
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamics of singlet fission and electron injection in self-assembled acene monolayers on titanium dioxide†
Natalie A.
Pace‡
 ab,
Dylan H.
Arias‡
a,
Devin B.
Granger
c,
Steven
Christensen
a,
John E.
Anthony
c and
Justin C.
Johnson
*a
ab,
Dylan H.
Arias‡
a,
Devin B.
Granger
c,
Steven
Christensen
a,
John E.
Anthony
c and
Justin C.
Johnson
*a
aNational Renewable Energy Laboratory, Golden, CO 80401, USA. E-mail: justin.johnson@nrel.gov
bDepartment of Chemistry and Biochemistry, University of Colorado, Boulder, CO 80309, USA
cDepartment of Chemistry, University of Kentucky, Lexington, KY 40506, USA
First published on 26th February 2018
Abstract
We employ a combination of linear spectroscopy, electrochemistry, and transient absorption spectroscopy to characterize the interplay between electron transfer and singlet fission dynamics in polyacene-based dyes attached to nanostructured TiO2. For triisopropyl silylethynyl (TIPS)-pentacene, we find that the singlet fission time constant increases to 6.5 ps on a nanostructured TiO2 surface relative to a thin film time constant of 150 fs, and that triplets do not dissociate after they are formed. In contrast, TIPS-tetracene singlets quickly dissociate in 2 ps at the molecule/TiO2 interface, and this dissociation outcompetes the relatively slow singlet fission process. The addition of an alumina layer slows down electron injection, allowing the formation of triplets from singlet fission in 40 ps. However, the triplets do not inject electrons, which is likely due to a lack of sufficient driving force for triplet dissociation. These results point to the critical balance required between efficient singlet fission and appropriate energetics for interfacial charge transfer.
Introduction
Solar energy conversion devices have the opportunity to surpass fundamental thermodynamic limits for Shockley–Queisser power conversion efficiency through multi-exciton generation (MEG), which generates multiple charges per incident photon.1,2 MEG has pushed external quantum efficiencies past 100% in solar fuel generation,3 quantum dot photovoltaics,4,5 and organic semiconductors.6,7 Singlet fission (SF) in particular holds great promise due to the molecular nature of the chromophore, offering potential for control of the SF process and tuning of electronic interactions. While much effort and progress has been made determining the mechanism of SF,8–22 devices typically feature low overall efficiencies.23–27 For devices based on bilayers or blends, the competitive interplay between SF, triplet transport, and exciton dissociation can be difficult to control because each depends crucially on the details of intermolecular coupling.28 For example, in tetracene polycrystalline films, exciton transport and SF are highly coupled, and both require specific molecular orientations for optimization that may not be compatible.29–31 These limitations are compounded by competition with singlet energy or charge transfer, and may be responsible for low triplet transfer efficiencies from tetracene to silicon.32 Two recent works have demonstrated triplet energy transfer from acenes to semiconductor nanocrystals.33,34 However, these studies utilized polycrystalline material and their eventual utility in device architectures may be limited by the same effects previously discussed. Recent work on covalent dimers35–40 and polymers41,42 offers a pathway to potentially overcome some limitations of morphology-dependent SF, but many of these systems have their own drawbacks, e.g. limited triplet lifetimes.Triplet diffusion issues can be eliminated in systems where SF chromophores are arranged in a monolayer on an electron acceptor surface. Wide band gap semiconducting electrodes can be used for this purpose, and although they are nanostructured in order to be practical, they allow for fundamental study of SF and charge injection. The commonly studied dye-sensitized solar cell (DSSC) features self-assembled chromophores covalently linked to transition metal oxide surfaces. DSSCs traditionally utilize ruthenium-based chromophores that benefit from significant triplet injection from intersystem crossing.43,44 Therefore, these systems are promising for triplet injection from singlet fission. A series of pentacene derivatives linked to titanium dioxide (TiO2) has been studied via DSSC device performance.45 Working devices were fabricated using this approach, but at best only 5% incident photons were converted to current. Weak overlap between relevant orbitals of the dyes and the TiO2 conduction band was blamed for the poor performance. No consideration was made for triplet excitons formed from SF. A later study utilizing 1,3-diphenylisobenzofuran (DPIBF) and TiO2 focused on a fundamental examination of the interplay between SF and charge injection dynamics.46 A small increase in photocurrent was observed upon reducing the electron injection rate with a zirconia barrier, which allowed SF to produce multiple triplet excitons that could dissociate. However, the intermolecular geometries appropriate for SF with DPIBF are restrictive,47 which most likely reduced the photocurrent enhancement.
Here, we specifically focus on spectroscopically characterizing the charge generation process, which has not been well-understood using full devices. We utilize the DSSC architecture with dyes based on pentacene (Pc) and tetracene (Tc) chromophores. Both pentacene and tetracene derivatives possess triplet energies capable of pushing solar cell efficiencies past 40% for a simple parallel tandem device, which is a significant gain over the singlet-junction thermodynamic efficiency limit of 33%.1,8 Pc and Tc are also ideal model systems because they display robust and efficient SF across a variety of solid-state and solution environments.29,48 They have similar molecular and crystal structures, but exhibit distinct SF rates, oxidation potentials, and triplet energies, allowing us to explore in detail how these parameters affect the interplay between electron injection and SF. We have chosen Pc and Tc modified with triisopropylsilylethynyl (TIPS) groups for increased solubility and stability, and carboxylic acid (COOH) groups for attachment to a TiO2 surface. Through the use of cyclic voltammetry (CV) and spectroelectrochemistry (SEC), we arrive at a model for the energetic landscapes of the two acene/TiO2 systems, including the charge-transfer species. In order to judge the influence of the local monolayer environment on SF, we compare TIPS Pc and TIPS Tc in polycrystalline thin films and TIPS Pc COOH and TIPS Tc COOH linked to TiO2. Transient absorption (TA) spectroscopy is then used to distinguish between charge and triplet generation processes, and to develop a kinetic model that is consistent with our picture of the energetics.
Experimental
Chromophore synthesis
TIPS Pentacene was purchased from Sigma Aldrich and used as received. TIPS Pc COOH and TIPS tetracene were synthesized according to previously reported procedures.45,49 The synthesis procedure for TIPS Tc COOH is detailed by Kroupa et al.50Film Preparation
An Angstrom Engineering Nexdep thermal evaporator was used to deposit 40 nm TIPS Pc and TIPS Tc polycrystalline thin films on glass substrates. A quartz crystal microbalance was used to deposit the films at a rate of 1.0 Å s−1. TIPS Tc films were annealed at 35 °C overnight to increase crystallinity. For TA measurements, the films were sealed using a 60 μm thick Surlyn frame melted between the substrate and a glass coverslip. Mesoporous TiO2 films were prepared using a paste of 20 nm TiO2 nanoparticles suspended in terpineol and ethylcellulose. The ∼1 micron thick films were screen-printed, annealed at 500 °C, then cooled to 70 °C, before they were soaked overnight in an ethanol solution saturated with TIPS Tc COOH or TIPS Pc COOH. The films were rinsed with pure ethanol to remove excess deposited molecules.Atomic layer deposition
Atomic layer deposition (ALD) was performed using a Beneq TFS 200. The ALD reactor was operated at 175 °C and 5 mbar pressure with a constant 550 sccm flow of ultra-high purity nitrogen (99.9999%) as the process carrier gas. Aluminum oxide was grown by alternating exposures, or ALD ‘cycles’, of trimethylaluminum and water. The duration of the precursor exposure for both trimethylaluminum and water was 0.2 s that was separated by a 3 s rest to allow vapors to purge from the reactor. Both precursors were held in stainless steel bubblers at 20 °C. The film thickness reported was calculated from a 0.11 nm per cycle rate obtained from regular calibration of the ALD of Al2O3 for this tool.Spectroelectrochemistry
Samples for SEC were prepared using the same acene/TiO2 assembly method detailed in “Film Preparation” on FTO substrates. SEC measurements were done using a 1 cm2 quartz cuvette and 0.1 M bis(trifluoromethane)sulfonimide lithium salt in dry acetonitrile. All measurements were done in an argon-filled glove box with electrical and fiber optic pass-through junctions. A Princeton Applied Research Model 263 potentiostat was used with a three-electrode configuration: a silver wire pseudoreference electrode, a Pt wire counter electrode, and the sample as the working electrode. Ferrocene was used as an internal standard. Potentiostatic bias was applied in either 50 or 100 mV increments. UV-VIS-NIR data was recorded with an OceanFX spectrometer (Ocean Optics, UV-VIS) and a NIRQuest 512-2.2 spectrometer (Ocean Optics, NIR), with an Ocean Optics DH-2000-BAL light source. The UV-VIS-NIR spectrum at open-circuit potential was subtracted from the spectrum at a given applied potential to find the change in absorbance with applied bias.Cyclic voltammetry
Cyclic voltammetry was performed by dissolving the acenes in a 0.1 M solution of tetrabutylammonium hexafluorophosphate in degassed dichloromethane with platinum working and counter electrodes and a Ag/AgNO3 reference electrode. Oxidation potentials were converted to ionization potentials for comparison with TiO2.51Spectroscopy
Solution UV-VIS linear absorption spectra were collected with a Cary 500 spectrometer. Film UV-VIS linear absorption spectra were acquired with a Varian Cary-6000i equipped with an integrating sphere. For films, the absorbance was calculated as A = −log10(R + T). Transient absorption datasets were acquired using a Coherent Libra Ti:sapphire laser, with an output of 800 nm at 1 kHz. A TOPAS-C OPA was used to generate the ∼150 fs pump pulse (center wavelength 530–650 nm). A small amount of 800 nm light was used to pump a CaF2 crystal to generate 330–850 nm probe light for UV-VIS TA or a 1 cm thick sapphire crystal to generate 750–1640 nm probe light for NIR TA. A second TOPAS-C OPA was used to generate ∼150 fs 5–6 μm probe pulses for MIR TA. The instrument response function was ∼150–250 fs depending on the probe wavelength. A Janis VPF 100 Cryostat was used for all TA measurements under vacuum. A solution cell was assembled with an o-ring and sapphire windows in a glovebox for measurements with solvent and electrolyte present. All datasets were analyzed using Surface Xplorer software. Charge injection yields were determined by comparing the maximum signal of a Z907/TiO2 sample to the maximum signal of an acene/TiO2 sample of interest under identical excitation conditions.Results
The mesoporous TiO2 films were found to be deeply colored after immersion in concentrated TIPS Pc COOH or TIPS Tc COOH solutions for 12 hours. Additional soaking time did not produce additional dye adhesion, and extensive washing in ethanol did not reduce the intensity of the dye stain, which is a strong indication of a covalently bound monolayer. The driving force for electron transfer between the covalently bound acenes and nanostructured anatase TiO2 was investigated using a combination of SEC, CV, and steady-state absorption spectroscopy. The acene ionization potentials, TiO2 electron affinity from the literature,53 exciton energies, and driving forces for charge transfer are shown in Table 1. Singlet energies were determined from the intersection between absorption and photoluminescence spectra (ESI Fig. S1†), while triplet energies were estimated from literature at 0.87 eV for pentacene10 and 1.25 eV for tetracene.12,54,55| E IP (acene)/eV vs. vacuum | E EA (TiO2)/eV vs. vacuum | E singlet/eV | E triplet/eV | ΔGsinglet/eV | ΔGtriplet/eV | |
|---|---|---|---|---|---|---|
| TIPS Pc COOH/TiO2 | −5.24 ± 0.05 | −4.3 ± 0.2 | 1.91 ± 0.01 | 0.87 | −1.0 ± 0.2 | 0.1 ± 0.2 |
| TIPS Tc COOH/TiO2 | −5.50 ± 0.05 | −4.3 ± 0.2 | 2.25 ± 0.01 | 1.25 | −1.1 ± 0.2 | −0.1 ± 0.2 |
The Gibbs free energy for electron transfer can be approximated using the equation:56
| ΔG = Efinal − Einitial ≈ EIP (D) − EEA (A) − Eexciton | (1) |
TIPS pentacene
We compare the photophysics of TIPS Pc with TIPS Pc COOH in both solution and solid-state in order to assign spectral features to singlets, triplets, and charges, as well as measure shifts that occur upon intermolecular interaction. The absorption spectra are shown in Fig. 1. In solution there is a redshift and slight broadening of TIPS Pc COOH relative to TIPS Pc. In a polycrystalline TIPS Pc thin film, the absorption is significantly broadened and red-shifted, with redistributed oscillator strength. These spectral changes reflect strong electronic coupling, which leads to new exciton states. In contrast, TIPS Pc COOH linked to TiO2 shows only slight broadening and almost no shift with respect to solution, indicating weak electronic coupling.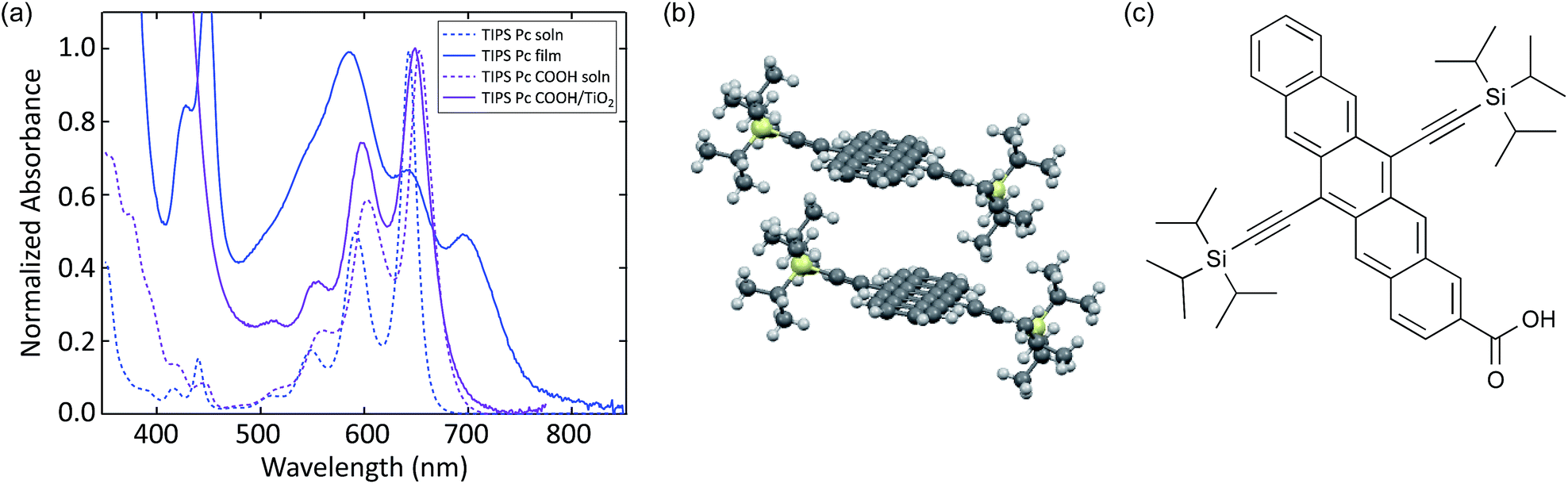 | ||
| Fig. 1 (a) UV-VIS absorption spectra of TIPS Pc in solution and polycrystalline film, as well as TIPS Pc COOH in solution and attached to TiO2. (b) Crystal structure of TIPS Pc film.52 (c) Molecular structure of TIPS Pc COOH. | ||
For both TIPS Pc and TIPS Pc COOH, solution-phase transient absorption (TA) spectroscopy (see ESI Fig. S2†) reveals an excited-state species that we assign as the lowest excited singlet S1. The spectra consist of a large photoinduced absorption (PIA) peaked near 450 nm that overlaps with the ground state bleach (GSB), and a stimulated emission (SE) feature near 700 nm. All features decay with 12–13 ns lifetimes in solution.
The TIPS Pc excited-state dynamics measured in the solid-state via TA are dramatically different from solution dynamics. Instead of a nanosecond lifetime, the singlet in polycrystalline TIPS Pc has a lifetime of ∼150 fs due to rapid and efficient SF, similar to previous results from TIPS Pc and derivatives (Fig. 2a and b).19,20,58,59 Specifically, the broad, low amplitude NIR singlet PIA decays in 150 fs (Fig. 2a) and is correlated with the growth of PIA features near 510 nm (Fig. 2a) and 750–950 nm (Fig. 2b). These latter features have previously been attributed to triplets in TIPS Pc thin films.58–60 In particular, the feature near 510 nm closely matches the sensitized TIPS Pc triplet spectrum in solution (dashed curves, Fig. 2a). The TIPS Pc cation spectrum, which was measured by TIPS Pc solution oxidation,61 features a relatively narrow PIA feature peaked near 970 nm. This feature is not evident in the TIPS Pc film raw spectra nor in the decay associated spectra (DAS) from global analysis (see ESI Fig. S3†), implying charge separation is a minority decay pathway for singlets and triplets in TIPS Pc thin films. After the initial sub-ps kinetics, there is a secondary growth of triplet PIA in the visible and NIR, which has been attributed to triplet–triplet separation.58
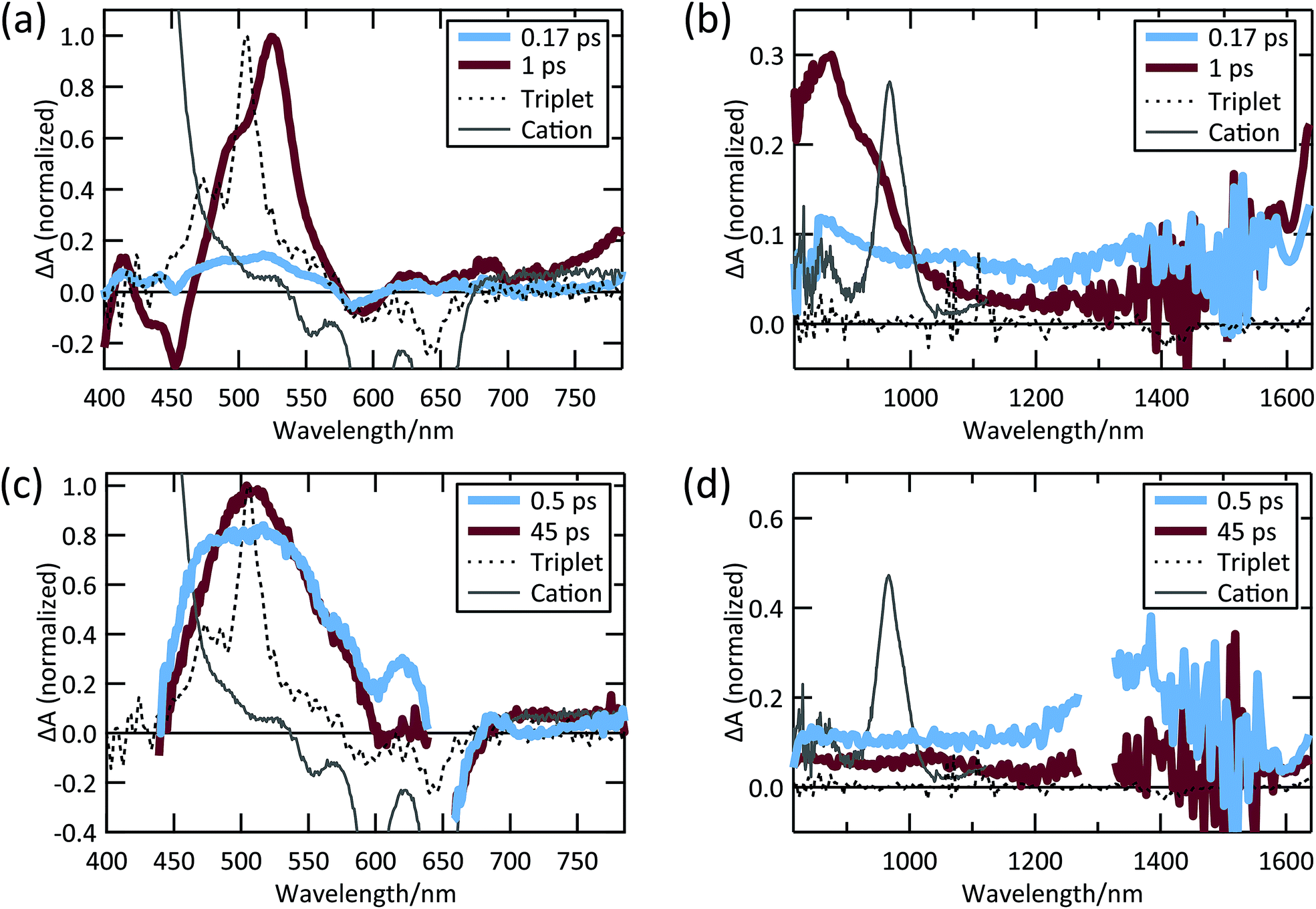 | ||
| Fig. 2 TIPS Pc thin film (a) visible and (b) NIR TA spectra; TIPS Pc COOH/TiO2 (c) visible and (d) NIR TA spectra. | ||
When TIPS Pc COOH is attached to TiO2, we observe spectral peaks that rise and decay with timescales distinct from both solution and TIPS Pc thin films (Fig. 2c and d). A SE feature near 700 nm decays in ∼6.5 ps, concurrent with a slight narrowing and growth of the PIA feature near 510 nm. There is a broad-low-amplitude NIR PIA feature in the 45 ps spectral slice covering the region where we would expect a cation PIA feature. However, the amplitude is very small compared to the cation peak observed in the TIPS Tc COOH/TiO2 system, vide infra, indicating that cationic TIPS Pc COOH is a minority species. Instead, the predominant species after singlet decay is most likely triplets formed via SF, as evidenced by the PIA feature near 510 nm.
TIPS tetracene
Similar to TIPS pentacene, we study TIPS Tc and TIPS Tc COOH in solution and the solid-state. Unlike the TIPS Pc system, which undergoes dramatic changes in linear absorption between solution and polycrystalline thin film, the TIPS Tc thin film absorption exhibits a small red-shift and two small additional shoulders between the 0–0 and 0–1 absorption peaks (Fig. 3a). The subtle changes indicate weak intermolecular electronic coupling resulting from reduced molecular overlap in the crystal structure (Fig. 3b) and limited film crystallinity compared to TIPS Pc (ESI Fig. S3†). The broadening and slight spectral red-shift in absorption between TIPS Tc COOH in solution and TIPS Tc COOH linked to TiO2 mirrors the behavior in the TIPS Pc COOH system with increased broadening and a slight spectral red-shift in the linked absorption spectrum (Fig. 3a).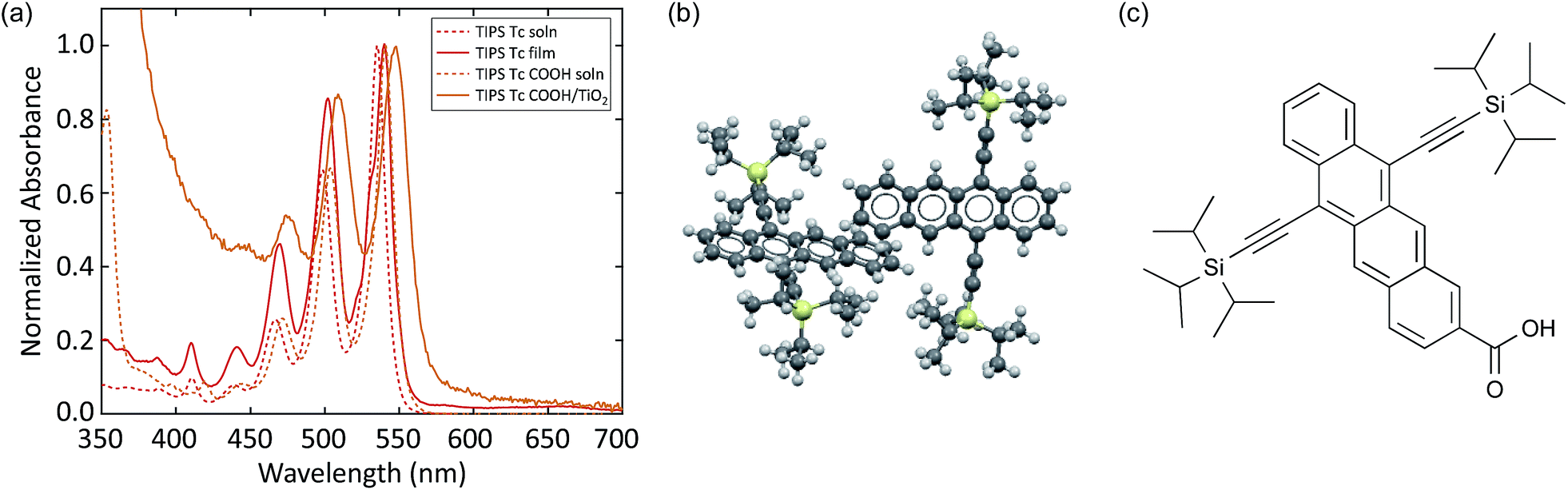 | ||
| Fig. 3 (a) UV-Vis absorption spectra of TIPS Tc in solution and polycrystalline film, as well as TIPS Tc COOH in solution and attached to TiO2. (b) Crystal structure of TIPS Tc film.62 (c) Molecular structure of TIPS Tc COOH. | ||
Similar to the TA spectra of pentacene derivatives in solution, both TIPS Tc and TIPS Tc COOH in solution show evidence of a singlet excited-state species with a large PIA feature from 450–550 nm and an SE feature near 585 nm that decay with 11–12 ns lifetimes (ESI Fig. S2†).
Despite weak coupling in TIPS Tc polycrystalline films, SF still occurs in 17.5 ps, which is faster than SF in polycrystalline tetracene.29,31 The TA spectra shown in Fig. 4a and b exhibit decay (17.5 ps) of a SE feature at 590 nm and PIA features in the 420–500 nm, 815–900 nm, and 1050–1500 nm regions, which we associate with the singlet state. The decay in these features is correlated with the rise of triplet PIA features at 515 nm, 860 nm, and 980 nm with the same 17.5 ps time constant (Fig. 4a and b).
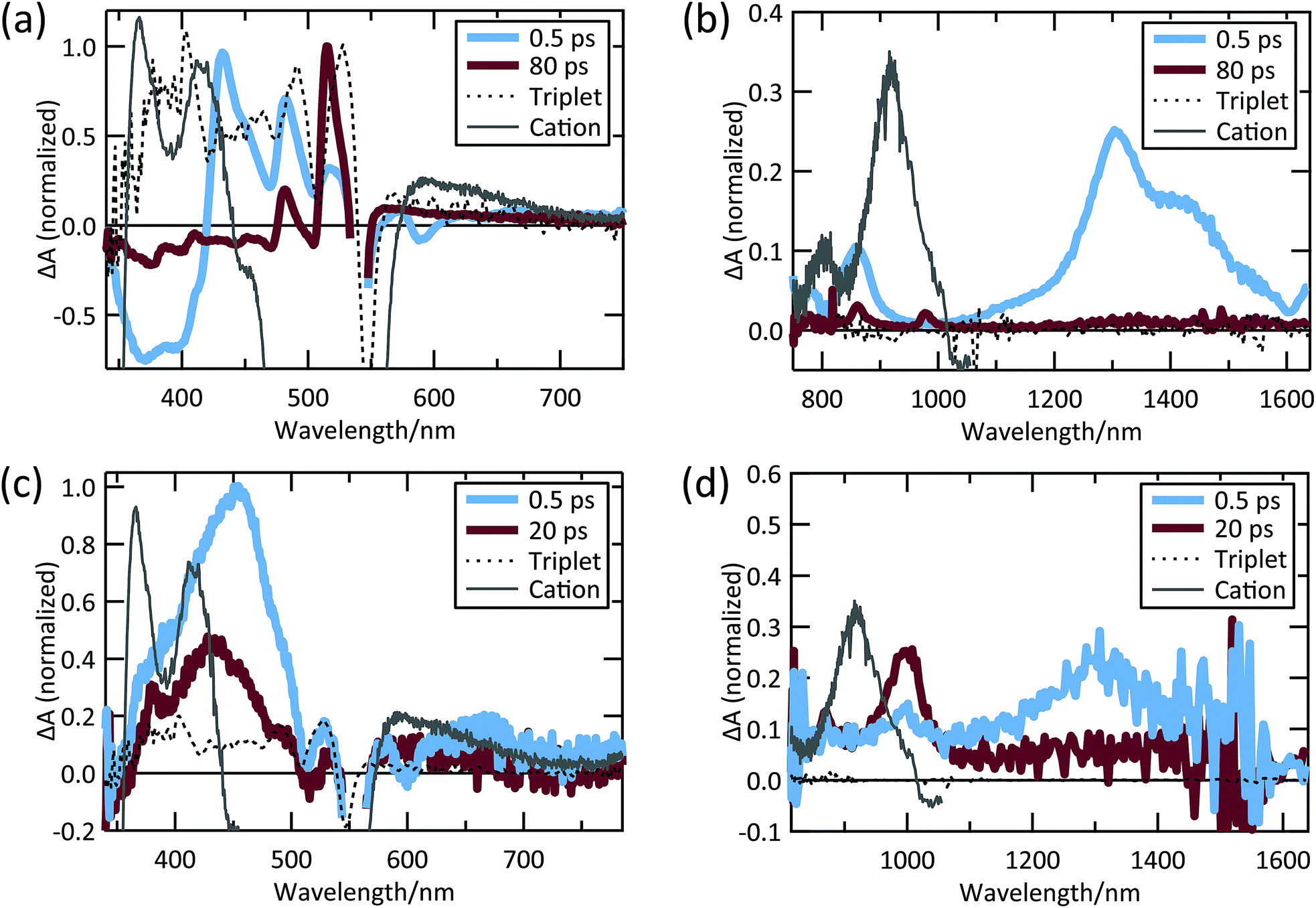 | ||
| Fig. 4 TIPS Tc thin film (a) visible and (b) NIR TA spectra; TIPS Tc COOH/TiO2 (c) visible and (d) NIR TA spectra. | ||
The PIA at 515 nm matches the sensitized triplet PIA in solution, whereas the peaks at 860 and 980 nm match peaks previously reported for TIPS Tc triplets.35–40,54,63 However, unlike Stern, et al.63 and Pun, et al.,48 we do not find evidence of an initial sub-ps SF mechanism. This could be due to the decreased time resolution in the measurements reported here or limited crystallinity of our thin films. We also detect no evidence of cation formation in TIPS Tc polycrystalline thin films.
The TIPS Tc COOH/TiO2 system produces a distinct set of dynamics (see Fig. 4c and d). TIPS Tc COOH/TiO2 spectra at 0.5 ps and 20 ps are overlaid with the sensitized TIPS Tc COOH triplet spectrum in solution and the cation spectrum from SEC. We track the singlet state through the SE feature at 600 nm and PIA peaks at 450 nm and 1300 nm, which decay on a ∼2 ps timescale. This decay is accompanied by the growth of a PIA peak at 1000 nm (Fig. 4d). Dynamics at later times involve subtle peak shifts and further peak decay, but no substantial changes. There are potential spectral matches to triplet peaks in the visible, but there is no significant growth of triplet PIA features seen in polycrystalline films nor sensitized TIPS Tc COOH solution spectra. This is likely because of a kinetic competition between charge injection and SF, which produces a small yield of triplets (<10%, estimated by comparing injection rate versus apparent SF rate on the nanoparticle surface, vide infra).
We assign the peak at 1000 nm to the TIPS Tc COOH cation, though it is offset from the SEC cation peak by ∼70 nm (compare Fig. 4d, 20 ps and gray spectra). In Fig. 5a, we show that the 1000 nm PIA feature shifts to match the PIA feature measured via SEC in the presence of the same solvent and electrolyte. This is consistent with previous observations of a strong correlation between solvent polarity and charge transfer energetics.64 While the cation peak noticeably shifts wavelength in the presence of solvent, the dynamics of charge transfer do not change dramatically. Samples both with and without solvent produce a charged species on a timescale of ∼2 ps (Fig. 5b). There is no evidence of significant additional charge injection on a longer timescale, either with or without solvent.
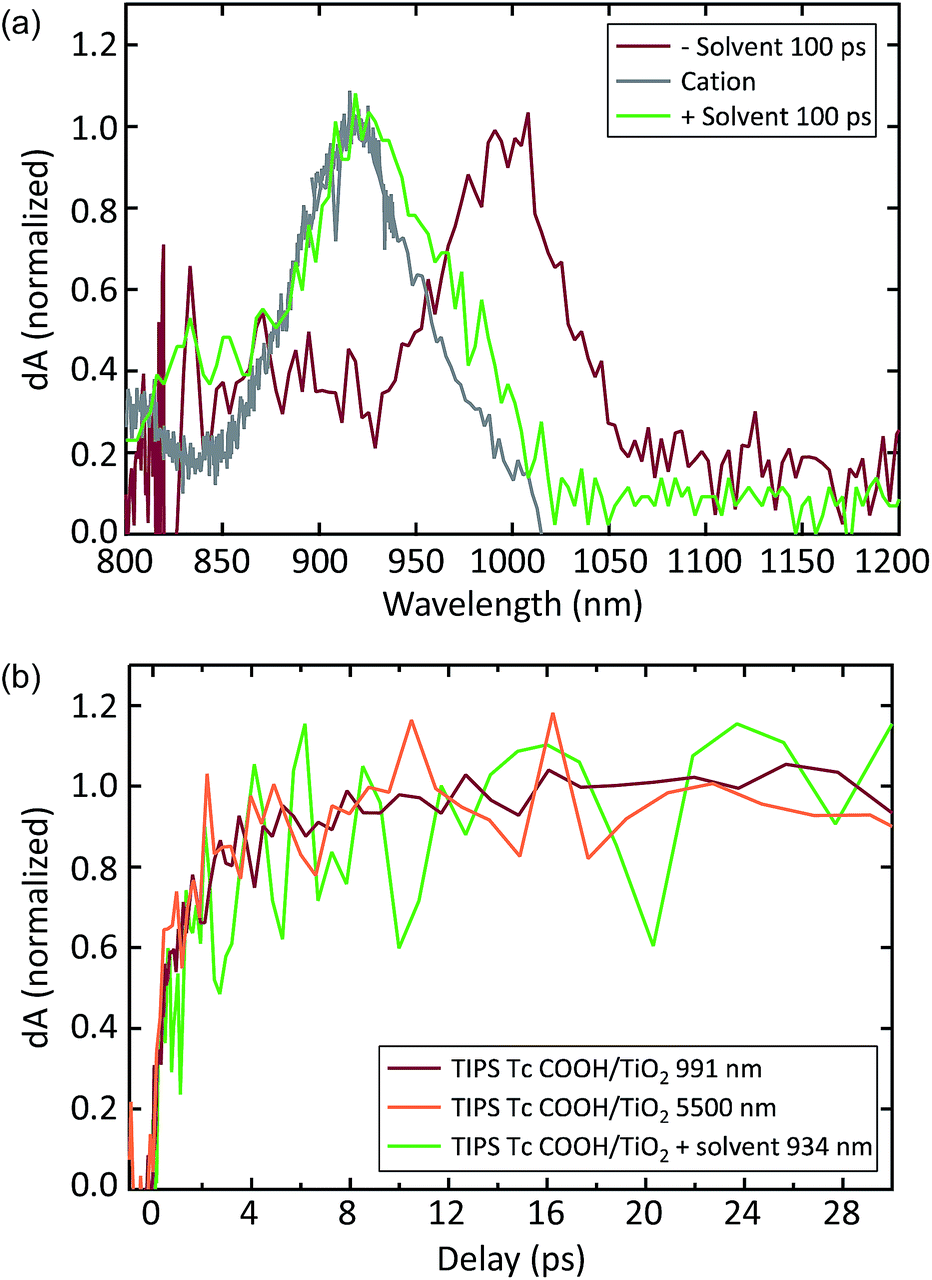 | ||
| Fig. 5 (a) NIR TIPS Tc COOH/TiO2 cation absorption. In vacuum, the cation PIA feature peaks near 1000 nm. In solution, the cation PIA shifts to match the SEC cation feature at 930 nm. (b) Kinetics associated with TiO2 electrons (5500 nm), TIPS Tc COOH cation under vacuum (991 nm), and TIPS Tc COOH cation in a solvent environment (934 nm). | ||
In order to quantify charge injection efficiency, we probed with mid-IR (MIR) pulses centered at ∼5500 nm after the initial photoexcitation near the acene absorption peak. The resulting PIA signal is associated with shallowly trapped electrons in the conduction band of anatase TiO2, and correlates linearly with extracted charge.65 Because we selectively excite the acene molecules in all of our experiments, this signal exclusively corresponds to electrons transferred from TIPS Tc COOH molecules to TiO2. Measurements on bare TiO2 films produce negligible signal. The MIR TA signal rises on the same ∼2 ps time scale as the NIR PIA, further corroborating the cationic nature of the NIR PIA. We used the ruthenium-based dye Z907 as a reference, which is known to inject electrons into TiO2 at a ∼85% yield.66 By comparing the MIR signal of Z907/TiO2 sample to that of the acene/TiO2 samples under identical excitation conditions, we find an injection efficiency of 66 ± 5% in the TIPS Tc COOH/TiO2 system.
To probe triplet formation kinetics in this system in the absence of fast charge injection, we deposited a 5 Å Al2O3 barrier onto TiO2via atomic layer deposition (ALD) before TIPS Tc COOH adsorption. This layer effectively slows charge generation (Fig. 6a) by over two orders of magnitude,67 which extends the singlet lifetime and allows SF to produce triplets. The much smaller population of charges formed with the alumina layer simplifies the spectral assignments in the visible region, which now closely matches the sensitized TIPS Tc COOH triplet spectrum at later times (Fig. 6b). This demonstrates the unequivocal formation of triplets via SF. We find a SF time constant of 40 ± 10 ps (ESI Fig. S4†).
 | ||
| Fig. 6 Addition of alumina layer slows electron injection and allows triplet formation via SF. (a) NIR showing singlet decay and slight charge formation, highlighted in grey. (b) Triplet formation (arrows). | ||
Discussion
The degree of intermolecular electronic coupling in TIPS Pc and TIPS Tc polycrystalline thin films determines the shifts and distortions of the absorption spectra relative to solution. TIPS Pc films exhibit characteristics of strong coupling, such as Davydov splitting. For TIPS Tc, the shifts and distortions are much smaller. Strong electronic coupling responsible for greater exciton delocalization has been correlated with faster SF in pentacene nanoparticles.21 In TIPS Pc, excited singlets undergo sub-ps SF, which is consistent with strong electronic coupling and an exoergic mechanism.8,9 Singlets in TIPS Tc also undergo SF, but at a slower rate than TIPS Pc. However, the coupling elements responsible for efficient SF are not always correlated with the coupling elements responsible for changes in absorption spectra, as evidenced by faster SF in disordered TIPS Tc films (∼17.5 ps) relative to more ordered Tc films (>70 ps).29When the acenes are covalently attached to TiO2 instead of deposited in a film, the dielectric environment changes along with the molecular packing. TIPS Pc COOH is particularly sensitive to this change in coupling because the TIPS Pc molecules in films are arranged with a high degree of order in a close, face-to-face arrangement (Fig. 1b). TIPS Pc COOH exhibits a significantly increased SF time constant of 6.5 ps when tethered to TiO2 (compared to 150 fs in a neat film), which is likely a product of reduced intermolecular coupling for chromophores with limited flexibility to arrange in tightly packed and ordered arrays. Some control over the TIPS-Pc SF rate has been achieved by inducing amorphous film growth,21 but the rate measured here is the slowest reported to our knowledge. The significant slowing may be assisted by the lack of fast singlet energy transfer to preferred SF sites for the quasi-2D assembly of dyes on the TiO2 surface compared to faster 3D transport in crystalline films. Interestingly, the secondary stage of triplet formation often observed for TIPS Pc films is absent in the TIPS Pc COOH/TiO2 samples, which may reflect the immediate formation of quasi-isolated triplets.
Curiously, SF outcompetes singlet charge injection in the TIPS Pc COOH/TiO2 film even with the increased SF time constant and a strong driving force for singlet injection. The low charge injection efficiency (<10%) in TIPS Pc COOH/TiO2 films may be due to the strong coupling between TIPS Pc COOH molecules relative to the coupling between TIPS Pc COOH and the TiO2 surface. Large intermolecular electronic coupling between molecular sensitizers is associated with strong intermolecular charge-transfer character, and thus, decreased power conversion efficiencies in DSSCs.68 The requirement to have some degree of molecular coupling in order to achieve singlet fission at a reasonable rate necessarily introduces intermolecular coupling pathways that may decrease injection efficiencies. Differences in intermolecular coupling between TIPS Pc COOH and TIPS Tc COOH may account for the differences in singlet injection efficiencies.
This explanation is further supported by the fact that the driving force for singlet charge injection is predicted to be nearly identical (∼1 eV) in both acene systems. However, unlike the TIPS Pc COOH/TiO2 system, TIPS Tc COOH singlets inject electrons efficiently into TiO2 with a time constant of ∼2 ps. The coupling between the singlet excited state of TIPS Tc COOH molecules and the conduction band states at the TiO2 surface may thus be more optimized compared to TIPS Pc COOH. The electron injection process outcompetes SF, which slows to 40 ps in the dye monolayer (compared to 17.5 ps in the thin film) as a likely result of reduced electronic coupling and increased disorder on the TiO2 surface compared to the thin film. The influence of disorder is not as large as in the pentacene case, which is probably a reflection of more weakly coupled and disordered molecular arrangements for SF in the TIPS Tc thin film. The link between intermolecular coupling, intermolecular charge transfer character, and charge injection efficiency in SF chromophores warrants future study.
There is no evidence of considerable cation growth for either film past the picosecond timescale, which makes significant injection from triplet states unlikely. In the TIPS Tc COOH case, there are likely few triplets remaining after 10 ps because of the dominant charge-transfer process from the singlet, whereas for TIPS Pc COOH, the triplets are present but likely have insufficient driving force for triplet injection. The excited state dynamics with the Al2O3 spacer layer between TIPS Tc COOH and TiO2 clearly demonstrates that singlet injection is hindered and that a significant population of triplets can be generated via SF. However, there is no evidence of further charge injection from these triplet states, suggesting that the main obstacle to utilizing SF in the present system is insufficient driving force for electron injection from the triplet. Triplet exciton dissociation at an interface has been rarely studied, and it is possible that knowledge of driving force alone as calculated by eqn (1) may be insufficient to predict triplet dissociation. The nature and density of states may help drive the dissociation process. For nanostructured oxides, there are a variety of procedures for adjusting the conduction band energetics, including the use of rutile TiO2,65 the addition of a Li salt,69 or the use of semiconductor dopants.70 Furthermore, turning to a semiconductor with a more negative conduction band position, such as SnO2, may also produce a significant driving force that allows the two triplet excitons generated after photon absorption to be dissociated and harvested.
Conclusions
We have systematically examined the changes in intermolecular electronic coupling and SF dynamics that accompany molecular dye assembly on high-surface area nanostructured metal oxide surfaces that are commonly used as photoelectrodes. We electrochemically characterized these systems and mapped relative driving forces for singlet and triplet dissociation. We found that both pentacene and tetracene derivatives are able to undergo SF in a heterogeneous, disordered environment, though neither are able to efficiently inject charges into TiO2 from their triplet states. Our experiments emphasize the importance of simultaneously optimizing SF rate, driving force for triplet injection, and molecular coupling with the acceptor surface. Though structurally similar, pentacene and tetracene display dramatically different singlet charge injection efficiencies independent of their SF rates. Therefore, subtleties in organic–inorganic electronic coupling likely play a crucial role in promoting electron transfer. Understanding the nature of this variation in rates and how it can be controlled through molecular structure or acceptor modification would provide an opportunity to independently tune injection rates while maintaining efficient SF, leading toward a novel route for harvesting multiple electron–hole pairs from a single photon.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Thomas J. Carey and Niels H. Damrauer for providing our TIPS Tc compound, Nedezhda V. Korovina for assistance with sample preparation for sensitization experiments, Mike Carroll for assistance with SEC measurements, and Garry Rumbles for stimulating discussions. N. A. P. and D. H. A. prepared and characterized solution and film samples, performed TA spectroscopy, and analyzed the data under the guidance of J. C. J. S. C. deposited alumina on TiO2 samples. D. B. G. synthesized TIPS Pc COOH and TIPS Tc COOH under the guidance of J. E. A. N. A. P. and D. H. A. wrote the manuscript with input from all the authors. N. A. P., D. H. A., S. C. and J. C. J. acknowledge the Solar Photochemistry Program supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences, Biosciences, and Geosciences under Contract No. DE-AC36-08GO28308 with NREL for support of transient absorption and steady-state spectroscopy, ALD, and electrochemistry. D. B. G. and J. E. A. thank the National Science Foundation (DMREF-1627428) for support of acene synthesis.Notes and references
- M. C. Hanna and A. J. Nozik, J. Appl. Phys., 2006, 100, 074510 CrossRef.
- M. C. Beard, J. C. Johnson, J. M. Luther and A. J. Nozik, Philos. Trans. R. Soc., A, 2015, 373, 20140412 CrossRef PubMed.
- Y. Yan, R. W. Crisp, J. Gu, B. D. Chernomordik, G. F. Pach, A. R. Marshall, J. A. Turner and M. C. Beard, Nat. Energy, 2017, 2, 17052 CrossRef CAS.
- O. E. Semonin, J. M. Luther, S. Choi, H.-Y. Chen, J. Gao, A. J. Nozik and M. C. Beard, Science, 2011, 334, 1530–1533 CrossRef CAS PubMed.
- M. L. Böhm, T. C. Jellicoe, M. Tabachnyk, N. J. L. K. Davis, F. Wisnivesky-Rocca-Rivarola, C. Ducati, B. Ehrler, A. A. Bakulin and N. C. Greenham, Nano Lett., 2015, 15, 7987–7993 CrossRef PubMed.
- D. N. Congreve, J. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. Van Voorhis and M. A. Baldo, Science, 2013, 340, 334–337 CrossRef CAS PubMed.
- L. M. Pazos-Outón, J. M. Lee, M. H. Futscher, A. Kirch, M. Tabachnyk, R. H. Friend and B. Ehrler, ACS Energy Lett., 2017, 2, 476–480 CrossRef PubMed.
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- M. B. Smith and J. Michl, Annu. Rev. Phys. Chem., 2013, 64, 361–386 CrossRef CAS PubMed.
- W.-L. Chan, M. Ligges, A. Jailaubekov, L. Kaake, L. Miaja-Avila and X. Y. Zhu, Science, 2011, 334, 1541–1545 CrossRef CAS PubMed.
- J. J. Burdett and C. J. Bardeen, J. Am. Chem. Soc., 2012, 134, 8597–8607 CrossRef CAS PubMed.
- W.-L. Chan, M. Ligges and X. Y. Zhu, Nat. Chem., 2012, 4, 840–845 CrossRef CAS PubMed.
- P. E. Teichen and J. D. Eaves, J. Phys. Chem. B, 2012, 116, 11473–11481 CrossRef CAS PubMed.
- T. C. Berkelbach, M. S. Hybertsen and D. R. Reichman, J. Chem. Phys., 2013, 138, 114102 CrossRef PubMed.
- T. C. Berkelbach, M. S. Hybertsen and D. R. Reichman, J. Chem. Phys., 2013, 138, 114103 CrossRef PubMed.
- T. C. Berkelbach, M. S. Hybertsen and D. R. Reichman, J. Chem. Phys., 2014, 141, 074705 CrossRef PubMed.
- J. C. Johnson, A. J. Nozik and J. Michl, Acc. Chem. Res., 2013, 46, 1290–1299 CrossRef CAS PubMed.
- D. Beljonne, H. Yamagata, J. L. Brédas, F. C. Spano and Y. Olivier, Phys. Rev. Lett., 2013, 110, 226402 CrossRef CAS PubMed.
- A. J. Musser, M. Liebel, C. Schnedermann, T. Wende, T. B. Kehoe, A. Rao and P. Kukura, Nat. Phys., 2015, 11, 352–357 CrossRef CAS.
- S. R. Yost, J. Lee, M. W. B. Wilson, T. Wu, D. P. McMahon, R. R. Parkhurst, N. J. Thompson, D. N. Congreve, A. Rao, K. Johnson, M. Y. Sfeir, M. G. Bawendi, T. M. Swager, R. H. Friend, M. A. Baldo and T. Van Voorhis, Nat. Chem., 2014, 6, 492–497 CrossRef CAS PubMed.
- R. D. Pensack, A. J. Tilley, S. R. Parkin, T. S. Lee, M. M. Payne, D. Gao, A. A. Jahnke, D. G. Oblinsky, P.-F. Li, J. E. Anthony, D. S. Seferos and G. D. Scholes, J. Am. Chem. Soc., 2015, 137, 6790–6803 CrossRef CAS PubMed.
- R. Tempelaar and D. R. Reichman, J. Chem. Phys., 2017, 146, 174703 CrossRef PubMed.
- P. J. Jadhav, A. Mohanty, J. Sussman, J. Lee and M. A. Baldo, Nano Lett., 2011, 11, 1495–1498 CrossRef CAS PubMed.
- B. Ehrler, M. W. B. Wilson, A. Rao, R. H. Friend and N. C. Greenham, Nano Lett., 2012, 12, 1053–1057 CrossRef CAS PubMed.
- J. Lee, P. Jadhav, P. D. Reusswig, S. R. Yost, N. J. Thompson, D. N. Congreve, E. Hontz, T. Van Voorhis and M. A. Baldo, Acc. Chem. Res., 2013, 46, 1300–1311 CrossRef CAS PubMed.
- J. Xia, S. N. Sanders, W. Cheng, J. Z. Low, J. Liu, L. M. Campos and T. Sun, J. Am. Chem. Soc., 2015, 137, 15980–15983 CrossRef PubMed.
- B. Ehrler, K. P. Musselman, M. L. Böhm, R. H. Friend and N. C. Greenham, Appl. Phys. Lett., 2012, 101, 153507 CrossRef.
- T. C. Wu, N. J. Thompson, D. N. Congreve, E. Hontz, S. R. Yost, T. Van Voorhis and M. A. Baldo, Appl. Phys. Lett., 2014, 104, 193901 CrossRef.
- G. B. Piland and C. J. Bardeen, J. Phys. Chem. Lett., 2015, 6, 1841–1846 CrossRef CAS PubMed.
- Y. Wan, Z. Guo, T. Zhu, S. Yan, J. Johnson and L. Huang, Nat. Chem., 2015, 7, 785–792 CrossRef CAS PubMed.
- D. H. Arias, J. L. Ryerson, J. D. Cook, N. H. Damrauer and J. C. Johnson, Chem. Sci., 2016, 7, 1185–1191 RSC.
- G. B. Piland, J. J. Burdett, T.-Y. Hung, P.-H. Chen, C.-F. Lin, T.-L. Chiu, J.-H. Lee and C. J. Bardeen, Chem. Phys. Lett., 2014, 601, 33–38 CrossRef CAS.
- N. J. Thompson, M. W. B. Wilson, D. N. Congreve, P. R. Brown, J. M. Scherer, T. S. Bischof, M. Wu, N. Geva, M. Welborn, T. V. Voorhis, V. Bulović, M. G. Bawendi and M. A. Baldo, Nat. Mater., 2016, 13, 1039–1043 CrossRef PubMed.
- M. Tabachnyk, B. Ehrler, S. Gelinas, M. L. Böhm, B. J. Walker, K. P. Musselman, N. C. Greenham, R. H. Friend and A. Rao, Nat. Mater., 2014, 13, 1033–1038 CrossRef CAS PubMed.
- A. M. Müller, Y. S. Avlasevich, W. W. Schoeller, K. Müllen and C. J. Bardeen, J. Am. Chem. Soc., 2007, 129, 14240–14250 CrossRef PubMed.
- J. C. Johnson, A. Akdag, M. Zamadar, X. Chen, A. F. Schwerin, I. Paci, M. B. Smith, Z. Havlas, J. R. Miller, M. A. Ratner, A. J. Nozik and J. Michl, J. Phys. Chem. B, 2013, 117, 4680–4695 CrossRef CAS PubMed.
- J. Zirzlmeier, D. Lehnherr, P. B. Coto, E. T. Chernick, R. Casillas, B. S. Basel, M. Thoss, R. R. Tykwinski and D. M. Guldi, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5325–5330 CrossRef CAS PubMed.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, M. T. Trinh, B. Choi, J. Xia, E. J. Taffet, J. Z. Low, J. R. Miller, X. Roy, X. Y. Zhu, M. L. Steigerwald, M. Y. Sfeir and L. M. Campos, J. Am. Chem. Soc., 2015, 137, 8965–8972 CrossRef CAS PubMed.
- J. D. Cook, T. J. Carey and N. H. Damrauer, J. Phys. Chem. A, 2016, 120, 4473–4481 CrossRef CAS PubMed.
- E. A. Margulies, C. E. Miller, Y. Wu, L. Ma, G. C. Schatz, R. M. Young and M. R. Wasielewski, Nat. Chem., 2016, 8, 1120–1125 CrossRef CAS PubMed.
- E. Busby, J. Xia, Q. Wu, J. Z. Low, R. Song, J. R. Miller, X. Y. Zhu, L. M. Campos and M. Y. Sfeir, Nat. Mater., 2015, 14, 426–433 CrossRef CAS PubMed.
- N. A. Pace, W. Zhang, D. H. Arias, I. McCulloch, G. Rumbles and J. C. Johnson, J. Phys. Chem. Lett., 2017, 8, 6086–6091 CrossRef CAS PubMed.
- S. E. Koops, B. C. O'Regan, P. R. F. Barnes and J. R. Durrant, J. Am. Chem. Soc., 2009, 131, 4808–4818 CrossRef CAS PubMed.
- J. B. Asbury, N. A. Anderson, E. Hao, X. Ai and T. Lian, J. Phys. Chem. B, 2003, 107, 7376–7386 CrossRef CAS.
- Z. Li, J. Photonics Energy, 2011, 1, 011106 CrossRef.
- J. N. Schrauben, Y. Zhao, C. Mercado, P. I. Dron, J. L. Ryerson, J. Michl, K. Zhu and J. C. Johnson, ACS Appl. Mater. Interfaces, 2015, 7, 2286–2293 CAS.
- J. L. Ryerson, J. N. Schrauben, A. J. Ferguson, S. C. Sahoo, P. Naumov, Z. Havlas, J. Michl, A. J. Nozik and J. C. Johnson, J. Phys. Chem. C, 2014, 118, 12121–12132 CAS.
- A. B. Pun, S. N. Sanders, E. Kumarasamy, M. Y. Sfeir, D. N. Congreve and L. M. Campos, Adv. Mater., 2017, 258, 1701416 CrossRef PubMed.
- S. A. Odom, S. R. Parkin and J. E. Anthony, Org. Lett., 2003, 5, 4245–4248 CrossRef CAS PubMed.
- D. M. Kroupa, D. H. Arias, J. L. Blackburn, G. M. Carroll, D. B. Granger, J. E. Anthony, M. C. Beard and J. C. Johnson, Nano Lett., 2018, 18, 865–873 CrossRef CAS PubMed.
- Y.-F. Lim, Y. Shu, S. R. Parkin, J. E. Anthony and G. G. Malliaras, J. Mater. Chem., 2009, 19, 3049 RSC.
- J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef CAS PubMed.
- J. Löberg, J. Perez Holmberg, I. Mattisson, A. Arvidsson and E. Ahlberg, Int. J. Dent., 2013, 2013, 1–14 CrossRef PubMed.
- H. L. Stern, A. J. Musser, S. Gelinas, P. Parkinson, L. M. Herz, M. J. Bruzek, J. Anthony, R. H. Friend and B. J. Walker, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7656–7661 CrossRef CAS PubMed.
- S. Reineke and M. A. Baldo, Sci. Rep., 2014, 4, 391 Search PubMed.
- D. C. Coffey, B. W. Larson, A. W. Hains, J. B. Whitaker, N. Kopidakis, O. V. Boltalina, S. H. Strauss and G. Rumbles, J. Phys. Chem. C, 2012, 116, 8916–8923 CAS.
- D. Cahen, G. Hodes, M. Grätzel, J. F. Guillemoles and I. Riess, J. Phys. Chem. B, 2000, 104, 2053–2059 CrossRef CAS.
- R. D. Pensack, E. E. Ostroumov, A. J. Tilley, S. Mazza, C. Grieco, K. J. Thorley, J. B. Asbury, D. S. Seferos, J. E. Anthony and G. D. Scholes, J. Phys. Chem. Lett., 2016, 7, 2370–2375 CrossRef CAS PubMed.
- J. Herz, T. Buckup, F. Paulus, J. Engelhart, U. H. F. Bunz and M. Motzkus, J. Phys. Chem. Lett., 2014, 5, 2425–2430 CrossRef CAS PubMed.
- B. J. Walker, A. J. Musser, D. Beljonne and R. H. Friend, Nat. Chem., 2013, 5, 1019–1024 CrossRef CAS PubMed.
- T. Sakanoue and H. Sirringhaus, Nat. Mater., 2010, 9, 736–740 CrossRef CAS PubMed.
- X. Xu, B. Shan, S. Kalytchuk, M. Xie, S. Yang, D. Liu, S. V. Kershaw and Q. Miao, Chem. Commun., 2014, 50, 12828–12831 RSC.
- H. L. Stern, A. Cheminal, S. R. Yost, K. Broch, S. L. Bayliss, K. Chen, M. Tabachnyk, K. Thorley, N. Greenham, J. M. Hodgkiss, J. Anthony, M. Head-Gordon, A. J. Musser, A. Rao and R. H. Friend, Nat. Chem., 2017, 1–8 CAS.
- E. O'Driscoll, J. D. Simon and K. S. Peters, J. Am. Chem. Soc., 1990, 112, 7091–7098 CrossRef.
- T. Berger, J. A. Anta and V. Morales-Flórez, Phys. Chem. Chem. Phys., 2013, 15, 13790 RSC.
- Y. Liu, J. R. Jennings, X. Wang and Q. Wang, Phys. Chem. Chem. Phys., 2013, 15, 6170 RSC.
- C. Prasittichai, J. R. Avila, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 16328–16331 CrossRef CAS PubMed.
- S. Feng, Q.-S. Li, P.-P. Sun, T. A. Niehaus and Z.-S. Li, ACS Appl. Mater. Interfaces, 2015, 7, 22504–22514 CAS.
- Y. Bai, J. Zhang, Y. Wang, M. Zhang and P. Wang, Langmuir, 2011, 27, 4749–4755 CrossRef CAS PubMed.
- B. Roose, S. Pathak and U. Steiner, Chem. Soc. Rev., 2015, 1–24 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Steady-state UV-VIS and PL, solution transient absorption, X-ray diffraction, decay associated spectra, and TIPS Tc COOH/Al2O3/TiO2 film kinetics. See DOI: 10.1039/c7sc04688j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |