
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium-catalyzed asymmetric hydrogenation of β-cyanocinnamic esters with the assistance of a single hydrogen bond in a precise position†
Xiuxiu
Li‡
a,
Cai
You‡
a,
Yusheng
Yang
a,
Yuhong
Yang
b,
Pan
Li
a,
Guoxian
Gu
b,
Lung Wa
Chung
 b,
Hui
Lv
*ac and
Xumu
Zhang
*ab
b,
Hui
Lv
*ac and
Xumu
Zhang
*ab
aKey Laboratory of Biomedical Polymers of Ministry of Education, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, Hubei 430072, P. R. China. E-mail: huilv@whu.edu.cn
bDepartment of Chemistry, Southern University of Science and Technology, Shenzhen, Guangdong 518055, P. R. China. E-mail: zhangxm@sustc.edu.cn
cEngineering Research Center of Organosilicon Compounds & Materials, Ministry of Education, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, 430072, China
First published on 4th January 2018
Abstract
With the assistance of hydrogen bonds, the first asymmetric hydrogenation of β-cyanocinnamic esters is developed, affording chiral β-cyano esters with excellent enantioselectivities (up to 99% ee). This novel methodology provides an efficient and concise synthetic route to chiral GABA-derivatives such as (S)-Pregabalin, (R)-Phenibut, (R)-Baclofen. Interestingly, in this system, the catalyst with a single H-bond donor performs better than that with double H-bond donors, which is a novel discovery in the metalorganocatalysis area.
Hydrogen bonding, an important noncovalent interaction, plays a crucial role in biosystem and enzyme catalysis. Inspired by enzyme catalysis, the strategy of hydrogen bonding has been elaborated and successfully applied in numerous cases of organocatalysis in the past two decades, which provides the synthetic community with many solutions for organic synthesis.1 Among the successful catalysts, thiourea is a special motif for hydrogen bonding. As a double hydrogen donor, thiourea could activate carbonyl compounds by lowering their LUMO energy.2 Their effective bonding with neutral functional groups, potent binding affinity, and high tunability make thiourea catalysts versatile for many types of organic reactions.3 Herein, using ferrocene–thiourea chiral bisphosphine ligands (ZhaoPhos series) and a combination of transition metal catalysis and organocatalysis, we focus on whether two hydrogen bonds from the thiourea group are essential in the transformation of β-cyanocinnamic esters, which are very challenging substrates. When the asymmetric hydrogenation of β-cyanocinnamic esters was carried out, the catalyst (L1) with only one H-bond donor in a precise position performed better than ZhaoPhos (possesses two H-bond donors) in both reactivity and enantioselectivity. This novel discovery provides new insight for the design of catalysts in the areas of organocatalysis and metalorganocatalysis.
Chiral γ-aminobutyric acids (GABA) are an essential class of compounds in neuroscience,4 and many of them are drugs or have potential biological activities in the area of neurotransmitters and brain science,5 such as Pregabalin,6 Phenibut7 and Baclofen8 (Fig. 1). In addition, many chiral GABA derivatives are common motifs in numerous drugs, such as Rolipram,9 Brivaracetam,10 Vernakalant11 and Enablex12 (Fig. 1). Thus, the synthesis of chiral GABA and their derivatives has attracted a great deal of interest; however, very limited approaches have been developed. The methods for their synthesis include the following: enzymatic kinetic resolution,13 biocatalytic asymmetric reduction,14 asymmetric conjugate reduction with polymethylhydrosiloxane (PMHS)15 and asymmetric Michael addition.16 However, asymmetric hydrogenation of β-cyanocinnamic esters, the most straightforward access to synthesize chiral GABA derivatives, has never been reported.
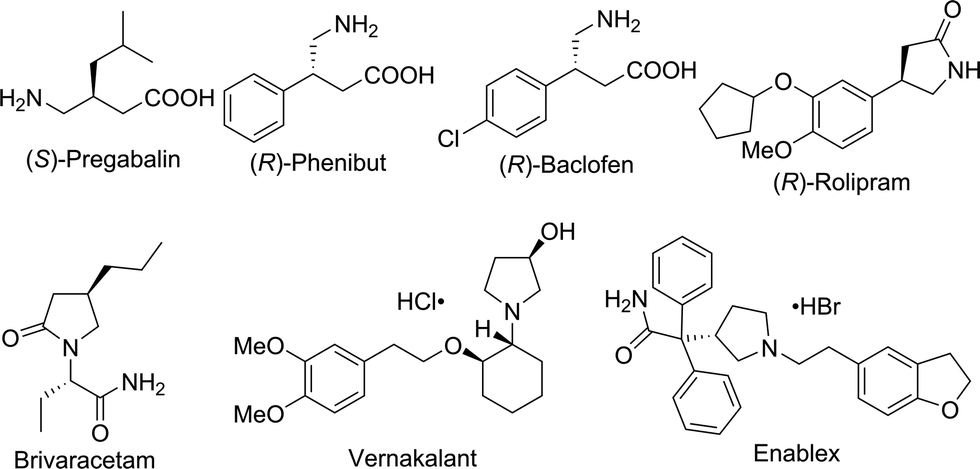 | ||
| Fig. 1 Chiral GABA derivatives in pharmaceuticals. | ||
Over the past decades, the transition metal catalyzed asymmetric hydrogenation of functionalized olefins has emerged as a powerful and environmentally friendly approach to generate chiral compounds.17 With the rapid development of catalytic systems, many types of olefins bearing functional groups such as carbonyl,18 sulfonyl,19 cyano group20 and nitryl,21 were hydrogenated in high ee and activities. However, the asymmetric hydrogenation of β-cyanocinnamic esters has not been reported, which is due to two reasons: (1) the electron-withdrawing ester group and nitrile group, which can sharply reduce the electron density of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, are disadvantageous for the CC bond coordination to the metal center, which leads to the low reactivity of these substrates and (2) the linearity of the nitrile group, which is not suitable for assisting the coordination between the CC bond and metal catalyst, makes it very difficult to achieve high enantioselectivities.20a,b As a result, it is a very difficult task to achieve satisfactory results from the asymmetric hydrogenation of β-cyanocinnamic esters with the classical bidentate diphosphine/Rh system, as summarized in Table 1.
C bond, are disadvantageous for the CC bond coordination to the metal center, which leads to the low reactivity of these substrates and (2) the linearity of the nitrile group, which is not suitable for assisting the coordination between the CC bond and metal catalyst, makes it very difficult to achieve high enantioselectivities.20a,b As a result, it is a very difficult task to achieve satisfactory results from the asymmetric hydrogenation of β-cyanocinnamic esters with the classical bidentate diphosphine/Rh system, as summarized in Table 1.
|
|
|||
|---|---|---|---|
| Entry | Ligand | Conv.b [%] | eec [%] |
a All reactions were carried out with an [Rh(NBD)2]BF4/ligand/substrate ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1:100, in 1 mL of methanol, at 20 °C, under hydrogen (30 atm) for 18 h.
b Determined by 1H NMR spectroscopy.
c Determined by HPLC analysis using a chiral stationary phase. The absolute configuration was assigned by comparing the sign of the optical rotation of the product 2a, methyl (R)-3-cyano-3-phenylpropanoate, with that reported in the literature; see ref. 14c. :1.1:100, in 1 mL of methanol, at 20 °C, under hydrogen (30 atm) for 18 h.
b Determined by 1H NMR spectroscopy.
c Determined by HPLC analysis using a chiral stationary phase. The absolute configuration was assigned by comparing the sign of the optical rotation of the product 2a, methyl (R)-3-cyano-3-phenylpropanoate, with that reported in the literature; see ref. 14c.
|
|||
| 1 | (Rc,Sp)-DuanPhos | 19 | rac |
| 2 | (S)-Binapine | Trace | — |
| 3 | (S,S)–Me-DuPhos | Trace | — |
| 4 | (R,R)-QuinoxP* | 23 | rac |
| 5 | (S)-BINAP | Trace | — |
| 6 | (S)-SegPhos | Trace | — |
| 7 | (S)-DTBM-SegPhos | Trace | — |
| 8 | (R,S)-tBu-JosiPhos | 25 | rac |
| 9 | (R)-TaniaPhos | 35 | −7 |
| 10 | (R)-WalPhos | Trace | — |
| 11 | ZhaoPhos | 28 | 84 |
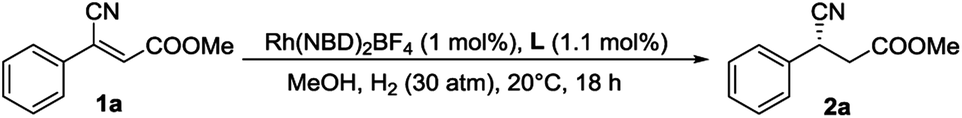
Methyl (Z)-3-cyano-3-phenylacrylate as a model substrate, a series of bidentate diphosphine ligands including P-chiral diphosphine ligands (entries 1–4), ligands having axial chirality (entries 5–7) and ferrocenyl chiral bisphosphorus ligands (entries 8–10) were tested, which gave low to even trace conversions and almost racemic products. This disclosed that the classical catalyst systems cannot complete this task. Recently, substrate orientation by hydrogen bonds as a strategy in asymmetric hydrogenation has been studied and some progress has been made.22 Therefore, we tried to utilize the ester group whose carbonyl is a very good H-bond acceptor to improve the reactivity of the electron-deficient substrate by employing ZhaoPhos-bearing H-bond donors. Although the preliminary result was moderate (entry 11, 28% conversion with 84% ee), it inspired us for further exploration.
In order to obtain the optimal reaction conditions, a serious of solvents were screened (summarized in the ESI†). When the reaction was carried out with 50 atm H2 gas at 35 °C in CF3CH2OH, full conversion was attained with 95% ee.
To obtain insight into this catalytic system, three other chiral ligands L1–L3 were employed and the results are summarized in Table 2. To our surprise, the N methylation of ZhaoPhos led to an increase in enantioselectivity (entry 2, 98% ee), which is quite different from the previous results we reported.18f,23 In comparison, when both N–H groups were replaced, almost no conversion was detected (entry 3), which indicated the importance of hydrogen bonds in this reaction. In addition, the ligand L3 without the thiourea group showed very low activity and enantioselectivity (entry 4). These results show that the thiourea motif efficiently activates the carbonyl group through hydrogen-bonding interaction and works as an excellent directing agent. Moreover, L1 is superior to ZhaoPhos, which indicates that one hydrogen bond in a precise position is better than two in this asymmetric transformation.
|
|
|||
|---|---|---|---|
| Entry | Ligand | Conv.b [%] | eec [%] |
|
a All reactions were carried out with an [Rh(NBD)2]BF4/ligand/substrate ratio of 1:1.1:100, in 1 mL of trifluoroethanol, at 35 °C, under hydrogen (50 atm) for 18 h.
b Determined by 1H NMR spectroscopy.
c Determined by HPLC analysis using a chiral stationary phase.
|
|||
| 1 | ZhaoPhos | 99 | 95 |
| 2 | L1 | 99 | 98 |
| 3 | L2 | Trace | — |
| 4 | L3 | 5 | 7 |
With an optimized set of conditions in hand, we explored the substrate scope and generality of this catalytic reaction. To confirm the universality of the phenomenon that L1 is superior to ZhaoPhos, almost each substrate was compared (Scheme 1). We thought that enhancement of the steric hindrance of the ester would influence this transformation; however, the results indicate that the changes in the substrate have a slight impact on the reaction (2a–2c). Many functional groups, such as methyl (2d), methoxy (2e), phenyl (2f), trifluoromethyl (2g), and halides (2h, 2i and 2j), at the para position of the phenyl group are compatible with this transformation. Using L1, substrates with meta- or ortho-substitution on the phenyl group are also tolerated and 97% ee and 99% ee values were obtained (2k and 2l, respectively). Generally, polyfluorinated compounds exist in drug molecules,24 thus a series of polyfluorinated substrates were examined. Substrates with 3,4-difluoro and 3,5-difluoro on the phenyl group were tested, and good yields with excellent enantioselectivities were achieved (2m and 2n, respectively). In comparison, when the 3,4,5-trifluoro compound 1o was employed, the adduct 2o was produced with slightly compromised ee values, which may be due to the electron-poor property of 1o, as discussed above. Other disubstituted substrates could also be accommodated, as exemplified by 2p and 2q. Moreover, good yields and excellent enantioselectivities were obtained with substrates containing other aromatic fragments, including naphthalenes and thiophenes (2r and 2s, respectively). To our delight, when the aryl substituent was changed to an alkyl group, such as isobutyl, the product 2t, which can be readily converted to (S)-Pregabalin, was produced with 98% ee. All the results reflect the fact that L1 bearing a single H-bond donor is more suitable than ZhaoPhos for this reaction. Moreover, when (E)-1a was employed, much lower reactivity and enantioselectivity were obtained, but the configuration of 2u supports our assumption that the enantioselectivity is inducted by the ester group of the substrates.
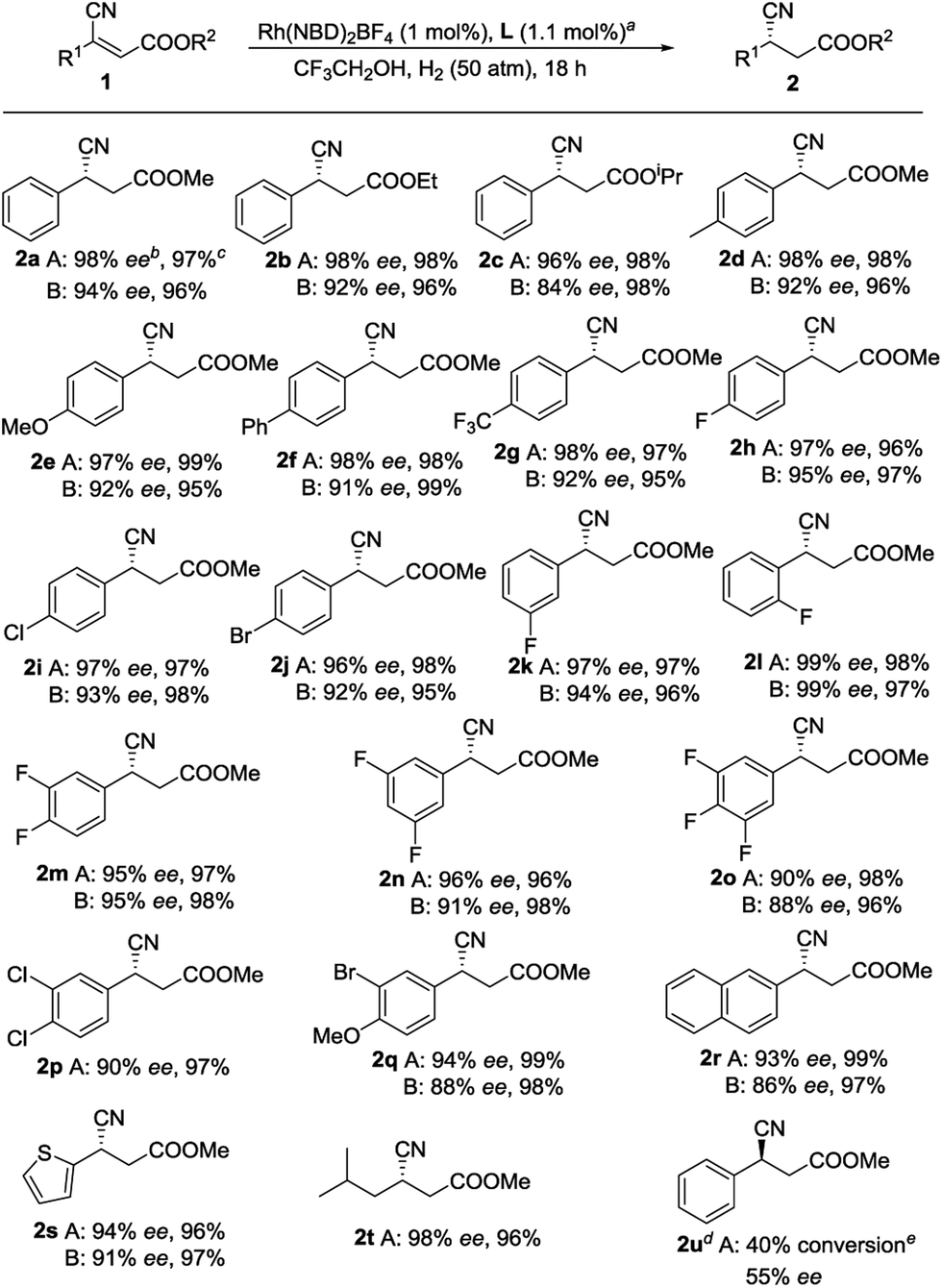 | ||
| Scheme 1 Substrate scope. Condition A: L = L1, 20 °C. Condition B: L = ZhaoPhos, 35 °C. aAll reactions were carried out with an [Rh(NBD)2]BF4/L/substrate ratio of 1:1.1:100, in 1 mL of trifluoroethanol, under hydrogen (50 atm) for 18 h. bDetermined by HPLC analysis using a chiral stationary phase. cIsolated yield. d(E)-1a was used. eDetermined by 1H NMR spectroscopy. | ||
To gain a better understanding of the performance of ZhaoPhos and L1, the free energies (kcal mol−1) of the hydrogen bonds between different ligands with 2a (ΔGHB) were evaluated using the B3LYP-GD3BJ/6-31G** method,25 as summarized in Table 3. To our surprise, the free energy of the hydrogen bond between 1a and ZhaoPhos is smaller than that between 1a and L1, even at different temperature, which can explain the experimental results very well. These results indicate that one H-bond can perform better than two H-bonds in this reaction, thus we proposed that the stronger hydrogen bond is important in making L1 a more selective catalyst. Both the L1 and ZhaoPhos systems are sensitive to the reaction temperature due to their weaker hydrogen-bonding interaction with substrates at higher temperature.
|
|
|||||
|---|---|---|---|---|---|
| Ligand | T (°C) | ||||
| 0 | 20 | 35 | 60 | ||
|
a All reactions were carried out with an [Rh(NBD)2]BF4/L/substrate ratio of 1:1.1:100, in 1 mL of trifluoroethanol, under hydrogen (50 atm) for 18 h.
b ee values were determined by HPLC analysis using a chiral stationary phase.
c Conversions were determined by 1H NMR spectroscopy.
|
|||||
| ZhaoPhos | ΔGHB (kcal mol−1) | −13.4 | −12.2 | −11.4 | −10.0 |
| ee | 97b (20c) | 95 (94) | 95 (99) | 92 (99) | |
| L1 | ΔGHB (kcal mol−1) | −16.4 | −15.3 | −14.5 | −13.2 |
| ee | 99 (60) | 98 (99) | 98 (99) | 93 (99) | |
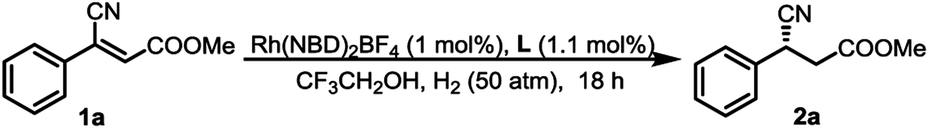
To verify the accuracy of the catalytic model, the reduction of 1a was performed using L1 of varying ee. As shown in Fig. 2a, a linear correlation between the ee of the ligand and that of the product was observed, which indicates that a 1:1 ratio of ligand to metal is present in the catalytic complex.26 Moreover, a Job plot was drawn and the curve suggests a 1:1 binding pattern between L1 and 1a (Fig. 2b), and a 1:1 binding pattern between ZhaoPhos and 1a was also suggested (summarized in the ESI†). These experimental data validate our hypothesis, and the catalytic model was illustrated, as shown in Fig. 2c.
 | ||
| Fig. 2 (a) Hydrogenation of 1a using L1 of varying ee, the line corresponds to the least-squares linear regression of the data with slope = 0.95, intercept = 1.77, and r2 = 0.99; (b) Job plot, x = c(L1)/[c(L1) + c(1a)], y = Δδ × 104 × c(L1)/[c(L1) + c(1a)] and (c) proposed hydrogen bonding between the reactive Rh species and 1a. | ||
In order to further demonstrate the synthetic utility of this methodology, several gram-scale transformations were achieved, as summarized in Scheme 2. First, upon decreasing the catalyst loading to 0.05 mol%, the asymmetric reaction was conducted on a gram scale, and 97% conversion (TON = 1940) with unchanged enantioselectivity was obtained (Scheme 2a). Then, a high-efficiency synthetic route for (S)-Pregabalin,13b (R)-Phenibut and (R)-Baclofen14a was developed. As shown in Scheme 2b, with a 0.1–0.125 mol% catalyst loading, three key intermediates were obtained on a gram scale with high yields (97–98% yield) and excellent ee values (97–98% ee). Thus far, this may be the most concise way to synthesize chiral Pregabalin, Phenibut and Baclofen. Next, gram-scale 2p, which can be readily converted to a chiral δ-amino alcohol whose enantiomer can be used in the synthesis of NK1 antagonists directly,27 was produced (Scheme 2c). Scheme 2d exhibits an efficient approach for the establishment of chiral pyrrolidines. The asymmetric hydrogenation product 2a was reduced by NiCl2/NaBH4 in MeOH and afforded the corresponding lactam 3 in high yield without any loss in enantioselectivity. By treating the lactam with lithium aluminium hydride (LAH) in THF, the chiral pyrrolidine can be readily obtained.28
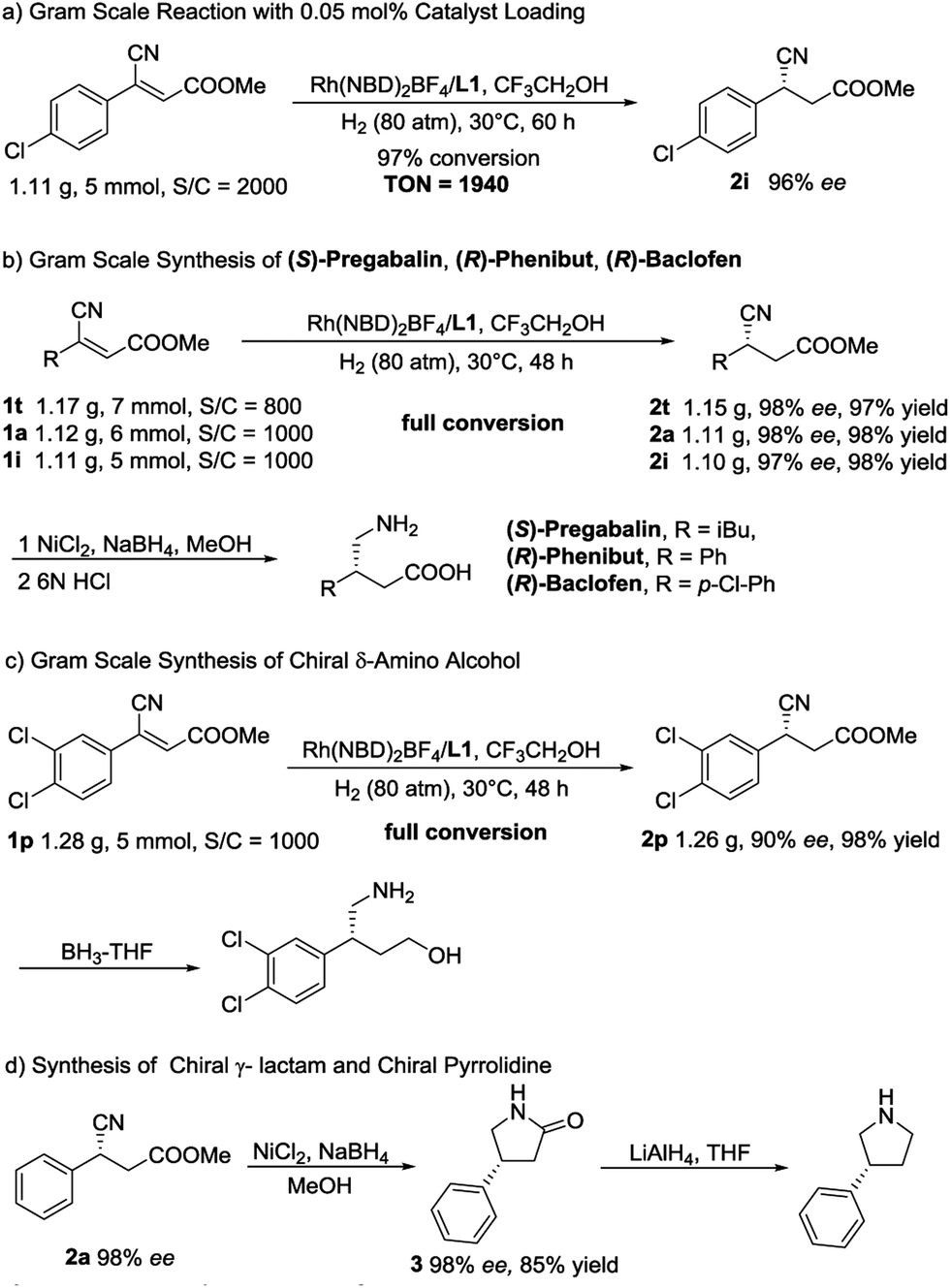 | ||
| Scheme 2 Synthetic transformations. | ||
Conclusions
In conclusion, the first asymmetric hydrogenation of β-cyanocinnamic esters has been reported, which provides an efficient approach for the synthesis of chiral GABA derivatives. This transformation exhibits excellent enantioselectivities (up to 99% ee) under mild reaction conditions with low catalyst loadings. Furthermore, this method provides a concise route for the synthesis of (S)-Pregabalin, (R)-Phenibut, (R)-Baclofen, chiral δ-amino alcohols, chiral γ-lactam and chiral pyrrolidines, which demonstrates the high synthetic utility of the current methodology. Notably, in our ferrocene–thiourea chiral bisphosphine ligand (ZhaoPhos series) system, the catalyst with a single H-bond donor in a precise position performed better than that with double H-bond donors, which is a novel discovery in the metalorganocatalysis area. This novel discovery provides a new way to design catalysts. Further investigations on the detailed mechanisms of the proposed key hydrogen bonds and the applications of the asymmetric hydrogenation strategy in organic synthesis are in progress in our lab.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (Grant Nos. 21402145, 21432007, and 21372179), the Youth Chen-Guang Science and Technology Project of Wuhan City (2015071704011640), the Natural Science Foundation of Hubei Province (2014CFB181), the Fundamental Research Funds for Central Universities (2042017kf0177), the Important Sci-Tech Innovative Project of Hubei Province (2015ACA058) and the “111” Project of the Ministry of Education of China.Notes and references
- (a) P. M. Pihko, Hydrogen Bonding in Organic Synthesis, Wiley, 2009 Search PubMed; (b) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS PubMed; (c) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520 CrossRef CAS PubMed; (d) P. Dydio and J. N. H. Reek, Chem. Sci., 2014, 5, 2135 RSC; (e) H. J. Davis and R. J. Phipps, Chem. Sci., 2017, 8, 864 RSC.
- Recent studies on thiourea in electrophile activation: (a) P. R. Schreiner and A. Wittkopp, Org. Lett., 2002, 4, 217 CrossRef CAS PubMed; (b) P. M. Pihko, Angew. Chem., Int. Ed., 2004, 43, 2062 CrossRef CAS PubMed; (c) P. N. H. Huynh, R. R. Walvoord and M. C. Kozlowski, J. Am. Chem. Soc., 2012, 134, 15621 CrossRef CAS PubMed; (d) R. R. Walvoord, P. N. H. Huynh and M. C. Kozlowski, J. Am. Chem. Soc., 2014, 136, 16055 CrossRef CAS PubMed.
- Reviews for recent advances on thiourea small-molecule catalysis: (a) S. J. Connon, Chem.–Eur. J., 2006, 12, 5418 CrossRef PubMed; (b) S. J. Connon, Chem. Commun., 2008, 2499 RSC; (c) X. Fang and C. J. Wang, Chem. Commun., 2015, 51, 1185 RSC.
- R. L. Macdonald and R. W. Olsen, Annu. Rev. Neurosci., 1994, 17, 569 CrossRef CAS PubMed.
- A. G. Malykh and M. R. Sadaie, Drugs, 2010, 70, 287 CrossRef CAS PubMed.
- R. B. Silverman, Angew. Chem., Int. Ed., 2008, 47, 3500 CrossRef CAS PubMed.
- I. Lapin, CNS Drug Rev., 2001, 7, 471 CrossRef CAS PubMed.
- B. A. Sachais, J. N. Logue and M. S. Carey, Arch. Neurol., 1977, 34, 422 CrossRef CAS PubMed.
- J. Zhu, E. Mix and B. Winblad, CNS Drug Rev., 2001, 7, 387 CrossRef CAS PubMed.
- B. M. Kenda, A. C. Matagne, P. E. Talaga, P. M. Pasau, E. Differding, B. I. Lallemand, A. M. Frycia, F. G. Moureau, H. V. Klitgaard, M. R. Gillard, B. Fuks and P. Michel, J. Med. Chem., 2004, 47, 530 CrossRef CAS PubMed.
- D. R. Boyd, N. D. Sharma, H. Dalton and D. A. Clarke, Chem. Commun., 1996, 45 RSC.
- P. E. Cross and A. R. MacKenzie, US Pat., 005096890 A, 1992.
- (a) H. Mukherjee and C. A. Martinez, ACS Catal., 2011, 1, 1010 CrossRef CAS; (b) C. A. Martinez, S. Hu, Y. Dumond, J. Tao, P. Kelleher and L. Tully, Org. Process Res. Dev., 2008, 12, 392 CrossRef CAS.
- (a) A. Fryszkowska, K. Fisher, J. M. Gardiner and G. M. Stephens, Org. Biomol. Chem., 2010, 8, 533 RSC; (b) C. K. Winkler, D. Clay, S. Davies, P. O'Neill, P. McDaid, S. Debarge, J. Steflik, M. Karmilowicz, J. W. Wong and K. Faber, J. Org. Chem., 2013, 78, 1525 CrossRef CAS PubMed; (c) E. Brenna, M. Crotti, F. G. Gatti, D. Monti, F. Parmeggiani, R. W. Powell III, S. Santangelo and J. D. Stewartc, Adv. Synth. Catal., 2015, 357, 1849 CrossRef CAS.
- Y. Hirata, A. Yada, E. Morita, Y. Nakao, T. Hiyama, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2010, 132, 10070 CrossRef CAS PubMed.
- (a) P. García-García, A. Ladépêche, R. Halder and B. List, Angew. Chem., Int. Ed., 2008, 47, 4719 CrossRef PubMed; (b) A. Leyva-Pérez, P. García-García and A. Corma, Angew. Chem., Int. Ed., 2014, 53, 8687 CrossRef PubMed; (c) T. Tsubogo, Y. Yamashita and S. Kobayashi, Angew. Chem., Int. Ed., 2009, 48, 9117 CrossRef CAS PubMed; (d) T. Tsubogo, H. Oyamada and S. Kobayashi, Nature, 2015, 520, 329 CrossRef CAS PubMed; (e) S. H. McCooey and S. J. Connon, Angew. Chem., Int. Ed., 2005, 44, 6367 CrossRef CAS PubMed; (f) K. Akagawa and K. Kudo, Angew. Chem., Int. Ed., 2012, 51, 12786 CrossRef CAS PubMed; (g) J. H. Sim and C. E. Song, Angew. Chem., Int. Ed., 2017, 56, 1835 CrossRef CAS PubMed; (h) O. V. Maltsev, A. S. Kucherenko, I. P. Beletskaya, V. A. Tartakovsky and S. G. Zlotin, Eur. J. Org. Chem., 2010, 2927 CrossRef CAS; (i) K. G. Lewis, S. K. Ghosh, N. Bhuvanesh and J. A. Gladysz, ACS Cent. Sci., 2015, 1, 50 CrossRef CAS PubMed; (j) D. A. Evans, S. Mito and D. Seidel, J. Am. Chem. Soc., 2007, 129, 11583 CrossRef CAS PubMed.
- For reviews, see: (a) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed; (b) J. H. Xie, S. F. Zhu and Q. L. Zhou, Chem. Rev., 2011, 111, 1713 CrossRef CAS PubMed; (c) J. H. Xie and Q. L. Zhou, Acta Chim. Sin., 2012, 70, 1427 CrossRef CAS; (d) S. F. Shou and Q. L. Zhou, Acc. Chem. Res., 2017, 50, 988 CrossRef PubMed; (e) Z. Zhang, N. A. Butt and W. Zhang, Chem. Rev., 2016, 116, 14769 CrossRef CAS PubMed; (f) D.-S. Wang, Q.-A. Chen, S.-M. Lu and Y.-G. Zhou, Chem. Rev., 2012, 112, 2557 CrossRef CAS PubMed; (g) S. J. Roseblade and A. Pfaltz, Acc. Chem. Res., 2007, 40, 1402 CrossRef CAS PubMed; (h) G. Erre, S. Enthaler, K. Junge, S. Gladiali and M. Beller, Coord. Chem. Rev., 2008, 252, 471 CrossRef CAS; (i) T. L. Church and P. G. Andersson, Coord. Chem. Rev., 2008, 252, 513 CrossRef CAS; (j) J. J. Verendel, O. Pàmies, M. Diéguez and P. G. Andersson, Chem. Rev., 2014, 114, 2130 CrossRef CAS PubMed.
- (a) M. Diéguez, J. Mazuela, O. Pàmies, J. J. Verendel and P. G. Andersson, J. Am. Chem. Soc., 2008, 130, 7208 CrossRef PubMed; (b) J. Mazuela, P.-O. Norrby, P. G. Andersson, O. Pàmies and M. Diéguez, J. Am. Chem. Soc., 2011, 133, 13634 CrossRef CAS PubMed; (c) M. Shevlin, M. R. Friedfeld, H. Sheng, N. A. Pierson, J. M. Hoyt, L.-C. Campeau and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 3562 CrossRef CAS PubMed; (d) W. Tang, W. Wang and X. Zhang, Angew. Chem., Int. Ed., 2003, 42, 943 CrossRef CAS PubMed; (e) D. Rageot, D. H. Woodmansee, B. Pugin and A. Pfaltz, Angew. Chem., Int. Ed., 2011, 50, 9598 CrossRef CAS PubMed; (f) Z. Han, P. Li, Z. Zhang, C. Chen, Q. Wang, X.-Q. Dong and X. Zhang, ACS Catal., 2016, 6, 6214 CrossRef CAS; (g) P. Li, X. Hu, X.-Q. Dong and X. Zhang, Chem. Commun., 2016, 52, 11677 RSC; (h) J. Wen, J. Jiang and X. Zhang, Org. Lett., 2016, 18, 4451 CrossRef CAS PubMed; (i) Y. Liu and W. Zhang, Angew. Chem., Int. Ed., 2013, 52, 2203 CrossRef CAS PubMed; (j) Y. Liu, I. D. Gridnev and W. Zhang, Angew. Chem., Int. Ed., 2014, 53, 1901 CrossRef CAS PubMed; (k) X. Liu, Z. Han, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2014, 53, 1978 CrossRef CAS PubMed; (l) J. Shang, Z. Han, Y. Li, Z. Wang and K. Ding, Chem. Commun., 2012, 48, 5172 RSC.
- (a) T. Zhou, B. Peters, M. F. Maldonado, T. Govender and P. G. Andersson, J. Am. Chem. Soc., 2012, 134, 13592 CrossRef CAS PubMed; (b) B. K. Peters, T. Zhou, J. Rujirawanich, A. Cadu, T. Singh, W. Rabten, S. Kerdphon and P. G. Andersson, J. Am. Chem. Soc., 2014, 136, 16557 CrossRef CAS PubMed.
- (a) M.-A. Müller and A. Pfaltz, Angew. Chem., Int. Ed., 2014, 53, 8668 CrossRef PubMed; (b) Q. Yan, D. Kong, M. Li, G. Hou and G. Zi, J. Am. Chem. Soc., 2015, 137, 10177 CrossRef CAS PubMed; (c) X. Li, C. You, Y. Yang, F. Wang, S. Li, H. Lv and X. Zhang, Chem. Commun., 2017, 53, 1313 RSC.
- (a) S. Li, K. Huang, B. Cao, J. Zhang, W. Wu and X. Zhang, Angew. Chem., Int. Ed., 2012, 51, 8573 CrossRef CAS PubMed; (b) Q. Zhao, S. Li, K. Huang, R. Wang and X. Zhang, Org. Lett., 2013, 15, 4014 CrossRef CAS PubMed.
- (a) P.-A. R. Breuil, F. W. Patureau and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 2162 CrossRef CAS PubMed; (b) J. Daubignard, R. J. Detz, A. C. H. Jans, B. Bruin and J. N. H. Reek, Angew. Chem., Int. Ed., 2017, 56, 13056 CrossRef CAS PubMed; (c) P. Dydio, C. Rubay, T. Gadzikwa, M. Lutz and J. N. H. Reek, J. Am. Chem. Soc., 2011, 133, 17176 CrossRef CAS PubMed; (d) L. Pignataro, M. Boghi, M. Civera, S. Carboni, U. Piarulli and C. Gennari, Chem.– Eur. J., 2012, 18, 1383 CrossRef CAS PubMed.
- (a) Q. Zhao, J. Wen, R. Tan, K. Huang, P. Metola, R. Wang, E. V. Anslyn and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 8467 CrossRef CAS PubMed; (b) J. Wen, R. Tan, S. Liu, Q. Zhao and X. Zhang, Chem. Sci., 2016, 7, 3047 RSC.
- (a) C. Warnier, C. Lemaire, G. Becker, G. Zaragoza, F. Giacomelli, J. Aerts, M. Otabashi, M. A. Bahri, J. Mercier, A. Plenevaux and A. Luxen, J. Med. Chem., 2016, 59, 8955 CrossRef CAS PubMed; (b) S. J. Finnema, N. B. Nabulsi, T. Eid, K. Detyniecki, S.-f. Lin, M.-K. Chen, R. Dhaher, D. Matuskey, E. Baum, D. Holden, D. D. Spencer, J. Mercier, J. Hannestad, Y. Huang and R. E. Carson, Sci. Transl. Med., 2016, 8, 348ra96 CrossRef PubMed.
- See computational details in ESI.†.
- C. Girard and H. B. Kagan, Angew. Chem., Int. Ed., 1998, 37, 2922 CrossRef.
- C. J. Ohnmacht, J. S. Albert, P. R. Bernstein, W. L. Rumsey, B. B. Masek, B. T. Dembofsky, G. M. Koether, D. W. Andisik and D. Aharony, Bioorg. Med. Chem., 2004, 12, 2653 CrossRef CAS PubMed.
- X. Bantreil, G. Prestat, A. Moreno, D. Madec, P. Fristrup, P.-O. Norrby, P. S. Pregosin and G. Poli, Chem.– Eur. J., 2011, 17, 2885 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra of compounds. See DOI: 10.1039/c7sc04639a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |