursor synthesis. This is feasible given the rapid growth of direct aliphatic C–H 19F fluorination over the past three years (Fig. 1b). In 2012, Lectka et al. and our group independently developed methods that can directly convert unactivated aliphatic C–H bonds to C–F bonds.4 Whereas Lectka's system relies on radical-based chemistry between a copper(I) promoter and an electrophilic fluorination reagent, Selectfluor,5 our method utilizes nucleophilic fluorides from AgF/TBAF as the sole fluorine source, in conjunction with a manganese porphyrin catalyst. Since then, a number of other elegant protocols have been reported that can selectively construct C sp3–F bonds by means of C–H activation.6 Whereas most of these newly developed fluorination methods utilize an electrophilic fluorine source, such as Selectfluor or NFSI, our Mn-based chemistry appears to be the only method that currently employs the fluoride ion, making it particularly suitable for 18F labeling methods.
The transition from 19F to 18F chemistry is always challenging, due largely to the short half-life of 18F, the requirement for short reaction time, the sub-stoichiometric concentrations of 18F fluoride and the unique 18F labeling conditions. As a result, the translation of promising fluorination reactions to radio-fluorination is often problematic. We have recently explored the value of a manganese salen catalyst for direct C–H 18F labeling at benzylic positions,7 which represents the first direct C sp3–H bond 18F labeling with no-carrier-added [18F]fluoride. Despite this success, direct labeling of unactivated aliphatic sites with stronger C–H bonds remained a challenge. In addition, there were no general methods that can incorporate 18F at tertiary carbon positions, since the traditional displacement approach usually leads to significant desaturation side reactions.8 Given these limitations, we sought to develop a more general method that can directly label unactivated aliphatic C–H bonds with [18F]fluoride. Radiolabeled alkyl fluorides are ubiquitous as PET tracers and incorporation of 18F at these positions hold promise to increase the metabolic stability of the target molecules, as the labeling positions are likely the sites of phase I metabolism by cytochrome P450 enzymes. In this context, we report herein a manganese porphyrin mediated 18F labeling strategy that can selectively replace an unactivated aliphatic sp3 hydrogen with an 18F atom. The resulting products can be used for the synthesis of labeled molecules suitable for PET imaging.
Results and discussion
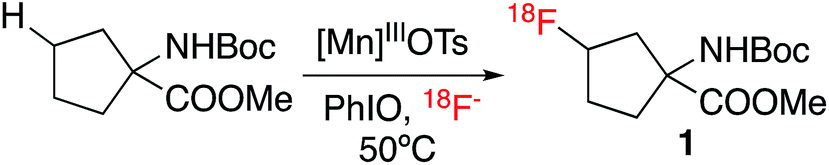
We chose Boc-protected 1-amino-cyclopentane-carboxylic acid methyl ester (Boc-ACPC-OMe) as a lead substrate for direct C–H 18F labeling. Fluorinated derivatives of this unnatural amino acid, 3- and 2-FACPC, target amino acid transporters and have shown uptake into brain tumors, but their preparation requires multi-step syntheses.9 After extensive screening, we found that a manganese(III)pentafluorophenyl porphyrin coordinated with a tosyl ligand, Mn(TPFPP)OTs, was the optimal catalyst. With Mn(TPFPP)OTs in conjunction with iodosylbenzene as the terminal oxidant, Boc-ACPC-OMe could be labeled efficiently with 18F to form 3-fluoro-Boc-ACPC-OMe (1) exclusively. As is illustrated in Table 1, various tosyl coordinated manganese(III)porphyrins and salen complexes were evaluated. Employment of the manganese salen complex, which effects selective benzylic C–H 18F labeling,7 afforded only trace amounts of aliphatic labeling product 1. Apparently, the intermediate O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) MnV (salen) species is either not reactive enough or too fleeting to abstract strong aliphatic C–H bonds effectively. We then turned our attention to different manganese porphyrin complexes, as they are efficient catalysts for aliphatic C–H halogenations.4b,10 Although Mn(TMP)OTs afforded only moderate radiochemical conversions, introducing electron-withdrawing meso-substituents into the porphyrin ring significantly increased the labeling efficiency, with Mn(TPFPP)OTs affording an RCC of 32%. After optimizing the solvent and oxidant loading, we found that protected 3-[18F]-FACPC could be prepared in an acetone/acetonitrile solution with radiochemical conversions up to 48% within only 10 minutes.
MnV (salen) species is either not reactive enough or too fleeting to abstract strong aliphatic C–H bonds effectively. We then turned our attention to different manganese porphyrin complexes, as they are efficient catalysts for aliphatic C–H halogenations.4b,10 Although Mn(TMP)OTs afforded only moderate radiochemical conversions, introducing electron-withdrawing meso-substituents into the porphyrin ring significantly increased the labeling efficiency, with Mn(TPFPP)OTs affording an RCC of 32%. After optimizing the solvent and oxidant loading, we found that protected 3-[18F]-FACPC could be prepared in an acetone/acetonitrile solution with radiochemical conversions up to 48% within only 10 minutes.
|
|
|||
|---|---|---|---|
| Lb | Solvents | PhIO | RCCc % |
| a Reactions were run with ∼200 μCi 18F fluoride. b Salen = Jacobsen's catalyst, TMP = tetramesitylporphyrin, TDClP = tetrakis(2,6-dichlorophenyl)porphyrin, TDFPP = tetrakis(2,6-difluorophenyl)porphyrin, TPFPP = tetrakis(pentafluorophenyl)porphyrin. c The radiochemical conversion (RCC%) is calculated by integration of radio-peaks from TLC and HPLC analysis of reaction mixtures; average of at least three experiments. | |||
| Salen | Acetone | 1.1 eq. | Trace |
| TMP | Acetone | 1.1 eq. | 6 ± 2 |
| TDClP | Acetone | 1.1 eq. | 24 ± 4 |
| TDFPP | Acetone | 1.1 eq. | 31 ± 2 |
| TPFPP | Acetone | 1.1 eq. | 32 ± 5 |
| TPFPP | CH3CN | 1.1 eq. | 36 ± 4 |
| TPFPP | 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH3CN/acetone :1 CH3CN/acetone |
1.1 eq. | 38 ± 2 |
| TPFPP | 2:1 CH3CN/acetone |
1.7 eq. | 48 ± 3 |
Using the optimized conditions, we then investigated the substrate scope of this direct aliphatic 18F labeling reaction. A variety of small molecules bearing multiple unactivated C sp3–H bonds were efficiently labeled with radiochemical conversions ranging from 12% to 67% (Scheme 1). Common functional groups, such as esters, amides and alcohols were well tolerated in this initial assessment. The labeling usually occurs at the least sterically hindered and most electron rich methylene or methine positions. For example, labeling of a series of substituted butyl benzoates (2–6) occurred mainly at the methylene positions remote from the electron-withdrawing benzoate group. In addition, substituted five-membered ring compounds (7–10, 12–14) afforded C3 labeled fluoride as the major products, probably due to a stereoelectronic effect. Notably, labeling of 1-phenylcyclohexanol (15) resulted in 18F incorporation exclusively at the positions cis to the hydroxyl group, suggesting the involvement of a directive effect in the fluorine transfer step, similar to that observed in the analogous Mn–19F fluorination reaction.4b Moreover, this labeling protocol can be used to prepare radiolabeled drug precursors, which can then be converted to the 18F labeled drug analog. For example, [18F]fluorotandospirone (20), which targets the 5-HT neurotransmitter system,11 was prepared via a two-step protocol in an overall 30% RCC. The specific activity of compound 2 was calculated to be 1.24 Ci μmol−1 (end of bombardment), which meets the imaging requirements for fluorine-18 labeling reactions.
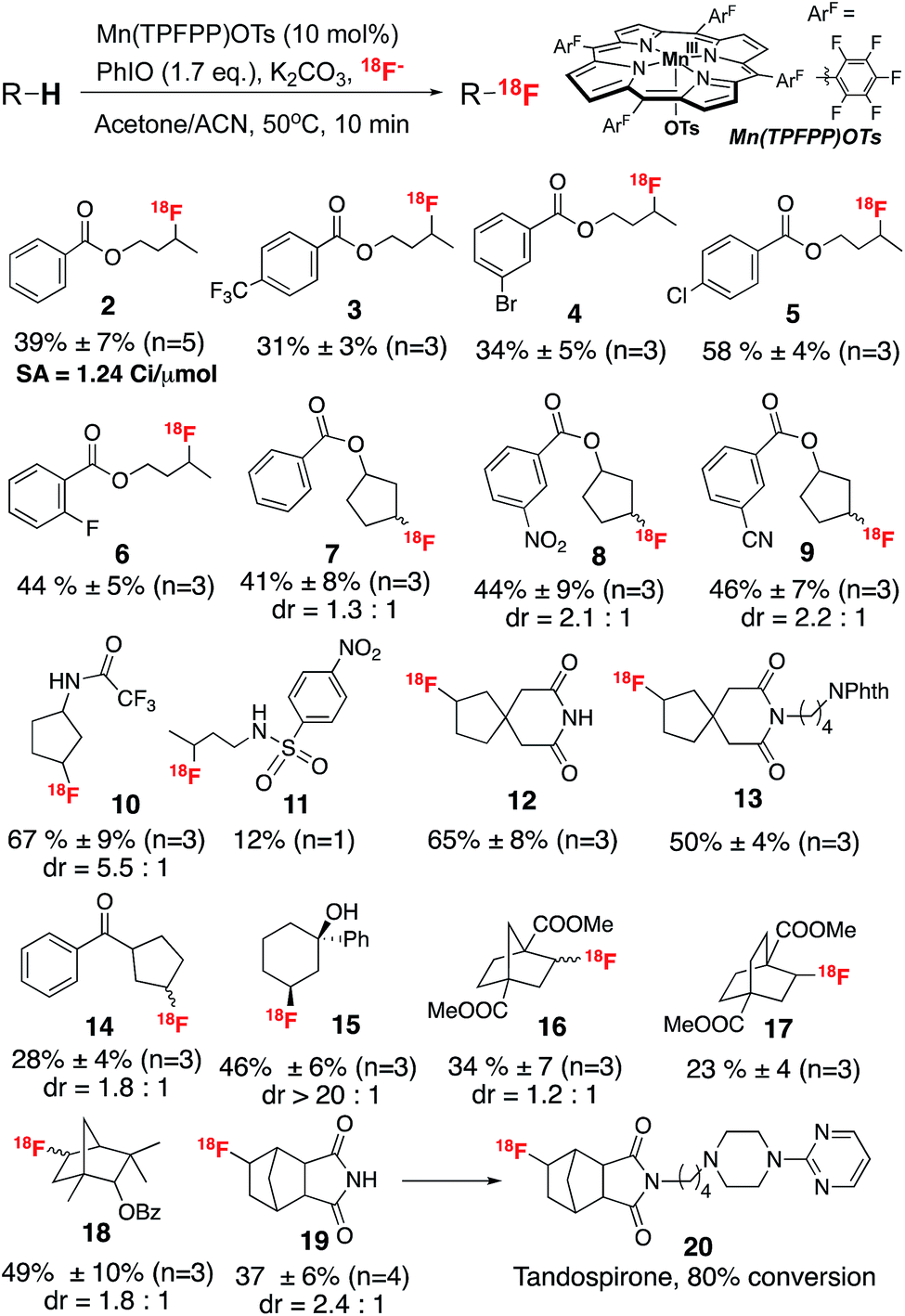 | ||
| Scheme 1 Substrate scope of the Mn-mediated aliphatic C–H 18F fluorination. Reactions were run with ∼400 μCi 18F fluoride. | ||
In order to demonstrate the utility of this method for late-stage radio-fluorination, we applied the reaction conditions to a variety of bioactive molecules (Scheme 2). 18F-labeled natural and unnatural amino acids are an important class of imaging agents for PET that target the increased rates of amino acid transport by many tumor cells.12 We anticipated that a direct fluorination method could significantly accelerate the development of amino acid based PET tracers. Although direct fluorination of leucine and valine have recently been achieved using electrophilic fluorine sources, Selectfluor or NFSI,13 we were surprised to find that 18F labeled leucine or valine analogs have never been reported, apparently due to the synthetic inaccessibility of their corresponding precursors. Gratifyingly, subjection of the protected analogues of these two amino acids to our standard labeling conditions afforded exclusively tertiary carbon-labeled compounds 21 and 22 with RCCs of 29% and 31%, respectively. Tertiary selectively was also observed for the labeling of a leucine containing dipeptide complex, Boc-Val-Leu-OMe, 23. In addition, 18F labeled tyrosine analogs, such as 2-[18F]-fluoro-L-tyrosine (FTYR) and O-(2′-[18F]fluoroethyl)-L-tyrosine (FET), are also promising tracers for brain imaging.14 Under our standard labeling reaction, a cyclopentane carboxylic ester derivative of tyrosine afforded the aliphatic ring labeled product 24 with an RCC of 40%. We anticipate that these compounds could have potential as tracers for amino acid transporter imaging.
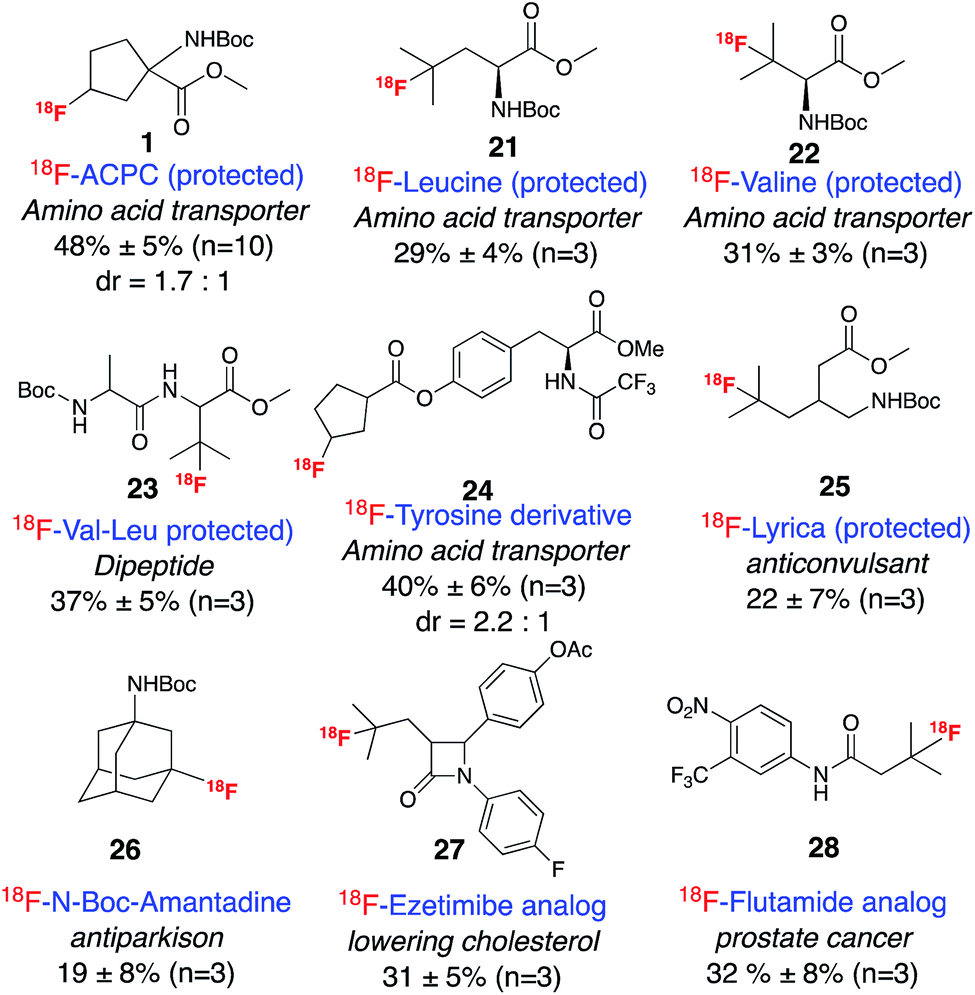 | ||
| Scheme 2 Selective radio-fluorination of bioactive molecules. Reactions were run with ∼400 μCi 18F fluoride. | ||
Another important application of PET imaging is in the area of pharmacokinetics as a noninvasive method for determining drug distribution and action.15 Thus, an efficient method for introducing 18F on a pre-existing drug motif may prove very useful in medicinal chemistry. In this context, several important drug molecules have been subjected to our standard labeling conditions. Radiolabeling of pregabalin, an anticonvulsant drug that has been commercialized by Pfizer under the trade name Lyrica,16 afforded mainly tertiary-labeled product 25. Analogs of flutamide,17 a prostate cancer drug, and ezetimibe, a cholesterol lowering drug, were both labeled efficiently at the tertiary positions, producing 27 and 28. Radio-fluorination of amantadine, an antiviral and anti-Parkinson disease drug, afforded mostly the tertiary product 26 with secondary labeled compounds as minor side products, which were easily separated by semi-preparative HPLC. The separation of these regio-isomers offers the potential for rapid testing of imaging properties of labeled analogs from a single late-stage synthetic product.
Finally, we have successfully translated this 18F-labeling reaction into a reliable and reproducible protocol that can afford radiolabeled molecules on a scale suitable for animal studies. The scale up of the labeling reaction was achieved by using a microwave-assisted “dry-down free” process. In this protocol, [18F]fluoride captured on an anion exchange cartridge was eluted using an acetone/acetonitrile solution of MnIII(TPFPP)OTs with over 90% elution efficiency of the 18F fluoride. Radiolabeling of protected FACPC to afford 3-[18F]-FACPC was accomplished within 10 min using the eluted Mn(TPFPP)18F solution in conjunction with the terminal oxidant, PhIO, with microwave irradiation. Starting with 20 mCi 18F−, we were able to complete the synthesis within 45 min and isolate 11.7 mCi 3-[18F]-FACPC as the final labeling product, with an overall 60% non-decay-corrected radio chemical yield (Scheme 3 and ESI† for details).
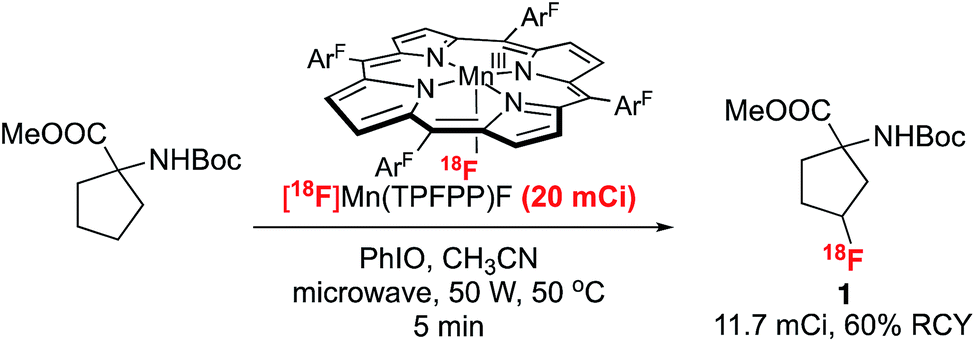 | ||
| Scheme 3 Scale-up synthesis of 3-[18F]-FACPC. | ||
Conclusions
We have developed a direct aliphatic C–H 18F labeling method using non-carrier-added [18F]fluoride that is suitable for inaccessible and unreactive sites. The value of this transformation has been highlighted via the radio-fluorination of biologically active molecules without the need for pre-activation. Our future investigation will focus on developing PET tracers using this methodology and applying them for animal and human studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the US National Science Foundation award CHE-1464578 (J. T. G.) and in part by the Center for Catalytic Hydrocarbon Functionalization, an Energy Frontier Research Center, U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Award No. DE SC0001298 (J. T. G.). Portions of this research were carried out by W. L. and X.H. at the Merck Research Laboratories, Rahway, NJ and the Martinos Center for Biomedical Imaging using resources provided by the Center for Functional Neuroimaging Technologies, P41EB015896, and shared instrumentation grants S10RR017208 and S10RR023452. We also thank Dr Hong Ren for assistance on the radiochemistry.Notes and references
- (a) M. E. Phelps, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9226–9233 CrossRef CAS PubMed; (b) S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS PubMed.
- M. Tredwell and V. Gouverneur, Angew. Chem., Int. Ed., 2012, 51, 11426–11437 CrossRef CAS PubMed.
- (a) T. L. Ross, J. Ermert, C. Hocke and H. H. Coenen, J. Am. Chem. Soc., 2007, 129, 8018–8025 CrossRef CAS PubMed; (b) C. Hollingworth, A. Hazari, M. N. Hopkinson, M. Tredwell, E. Benedetto, M. Huiban, A. D. Gee, J. M. Brown and V. Gouverneur, Angew. Chem., Int. Ed., 2011, 50, 2613–2617 CrossRef CAS PubMed; (c) Z. H. Gao, Y. H. Lim, M. Tredwell, L. Li, S. Verhoog, M. Hopkinson, W. Kaluza, T. L. Collier, J. Passchier, M. Huiban and V. Gouverneur, Angew. Chem., Int. Ed., 2012, 51, 6733–6737 CrossRef CAS PubMed; (d) E. Lee, A. S. Kamlet, D. C. Powers, C. N. Neumann, G. B. Boursalian, T. Furuya, D. C. Choi, J. M. Hooker and T. Ritter, Science, 2011, 334, 639–642 CrossRef CAS PubMed; (e) E. Lee, J. M. Hooker and T. Ritter, J. Am. Chem. Soc., 2012, 134, 17456–17458 CrossRef CAS PubMed; (f) M. Huiban, M. Tredwell, S. Mizuta, Z. H. Wan, X. M. Zhang, T. L. Collier, V. Gouverneur and J. Passchier, Nat. Chem., 2013, 5, 941–944 CrossRef CAS PubMed; (g) T. J. A. Graham, R. F. Lambert, K. Ploessl, H. F. Kung and A. G. Doyle, J. Am. Chem. Soc., 2014, 136, 5291–5294 CrossRef CAS PubMed; (h) S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719–766 CrossRef CAS PubMed; (i) F. Buckingham and V. Gouverneur, Chem. Sci., 2016, 7, 1645–1652 RSC; (j) N. Ichiishi, A. F. Brooks, J. J. Topczewski, M. E. Rodnick, M. S. Sanford and P. J. H. Scott, Org. Lett., 2014, 16, 3224–3227 CrossRef CAS PubMed; (k) S. H. Liang and N. Vasdev, Angew. Chem., Int. Ed., 2014, 53, 11416–11418 CrossRef CAS PubMed; (l) G. Tarantino, L. Botti, N. Dimitratos and C. Hammond, RSC Adv., 2017, 7, 30185–30190 RSC; (m) M. S. McCammant, S. Thompson, A. F. Brooks, S. W. Krska, P. J. H. Scott and M. S. Sanford, Org. Lett., 2017, 19, 3939–3942 CrossRef CAS PubMed; (n) M. D. Levin, T. Q. Chen, M. E. Neubig, C. M. Hong, C. A. Theulier, I. J. Kobylianskii, M. Janabi, J. P. O'Neil and F. D. Toste, Science, 2017, 356, 1272–1275 CrossRef CAS PubMed; (o) M. G. Campbell, J. Mercier, C. Genicot, V. Gouverneur, J. M. Hooker and T. Ritter, Nat. Chem., 2017, 9, 1–3 CrossRef CAS PubMed; (p) M. S. Sanford and P. J. H. Scott, ACS Cent. Sci., 2016, 2, 128–130 CrossRef CAS PubMed; (q) C. N. N. Eumann, J. M. Hooker and T. Ritter, Nature, 2016, 534, 369–373 CrossRef PubMed; (r) M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612–633 CrossRef CAS PubMed.
- (a) S. Bloom, C. R. Pitts, D. C. Miller, N. Haselton, M. G. Holl, E. Urheim and T. Lectka, Angew. Chem., Int. Ed., 2012, 51, 10580–10583 CrossRef CAS PubMed; (b) W. Liu, X. Y. Huang, M. J. Cheng, R. J. Nielsen, W. A. Goddard and J. T. Groves, Science, 2012, 337, 1322–1325 CrossRef CAS PubMed; (c) X. Huang and J. T. Groves, J. Biol. Inorg Chem., 2017, 22, 185–207 CrossRef CAS PubMed; (d) W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727–1735 CrossRef CAS PubMed; (e) C. R. Pitts, D. D. Bume, S. A. Harry, M. A. Siegler and T. Lectka, J. Am. Chem. Soc., 2017, 139, 2208–2211 CrossRef CAS PubMed.
- C. R. Pitts, S. Bloom, R. Woltornist, D. J. Auvenshine, L. R. Ryzhkov, M. A. Siegler and T. Lectka, J. Am. Chem. Soc., 2014, 136, 9780–9791 CrossRef CAS PubMed.
- (a) Y. Amaoka, M. Nagatomo and M. Inoue, Org. Lett., 2013, 15, 2160–2163 CrossRef CAS PubMed; (b) S. Bloom, C. R. Pitts, R. Woltornist, A. Griswold, M. G. Holl and T. Lectka, Org. Lett., 2013, 15, 1722–1724 CrossRef CAS PubMed; (c) S. Bloom, S. A. Sharber, M. G. Holl, J. L. Knippel and T. Lectka, J. Org. Chem., 2013, 78, 11082–11086 CrossRef CAS PubMed; (d) M. G. Braun and A. G. Doyle, J. Am. Chem. Soc., 2013, 135, 12990–12993 CrossRef CAS PubMed; (e) W. Liu and J. T. Groves, Angew. Chem., Int. Ed., 2013, 52, 6024–6027 CrossRef CAS PubMed; (f) W. Liu, X. Y. Huang and J. T. Groves, Nat. Protoc., 2013, 8, 2348–2354 CrossRef CAS PubMed; (g) J. B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494–17500 CrossRef CAS PubMed; (h) S. Bloom, J. L. Knippel and T. Lectka, Chem. Sci., 2014, 5, 1175–1178 RSC; (i) S. Bloom, M. McCann and T. Lectka, Org. Lett., 2014, 16, 6338–6341 CrossRef CAS PubMed; (j) C. W. Kee, K. F. Chin, M. W. Wong and C. H. Tan, Chem. Commun., 2014, 50, 8211–8214 RSC; (k) J. B. Xia, Y. Ma and C. Chen, Org. Chem. Front., 2014, 1, 468–472 RSC; (l) P. Xu, S. Guo, L. Y. Wang and P. P. Tang, Angew. Chem., Int. Ed., 2014, 53, 5955–5958 CrossRef CAS PubMed; (m) J. M. Miao, K. Yang, M. Kurek and H. B. Ge, Org. Lett., 2015, 17, 3738–3741 CrossRef CAS PubMed; (n) Q. Zhang, X. S. Yin, K. Chen, S. Q. Zhang and B. F. Shi, J. Am. Chem. Soc., 2015, 137, 8219–8226 CrossRef CAS PubMed; (o) R. Y. Zhu, K. Tanaka, G. C. Li, J. He, H. Y. Fu, S. H. Li and J. Q. Yu, J. Am. Chem. Soc., 2015, 137, 7067–7070 CrossRef CAS PubMed; (p) X. Y. Huang, W. Liu, J. M. Hooker and J. T. Groves, Angew. Chem., Int. Ed., 2015, 54, 5241–5245 CrossRef CAS PubMed.
- X. Y. Huang, W. Liu, H. Ren, R. Neelamegam, J. M. Hooker and J. T. Groves, J. Am. Chem. Soc., 2014, 136, 6842–6845 CrossRef CAS PubMed.
- T. Nishikata, S. Ishida and R. Fujimoto, Angew. Chem., Int. Ed., 2016, 55, 10008–10012 CrossRef CAS PubMed.
- N. Jarkas, R. J. Voll, L. Williams, V. M. Camp and M. M. Goodman, J. Med. Chem., 2010, 53, 6603–6607 CrossRef CAS PubMed.
- W. Liu and J. T. Groves, J. Am. Chem. Soc., 2010, 132, 12847–12849 CrossRef CAS PubMed.
- S. Endo, CNS Drugs, 1996, 5, 154 CrossRef.
- (a) J. McConathy, L. Martarello, E. J. Malveaux, V. M. Camp, N. E. Simpson, C. P. Simpson, G. D. Bowers, J. J. Olson and M. M. Goodman, J. Med. Chem., 2002, 45, 2240–2249 CrossRef CAS PubMed; (b) N. Galldiks and K. J. Langen, The quarterly journal of nuclear medicine and molecular imaging: official publication of the Italian Association of Nuclear Medicine, 2014 Search PubMed.
- S. D. Halperin, D. Kwon, M. Holmes, E. L. Regalado, L. C. Campeau, D. A. DiRocco and R. Britton, Org. Lett., 2015, 17, 5200–5203 CrossRef CAS PubMed.
- L. M. Wang, W. C. Qu, B. Lieberman, K. Ploessl and H. F. Kung, Bioorg. Med. Chem. Lett., 2010, 20, 3482–3485 CrossRef CAS PubMed.
- A. J. Fischman, N. M. Alpert and R. H. Rubin, Clin. Pharmacokinet., 2002, 41, 581–602 CrossRef CAS PubMed.
- R. H. Dworkin and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2005, 4, 455–456 CrossRef CAS PubMed.
- J. W. Baker, G. L. Bachman, I. Schumach, D. P. Roman and A. L. Tharp, J. Med. Chem., 1967, 10, 93–95 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and spectroscopic data for all new compounds. See DOI: 10.1039/c7sc04545j |
| This journal is © The Royal Society of Chemistry 2018 |