
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNHC and nucleophile chelation effects on reactive iron(II) species in alkyl–alkyl cross-coupling†
Valerie E.
Fleischauer‡
,
Salvador B.
Muñoz III‡
,
Peter G. N.
Neate
,
William W.
Brennessel
 and
Michael L.
Neidig
*
and
Michael L.
Neidig
*
Department of Chemistry, University of Rochester, Rochester, New York 14627, USA. E-mail: neidig@chem.rochester.edu
First published on 8th January 2018
Abstract
While iron–NHC catalysed cross-couplings have been shown to be effective for a wide variety of reactions (e.g. aryl–aryl, aryl–alkyl, alkyl–alkyl), the nature of the in situ formed and reactive iron species in effective catalytic systems remains largely undefined. In the current study, freeze-trapped Mössbauer spectroscopy, and EPR studies combined with inorganic synthesis and reaction studies are utilised to define the key in situ formed and reactive iron–NHC species in the Kumada alkyl–alkyl cross-coupling of (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide and 1-iodo-3-phenylpropane. The key reactive iron species formed in situ is identified as (IMes)Fe((1,3-dioxan-2-yl)ethyl)2, whereas the S = 1/2 iron species previously identified in this chemistry is found to be only a very minor off-cycle species (<0.5% of all iron). Reaction and kinetic studies demonstrate that (IMes)Fe((1,3-dioxan-2-yl)ethyl)2 is highly reactive towards the electrophile resulting in two turnovers with respect to iron (kobs > 24 min−1) to generate cross-coupled product with overall selectivity analogous to catalysis. The high resistance of this catalytic system to β-hydride elimination of the alkyl nucleophile is attributed to its chelation to iron through ligation of carbon and one oxygen of the acetal moiety of the nucleophile. In fact, alternative NHC ligands such as SIPr are less effective in catalysis due to their increased steric bulk inhibiting the ability of the alkyl ligands to chelate. Overall, this study identifies a novel alkyl chelation method to achieve effective alkyl–alkyl cross-coupling with iron(II)–NHCs, provides direct structural insight into NHC effects on catalytic performance and extends the importance of iron(II) reactive species in iron-catalysed cross-coupling.
Introduction
Transition-metal-catalysed cross-coupling chemistry has solved countless problems in total synthesis, pharmaceutical chemistry, and the production of fine chemicals. Traditionally these reactions have been carried out with precious metals, but lower cost, low toxicity, and rich redox chemistry make iron-catalysed C–C cross-coupling an attractive alternative.1–4 Towards this goal, systems using iron and N-heterocyclic carbenes (NHCs) have been shown to facilitate a wide variety of cross-coupling reactions5–7 including biaryl,8–11 alkyl–aryl,12–22 alkyl–alkyl,23 and aryl–alkenyl (Scheme 1).24 A key example is early work by Bedford and co-workers which demonstrated the use of monodentate NHCs in the catalytic cross-coupling of aryl Grignard reagents and alkyl halides in high yield using well-defined iron–NHC complexes (Scheme 1d).25 Also of note is the selective biaryl cross-coupling system using SIPr·HCl and FeF3·3H2O developed by Nakamura and co-workers (Scheme 1b).8,26 In this case, other NHC complexes and iron starting materials resulted in poor yields of cross-coupled product with the exception of FeCl3 in the presence of KF.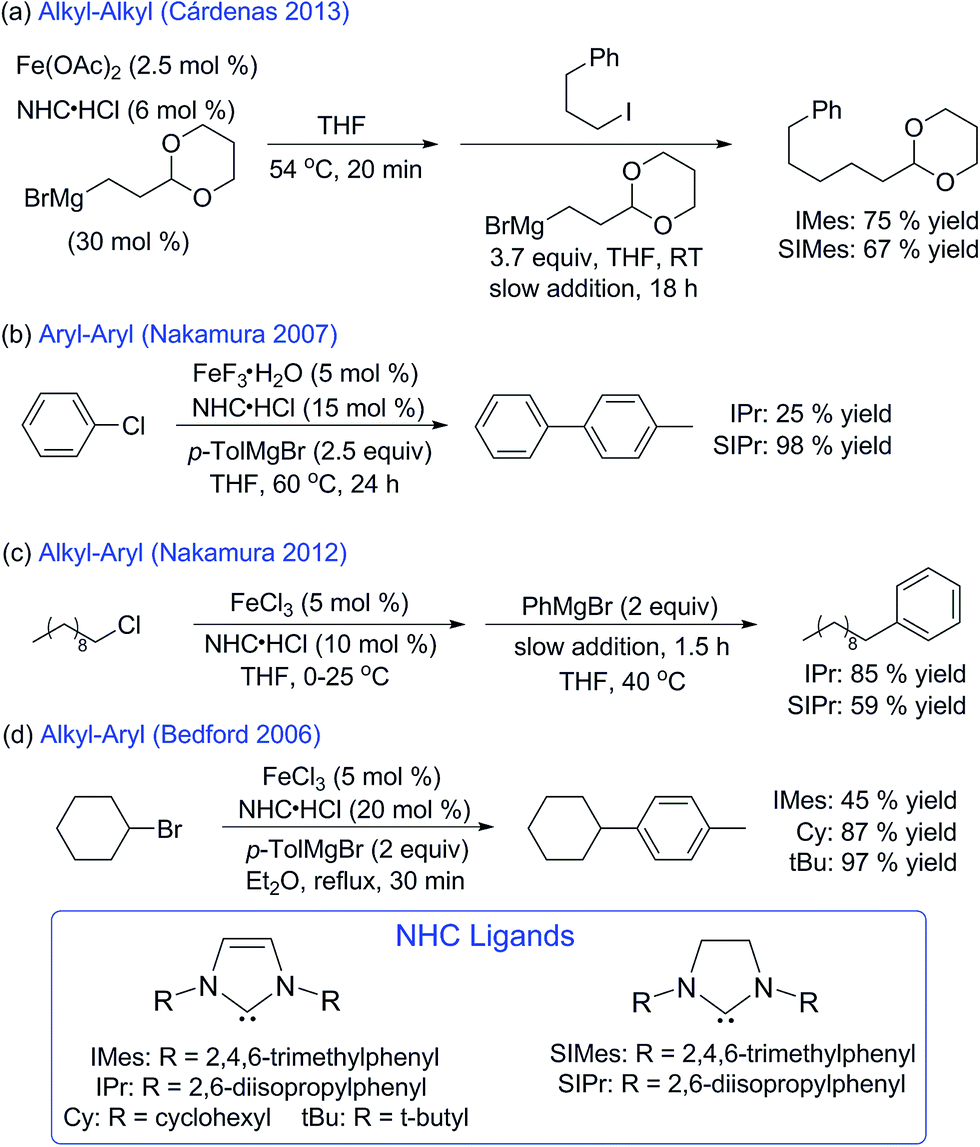 | ||
| Scheme 1 Examples of iron–NHC catalysed cross-coupling reactions. | ||
Despite these recent advances in the development of effective iron–NHC systems for cross-coupling and isolation of transmetalated iron–NHC's,27,28 active catalyst structure and mechanism in these reactions remain poorly defined. In fact, the most detailed insight available to date on iron–NHCs for cross-coupling has focused on stoichiometric reactions of well-defined iron–NHC complexes. For example, Deng and co-workers reported some of the first mechanistic investigations of iron-catalysed cross-coupling reactions with NHCs.29–32 These studies included the synthesis of well-defined (NHC)2FeR2 (R = alkyl or aryl) complexes and reactivity of well-defined (NHC)2Fe(aryl)2 complexes with alkyl halides, resulting in a proposed iron(II/III) cycle with a bis-arylated iron–NHC intermediate as the likely reactive species towards electrophile.33 While these stoichiometric reactions provide initial insight into the cross-coupling reactivity of well-defined iron–NHCs, it is unclear if similar reactive species are formed in situ in effective catalytic reactions.
While studies across the breadth of iron–NHC catalysed cross-couplings are required to define the variation in iron–NHC active species that might exist, an important system for initial investigation is the iron–NHC catalysed alkyl–alkyl cross-coupling reaction reported by Cárdenas and co-workers due to the challenges that exist for effective alkyl–alkyl cross-couplings.23 In this reaction, 1-iodo-3-phenylpropane is coupled to an alkyl Grignard reagent containing an acetal group using Fe(OAc)2 and IMes·HCl with 75% product yield. In contrast to the use of NHC ligands, Xantphos as a ligand system only results in 27% product yield.23 This is a dramatic contrast to the other two existing alkyl–alkyl cross-coupling systems with iron by Chai34 and Nakamura35 where Xantphos is uniquely effective as a ligand additive for the production of cross-coupled product. Based upon GC monitoring of homocoupled nucleophile upon pre-catalyst quenching, radical clock substrate experiments, and electron paramagnetic resonance spectroscopy (EPR), Cárdenas proposed an iron(I/III) catalytic cycle with an iron(I) active species. While these initial mechanistic studies by Cárdenas represent the most detailed study of a catalytic iron–NHC cross-coupling system to date, several issues remain. For example, no spin quantification of the EPR studies was reported, and it is therefore unclear what amount of S = 1/2 iron is formed in situ. Additionally, no direct structural evidence was provided in support of the proposed catalytic cycle, nor was any direct reactivity of a S = 1/2 species with electrophile evaluated to determine if it reacts selectively or at a catalytically competent rate. More studies are required to unambiguously determine the nature of the key iron species in iron–NHC catalysed alkyl–alkyl cross-coupling.
Recently, our group has applied a series of inorganic spectroscopic techniques to study the mechanism of iron–bisphosphine catalysed cross-coupling reactions involving the coupling of alkyl electrophiles with aryl and alkynyl Grignard reagents using FeCl2(SciOPP) as a pre-catalyst.36–38 Magnetic circular dichroism (MCD), EPR, and Mössbauer spectroscopic studies of freeze-trapped solutions of in situ generated iron species were able to identify the catalytically active iron complexes in these reactions. Importantly, EPR and Mössbauer spectroscopies allow for the quantification of iron species during catalysis.39 The combination of these techniques with inorganic synthesis, GC analysis, and density functional theory (DFT) investigations is a powerful method for identifying the active iron species in catalysis, providing insight into the mechanism of catalysis and defining the role of reaction protocols in promoting high cross-coupling yields.
In the current study, this physical-inorganic approach has identified the key in situ formed reactive iron(II)–NHC species in this alkyl–alkyl cross-coupling as (IMes)Fe((1,3-dioxan-2-yl)ethyl)2. While a S = 1/2 iron species was previously proposed as the reactive species in this chemistry, spin quantitated EPR has shown that it is only present as <0.5% of all iron in solution and represents an off-cycle species. In (IMes)Fe((1,3-dioxan-2-yl)ethyl)2, the acetal moieties of the alkyls create two weakly coordinating metallacycles, protecting against β-hydride elimination. In turn, further studies provided direct structural insight into NHC effects on cross-coupling performance and explore alternative nucleophiles with potential chelating substituents in order to broaden the accessible alkyl nucleophiles for effective cross-coupling.
Experimental
General considerations
All general experimental methods, NMR, characterisation including collection and refinement of X-ray data, and synthesis of alternative nucleophiles may be found in the ESI.†Preparation of (IMes)Fe((1,3-dioxan-2-yl)ethyl)2 (1)
To a 20 mL scintillation vial was added Fe(OAc)2 (75 mg, 0.43 mmol) and 1,3-dimesitylimidazolium chloride (150 mg, 0.44 mmol) along with a stir bar. To the solid mass was added (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide in THF (0.5 M, 0.86 mL, 0.43 mmol) dropwise while stirring at room temperature. After 10 min an additional equivalent of the Grignard reagent in THF (0.5 M, 0.86 mL, 0.43 mmol) was added to the tan slurry. A third equivalent of Grignard reagent in THF (0.5 M, 0.86 mL, 0.43 mmol) was added to the yellow solution 10 minutes later. The final equivalent of Grignard reagent was added after 3 more minutes and the yellow reaction mixture stirred at room temperature for 2 min followed by cooling to −78 °C with continued stirring. The yellow slurry was then warmed on a room temperature stir plate for five min after which room temperature pentane (ca. 14 mL) was added to the mixture and the vial cooled back to −78 °C. The resulting solution was filtered through Celite (precooled to −78 °C) and the collected filtrate was stored at −80 °C for one week rendering yellow crystals. Yellow crystals were carefully selected at low temperature in batches from several reactions with varying crystallisation yields. Evans method analysis of crystalline 1 redissolved in THF-d8 indicates μeff = 5.2(3) μb. Elemental Anal. Calcd: C, 67.11; H, 7.85; N, 4.74. Found: C, 67.09; H, 8.023; N, 4.692.Preparation of (IMes)FeBr((1,3-dioxan-2-yl)ethyl) (2) and (IMes)FeBr2(THF) (3)
To a solution of Fe(OAc)2 (80 mg, 0.46 mmol) and 1,3-dimesitylimidizolium chloride (346 mg, 1.02 mmol) in THF was added (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide (0.5 M in THF, 0.39 mL, 1.94 mmol) at 55 °C in THF and stirred 20 min. The yellow solution was cooled to room temperature and solvent removed in vacuo. A 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diethyl ether and pentane was added to the pale yellow solid residue and stirred for 3 min. The solution was filtered through Celite resulting in a yellow clear solution and was stored at −30 °C for one week after which yellow crystals were isolated. Yellow crystals were carefully selected at low temperature in batches from several reactions with varying crystallisation yields. Evans method analysis of the crystalline mixture redissolved in THF-d8 yields μeff = 5.1 μb. Elemental Anal. Calcd (for 86:14 mixture of 2-Br and 3 with co-crystallised ether and pentane): C, 57.96; H, 6.96; N, 4.48. Found: C, 57.84; H, 6.52; N, 4.60.
:1 mixture of diethyl ether and pentane was added to the pale yellow solid residue and stirred for 3 min. The solution was filtered through Celite resulting in a yellow clear solution and was stored at −30 °C for one week after which yellow crystals were isolated. Yellow crystals were carefully selected at low temperature in batches from several reactions with varying crystallisation yields. Evans method analysis of the crystalline mixture redissolved in THF-d8 yields μeff = 5.1 μb. Elemental Anal. Calcd (for 86:14 mixture of 2-Br and 3 with co-crystallised ether and pentane): C, 57.96; H, 6.96; N, 4.48. Found: C, 57.84; H, 6.52; N, 4.60.
Preparation of (SIPr)Fe((1,3-dioxan-2-yl)ethyl)2 (4)
To a scintillation vial was added Fe(OAc)2 (33 mg, 0.19 mmol) and 1,3-bis(2,6-diisopropylphenyl)imidazolinium chloride (81 mg, 0.19 mmol) along with a stir bar. To the solid mass was added (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide in THF (0.5 M, 2.28 mL, 1.14 mmol) over the course of two minutes while stirring at room temperature. The yellow reaction mixture was stirred at room temperature for an additional 10 minutes followed by cooling to −78 °C. The yellow slurry was then warmed on a room temperature stir plate for five minutes while stirring after which room temperature pentane (ca. 18 mL) was added to the mixture and the vial cooled back to −78 °C. The resulting solution was filtered through Celite (precooled to −78 °C) and the yellow filtrate stored at −80 °C for one week rendering yellow crystals. Yellow crystals were carefully selected at low temperature in batches from several reactions with varying crystallisation yields. Evans method analysis of crystalline 4 redissolved in THF-d8 indicates μeff = 5.0(1) μb. Elemental Anal. Calcd for C39H60FeN2O4·0.1MgBr2: C 67.38, H 8.70, N 4.03. Found: C 67.39, H 8.97, N 3.98.Mössbauer spectroscopy
Solid samples for 57Fe Mössbauer spectroscopy were made with isolated complexes based on natural abundance of 57Fe or from 57Fe-enriched samples of isolated complexes. Frozen solution samples for 57Fe Mössbauer spectroscopy were prepared from 57Fe(OAc)2 or isolated 57Fe-enriched complexes. All samples were prepared in an inert atmosphere glovebox with a liquid nitrogen fill port to freeze each sample at 77 K within the glovebox. Each sample was loaded in to a Delrin Mössbauer sample cup for measurements and loaded under liquid nitrogen. Low temperature 57Fe Mössbauer measurements were performed using a See Co. MS4 Mössbauer spectrometer integrated with a Janis SVT-400T He/N2 cryostat for measurements at 80 K or 5 K. Isomer shifts were determined relative to α-Fe at 298 K. All Mössbauer spectra were fit using the program WMoss (See Co).Electron paramagnetic resonance
Catalytic reaction solution samples for EPR spectroscopy were prepared directly from the reaction mixture as reported in the literature23 under N2 atmosphere. In situ reaction samples were prepared directly from the reactions as described. All samples for EPR spectroscopy were prepared in an inert atmosphere glovebox equipped with a liquid nitrogen fill port to enable sample freezing to 77 K within the glovebox. EPR samples were prepared in 4 mm OD Suprasil quartz EPR tubes from Wilmad Labglass. Samples for spin integration utilised high precision suprasil quartz tubes to allow for direct comparison of intensities between different samples. X-band EPR spectra were recorded on a Bruker EMXplus spectrometer equipped with a 4119HS cavity and an Oxford ESR-900 helium flow cryostat. The instrumental parameters employed for all samples were as follows: 1 mW power; time constant 41 ms; modulation amplitude 8 G; 9.38 GHz; modulation frequency 100 kHz. Samples exhibiting S = 1/2 EPR spectra were spin integrated using a 3 mM CuSO4 standard under non-saturating conditions. Identical instrumentation parameters were used for both the iron and standard samples.Electronic structure calculations
Spin unrestricted density functional theory (DFT) calculations were performed with the Gaussian 09 package.40 All geometry optimisation calculations were performed with the PBEPBE functional with the TZVP basis set on all atoms and inclusion of solvation effects using the polarised continuum model (PCM) with THF as the solvent.41–43 The dispersion correction of Grimme (GD2) was used in geometry optimisations of all complexes.44 The geometries of all complexes were fully optimised starting from X-ray crystal structures. All optimised geometries had frequencies found to be positive. Energies are given in the ESI† and include zero-point and thermal corrections.Further calculations of molecular orbitals (MOs) and TD-DFT used the B3LYP functional with the TZVP basis set on all atoms. The analysis of MO compositions and Mayer bond orders were performed using the AOMix program.45,46 Orbitals from the Gaussian calculations were plotted with the ChemCraft program.
Reactions of Fe(OAc)2 with IMesHCl, Grignard reagent, and electrophile for pre-catalyst and in situ reaction studies
As an example of the general procedure performed; 57Fe(OAc)2 (0.010 mmol) was combined with IMes·HCl (0.021 mmol) in THF at 55 °C in THF (2 mL) and alkyl Grignard reagent was added quickly (0.122 mmol, 12 equiv. to iron), dropwise, to form the pre-catalyst. The reaction was cooled to 23 °C after 20 min and 1-iodo-3-phenylpropane was added (0.400 mmol). Mössbauer or EPR samples were then prepared at various time points (1 min, 20 min, etc.) and frozen in liquid nitrogen to freeze quench the reaction at the desired time. When GC studies were performed, dodecane was added to the reaction to match the molarity of electrophile after it was cooled to room temperature. Reactions were quenched using 0.1 mM HCl in THF and filtered through a Florisil pad. Cross-coupled product yields were determined from quantitative GC analysis.Reactions of (IMes)57Fe((1,3-dioxan-2-yl)ethyl)2 with alkyl iodide
(IMes)57Fe((1,3-dioxan-2-yl)ethyl)2·2(THF) (7.3 mg, 0.01 mmol) was reacted with x equiv. of 1-iodo-3-phenylpropane in 2 mL THF at 23 °C (x = 0.7, 20). Time points for GC were quenched using 0.5 M HCl in THF using dodecane as an internal standard. Freeze trapped Mössbauer samples were prepared as previously described.Reactions of (IMes)57FeBr((1,3-dioxan-2-yl)ethyl) with Grignard reagent and alkyl iodide for in situ spectroscopic studies
(IMes)57FeBr((1,3-dioxan-2-yl)ethyl) (2.80 mg, 0.005 mmol) was reacted with 1.0 equiv. of (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide (0.5 M in THF) and 20 equiv. of 1-iodo-3-phenylpropane in 1 mL THF at 23 °C. After stirring at room temperature the reaction was freeze quenched in liquid nitrogen in inert atmosphere as previously described at various time points for Mössbauer spectroscopy. GC studies were also performed with addition of dodecane and workup as previously described.Results and analysis
Pre-catalyst formation and characterisation
The iron–NHC catalysed Kumada alkyl–alkyl cross-coupling reaction is composed of three major steps: (1) in situ formation of a pre-catalyst at elevated temperature, (2) addition of electrophile at room temperature, and (3) slow addition of Grignard reagent at room temperature. To complete a rigorous mechanistic investigation of this reaction each step was evaluated using in situ freeze-trapped EPR and 57Fe Mössbauer spectroscopy. Initial investigations focused on the identification of the iron species formed in the pre-catalyst formation step, where Cárdenas and co-workers previously reported that a S = 1/2 iron(I) active species is generated.23 Following the reported procedure, 12 equiv. of (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide were added to Fe(OAc)2 and IMes·HCl in THF (1.0 and 2.1 equiv. respectively) at 54 °C and stirred for 20 min, forming a bright yellow solution. The 5 K Mössbauer spectrum of the reaction mixture freeze-trapped at the end of the 20 min pre-catalyst reaction indicated the presence of one major iron species in solution (1), with Mössbauer parameters of δ = 0.57 mm s−1 and ΔEQ = 2.33 mm s−1 (78%, blue, Fig. 1A) that is stable at this elevated temperature for over an hour before any significant decomposition occurs. A minor species is also present in the pre-catalyst Mössbauer spectrum with parameters of δ = 0.50 mm s−1 and ΔEQ = 2.70 mm s−1 (22%, orange, Fig. 1A). Spin quantitated X-band EPR indicates that the same S = 1/2 species reported by Cárdenas also forms in the pre-catalyst reaction (see ESI†) but represents <0.5% of all iron in solution and, hence, is not observable by Mössbauer spectroscopy. | ||
| Fig. 1 Iron–NHC pre-catalyst formation. (A) 5 K Mössbauer spectrum of the pre-catalyst reaction iron products in frozen THF solution. The data is shown as black dots and individual fit components are given by blue (1) and orange (1-THF) lines. (B) Single crystal X-ray crystal structure of (IMes)Fe(1,3-dioxan-2-ylethyl)2 (1) with thermal ellipsoids shown at the 50% probability level and selected bond distances and angles given, H-atoms were omitted for clarity. | ||
Single crystal X-ray crystallographic studies were pursued in order to unambiguously assign the structure of the major iron species 1 formed in the pre-catalyst reaction. Slow addition of 4 equiv. of (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide to a mixture of Fe(OAc)2 and IMes·HCl (1 equiv. of each) in THF at room temperature yielded yellow crystals suitable for single crystal X-ray diffraction. The single crystal X-ray structure of 1 corresponds to (IMes)Fe((1,3-dioxan-2-yl)ethyl)2 (Fig. 1B). The structure can be described as a trigonal bipyramidal iron(II) complex with an Fe–IMes bond length of 2.1222(12) Å. The iron–carbon bond lengths to the alkyl groups are 2.0994(13) and 2.1062(12) Å. Additional iron–oxygen interactions from the acetal on the alkyl group are at distances of 2.4632(9) and 2.4428(9) Å, forming a set of five membered rings with iron. Evans method analysis of crystalline 1 dissolved in THF-d8 indicates the presence of high-spin, S = 2 iron(II) for the solution (μeff = 5.2(3) μb), consistent with the Mössbauer parameters of 1 (vide infra).
Spin unrestricted DFT calculations were utilised to further analyse the bonding in 1. Geometry optimisations with the PBEPBE functional and TZVP basis set yielded calculated structures in good agreement with crystallographic data in gas phase and THF solvent model cases (see Table 1 and ESI†). Additionally, molecular orbitals were evaluated using B3LYP/TZVP in the gas phase (see ESI†), placing emphasis on the occupied and unoccupied frontier molecular orbitals (FMOs) of the β manifold to describe the major contributions to bonding. Complex 1 exhibits dominant Fe d orbital character in β156 (HOMO, 84% dxy), β162 (LUMO + 5, 80% dxz), β167 (LUMO + 7, 68% dz2) and β169 (LUMO + 12, 42% dx2−y2). β157 (LUMO) and β161 (LUMO + 4) both show diffuse Fe dyz orbital character (16 and 12% respectively) with a strong π-bonding interaction to the carbene carbon of the IMes ligand. The highest occupied ligand based FMO is β155 (HOMO − 1), exhibiting σ bonding interactions between the alkyl carbons and dxy contributions. Mayer bond order analysis was carried out from optimised gas phase, solvent model, and crystal structure coordinates to quantify the interaction between iron and oxygen of the acetal alkyl substrate. In each of the three cases, very small bond orders between iron and the coordinating oxygen of the alkyl ligand (e.g. 0.071 and 0.099 for the gas phase model, Table 1) were found. This indicates the presence of very weak Fe–O interactions, consistent with the elongated Fe–O distances observed crystallographically.
| Complex | Bond lengths (Å) | Mayer bond order | ||||
|---|---|---|---|---|---|---|
| Fe–O interaction (Å) | Gas phase calcd | Solution calcd | Exptla | Gas phase calcd | Solution calcd | |
| a Mayer bond order calculated directly from crystallographic coordinates. | ||||||
| 1 | 2.4632(9) | 2.4193 | 2.4052 | 0.042 | 0.071 | 0.042 |
| 2.4428(9) | 2.3939 | 2.4251 | 0.046 | 0.099 | 0.047 | |
| 2-Br | 2.181(4) | 2.2468 | 2.2555 | 0.224 | 0.224 | 0.176 |
Mössbauer spectroscopy of crystalline (IMes)Fe((1,3-dioxan-2-yl)ethyl)2 confirms that this complex represents the major iron species 1 observed to form in situ in the pre-catalyst reaction. The 5 K Mössbauer spectrum of solid 1 is represented by a single quadrupole doublet with parameters δ = 0.57 mm s−1 and ΔEQ = 2.43 mm s−1, where the small change in ΔEQ in the solid state spectrum suggests a slight structural distortion between solid and solution states (see ESI†). Consistent with this hypothesis, dissolution of 57Fe-enriched 1 in THF solution results in a Mössbauer spectrum (see ESI†) analogous to that previously observed for the pre-catalyst reaction mixture containing two major iron species: δ = 0.57 mm s−1 and ΔEQ = 2.33 mm s−1 (1, 81%, blue), and δ = 0.50 mm s−1 and ΔEQ = 2.70 mm s−1 (19%, green). Dissolution of 57Fe-enriched 1 in 2-MeTHF results in only a single major iron species in frozen solution by Mössbauer spectroscopy with parameters corresponding to 1 (see ESI†), enabling the assignment of the minor δ = 0.50 mm s−1 and ΔEQ = 2.70 mm s−1 component as the THF adduct of 1 (1-THF). Thus, the pre-catalyst reaction results in the formation of 1 and 1-THF with only a trace amount of S = 1/2 iron also present.
Due to the elevated temperature utilised in the reported pre-catalyst reaction step by Cárdenas and co-workers, it was also important to consider whether 1 and 1-THF could be formed in situ at room temperature or at shorter time points. In fact, the addition of 12 equiv. of (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide to Fe(OAc)2 and IMes·HCl in THF (1.0 and 2.1 equiv. respectively) at 54 °C for 1 min or room temperature with a 1 h reaction time generates an analogous mixture of 1 and 1-THF (Fig. 2) and <0.5% of S = 1/2 iron by spin quantitated EPR (see ESI†) demonstrating that elevated temperature is not required to form the key iron(II) species. Even after 2 h of reaction at room temperature, the distribution of iron species remains constant with no additional S = 1/2 demonstrating the stability of these complexes.
 | ||
| Fig. 2 5 K Mössbauer spectrum of the pre-catalyst reaction iron products in frozen THF solution following formation at RT. The data is shown as black dots and individual fit components are given by blue (1, 76% of total iron) and orange (1-THF, 24% of total iron) lines. | ||
This result indicated that effective cross-coupling might be achieved by performing all reaction steps at room temperature. Pre-catalyst reaction at room temperature for 1 h followed by the addition of electrophile and subsequent slow addition of Grignard reagent was found to give 81% cross-coupled product. This yield is analogous to those achieved when pre-catalyst treatment was carried out at 54 °C for either 1 or 20 min (∼79%, Table 2). Thus, the elevated temperature pre-catalyst treatment in the original method is clearly not required. It should also be noted that further evaluation of the original catalytic protocol determined slow addition of 1.23 equiv. of Grignard reagent (with respect to electrophile) over 6.25 h, as opposed to 3.7 equiv. over 18 h, gave analogous cross-coupled product yields to the originally reported protocol.23
|
|
|||||
|---|---|---|---|---|---|
| NHC | Pre-treatment T (°C) | Pre-treatment time (min) | A (%) | B (%) | C (%) |
| IMes·HCl | 54 | 1 | 80 | 14 | 6 |
| IMes·HCl | 54 | 20 | 78 | 15 | 7 |
| IMes·HCl | RT | 60 | 81 | 14 | 6 |
| SIPr·HCl | 54 | 1 | 15 | 59 | 26 |
| SIPr·HCl | 54 | 20 | 23 | 56 | 21 |
While 1 and 1-THF represent nearly all iron species formed in the pre-catalyst reaction (>99.5% at either RT or 54 °C) and spin quantitated EPR indicated a very minor S = 1/2 component (<0.5%), Cárdenas and co-workers also reported that a large amount of homocoupled nucleophile was also formed in the pre-catalyst reaction. Previous studies from our group on iron–SciOPP catalysed phenyl-alkyl cross-coupling have demonstrated that homocoupled nucleophile can form as a result of the chemical quench of transmetalated iron(II)–SciOPP species with the sample preparation used for GC analysis.36 To test whether a similar issue might be present in the current system, isolated crystalline 1 was re-dissolved in THF at 54 °C and quickly quenched for GC analysis using the same procedure reported by Cárdenas in his homocoupling experiment. Large amounts of homocoupled alkyl nucleophile were observed by GC analysis following the chemical quenching procedure (∼0.7 equiv. homocoupled nucleophile with respect to 1). Consistent with previous iron–SciOPP studies, these results demonstrate that correlation of homocoupled nucleophile to iron oxidation states in solution can be unreliable as the chemical quenching of alkylated iron(II) species can result in the formation of homocoupled nucleophile. As expected, quantitated spectroscopic methods that directly assess iron speciation and electronic structure are much more reliable means for evaluating in situ formed iron speciation than quantitation of organic products that can artificially form because of the quenching procedures used for GC analysis.
Formation and characterisation of undertransmetalated iron–NHCs
Following the identification of the bis-transmetalated iron(II)–IMes complex 1 as the major iron species formed in the pre-catalyst reaction, it was clear that a related mono-transmetalated iron(II)–IMes complex could also be accessible. In order to form this type of species, a minimal amount of alkyl Grignard reagent was utilised. 4.2 equiv. alkyl nucleophile was added to a mixture of 1 equiv. 57Fe(OAc)2 and 2.2 equiv. IMes·HCl in THF at 54 °C followed by removal of solvent in vacuo. The residues were extracted with 1:1 ether:pentane, filtered and stored at −30 °C yielding yellow crystals suitable for X-ray analysis. X-ray crystallography confirmed the formation of (IMes)FeBr((1,3-dioxan-2-yl)ethyl) (2-Br) (Fig. 3A) co-crystallised with (IMes)FeBr2(THF) (3) in an ∼86:14 ratio (Fig. 3B). For both species, the bromide ligand derives from the magnesium salt formed upon transmetalation with the Grignard reagent via anion exchange. The solid state structure of 2-Br is described as having an Fe–IMes bond length of 2.096(4) Å, slightly decreased compared to the Fe–IMes bond length observed for 1 (2.1222(12) Å). The Fe–C bond to the alkyl in 2-Br has a length of 2.016(12) Å (compared to 2.0994(13) and 2.1062(12) Å for Fe–Calkyl interactions in 1). Notably, the Fe–O bond length to the oxygen on the acetal for 2-Br is 2.181(4) Å, approximately 0.25 Å shorter than that of the Fe–O interactions in the same 5-membered metallacycle of 1. The bishalide complex 3 is also well-described as a distorted tetrahedron with Fe–Br bond lengths of 2.4184(8) and 2.329(6), which are slightly elongated compared to other isolated bishalide iron(II)–NHC complexes (e.g. 2.310 and 2.992 Å in (IMes)2FeCl2).29,47,48
 | ||
| Fig. 3 X-ray structural and Mössbauer analysis of 2-Br and 3. X-ray crystal structures of (A) (IMes)FeBr(1,3-dioxan-2-ylethyl) (2-Br) and (B) (IMes)FeBr2(THF) (3). For both structures, thermal ellipsoids shown at the 50% probability level and selected bond distances and angles given, H atoms are omitted for clarity. Note that these species co-crystallise in an ∼86:14 ratio of 2-Br:3. (C) 5 K Mössbauer spectrum of 57Fe-enriched crystals containing 2-Br and 3 redissolved in THF. The data is shown as black dots and individual fit components are given by green (2-Br) and red (3) lines. | ||
The 5 K frozen solution Mössbauer spectrum of 57Fe-enriched crystals used for X-ray crystallography reflected the co-crystallised iron complexes when re-dissolved in THF (Fig. 3C) with Mössbauer parameters of δ = 0.67 mm s−1, ΔEQ = 3.00 mm s−1 (88%, green) and δ = 0.90 mm s−1, ΔEQ = 3.18 mm s−1 (12% red). These are similar to the ratios observed in the X-ray data and, hence, are assigned to 2-Br and 3, respectively (see Table 3 for comparison to previously reported bishalide and mono- and bis-transmetalated iron–NHCs). Importantly, 2-Br is stable in solution and does not disproportionate in solution to form 1 and 3. Solid state Mössbauer spectroscopy of a sample of crystalline material also shows a mixture of 2-Br and 3 (see ESI†). Evans method analysis of the crystals redissolved in THF-d8 yields μeff = 5.1(3) μb. Thus, both 2 and 3 are high-spin, S = 2 iron(II) complexes, consistent with their Mössbauer parameters and distorted tetrahedral structures.
| Complex | Sample | δ (mm s−1) | ΔEQ (mm s−1) |
|---|---|---|---|
| This work (5 K) | |||
| 1 | Frozen soln | 0.57 | 2.34 |
| Solid | 0.57 | 2.42 | |
| 1-THF | Frozen soln | 0.50 | 2.70 |
| 2-Br | Frozen soln | 0.67 | 2.90 |
| Solid | 0.67 | 2.90 | |
| 3 | Frozen soln | 0.90 | 3.18 |
| Solid | 0.90 | 3.18 | |
| 4 | Frozen soln | 0.45 | 2.87 |
| Solid | 0.44 | 2.91 | |
|
|||
| Previously reported (80 K) | |||
| (IMes)2FeCl2 | Solid47 | 0.80 | 2.12 |
| (MeIPr)2FePhBr | Solid17 | 0.58 | 3.10 |
| (MeIPr)2FePh2 | Solid17 | 0.47 | 2.48 |
| (IPr)Fe(CH2TMS)2 | Solid47 | 0.34 | 1.04 |
The resulting gas phase and solvent model calculations of 2-Br produced structures with bond lengths and angles in good agreement with crystallographic data (see ESI†). FMO analysis reveals a ground state consisting of one occupied FMO, β142, with 84% Fe d orbital character (HOMO, dxy) and a weak π*-bonding interaction with the oxygen of the alkyl ligand. Fe d character is also observed in β143 (LUMO, 43% dyz), β148 (LUMO + 5, 79% dxz), β149(LUMO + 6, 63% dz2), and β151(LUMO + 8, 52% dx2−y2). Strong π interactions with the carbene of the IMes ligand are observed as shown in β143, matching the Fe dyz orbital observed in the LUMO of 1. Similar to 1, the highest occupied ligand based orbital in 2-Br is composed of a Fe d σ- bonding interaction with the alkyl carbon. Lastly, Mayer bond order analysis of the Fe–O interaction in 2-Br yields a MBO of 0.224 in the gas phase model (see Table 1 for other models), more than twice that observed in 1 for all models calculated, demonstrating a much stronger Fe–O interaction is present in the mono-alkyl complex (Table 1) consistent with the shorter Fe–O distance observed crystallographically in 2-Br.
Reactivity of (IMes)Fe((1,3-dioxan-2-yl)ethyl)2 (1) with electrophile
Since the bis-alkylated iron(II)–IMes complex 1 is the major iron species formed in the pre-catalyst step, it was important to evaluate its reactivity with electrophile. Following the reported catalytic protocol, after pre-catalyst formation the second reaction step involves the addition of excess electrophile (40 equiv. 1-iodo-3-phenylpropane) at 23 °C directly to the in situ formed pre-catalyst reaction mixture. The freeze trapped 57Fe Mössbauer spectrum of the reaction 1 min after electrophile addition shows that in situ formed 1 has completely reacted to form one major iron species with parameters of δ = 0.90 mm s−1, ΔEQ = 3.18 mm s−1 (100%, red) (see ESI†), similar to those for the iron bis-halide complex 3 (vide supra). GC analysis of the reaction mixture at the same time point after electrophile addition indicates the formation of 9% cross-coupled product and 8% allylbenzene with respect to electrophile, corresponding to a total of 8 turnovers with respect to iron. This is consistent with complete consumption of the excess Grignard reagent used in the formation step. Notably, spin quantitated EPR analysis indicates no consumption of the trace S = 1/2 species during this electrophile reaction (see ESI†). Thus, the S = 1/2 iron present following the pre-catalyst reaction is either unreactive or significantly less reactive than 1 and, hence, represents an off-cycle iron species in catalysis.Reactivity studies of isolated 1 with excess alkyl iodide (20 equiv.) in THF at 23 °C were also performed to determine the inherent reaction rate and selectivity in the absence of excess Grignard reagent in pseudo first order reaction conditions. Two turnovers are observed with respect to 1, resulting in a product distribution of 160% cross-coupled product (2-(5-phenylpentyl)-1,3-dioxane) (A), 20% allylbenzene (B) and 20% propylbenzene (D) (yields are with respect to iron, see Table 4). The observed product distribution for the direct reaction of 1 with excess electrophile (80% cross-coupled product with respect to electrophile) is consistent with that observed in catalysis (75% 2-(5-phenylpentyl)-1,3-dioxane, 12% allylbenzene, 9% propylbenzene, along with trace amounts of 1,6-diphenylhexane and 1-phenyl-1-propene). Both alkyl groups of 1 are ultimately consumed in this reaction, consistent with the formation of an Fe(IMes)X2 complex by Mössbauer spectroscopy with an isomer shift of 0.88 mm s−1 and quadrupole splitting of 3.32 mm s−1 (Fig. 4A). These parameters are similar to those observed for 3, consistent with a variation in halide from Br to I due to reaction in the absence of excess Br. The reaction is complete within five seconds, setting the lower limit of rate of reaction at 24 min−1 for each turnover. In the reported catalytic reaction, electrophile addition is followed by slow addition of the remaining Grignard reagent at a rate of only 8 equiv. to iron per hour, much slower than the rate of reaction of 1 with electrophile and, hence, rate limiting for catalysis.
|
|
||||||
|---|---|---|---|---|---|---|
| Formation of 1 | x equiv. R–I | TON | % yield with respect to iron | |||
| A | B | C | D | |||
| a 1 formed from the reaction of Fe(OAc)2 with 2.1 equiv. of IMes·HCl and 12 equiv. of (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide at 54 °C and cooled to 23 °C before addition of R–I. b 2-Br first reacted with 1 equiv. (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide at 23 °C for 1 min. | ||||||
| In situ | 40 | 8 | 360 | 320 | 0 | 0 |
| 2-Br +1 equiv. RMgBrb | 20 | 2 | 162 | 30 | 0 | 10 |
| Isolated | 20 | 2 | 160 | 20 | 0 | 20 |
| Isolated | 0.7 | 0.7 | 40 | 0 | 12 | 18 |
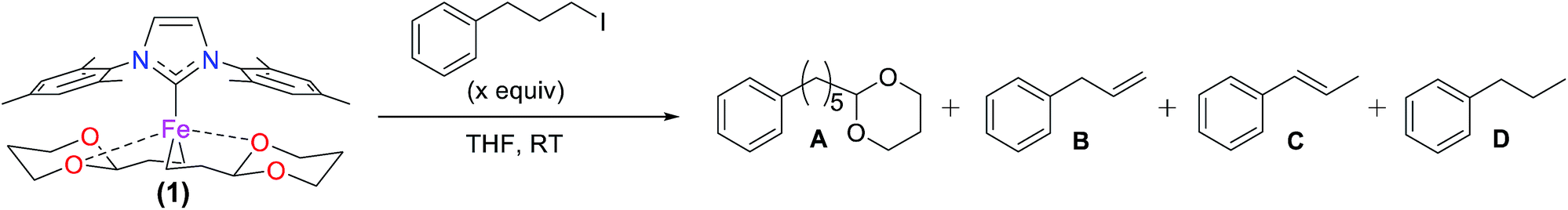
 | ||
| Fig. 4 In situ freeze trapped Mössbauer spectra of reactions of isolated 1 with excess and substoichiometric alkyl iodide. 5 K Mössbauer spectra of the iron speciation in solution following addition of (A) 20 equiv. of 1-iodo-3-phenylpropane to isolated 1 and (B) 0.7 equiv. of 1-iodo-3-phenylpropane to 1 at room temperature. | ||
Since the reaction of 1 with electrophile results in two total turnovers, it was important to evaluate whether a mono-alkylated iron species might form after an initial turnover as well as the relative selectivity of the first and second turnovers of 1 to generate product. Reaction of isolated 1 with 0.7 equiv. 1-iodo-3-phenylpropane for 1 min results in the complete consumption of electrophile and production of 60% cross-coupled product (A), 15% propylbenzene (D), and 24% phenylpropene (C) (with respect to electrophile) as determined by GC analysis, a lower product selectivity than observed at the end of both turnovers (∼80%, vide supra). In the same reaction, freeze-trapped in situ Mössbauer spectroscopy shows a distribution of two iron species in solution (Fig. 4B) with parameters of δ = 0.57 mm s−1 and ΔEQ = 2.33 mm s−1 (33%, blue, 1), δ = 0.67 mm s−1 and ΔEQ = 2.70 mm s−1 (67%, purple), the latter consistent with the iodine analogue of 2-Br due to halide exchange (denoted 2-I; note that when 0.7 equiv. of 1-bromo-3-phenylpropane are added to 1, 2-Br is observed to form by Mössbauer spectroscopy though at reduced rate, see ESI†). Thus, the two turnovers of 1 proceed through an intermediate mono-alkylated iron(II)–IMes species. Furthermore, the substoichiometric reaction with electrophile indicates that the first turnover of 1 is only ∼60% selective towards cross-coupled product. Since reaction of 1 with excess electrophile is ∼80% selective to cross-coupled product following two total turnovers (vide infra), the second turnover is highly selective.
Reactivity of (IMes)FeBr((1,3-dioxan-2-yl)ethyl) (2-Br) with alkyl iodide
After substoichiometric reactions of 1 with electrophile indicated the formation and reactivity of a mono-alkylated intermediate, the direct reactivity of 2-Br with 1-iodo-3-phenylpropane was evaluated. Following the addition of 20 equiv. of 1-iodo-3-phenylpropane in THF at 23 °C, no reactivity was observed by GC analysis over the course of 5 min. Consistent with this lack of reactivity, both in situ formed 2-Br during catalysis and redissolved crystalline mixtures of 2-Br and 3 show no ligand redistribution to form 1 and 3 from freeze-trapped solution Mössbauer. While 2-X species (X = Br, I) can be freeze-trapped as intermediates in the two observed turnovers of 1 to yield product, they are not the higher selectivity intermediate mono-alkylated species. It is likely that there is a different form of the mono-alkylated iron species that is reactive in solution but is transformed to 2-X upon freezing.One possibility would be an open form of 2-X resulting from reaction of 1 with electrophile, where the alkyl group from the Grignard reagent is not chelated to iron through oxygen on the acetal. However, all attempts to isolate this mono-alkylated species (by freeze-trapped analysis or crystallisation) have only resulted in the isolation of 2-Br.
Despite the lack of reactivity of 2-Br towards electrophile, 2-Br can react with additional Grignard reagent to generate the highly reactive bis-alkyl complex 1. Addition of 1 equiv. of (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide in THF at 23 °C to 2-Br results in an immediate colour change of the solution from pale to bright yellow, and Mössbauer spectroscopy indicates the formation of the pre-catalyst mixture of 1 (85%) and 1-THF (15%) (see ESI†).
In situ iron speciation with slow Grignard reagent addition
After identifying the key reactive species in the pre-catalyst reaction as the bis-alkylated iron(II)–IMes complex, the catalytic distribution of iron species present during catalysis was evaluated in order to determine the degree of transmetalation during catalysis and to probe for the potential formation of additional off cycle iron species. After formation of 1in situ, and addition of electrophile, 0.5 M (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide is slowly added at a rate of 0.16 mL h−1. Consistent with slow addition of nucleophile, 1 is not observed to reform during the catalytic reaction; Fig. 5 shows the presence of only 2-Br and 3 (18% and 82%, respectively) after the first 2 h of Grignard reagent addition. Spin quantified EPR shows the persistence of <0.5% S = 1/2 iron in solution during catalysis (see ESI†). Analogous iron speciation during slow nucleophile addition is also observed for catalysis performed using room temperature pre-catalyst formation (see ESI†). GC results indicate that upon completion of slow addition of Grignard reagent, 75% cross-coupled product, 9% allylbenzene, 9% 1-phenylpropene, and 7% n-propylbenzene are produced.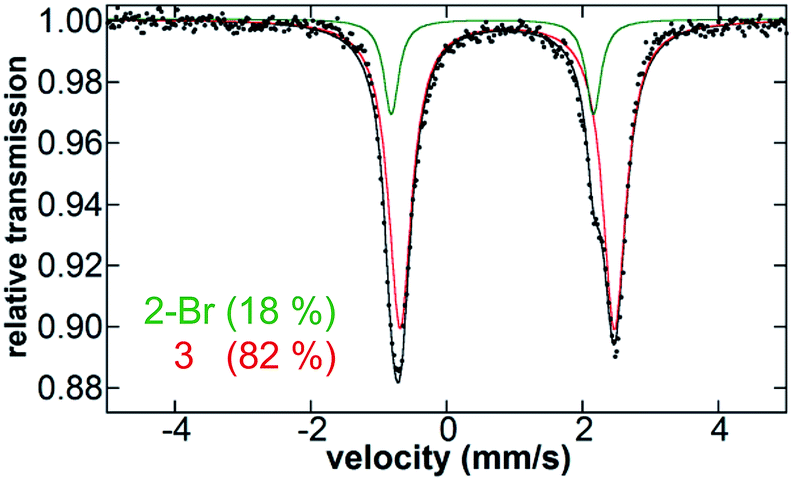 | ||
| Fig. 5 5 K Mössbauer spectrum of in situ iron species present in solution 2 h into the slow Grignard reagent addition step. The individual components of the fit are shown which include 3 (red, 82%) and 2-Br (green, 18%). | ||
Catalytic reactions with alternative nucleophiles
The catalytic reaction reported by Cárdenas utilises (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide. After the isolation of 1 and 2-Br, which both contain oxygen interactions from this substrate to form chelating interactions to iron, it was important to evaluate whether the presence of the acetal moiety (and resulting Fe metallacycle) was essential for effective catalysis. Therefore, the reported catalytic reaction was performed using (2-cyclohexylethyl)magnesium bromide (i.e. the cyclohexyl analogue of the acetal moiety) and 1-iodo-3-phenylpropane (Scheme 2). For this reaction, only 9% of cross-coupled product was observed along with 1-phenylpropene and propylbenzene as the major side products. Thus, the inability to achieve effective cross-coupling upon substitution of the acetal moiety for the cyclohexyl analogue demonstrates that the acetal moiety (and likely the ability to form an iron metallacycle through Fe–O interactions) is essential for obtaining high yields of cross-coupled product. | ||
| Scheme 2 Iron–IMes catalysed cross-coupling reactions with alkyl nucleophiles containing alternative terminal moieties. | ||
Due to the importance of the acetal moiety of the alkyl nucleophile in coordinating to iron(II) to disfavour β-H elimination from the alkyl nucleophile and promote cross-coupled product formation, we hypothesised that the introduction of alternative moieties on the alkyl nucleophile with the potential to coordinate to iron (e.g. ether, pyridine) might also be effective for achieving alkyl–alkyl cross-coupling. The ether based nucleophiles (2-methoxypropyl)magnesium bromide and (3,3-dimethoxypropyl)magnesium bromide were ineffective in the formation of cross-coupled product in reactions with 1-iodo-3-phenylpropane (Scheme 2), instead side-products arising from β-hydride elimination were observed. This indicates the sensitivity of nucleophile chelation to the rigidity and possibly steric encumbrance of the chelating motif. In contrast, (2-(pyridine-2-yl)ethyl)magnesium bromide was found to successfully yield cross-coupled product without optimising reaction conditions and with a yield comparable to those obtained using more functionalised electrophiles in the original study (∼50%).23 The ability to utilise alternative ligating moieties in the alkyl nucleophile not only reinforces the critical importance of alkyl chelation in forming cross-coupled product, but also provides initial insight into how nucleophile scope for iron–NHC alkyl–alkyl cross-couplings might be further broadened through the use of different chelating groups on the nucleophile. Based on these results, future studies will focus on optimising the use of varied directing groups to maximise the yield and scope of such cross-coupling reactions.
Molecular level insight into the poor selectivity of other NHC additives
While (IMes)Fe((1,3-dioxan-2-yl)ethyl)2 (1) was identified as the key reactive iron species for alkyl–alkyl cross-coupling, interesting NHC dependences exist for this reaction. For example, while IMes is a highly effective NHC ligand additive for catalysis (80% product yield), the use of SIPr as the ligand additive results in a significant reduction in cross-coupled product formation (23%) with increased amounts of allylbenzene and n-propylbenzene side product formation compared to reactions with IMes as the ligand additive (see ESI†). Thus, molecular-level insight into the effects of SIPr versus IMes on iron speciation and reactivity could provide critical insight into ligand design for catalysis. Since the key reactive iron species 1 forms in the pre-catalyst formation step in the presence of IMes, an analogous pre-catalyst reaction was performed using SIPr as the ligand additive. Addition of 12 equiv. of (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide to Fe(OAc)2 and SIPr·HCl in THF (1.0 and 2.1 equiv. respectively) at 54 °C followed by stirring for 20 min led to formation of a yellow solution. The 5 K Mössbauer spectrum of the reaction mixture freeze-trapped at the end of the 20 min pre-catalyst reaction indicated the presence of one major iron species in solution (4), with Mössbauer parameters of δ = 0.45 mm s−1 and ΔEQ = 2.87 mm s−1 (Fig. 6A). The significant reduction in the isomer shift and increase in the quadrupole splitting of 4 compared to 1 is consistent with a significant structural difference in the pre-catalyst formed as a function of the NHC ligand employed. Analogous to the pre-catalyst reaction with IMes, spin quantitated EPR indicated the additional presence of a very minor S = 1/2 component (<0.5%, see ESI†).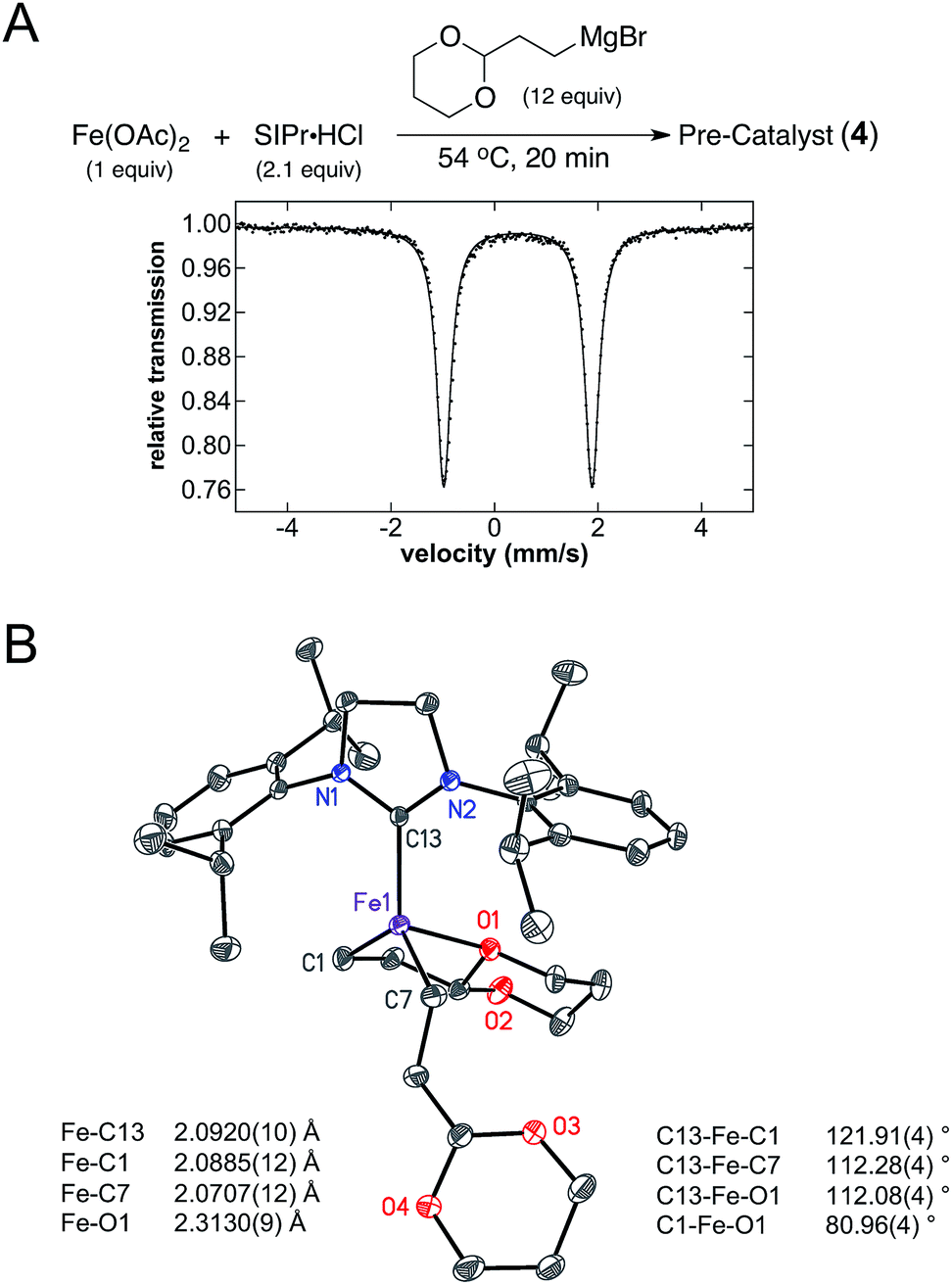 | ||
| Fig. 6 Iron–SIPr pre-catalyst formation. (A) 5 K Mössbauer spectrum of the pre-catalyst reaction iron products using SIPr·HCl as the ligand additive in frozen THF solution. The data is shown as black dots and the fit as a solid line. (B) Single crystal X-ray crystal structure of (SIPr)Fe(1,3-dioxan-2-ylethyl)2 (4) with thermal ellipsoids shown at the 50% probability level and select bond distances and angles given, H atoms are omitted for clarity. | ||
Single crystal X-ray crystallographic studies were pursued in order to unambiguously assign the structure of the major iron species 4 formed in the pre-catalyst reaction with SIPr. Slow addition of 6 equiv. of (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide to a mixture of Fe(OAc)2 and SIPr·HCl (1 equiv. of each) in THF at room temperature followed by cooling to −78 °C yielded yellow crystals suitable for single crystal X-ray diffraction. The single crystal X-ray structure of 4 corresponds to (SIPr)Fe((1,3-dioxan-2-yl)ethyl)2 (Fig. 6B). The structure of this iron(II) complex has an Fe–SIPr bond length of 2.0920(10) Å as well as iron–carbon bond lengths to the alkyl groups of 2.0885(12) and 2.0707(12) Å. In contrast to 1, only one iron–oxygen interaction from the acetal on one of the alkyl groups is present at a distance of 2.3130(9) Å, over 0.1 Å shorter than the corresponding Fe–O interactions in 1. The other alkyl group in 4 is in an open conformation, likely the result of the increased steric bulk of the 2,6-diisopropyl substituents present in SIPr. DFT studies further support steric bulk as a key mode for decrease in reactivity with use of the SIPr ligand as analogous d-orbital configurations to those of the IMes structure are found (see ESI† for further orbital descriptions). In addition, Mayer bond order analysis revealed that the coordinated Fe–O bond has a bond order of 0.074. This is on the same order of interaction as found in 1 despite the shortened bond length.
Mössbauer spectroscopy of crystalline (SIPr)Fe((1,3-dioxan-2-yl)ethyl)2 confirms that this complex represents the major iron species 4 observed to form in situ in the pre-catalyst reaction. The 5 K Mössbauer spectrum of solid 4 is represented by a single quadrupole doublet with parameters δ = 0.44 mm s−1 and ΔEQ = 2.91 mm s−1 (see ESI†), where the small change in ΔEQ in the solid state spectrum suggests a slight structural distortion between solid and solution states.
The significant structural differences between the iron–NHC pre-catalysts formed with IMes and SIPr provide direct insight in to their differences in catalytic performance. The IMes derivative, 1, contains two alkyl ligands with oxygen chelation which protects each alkyl group from β-hydride elimination reactions. In the case of SIPr, 4, only a single chelating alkyl ligand is observed with the second being in an open configuration. This leaves 4 more susceptible to β-hydride elimination reactions, shown through its significantly decreased catalytic performance (see Table 2).
Discussion
The determination of the reactive iron species and mechanism of iron-catalysed cross-coupling reactions provides a fundamental framework for the development of novel iron cross-coupling methods. In the present study, the use of rigorous physical inorganic methodology combining 57Fe Mössbauer spectroscopy, EPR, and inorganic synthesis with GC analysis defines the in situ formed and reactive iron species in iron–NHC catalysed alkyl–alkyl cross-coupling. These studies are the first determination of the key reactive species in an iron-catalysed alkyl–alkyl cross-coupling system.Detailed spectroscopic and synthetic studies enable identification of the key alkylated iron–NHC species formed in situ during the pre-catalyst generation reaction with Fe(OAc)2, 1,3-dimesitylimidizolium chloride and excess (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide. The bis-alkylated iron(II)–IMes complex (IMes)Fe((1,3-dioxan-2-yl)ethyl)2 (1) and its THF adduct are the major iron species formed in situ during the pre-catalyst reaction, representing >99% of all iron in solution. Independent synthetic studies also demonstrate that the mono-alkylated complex (IMes)FeBr((1,3-dioxan-2-yl)ethyl) (2-Br) is also accessible in this chemistry, which can readily be converted to 1 upon reaction with additional Grignard reagent.
Previously, a S = 1/2 iron(I) active species was proposed to form in the pre-catalyst reaction based upon EPR spectroscopy and GC monitoring of homocoupled Grignard reagent. However, this study shows that upon oxidation of iron during acid quench of isolated 1, homocoupled product is formed, demonstrating that GC counting of electrophile can be an unreliable way to identify metal oxidation state. This observation is consistent with previous work by Kochi and co-workers where reductive elimination of two alkyl groups on iron to form homocoupled product can be promoted by formation of more highly oxidised iron species.49 Additionally, spin quantified EPR studies show that this reported S = 1/2 iron species represents <0.5% of all the iron formed in situ even after addition of electrophile and throughout the subsequent slow addition of Grignard reagent.
Reaction studies combining GC analysis and freeze-trapped Mössbauer spectroscopy enable identification of the key reactive iron–NHC species for the generation of cross-coupled product. While 2-Br exhibits no reactivity towards 1-iodo-3-phenylpropane, 1 reacts rapidly to generate cross-coupled product and an overall product distribution analogous to that observed in catalysis. Specifically, 1 is observed to react with excess electrophile to undergo two rapid turnovers at rates that are catalytically relevant (kobs > 24 min−1), resulting in the formation of bishalide iron(II)–IMes species. When 1 is formed in situ the previously reported S = 1/2 species is also present, but the consumption of the S = 1/2 species is not observed by spin quantitated EPR analysis. This is consistent with the S = 1/2 species being either non-reactive or much less reactive towards electrophile than 1 and, therefore, an off-cycle species in catalysis.
Combined with radical clock substrate experiments previously reported,12 the observed reactivity of 1 is consistent with an Fe(II)/Fe(III) mechanistic cycle involving radical formation from the alkyl iodide. Two total turnovers of 1 are observed, suggesting the formation of an intermediate mono-alkyl iron(II)–IMes intermediate as summarised in Scheme 3. In fact, substoichiometric reaction studies of 1 with electrophile result in the formation of a mono-alkylated iron(II)–IMes complex and ∼60% selectivity towards cross-coupled product by GC analysis. When this reaction mixture is subsequently exposed to excess electrophile, overall selectivity increases to that observed in catalysis (∼80% cross-coupled product) and the second turnover is highly selective for cross-coupled product. This selectivity suggests that 1 is a key reactive species in catalysis rather than reactivity deriving solely from a transient mono-alkylated iron–NHC which would have a much higher overall selectivity, like that of the second turnover described above. Lastly, while reactivity of this intermediate mono-alkylated iron with alkyl radical generated by 1 could be envisioned, no organic radical is observed in in situ freeze trapped EPR.
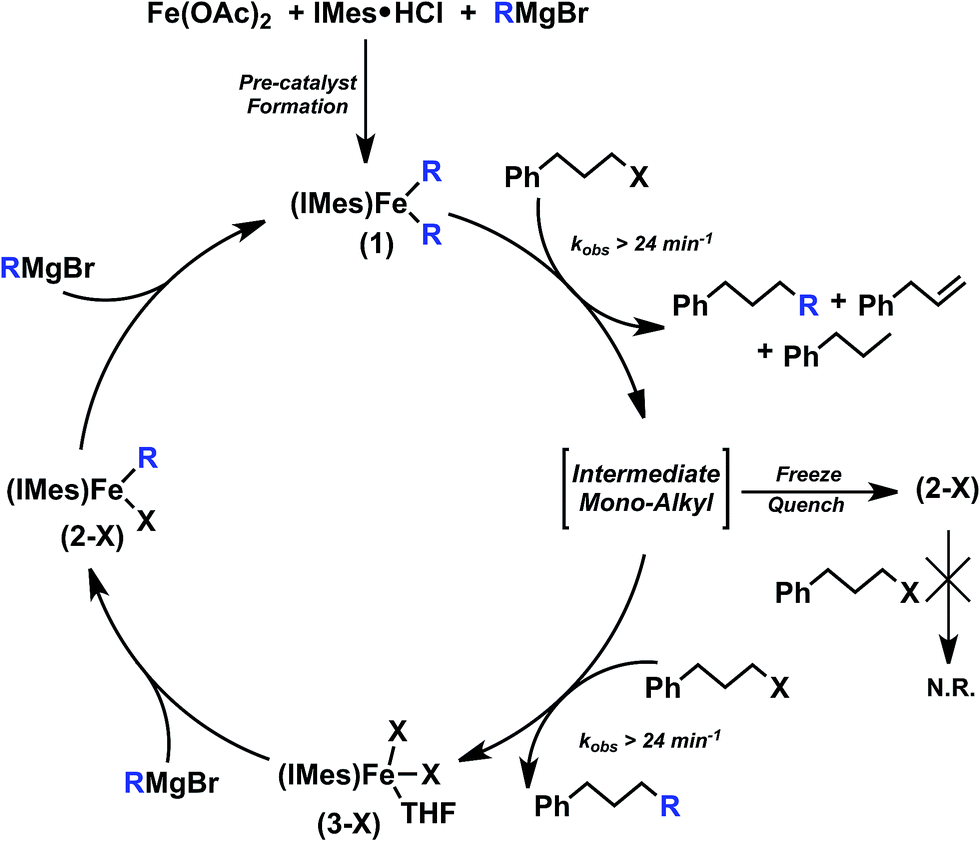 | ||
| Scheme 3 Summary of observed iron(II) species and their reactive transformations in the Cárdenas iron–NHC catalysed alkyl–alkyl cross-coupling system (note: X = Br or I). | ||
The isolated mono-alkylated species observed by Mössbauer spectroscopy in these studies corresponds to 2-X (X = Br, I); however, the lower oxidizing ability of 2-Br compared with 1 may be responsible for its lack of reactivity with electrophile. Even after heating at 60 °C for 5 min to encourage dissociation of the acetal chelate in the presence of excess 1-iodo-3-phenylpropane, 100% of the alkyl iodide is recovered by GC analysis. Notably, catalysis remains effective with the use of an alkyl bromide electrophile12 indicating that a halide effect is not present. Instead, these studies suggest that the intermediate mono-alkylated iron(II)–IMes species likely represents a perturbed form of 2-X, perhaps one in which the hemi-labile acetal is not coordinated to iron. However, all attempts at freeze-trapping or isolation of this intermediate mono-alkyl species only result in the isolation of unreactive 2-Br. Importantly, the observed product yields and selectivity in catalysis is consistent with the 80:20 (C–C product:side product) distribution resulting from reaction of 1 with excess electrophile. Therefore, catalysis most likely proceeds through two total turnovers of 1 rather than the proposed mono-alkylated iron(II)–IMes intermediate which would produce a much higher overall selectivity. This is further supported by the observation of mono-alkylated 2-Br during steady state catalysis.
The requirement for slow addition of Grignard reagent after electrophile addition to achieve high cross-coupling yields results from the low selectivity observed during the first turnover of 1. First of all, simply doubling the Grignard reagent addition rate results in product distributions containing only ∼50% cross-coupled product.23 Additionally, the reported reaction of in situ generated 1 with electrophile occurs in the presence of excess Grignard reagent. As a direct result, product yields are only ∼50% selective towards cross-coupled product with excess Grignard reagent present in solution prior to the subsequent slow addition of the remaining Grignard reagent. Together, these observations show that high product selectivity requires that 1 react twice with 1-iodo-3-phenylpropane prior to re-transmetalation with Grignard reagent. In cases where excess Grignard reagent is present in solution (i.e. from rapid addition of Grignard reagent to form 1), the resulting mono-alkylated intermediate from the first turnover of 1 with electrophile could be re-transmetalated to 1 faster than its reaction with electrophile to form product. In turn, cross-coupled product yields consistent with only the first turnover of 1 with electrophile would be generated, consistent with our observations in the current study. Thus, slow Grignard reagent addition is required in order to enable two turnovers of 1 prior to any transmetalation event, ensuring that the more highly selective second turnover reaction can occur.
The relatively low amount of β-hydride elimination product derived from the nucleophile, a common problem for reactions with alkyl nucleophiles, is another key aspect of the Cárdenas iron–NHC alkyl–alkyl cross-coupling system. The ability to isolate and structurally characterize the key alkylated iron(II)–IMes species formed in situ provides direct insight into the origins of minimised β-hydride eliminated product derived from the alkyl nucleophile. Both the bis-alkylated and mono-alkylated iron(II)–IMes complexes exhibit chelation of the ligated alkyl via coordination to carbon and interaction with one oxygen atom of the acetal moiety of the nucleophile to generate a 5-membered ring chelate to iron(II). This chelation appears to lock the alkyl substrate into a configuration that is less prone to β-hydride elimination, orienting the β-hydrogens away from iron while also increasing the barrier to geometric rearrangement to achieve the required orientation for elimination. As a result, even at elevated temperature (T > 50 °C) these alkylated iron(II)–IMes complexes exhibit significant stability over the course of many tens of minutes. Consistent with this hypothesis, the use of an analogous alkyl Grignard reagent where the acetal moiety is replaced by a cyclohexyl group leads to substantially reduced cross-coupling activity, likely the result of the instability of IMes–iron(II)–alkyl species formed with the cyclohexyl substituted nucleophile as indicated by the complex iron speciation observed to form in situ with this nucleophile (see ESI†). Thus, the current system is highly dependent on coordination of the acetal moiety in order to achieve high alkyl–alkyl cross-coupling yields.
The isolation and reactivity of these iron(II) chelate intermediates suggests that the development of future cross-coupling systems could benefit from the use of reagents capable of chelate formation to increase selectivity with alkyl nucleophiles. Specifically, the use of alternative alkyl nucleophiles such as (2-(pyridine-2-yl)ethyl)magnesium bromide as an effective alkyl nucleophile for alkyl–alkyl cross-coupling, demonstrates how the insight into alkyl chelation can be leveraged to broaden the scope of nucleophile partners for alkyl–alkyl cross-coupling.
The specific NHC used in this catalytic system also has an effect on the productivity of catalysis. Specifically, SIPr is less effective as a ligand additive in this system compared to IMes. This could be directly correlated to structural effects of the NHC ligand on the ability of the alkyl nucleophiles to chelate. Additionally, the added steric bulk of the SIPr compared with IMes could lead to a slower or less efficient rebound of the substrate radical, leading to decreased cross-coupling performance. In fact, the d-orbital configuration and energies of 1 and 4 are remarkably similar (see ESI†), further demonstrating the importance of steric effects of NHC ligands over electronic structure changes.
The observation of a highly reactive iron(II)–IMes species in iron–NHC alkyl–alkyl cross-coupling further expands the catalytic relevance of iron(II) from previously reported iron–SciOPP cross-coupling to both alkyl nucleophiles and NHC supporting ligands. While it remains unlikely that a conserved overall mechanism exists for all iron-catalysed cross-coupling reactions, the current studies demonstrate that iron(II) complexes must be considered as potential reactive species across the breadth of iron cross-couplings. Potential iron(I) active species have been proposed in other cross-couplings, notably in Negishi cross-coupling using dppe and dpbz ligands,50,51 and it will be exciting to determine the role of iron(I) versus iron(II) in those systems in order to more broadly define the role of these oxidation states in iron-catalysed cross-coupling.
Conclusions
In this study, inorganic spectroscopic methods (i.e. Mössbauer, EPR) combined with inorganic synthesis and reaction studies have been utilised to determine the in situ formed and reactive iron species in iron–NHC catalysed alkyl–alkyl cross-coupling. (IMes)Fe((1,3-dioxan-2-yl)ethyl)2 is identified as the key reactive iron species formed in situ, while the S = 1/2 iron species previously identified in this chemistry is found to be only a minor (<0.5% of all iron) off-cycle species. The high resistance of this catalytic system to β-hydride elimination is attributed to chelation of the alkyl nucleophile through carbon and one oxygen of the nucleophile, which could be disrupted by the use of more bulkily NHC ligands such as SIPr resulting in reduced cross-coupling yields. These studies extend the importance of iron(II) active species to systems utilizing NHC ligands and alkyl nucleophiles, demonstrating the broader importance of this oxidation state of iron for generating highly reactive species for effective cross-coupling catalysis, and identifying a novel alkyl chelation method to achieve effective alkyl–alkyl cross-coupling with iron–NHCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the National Institutes of Health (R01GM111480 to M. L. N.) and by a Sloan Research Fellowship (M. L. N.). S. B. M. III also acknowledges a Ruth L. Kirchenstein National Research Service award (F32GM120823).Notes and references
- R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS PubMed.
- B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500–1511 CrossRef CAS PubMed.
- R. B. Bedford and P. B. Brenner, Top. Organomet. Chem., 2015, 50, 19–46 CrossRef CAS.
- A. Fuerstner and R. Martin, Chem. Lett., 2005, 34, 624–629 CrossRef CAS.
- D. Bezier, J.-B. Sortais and C. Darcel, Adv. Synth. Catal., 2013, 355, 19–33 CrossRef CAS.
- M. J. Ingleson and R. A. Layfield, Chem. Commun., 2012, 48, 3579–3589 RSC.
- K. Riener, S. Haslinger, A. Raba, M. P. Högerl, M. Cokoja, W. A. Herrmann and F. E. Kühn, Chem. Rev., 2014, 114, 5215–5272 CrossRef CAS PubMed.
- T. Hatakeyama and M. Nakamura, J. Am. Chem. Soc., 2007, 129, 9844–9845 CrossRef CAS PubMed.
- Y.-Y. Chua and H. A. Duong, Chem. Commun., 2014, 50, 8424–8427 RSC.
- W. Wu, Q. Teng, Y.-Y. Chua, H. V. Huynh and H. A. Duong, Organometallics, 2017, 36, 2293–2297 CrossRef CAS.
- T. Agrawal and S. P. Cook, Org. Lett., 2014, 16, 5080–5083 CrossRef CAS PubMed.
- K. Bica and P. Gaertner, Org. Lett., 2006, 8, 733–735 CrossRef CAS PubMed.
- H.-h. Gao, C.-h. Yan, X.-P. Tao, Y. Xia, H.-M. Sun, Q. Shen and Y. Zhang, Organometallics, 2010, 29, 4189–4192 CrossRef CAS.
- H.-n. Deng, Y.-l. Xing, C.-l. Xia, H.-m. Sun, Q. Shen and Y. Zhang, Dalton Trans., 2012, 41, 11597–11607 RSC.
- S. K. Ghorai, M. Jin, T. Hatakeyama and M. Nakamura, Org. Lett., 2012, 14, 1066–1069 CrossRef CAS PubMed.
- C.-L. Xia, C.-F. Xie, Y.-F. Wu, H.-M. Sun, Q. Shen and Y. Zhang, Org. Biomol. Chem., 2013, 11, 8135–8144 CAS.
- Y. Liu, J. Xiao, L. Wang, Y. Song and L. Deng, Organometallics, 2015, 34, 599–605 CrossRef.
- A. L. Silberstein, S. D. Ramgren and N. K. Garg, Org. Lett., 2012, 14, 3796–3799 CrossRef CAS PubMed.
- M. C. Perry, A. N. Gillett and T. C. Law, Tetrahedron Lett., 2012, 53, 4436–4439 CrossRef CAS.
- T. Agrawal and S. P. Cook, Org. Lett., 2013, 15, 96–99 CrossRef CAS PubMed.
- R. Agata, T. Iwamoto, N. Nakagawa, K. Isozaki, T. Hatakeyama, H. Takaya and M. Nakamura, Synthesis, 2015, 47, 1733–1740 CrossRef CAS.
- J. N. Sanderson, A. P. Dominey and J. M. Percy, Adv. Synth. Catal., 2017, 359, 1007–1017 CrossRef CAS.
- M. Guisán-Ceinos, F. Tato, E. Buñuel, P. Calle and D. J. Cárdenas, Chem. Sci., 2013, 4, 1098 RSC.
- B.-J. Li, L. Xu, Z.-H. Wu, B.-T. Guan, C.-L. Sun, B.-Q. Wang and Z.-J. Shi, J. Am. Chem. Soc., 2009, 131, 14656–14657 CrossRef CAS PubMed.
- R. B. Bedford, M. Betham, D. W. Bruce, A. A. Danopoulos, R. M. Frost and M. Hird, J. Org. Chem., 2006, 71, 1104–1110 CrossRef CAS PubMed.
- T. Hatakeyama, S. Hashimoto, K. Ishizuka and M. Nakamura, J. Am. Chem. Soc., 2009, 131, 11949–11963 CrossRef CAS PubMed.
- A. A. Danopoulos, P. Braunstein, M. Wesolek, K. Y. Monakhov, P. Rabu and V. Robert, Organometallics, 2012, 31, 4102–4105 CrossRef CAS.
- A. A. Danopoulos, N. Tsoureas, J. A. Wright and M. E. Light, Organometallics, 2004, 23, 166–168 CrossRef CAS.
- L. Xiang, J. Xiao and L. Deng, Organometallics, 2011, 30, 2018–2025 CrossRef CAS.
- Q. Zhang, L. Xiang and L. Deng, Organometallics, 2012, 31, 4537–4543 CrossRef CAS.
- Y. Liu, L. Wang and L. Deng, Organometallics, 2015, 34, 4401–4407 CrossRef CAS.
- X. Wang, J. Zhang, L. Wang and L. Deng, Organometallics, 2015, 34, 2775–2782 CrossRef CAS.
- J. A. Przyojski, K. P. Veggeberg, H. D. Arman and Z. J. Tonzetich, ACS Catal., 2015, 5, 5938–5946 CrossRef CAS.
- K. G. Dongol, H. Koh, M. Sau and C. L. L. Chai, Adv. Synth. Catal., 2007, 349, 1015–1018 CrossRef CAS.
- T. Hatakeyama, T. Hashimoto, K. K. Kathriarachchi, T. Zenmyo, H. Seike and M. Nakamura, Angew. Chem., Int. Ed., 2012, 51, 8834–8837 CrossRef CAS PubMed.
- S. L. Daifuku, J. L. Kneebone, B. E. R. Snyder and M. L. Neidig, J. Am. Chem. Soc., 2015, 137, 11432–11444 CrossRef CAS PubMed.
- S. L. Daifuku, M. H. Al-Afyouni, B. E. R. Snyder, J. L. Kneebone and M. L. Neidig, J. Am. Chem. Soc., 2014, 136, 9132–9143 CrossRef CAS PubMed.
- J. L. Kneebone, W. W. Brennessel and M. L. Neidig, J. Am. Chem. Soc., 2017, 139, 6988–7003 CrossRef CAS PubMed.
- R. Aasa and T. Vanngard, J. Magn. Reson., 1975, 19, 308–315 CAS.
- M. J. T. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Wallingford, CT, 2009 Search PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 5829–5835 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 3865–3868 CrossRef CAS PubMed.
- J. Tomasi, B. Menucci and R. Cammi, Chem. Rev., 2011, 2999–3094 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. I. Gorelsky and A. B. P. Lever, J. Organomet. Chem., 2001, 635, 187–196 CrossRef CAS.
- S. I. Gorelsky, AOMix: Program for Molecular Orbital Analysis 2014, version 6.8.5, 2014, http://www.sg-chem.net/ Search PubMed.
- K. L. Fillman, J. A. Przyojski, M. H. Al-Afyouni, Z. J. Tonzetich and M. L. Neidig, Chem. Sci., 2015, 6, 1178–1188 RSC.
- J. A. Przyojski, H. D. Arman and Z. J. Tonzetich, Organometallics, 2012, 31, 3264–3271 CrossRef CAS.
- W. Lau, J. C. Huffman and J. K. Kochi, J. Organomet. Chem., 1982, 1, 155–169 CrossRef CAS.
- C. J. Adams, R. B. Bedford, E. Carter, N. J. Gower, M. F. Haddow, J. N. Harvey, M. Huwe, M. Á. Cartes, S. M. Mansell, C. Mendoza, D. M. Murphy, E. C. Neeve and J. Nunn, J. Am. Chem. Soc., 2012, 134, 10333–10336 CrossRef CAS PubMed.
- R. B. Bedford, E. Carter, P. M. Cogswell, N. J. Gower, M. F. Haddow, J. N. Harvey, D. M. Murphy, E. C. Neeve and J. Nunn, Angew. Chem., Int. Ed., 2013, 52, 1285–1288 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1552137, 1552138 and 1583650. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04750a |
| ‡ Valerie E. Fleischauer and Salvador B. Muñoz III contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |