
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photochemical syntheses, transformations, and bioorthogonal chemistry of trans-cycloheptene and sila trans-cycloheptene Ag(I) complexes†
Yinzhi
Fang
a,
Han
Zhang‡
a,
Zhen
Huang‡
b,
Samuel L.
Scinto
a,
Jeffrey C.
Yang
b,
Christopher W.
am Ende
c,
Olga
Dmitrenko
a,
Douglas S.
Johnson
b and
Joseph M.
Fox
 *a
*a
aBrown Laboratories, Department of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716, USA. E-mail: jmfox@udel.edu
bPfizer Worldwide Research and Development, Cambridge, Massachusetts 02139, USA
cPfizer Worldwide Research and Development, Groton, Connecticut 06340, USA
First published on 8th January 2018
Abstract
A photochemical synthesis of AgNO3 complexes of trans-cycloheptene (TCH) and trans-1-sila-4-cycloheptene (Si-TCH) derivatives is described. A low temperature flow photoreactor was designed to enable the synthesis of carbocyclic TCH derivatives due to their thermal sensitivity in the absence of metal coordination. Unlike the free carbocycles, TCH·AgNO3 complexes can be handled at rt and stored for weeks in the freezer (−18 °C). Si-TCH·AgNO3 complexes are especially robust, and are bench stable for days at rt, and for months in the freezer. X-ray crystallography was used to characterize a Si-TCH·AgNO3 complex for the first time. With decomplexation of AgNO3in situ, metal-free TCO and Si-TCH derivatives can engage in a range of cycloaddition reactions as well as dihydroxylation reactions. Computation was used to predict that Si-TCH would engage in bioorthogonal reactions that are more rapid than the most reactive trans-cyclooctenes. Metal-free Si-TCH derivatives were shown to display good stability in solution, and to engage in the fastest bioorthogonal reaction reported to date (k2 1.14 × 107 M−1 s−1 in 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O:MeOH). Utility in bioorthogonal protein labeling in live cells is described, including labeling of GFP with an unnatural tetrazine-containing amino acid. The reactivity and specificity of the Si-TCH reagents with tetrazines in live mammalian cells was also evaluated using the HaloTag platform. The cell labeling experiments show that Si-TCH derivatives are best suited as probe molecules in the cellular environment.
:1 H2O:MeOH). Utility in bioorthogonal protein labeling in live cells is described, including labeling of GFP with an unnatural tetrazine-containing amino acid. The reactivity and specificity of the Si-TCH reagents with tetrazines in live mammalian cells was also evaluated using the HaloTag platform. The cell labeling experiments show that Si-TCH derivatives are best suited as probe molecules in the cellular environment.
Introduction
For nearly seventy years,1–3 the unusual bonding, reactivity and planar chirality of trans-cycloalkenes have captured the imagination of scientists. The unique reactivity of trans-cycloalkenes has produced an impressive collection of applications in synthesis, including reactions with dienes,4–6 1,3-dipoles7 and ketenes.8–10 Additionally, strained trans-cycloalkenes can serve as excellent ligands for transition metals. In the field of bioorthogonal chemistry,11–19trans-cycloalkenes hold special significance due to their particularly fast kinetics in cycloaddition reactions.20–23 Relative to trans-cyclooctene, the chemistry of the homolog trans-cycloheptene is less explored. trans-Cycloheptene was first trapped with diphenylisobenzofuran by Corey and Winter from trans-1,2-cycloheptenethionocarbonate through treatment with P(OMe)3.24 Studies by Marshall,25 Kropp26 and Beauchemin27 on the photoprotonation reactions of cyclic alkenes, including cycloheptene, have shown that cis/trans equilibria could be driven by selective addition reactions of trans-cycloalkenes.trans-Cycloheptene was first spectroscopically characterized by Inoue via singlet sensitized photoisomerization of cis-cycloheptene at −35 °C.28,29 Unlike trans-cyclooctene, which is stable at room temperature, trans-cycloheptene undergoes rapid isomerization under ambient conditions via a proposed ‘interrupted dimerization’ mechanism.30trans-Cycloheptene has also been prepared via ligand exchange from a trans-cycloheptene·CuOTf complex.31 1-Phenyl-trans-cycloheptene and trans-cycloheptenone derivatives are also known to be thermally unstable at ambient temperature, but can be trapped in situ.32–38
While the parent trans-cycloheptene is thermally labile, it has been demonstrated in several studies that metal complexes can be isolated. CuOTf has been proposed to catalyze photodimerization reactions of cyclic olefins via photoinduced cis–trans isomerization,39 with predominant formation of a cyclotrimer from cycloheptene.40 A stable trans-cycloheptene·CuOTf complex has been prepared through irradiation of cis-cycloheptene·CuOTf, but a yield for the process was not reported.41 A pybox–RuCl2 complex of trans-cycloheptene has been prepared by irradiation of the corresponding ethylene complex in the presence of cis-cycloheptene under singlet sensitized conditions.42 Jendralla described the preparation of AgOTf and AgClO4 complexes of 3-methoxy-trans-cycloheptene and 6-methoxy-(Z),4(E)-cycloheptadiene.43,44 These compounds can be prepared through the Ag-mediated ring opening of a nitrosourea derivative of bicyclo[4.1.0]heptane. In unspecified yields, the AgClO4·3-methoxy-trans-cycloheptene complex was combined with a number of dienes to give the products of metal decomplexation and [4 + 2] cycloaddition.43,44
Because C–Si bonds are long, the inclusion of silicon into the cyclic backbone can alleviate olefinic strain and impart stability to trans-cycloalkenes.45–52 In 1997, (E)-1,1,3,3,6,6-hexamethyl-1-sila-4-cycloheptene was synthesized, resolved, and characterized crystallographically.46,47 Here, the exhaustive allylic substitution imparts a high degree of stability to the trans-alkene. Recent studies by Woerpel48–51 and Tomooka52 have provided demonstrations of the utility of backbone heteroatom-containing trans-cycloalkene derivatives in synthesis. Woerpel has elegantly developed a general method for the preparation of trans-oxasilacycloheptenes—seven membered rings that contain trans-alkenes and siloxy bonds in the backbone.48–51
There has been little investigation into the reaction chemistry of trans-cycloheptene derivatives. In 1980, Jendralla showed that silver complexes of 3-methoxy-trans-cycloheptene are isolable, and can reversibly dissociate and in unspecified yields undergo cycloaddition reactions.43,44 Woerpel has described selective addition reactions and difunctionalization reactions of trans-oxasilacycloheptenes, and recently has reported the Diels–Alder reactions of trans-oxasilacycloheptenes with furan and tetrazine derivatives.48–51 At 25 °C in benzene, an oxasilacycloheptene was 7-fold more reactive toward 2,5-diphenylisobenzofuran than a conformationally strained trans-cyclooctene (‘s-TCO’) derivative.48 An oxasilacycloheptene derivative was shown to react with 3,6-diphenyl-s-tetrazine in benzene at rt in less than 10 min and in 90% NMR yield.48
Our group has described a closed-loop flow reactor for the synthesis of trans-cyclooctene derivatives, whereby selective complexation with AgNO3 is used to drive the formation of trans-isomer from cis-cyclooctene.21,53–55 Described herein is an approach to the synthesis of trans-cycloheptene (TCH) and trans-1-sila-4-cycloheptene (Si-TCH) derivatives via flow photochemical synthesis. The derivatives described are especially stable as their AgNO3 metal complexes, which can be stored neat or in solution for long periods. With decomplexation of AgNO3in situ, metal-free TCH and Si-TCH derivatives can engage in a range of cycloaddition reactions as well as dihydroxylation reactions. Unlike the carbocycles, Si-TCH derivatives display good stability in solution and are shown to engage in the fastest bioorthogonal reaction reported to date. Decomplexation of AgNO3 can be carried out in situ directly in cell media for bioorthogonal protein labeling in live cells.
Results and discussion
For the photochemical synthesis of trans-cyclooctene derivatives, the trans-cycloalkene is first scavenged on AgNO3/SiO2 and then liberated through treatment with aqueous or methanolic ammonia.21,53–55 However, our attempts to directly apply this procedure to the synthesis of carbocyclic TCHs was unsuccessful, most likely due to the susceptibility of carbocyclic TCHs to readily isomerize to their cis-isomers.30 Recently, we demonstrated that the shelf-life of conformationally strained trans-cyclooctenes could be enhanced by storing the cycloalkenes as their AgNO3 complexes, and that the free alkenes could be liberated in situ through treatment with NaCl in aqueous solution or cell media.56 We reasoned that TCH and Si-TCH derivatives may also be isolable and better stored as their Ag-complexes, and that the corresponding free-alkenes could be liberated at later time points as required.Photoisomerizations to form Si-TCH·AgNO3 derivatives were carried out at rt using the previously described flow-photoisomerization apparatus,21,53,54 with the modification that Si-TCH·AgNO3 complexes were directly isolated from SiO2 without Ag-decomplexation. The metal complexes that were obtained were stable in neat form for >1 month in the freezer. However, it was necessary to alter our reactor design for the synthesis of carbocyclic trans-cycloheptenes due to their thermal lability (Fig. 1). As with the standard photoreactor, this system uses methyl benzoate as a sensitizer and a metering pump to pass a solution of substrate at a rate of 100 mL min−1 through a photowell and then through a column of AgNO3·SiO2, where the trans-cycloalkene is selectively captured as a AgNO3 complex. In the reactor for carbocyclic TCH synthesis, a reservoir of solvent chilled in a cold bath (−50 °C) was positioned before the photowell, and the photoisomerization was conducted in a coil of optically transparent FEP tubing.57 The fluoropolymer tubing provides a high surface area and minimal volume (only 30 mL for 8 m tubing) thereby minimizing the residence time before product adsorption on AgNO3/SiO2. In our standard setup, an inline thermometer was included to measure the temperature for the flowing mixture either before or just after the UV lamp. The temperature was measured as 0 °C before entering the Rayonet photoreactor, and as 20 °C after exiting the photoreactor. With this apparatus, TCH·AgNO3 complexes were eluted from the column, and isolated as semisolids that are moderately stable at rt but stable for weeks in the freezer.
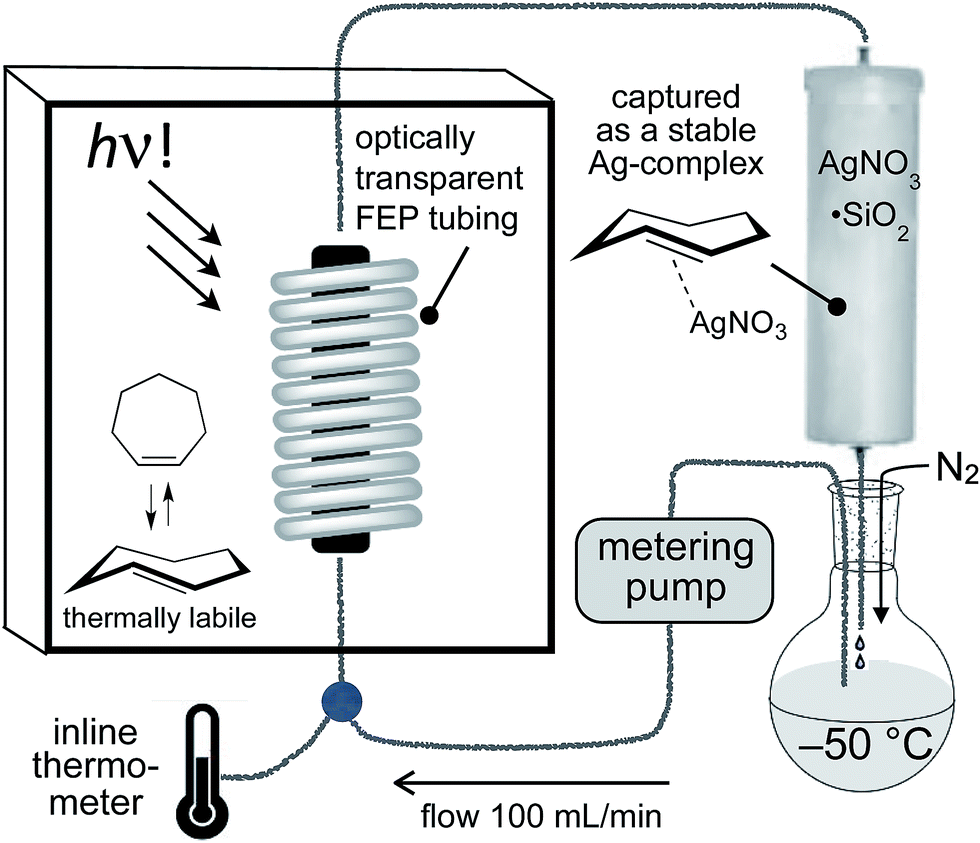 | ||
| Fig. 1 The apparatus for photoisomerization of carbocyclic trans-cycloheptene derivatives was designed to minimize loss of trans-cycloheptene due to thermal isomerization by using FEP tubing and inline cooling. For the synthesis of trans-1-sila-4-cycloheptenes, photoisomerizations could be carried out at rt using a conventional flow photoisomerization setup. | ||
The scope of TCH and Si-TCH synthesis is shown in Scheme 1. With the exception of cycloheptene itself, the carbocyclic and sila-cycloheptene precursors were prepared in 2–7 steps using olefin metathesis as a key step (ESI†). Silver nitrate complexes of trans-cycloheptene (1a) and trans-5-hydroxymethylcycloheptene (1b) were prepared in 53% and 64% yields, respectively. These TCH·AgNO3 complexes are stable enough to handle on the bench for modest periods (hours), and to longer-term storage in the freezer (−18 °C). NMR monitoring showed 90% fidelity for a CD3OD solution of 1a after 10 days storage in freezer (−18 °C), and 92% fidelity for a CD3OD solution of 1a after 10 hours at rt on the bench.
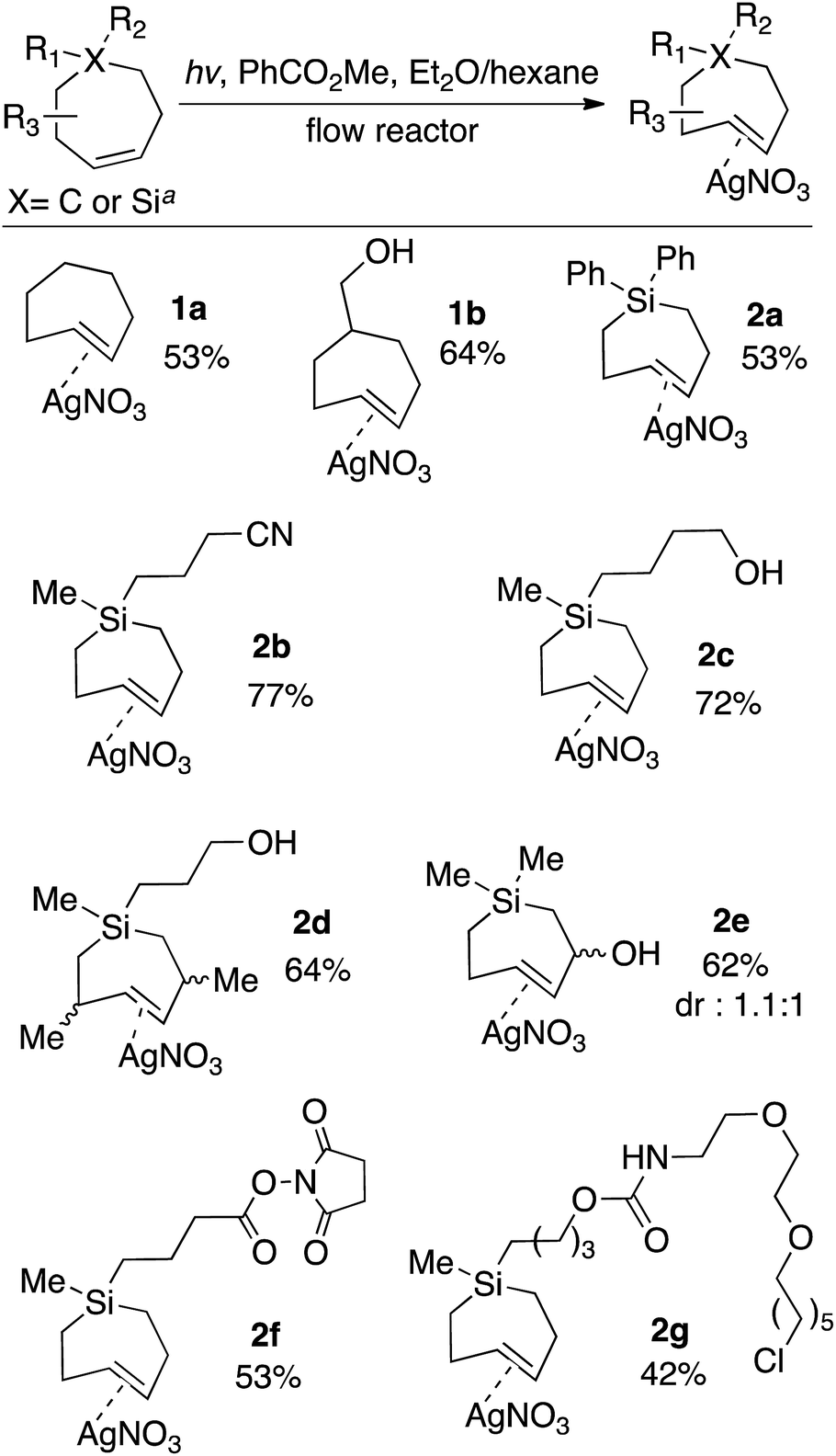 | ||
| Scheme 1 Flow-photochemical synthesis of AgNO3 complexes of trans-cycloheptenes and trans-1-sila-4-cycloheptenes. TCH·AgNO3 and Si-TCH·AgNO3 complexes were isolated as semisolids that contained 20–30% free AgNO3. Yields are the average of two runs, and were corrected by measuring the 1H NMR against an internal standard. aPhotoisomerization of carbocycles were conducted at low temperature using the flow apparatus described in Fig. 1. Photoisomerizations of silicycles were conducted at room temperature using the flow apparatus previously described for trans-cyclooctene synthesis. | ||
The silver complexes of Si-TCH are much more stable. NMR monitoring showed 93% fidelity for a CD3OD solution of 2a after 8 days at rt, and 96% fidelity for 2a after storage for 1 month in the freezer (−18 °C, neat). The photoisomerization method could be used to produce diphenyl (2a) or dialkyl (2b–g) substituted silacycles. Cyano (2b) and hydroxyl (2c–e) groups were tolerated, as were NHS ester (2f) and chloroalkane (2g) groups that could be used to enable conjugation to fluorophores and HaloTag58,59 fusion proteins, respectively. TCH·AgNO3 and Si-TCH·AgNO3 complexes were isolated as semisolids that contained 20–30% free AgNO3. The isolated yields were corrected by measuring the 1H NMR against an internal standard.
Previously, a crystal structure showed that trans-cyclooctene coordinates with AgNO3 as a 1:1 complex.60 X-ray quality crystals of silver complex 2a were grown from ethyl acetate/methanol. Selected bond lengths and angles are displayed in Fig. 2. The coordination environment at silver is distorted trigonal, with bridging coordination of nitrite in an extended polymeric structure in the solid state. For comparison, we also grew crystals of the silver(I) nitrate complex of the equatorial diastereomer of 5-hydroxy-trans-cyclooctene 3 (Fig. 2). Here, the coordination environment at silver is distorted tetrahedral due to the ability of the hydroxyl to serve as a second bridging ligand. The C–Ag bond lengths and bond angles were similar for 2a and 3. As expected, the C–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–C dihedral angle for 3 (136.7°) is smaller than metal-free TCO 4 (139.1°).53 Similarly, the C–CC–C dihedral angle for 2a (126.3°) was smaller than that of metal free Si-trans-cycloheptene 5 (130.9°),46 but comparable to 6 (126.1 °C)50—a compound with additional strain due to relatively short C–O and Si–O bonds in the cyclic backbone. Reflecting the low level of metal backbonding that is common for Ag(I) alkene complexes, the CC bond length for the Ag(I) complexes (1.329 Å for 2a, 1.366 Å for 3) was very similar to that of the metal-free complexes 4–6 (1.331–1.335 Å).
C–C dihedral angle for 3 (136.7°) is smaller than metal-free TCO 4 (139.1°).53 Similarly, the C–CC–C dihedral angle for 2a (126.3°) was smaller than that of metal free Si-trans-cycloheptene 5 (130.9°),46 but comparable to 6 (126.1 °C)50—a compound with additional strain due to relatively short C–O and Si–O bonds in the cyclic backbone. Reflecting the low level of metal backbonding that is common for Ag(I) alkene complexes, the CC bond length for the Ag(I) complexes (1.329 Å for 2a, 1.366 Å for 3) was very similar to that of the metal-free complexes 4–6 (1.331–1.335 Å).
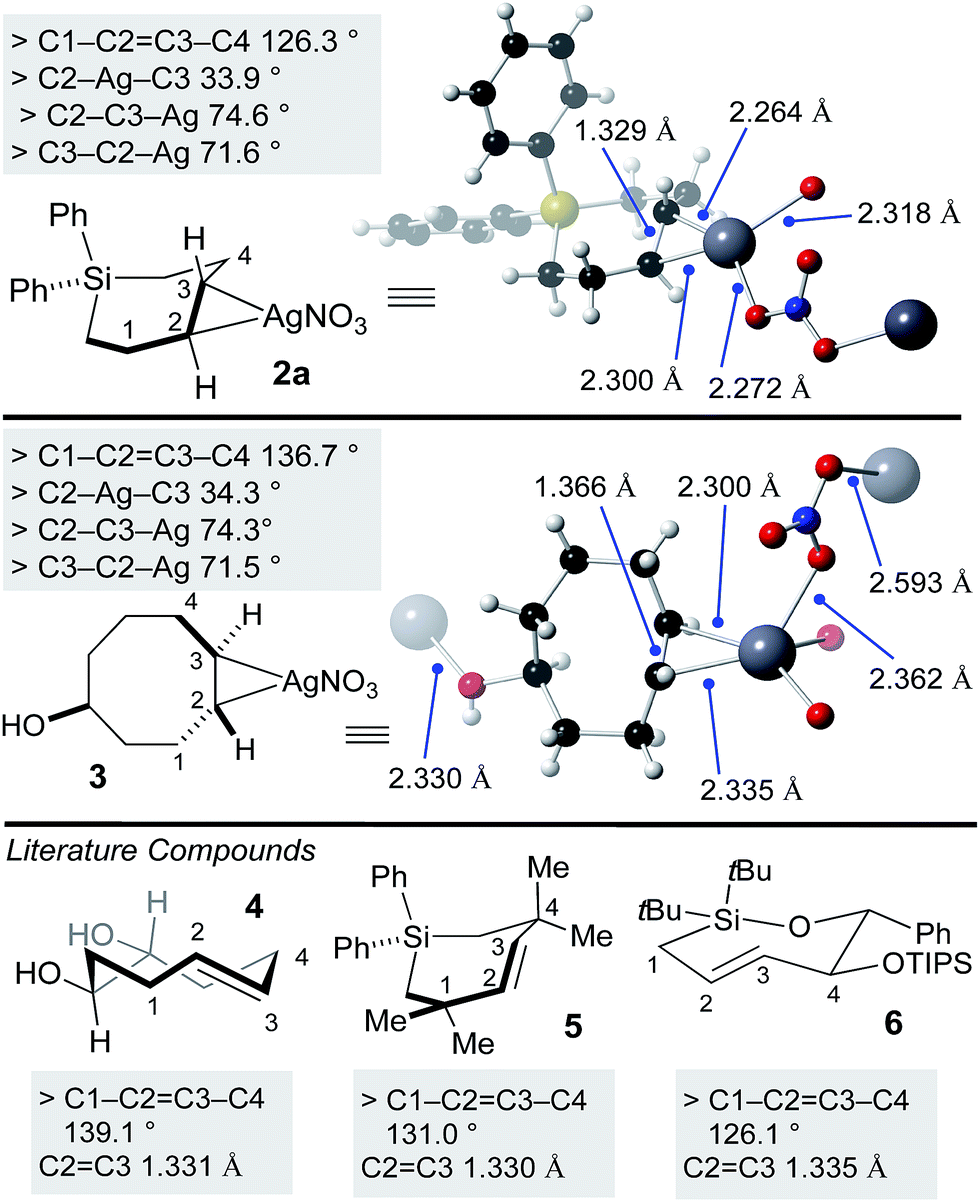 | ||
| Fig. 2 X-ray crystal structures of SiTCH·AgNO3 and TCO·AgNO3 complexes, with comparisons to known metal-free trans-cycloalkenes that have been crystallographically characterized. | ||
Silver-free Si-TCH compounds could be prepared by treating their corresponding silver nitrate complexes with an excess of aq. NH4OH or aq. NaCl, followed by extraction with organic solvent. For example, silver complex 2a upon treatment with aq. NH4OH was extracted with C6D6 to give a solution of Si-TCH 7a (98% trans isomer). Consistent with previous reports on a biomolecular mechanism for TCH isomerization,30 variable amounts (20–30%) of the cis-isomer of 7a was observed when 7a was concentrated to dryness on the rotovap. However, 7a displayed high stability when maintained in solution, with only 8% isomerization observed for a 100 mM solution of 7a that was stored for 24 hours at room temperature, and <5% isomerization for a similar solution that was stored for 24 hours in a freezer (−20 °C).
Ag(I)-complex 2d could also be freed of metal to give alkene 7d as a mixture of diastereomers (Fig. 3A). The allylic substituents of 7d protect the alkene from biomolecular chemistry, and unlike most other metal-free Si-TCH compounds, 7d is stable when stored in neat form and can be characterized by FT-IR (Fig. 3B). The weak CC double bond stretch of 7d at 1624 cm−1 is shifted to 1559 cm−1 for the Ag(I) complex 2d. This 65 cm−1 shift is consistent both in magnitude and direction for a Ag(I) alkene complex.61 Finally, we noted that Ag(I)-complexation leads to signature shifts of alkene resonances in both the 1H and 13C NMR spectra (Fig. 3C). For example, alkene resonances in the 1H NMR spectrum of metal complex 2b were shifted downfield relative to metal-free 7b by 0.15 ppm, while in 13C NMR spectra alkene resonances of 2b were shifted up field by ∼16 ppm. In the 1H NMR spectra, complexity arises due to the higher order effects for a ddd couplet.
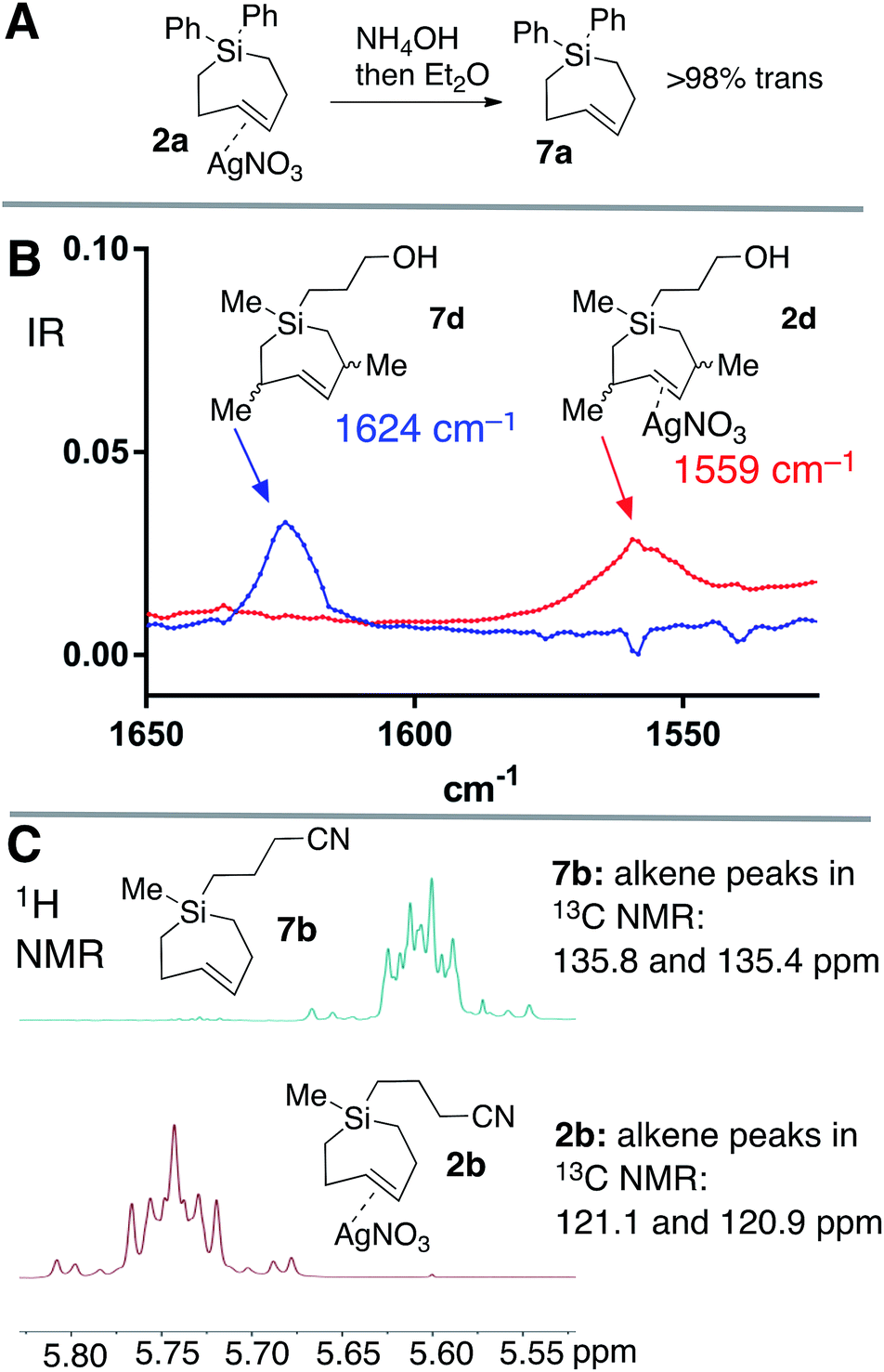 | ||
| Fig. 3 (a) Decomplexation of AgNO3 with NH4OH gives metal-free Si-TCH 7a which is stable to overnight storage in solution. Aq. NaCl can also be used to liberate metal from SiTCH·AgNO3 complexes. The metal-free complexes are stable in solution, but give variable amounts of isomerization when concentrated to dryness. (b) FT-IR spectrum of the CC stretches of metal complex 2d and free alkene 7d. (c) Diagnostic shifts in alkene peaks in the 1H and 13C NMR spectra of metal complex 2b and metal-free 7b. | ||
TCH 1a and Si-TCH 2a were shown to engage in a range of reactions as shown in Scheme 2. Metal complex 1a was directly combined with 3,6-diphenyl-1,2,4,5-tetrazine to give pyridazine 8—the product of metal dissociation, Diels–Alder/retro-Diels–Alder, and oxidation, in 98% yield. Cyclopenta-1,3-diene was also used to trap trans-cycloheptene, delivering the [4 + 2] cycloaddition adduct 9 in 81% yield as a single diastereomer. We also investigated the vicinal dihydroxylation of 1a, and found that catalytic OsO4 and NMO gave 10 in 82% yield as a single diastereomer. The observation that the dihydroxylation of 1a is stereospecific is in line with earlier observations by Cope with trans-cyclooctene.2
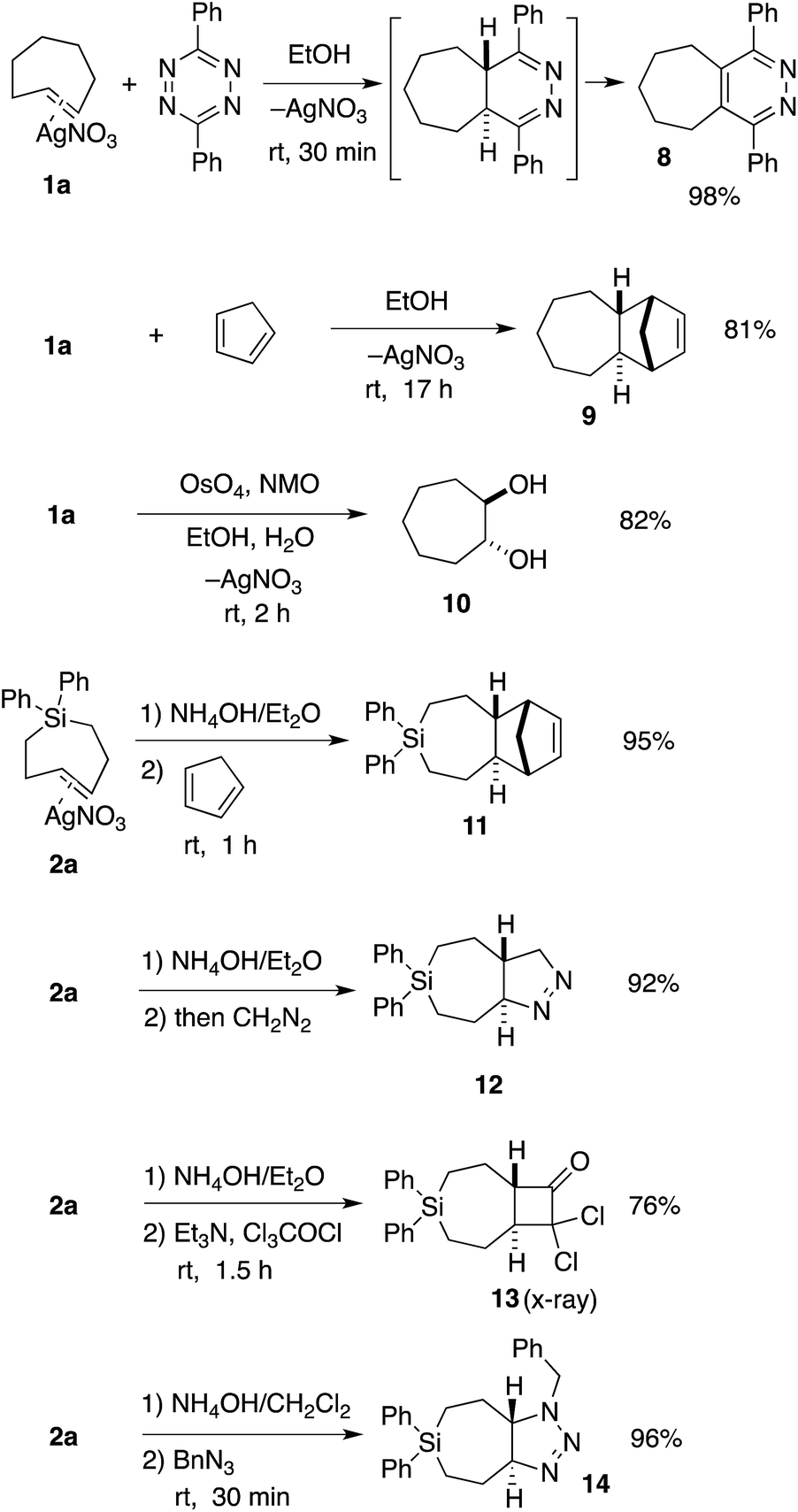 | ||
| Scheme 2 Reactions of TCH 1a and Si-TCH 2a. aYields represent isolated yields as average of two runs. | ||
Si-TCH·AgNO3 complex 2a could be freed from silver by treatment with NH4OH (Scheme 2), and subsequently combined with cyclopentadiene, diazomethane, dichloroketene, and benzyl azide to provide the cycloadducts 11–14 in 76–96% yields (Scheme 2). In each case, a single diastereomer was obtained. Attempts to combine cis-1-diphenylsila-4-cycloheptene (the cis-isomer of 7a) with dichloroketene, benzylazide or diazomethane only returned unreacted starting material. An X-ray structure was obtained for the dichloroketene adduct 13 (ESI†).
We sought to demonstrate that Si-TCH cycloadducts could be oxidized to give formal cycloadducts of 1,2,-dialkylolefins—which are recalcitrant substrates in intermolecular Diels–Alder reactions.62 With the cycloadduct 11, we demonstrated that the diphenylsila-group could be oxidized to a diol-product. Thus 11 was subjected to Tamao–Fleming reaction under Woerpel's conditions63 to give the unknown diol product 15 in 76% yield (Scheme 3).
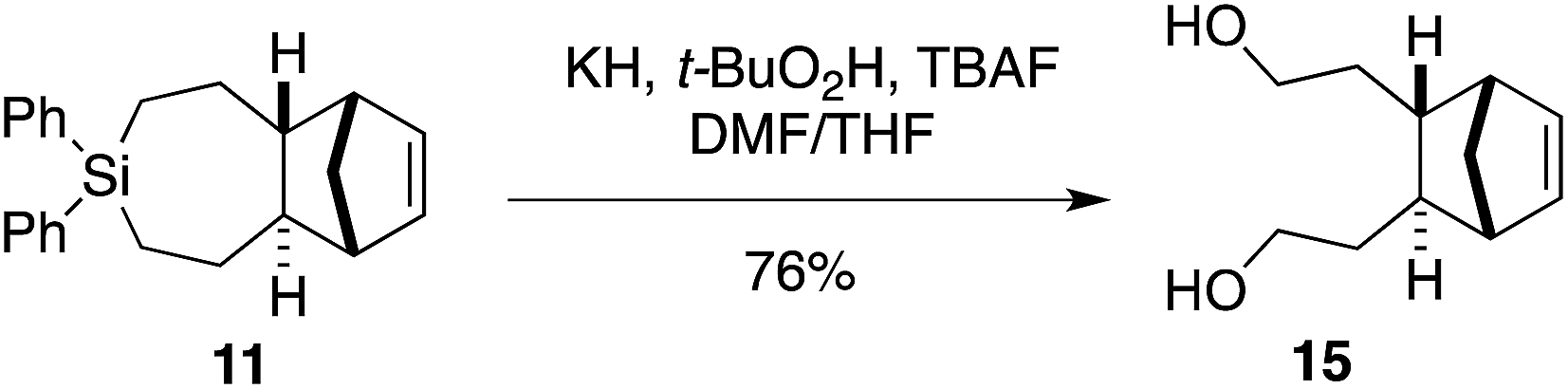 | ||
| Scheme 3 Fleming–Tamao oxidation. | ||
We further demonstrated that NHS-ester 2f could be modified though coupling to a BODIPY-fluorophore conjugate. As shown in Scheme 4, NHS ester 2f could be decomplexed from AgNO3 by treatment with brine and extraction into CH2Cl2. The resulting free Si-TCH was then conjugated to an aminohexyl BODIPY 16, and the resulting conjugate was isolated and stored as a AgNO3 complex (AgSiTCH-BODIPY). As discussed below, this fluorophore conjugate finds utility for bioorthogonal labeling in live cells. We also demonstrated that the equatorial allylic alcohol 17(eq), derived from Ag-complex 2e, could be elaborated to the carbamate 18 through treatment with benzylisocyanate (Scheme 4). trans-Cyclooctenes with allylic carbamate leaving groups have been used by Robillard64,65 and Chen66,67 for the tetrazine-ligation initiated decaging of doxorubicin and other cargo molecules. The 7-membered analog 18 is particularly stable as the Ag-free trans-cycloalkene, and can be handled neat and stored without AgNO3 in the freezer for long periods. Efforts to synthesize and explore the ability of analogs of 18 to function for payload release is a topic of ongoing study.
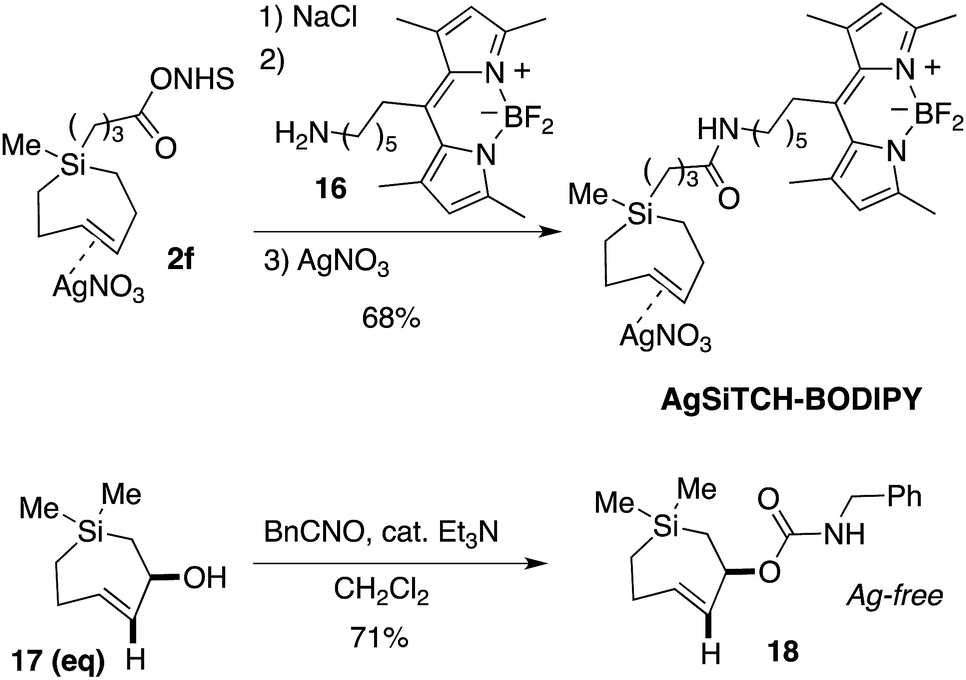 | ||
| Scheme 4 Transformations of Si-TCH derivatives. | ||
Anticipating that Si-TCH compounds may be useful in bioorthogonal chemistry, computation was used to study the structure and Diels–Alder reactivity of Si-TCH 19 (Scheme 5). We compared 19 to the parent trans-cyclooctene and a conformationally strained ‘s-TCO’ derivative 20 that was previously studied in our labs.21,54 We reasoned that the 7-membered ring in the backbone of 19 would augment the olefinic strain of the trans-cycloalkene, and thereby increase the reactivity in tetrazine ligation. Previously, thiacycloheptynes and dibenzoselenocycloheptynes have been studied in bioorthogonal chemistry.68 As shown in Scheme 5A, ground state calculations were carried out for Si-TCH 19 at the M06L/6-311+G(d,p) level, and compared to previous calculations on trans-cyclooctene and s-TCO 20.21,54 The calculated C–CC–C dihedral angle for 19 (128.8°) is smaller than that for trans-cyclooctene (137.7°) or 20 (131.9°). M06L/6-311+G(d,p) calculations were also carried out to compare the Diels–Alder reactivity of Si-TCH 19 to trans-cyclooctene and s-TCO 20 (Scheme 5B). These calculations were carried out with diphenyl-s-tetrazine so that they could be benchmarked against previous calculations.21,54 For Si-TCH 19, the calculated barriers relative to a pre-reaction complex for the Diels–Alder reaction with 3,6-diphenyl-s-tetrazine are ΔG‡ 12.48 kcal mol−1; ΔE‡ 9.27 kcal mol−1, ΔE‡ (ZPE) 9.91 kcal mol−1 and ΔH‡ 8.87 kcal mol−1. The barrier is significantly lower than that of trans-cyclooctene with 3,6-diphenyl-s-tetrazine (ΔΔG‡ −3.61 kcal mol−1, ΔΔE‡ −4.02 kcal mol−1, ΔΔE‡ (ZPE) −3.99 kcal mol−1, ΔΔH‡ 4.06 kcal mol−1). The barrier is also lower than that calculated for s-TCO 20 (ΔΔG‡ −0.26 kcal mol−1, ΔΔE‡ −0.92 kcal mol−1, ΔΔE‡ (ZPE) −0.59 kcal mol−1, ΔΔH‡ −0.72 kcal mol−1). The computations prompted us to conduct experimental investigations into the utilization of Si-TCH complexes for applications in bioorthogonal chemistry.
 | ||
| Scheme 5 (A) Ground state calculations on Si-TCH 19 show that the <C–CC–C dihedral angle is more distorted than that of trans-cyclooctene or the conformationally constrained 20. (B) Transition state calculations predict that Si-TCH 19 would be more reactive than trans-cyclooctene and 20. | ||
Previously, we had studied the reaction of diphenyl-s-tetrazine in MeOH at 25 °C with s-TCO,21,54 d-TCO,21,22 and trans-cyclooctene,69 with second order rate constants of 3100 (+/−50), 520 (+/−3) and 19.1 (+/−0.2) M−1 s−1, respectively. Si-TCH 7c was liberated from AgNO3, and in agreement with computational prediction, was found to react with diphenyl-s-tetrazine with a rate constant of 4360 (+/−430) M−1 s−1—1.4 times faster than sTCO, 8.4 times faster than dTCO, and 228 times faster than trans-cyclooctene itself (Scheme 6A).
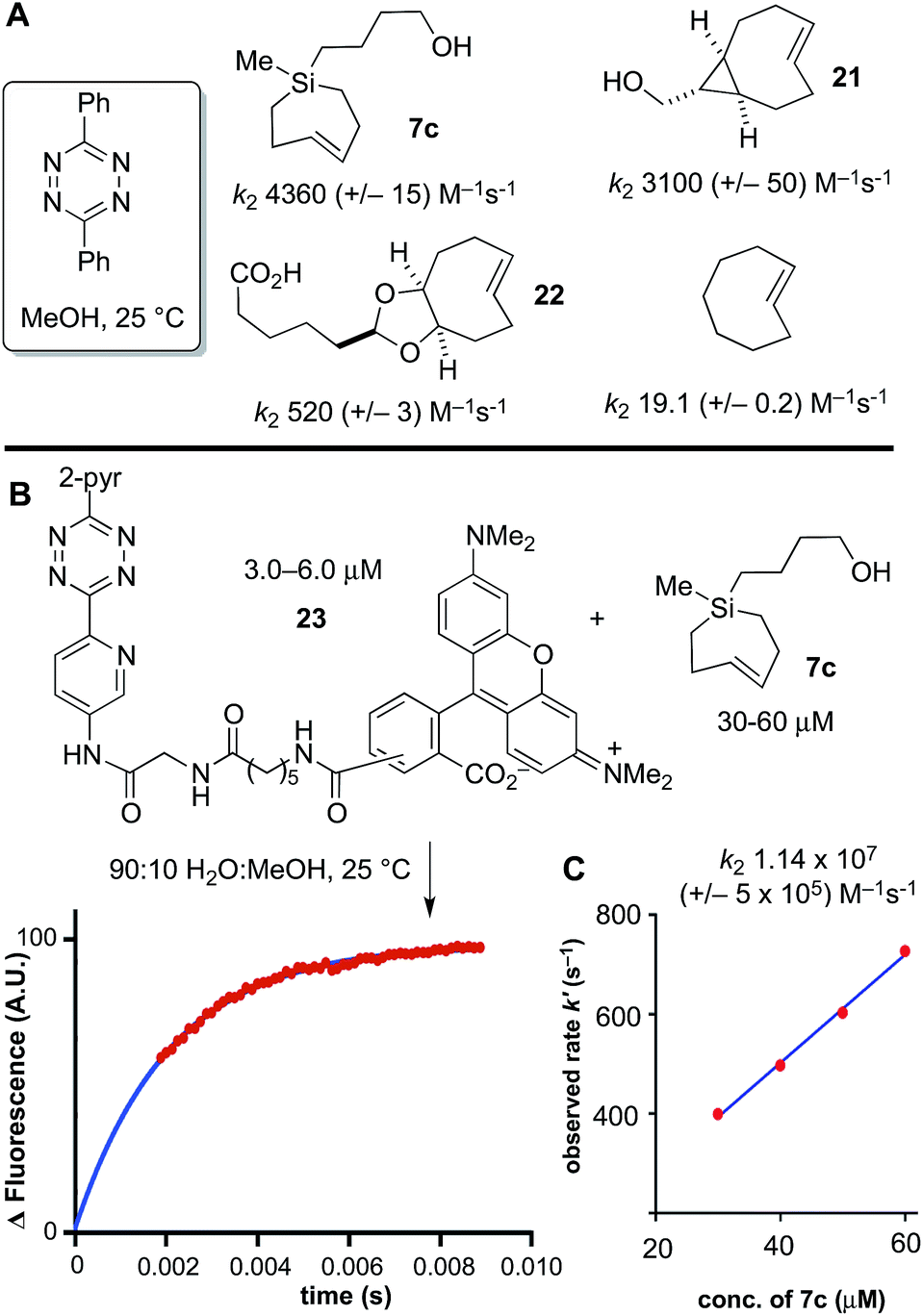 | ||
| Scheme 6 Second order rate constants (k2) were determined with a stopped-flow spectrophotometer under pseudo-first order conditions using excess trans-cycloalkene and limiting tetrazine. (A) The kinetics of the cycloaddition of 3,6-diphenyl-s-tetrazine in MeOH at 25 °C with Si-TCH 7c was measured by stopped flow spectroscopy with UV-vis monitoring at 295 nm, and compared to previously reported rate constants for s-TCO 21, d-TCO 22 and trans-cyclooctene. (B) Stopped flow kinetics with a TAMRA-3,6-dipyridyl-s-tetrazine conjugate were monitored in 9:1 H2O:MeOH with monitoring by fluorescence (Ex: 556 nm; Em: 576 nm). Data points are shown in red, and the fit is shown in blue. (C) Second order rate constants (k2) were determined by plotting kobsvs. the concentration of Si-TCH 7c. | ||
Under aqueous conditions, tetrazine ligations are accelerated due to the hydrophobic effect. We previously had studied the reactions of s-TCO, d-TCO and trans-cyclooctene with a water soluble dipyridyl-s-tetrazine derivative under stopped flow conditions in water with UV-vis monitoring.21 In the most rapid example, a rate constant of 3.3 × 106 M−1 s−1 was measured for an s-TCO derivative.21 However, our attempts to conduct a similar measurement using Si-TCH 7c were complicated because the reaction was complete before we could collect data even with stopped flow monitoring. To enable the measurement, we synthesized a fluorescent tetrazine-TAMRA conjugate 23, and used fluorescence ‘turn-on’70 to monitor reaction progress. The fluorogenic reaction enabled reaction monitoring at much lower concentrations (down to 3 μM in tetrazine). As shown in Scheme 6B and C, the reaction of 23 with Si-TCH 7c in 9:1 water:MeOH proceeds at 25 °C with a second order rate constant k2 1.14 × 107 (+/−5 × 105) M−1 s−1. This is the fastest rate constant reported to date for a bioorthogonal reaction.
We also studied the in vitro and in vivo cycloaddition of SiTCH and a green fluorescent protein with an unnatural tetrazine-containing amino acid (sfGFP-150Tet-v.2.0, referred to as GFP-Tet), encoded via the procedure of Mehl and coworkers.71 Thus, 4-(6-methyl-s-tetrazin-3-yl)phenylalanine was site-specifically introduced into a C-terminally hexahistidine-tagged GFP (sfGFP-150TAG-His6) via orthogonal translation using the evolved aminoacyl-tRNA synthetase MjRS/tRNACUA pair. Co-expression of these components in E. coli resulted in the amino acid-dependent synthesis of full-length recombinant GFP-Tet (Scheme 7A). The reaction of GFP-Tet with dienophiles is fluorogenic, and it is therefore possible to determine the reaction kinetics by monitoring the increase in GFP fluorescence.
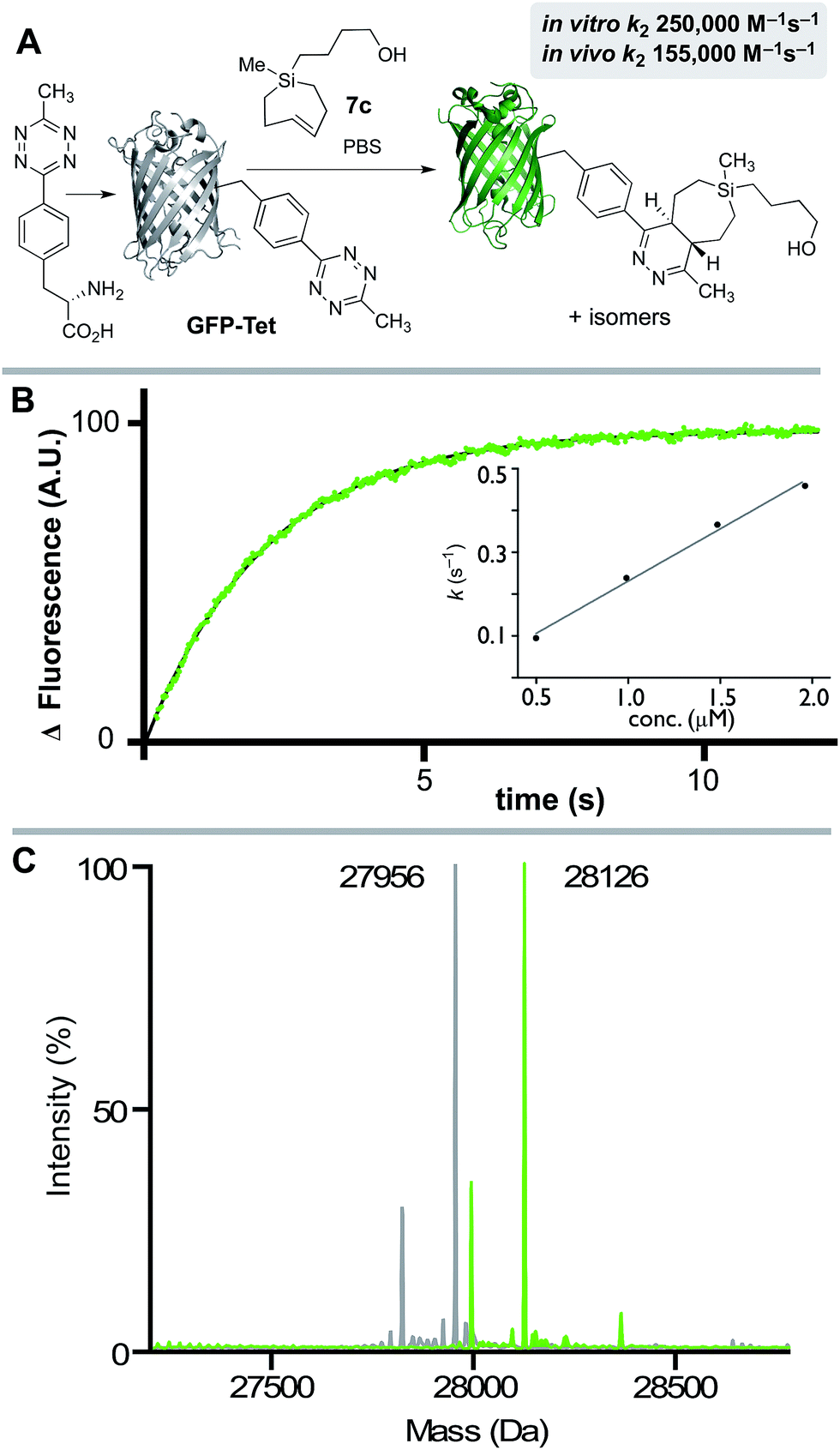 | ||
| Scheme 7 (A) GFP-Tet reacts with 7c to give a Diels–Alder adduct. Because the tetrazine quenches GFP fluorescence, the reaction progress can be measured by monitoring fluorescence increase. (B) Stopped-flow kinetic data for the reaction of GFP-Tet (50 nM) with 7c (1.96 μM) at 25 °C in PBS with fluorescence monitoring (Ex: 488 nm; Em: 506 nm). Data points are shown in green, and the fit is shown in black. A second order rate constant (k2) was determined by plotting kobsvs. the concentration of Si-TCH 7c. (C) Quantitative determination of the cycloadduct was confirmed by ESI-MS. Deconvoluted mass spectra of GFP-Tet (grey) and the cycloadduct (green) are displayed and show the expected mass shift of 170 Da. Smaller peaks in each spectrum correspond to proteins that are truncated by a methionine. | ||
Although GFP-Tet is highly reactive, with in vitro rates as fast as 87000 M−1 s−1 toward sTCO, GFP-Tet is not as rapid as 23 due to the less reactive nature of the tetrazine. Kinetic measurements were carried out with silver free Si-TCH 7c, which was obtained by treating Ag-complex 2c with NH4OH and extracting with ether. The second order rate constant of the reaction between Si-TCH 7c and GFP-Tet was determined to be 250000 ± 15000 M−1 s−1 in PBS at rt (Scheme 7B). The reaction was quantitative under these conditions as determined by ESI-MS (Scheme 7C), and is the fastest rate measured to date for GFP-Tet—2.9 times faster than previously measured rate constant for sTCO.71 Si-TCH 18 was also shown to display very rapid labeling of GFP-Tet when carried out in live bacteria. The kinetics of the in vivo tetrazine ligation were monitored in a suspension (PBS) of E. coli overexpressing GFP-Tet by measuring the increase in whole-cell fluorescence upon addition of 7c. At room temperature, a second-order rate constant of 155000 ± 20000 M−1 s−1 was measured for the in vivo reaction, which is 62% as rapid as the in vitro ligation. The modest reduction in rate is in line with previous observations with TCO-based dienophiles.21,71,72 Quantitative determination of the bioorthogonal reaction was verified by cell washing, lysis, purification by IMAC, and analysis by ESI-MS. Thus, Si-TCH 7c is capable of crossing the bacterial cell membrane and engaging in rapid, high yielding conjugation inside a living cell.
The reactivity and specificity of the SiTCH reagent with tetrazines in live mammalian cells was evaluated using the HaloTag platform,56 which we have previously used to benchmark the efficiency of various bioorthogonal reactions in the cellular environment. We have previously shown that trans-cycloalkene–AgNO3-complexes liberate AgNO3 immediately in cell media due to the high NaCl content, and perform identically to their metal-free analogs in cell labeling experiments.56 Thus, we synthesized chloroalkane derivatives of SiTCH (AgSiTCH-Halo) and methyl-tetrazine (MeTz-Halo) and used these clickable HaloTag ligands to covalently label HaloTag protein expressed in HEK293T cells with the clickable tag. In a competitive pulse-chase experiment, it was shown that 10 μM of these HaloTag ligands completely blocked incorporation of BODIPY-Halo substrate (Fig. S25†), and therefore this concentration was used for subsequent experiments. Next we evaluated the tetrazine ligation of SiTCH in mammalian cells by reacting the AgSiTCH-Halo and MeTz-Halo protein conjugates with the corresponding MeTz-BODIPY or AgSiTCH-BODIPY fluorescent probes (300 nM) for different times (2–90 min) (Scheme 8 and Fig. S26†). The reaction was quenched at the various time points by chasing with excess non-fluorescent tetrazine-amine (method 1) or TCO-amine (method 2) (Fig. S27†), and in-gel fluorescence was used to quantify conversion vs. time.
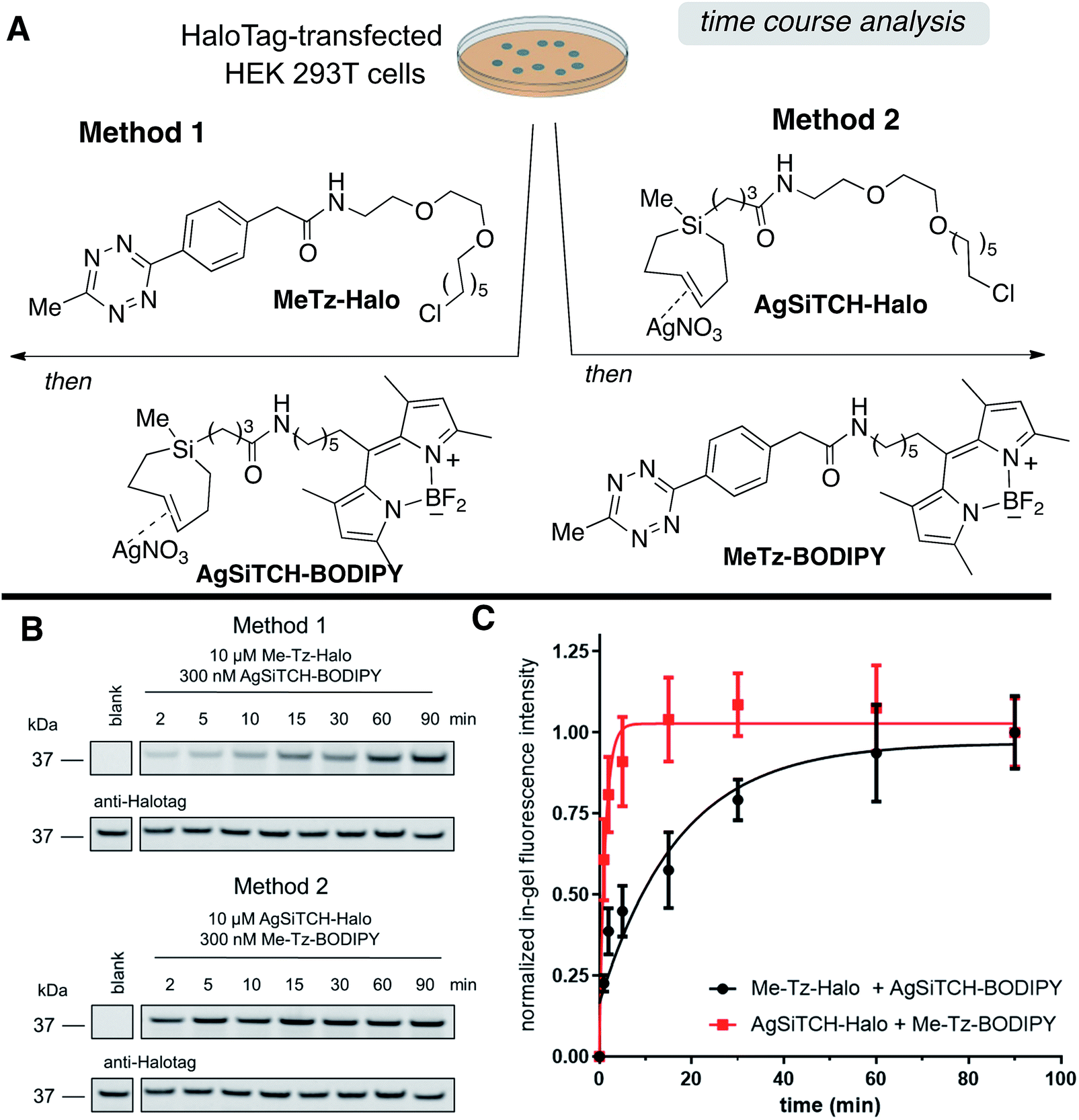 | ||
| Scheme 8 Reactions of AgSiTCH with 3-methyl-6-s-tetrazine derivatives in live HEK293T cells expressing HaloTag. (A) Cells were treated with 10 μM MeTz-Halo (method 1) or AgSiTCH-Halo (method 2) for 30 min, followed by two 30 min washings and treatment with 300 nM AgSiTCH-BODIPY (method 1) or 300 nM MeTz-BODIPY (method 2) for the indicated time. (B) Analyzed by in-gel fluorescence and western blot for each method: in-gel fluorescence is shown on the top panel, and western blot with an anti-HaloTag antibody is on the bottom panel. (C) Time course for the two reactions in (B). In-gel fluorescence intensity from n = 3 independent replicates was normalized by the corresponding western blot signal, and was fit to a one-phase exponential equation. Data were plotted as mean ± SEM. | ||
We found that the reaction of SiTCH-Halo with MeTz-BODIPY was complete within 15 minutes (Scheme 8B, method 2). Interestingly, the reaction appeared to be slower with the reverse pairing where SiTCH-BODIPY was reacted with MeTz-Halo and did not reach saturation until 90 minutes (Scheme 8B, method 1). We believe this is due to the suboptimal permeability and nonspecific protein binding of AgSiTCH-BODIPY which results in lower free concentrations available for the reaction. Compared to MeTz-BODIPY or fluorescent 5-hydroxy-trans-cyclooctenes,56AgSiTCH-BODIPY also resulted in more nonspecific protein labeling (Fig. S26†). However, compared to in-gel fluorescence with a TAMRA-labeled bicyclononyne (BCN),56AgSiTCH-BODIPY appears to be more selective.
To investigate in vivo stability of incorporated SiTCH probes in the intracellular environment, we compared HaloTag protein tagging by AgSiTCH-Halo to the previously described Ag-sTCO-Halo.56 Subsequently the TAMRA-Tz fluorescent probe was attached via the tetrazine ligation (Scheme 9A). As a benchmark, we also compared directly incorporated TAMRA-Halo without a second bioorthogonal step. In these experiments, HaloTag-transfected cells were treated with 10 μM HaloTag ligands for 30 min, followed by two 30 min wash periods. After this initial 90 min exposure to cells, time course experiments were carried out to test the intracellular stability of the sTCO- and SiTCH-labeled HaloTag proteins. Thus, cells were allowed to incubate for up to 24 additional hours, and cells initially tagged by AgSiTCH-Halo or Ag-sTCO-Halo were then treated with TAMRA-Tz at the indicated time points. Loss of fluorescence intensity relative to the TAMRA-Halo benchmark indicated instability of the bioorthogonal protein tag.
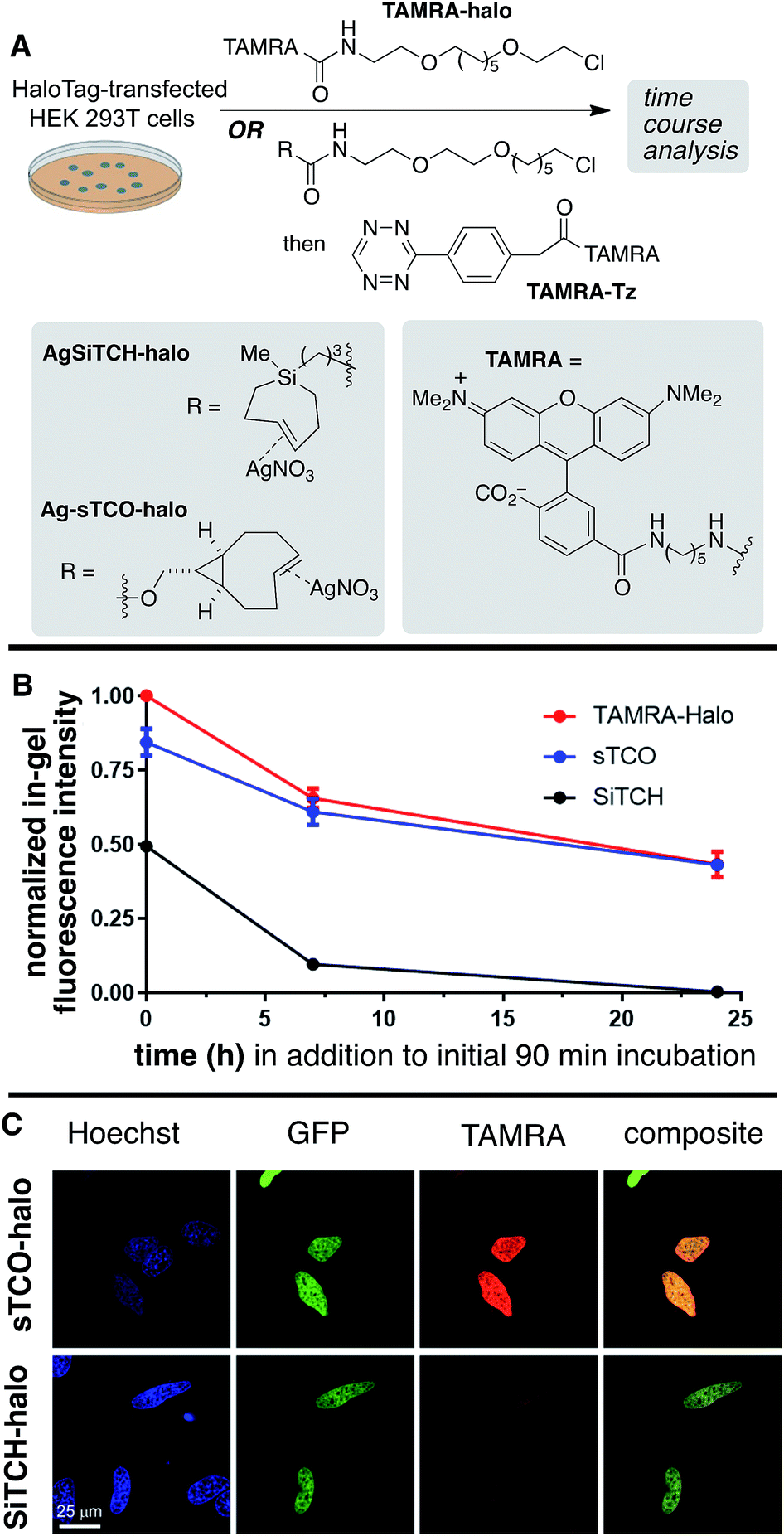 | ||
| Scheme 9 Live cell stability and imaging studies of SiTCH-Halo. (A) In the stability experiment, HEK293T cells expressing HaloTag were treated with 10 μM Ag-sTCO-Halo or AgSiTCH-Halo for 30 min, followed by two 30 min wash periods. Control cells were treated with 2 μM TAMRA-Halo. At 0, 7, and 24 h time points, 2 μM TAMRA-Tz was added and allowed to react for 1 h before analysis. (B) Cellular stability of Ag-sTCO-Halo and AgSiTCH-Halo. In-gel fluorescent intensities were normalized by the corresponding western blot signals, and were subsequently normalized by the value of TAMRA-Halo at time 0. Data were plotted as mean ± SEM. (C) Live cell images of HeLa cells expressing Halo-H2B-GFP. Cells were labeled with 10 μM Ag-sTCO-Halo or AgSiTCH-Halo for 30 min, followed by two 30 min wash periods, before the treatment with 1 μM TAMRA-Tz for 5 min. The reactions were quenched with 100 μM TCO-amine for 10 min, followed by two 30 min wash periods prior to addition of 8 μM Hoechst 33342 to visualize the nuclei and confocal imaging. Signals of Hoechst, GFP, and TAMRA were shown in blue, green, and red, respectively. Colocalization was demonstrated in composite images of GFP and TAMRA. Scale bar = 25 μm. | ||
Compared to the TAMRA-Halo ligand, the Ag-sTCO-Halo tagged protein displayed 84% fluorescence intensity when the TAMRA-Tz probe was attached immediately after the 30 min HaloTag-labeling and two 30 min wash periods (Scheme 9B, Fig. S28 and S29†). By contrast, AgSiTCH-Halo showed only 50% fluorescence intensity at this initial timepoint. After incubation for 7 h and 24 h, Ag-sTCO-Halo tagged protein showed 61% and 43% fluorescence intensity—very similar to the TAMRA-Halo benchmark. However, the AgSiTCH-Halo tagged protein displayed only 10% fluorescence after 7 h, and negligible fluorescence after 24 h. The reduced cellular labeling efficiency with AgSiTCH-Halo could also be observed in live cell images according to the published protocol (Scheme 9C and Fig. S30†).56 Specifically, HeLa cells overexpressing Halo-H2B-GFP were labeled with AgSiTCH-Halo or Ag-sTCO-Halo, and after two 30 min wash periods, treated with TAMRA-Tz for imaging. In both cases, specific fluorescent signals were observed in the nucleus with good colocalization with GFP signals. When AgSiTCH-Halo was used, however, the fluorescent intensity was only about half of that achieved with Ag-sTCO-Halo, confirming that the reduced stability of SiTCH in cells resulted in lower labeling efficiency. The lower labelling efficiency with AgSiTCH-Halo is likely due to trans-to-cis deactivation of the cycloalkane in the intracellular environment. Previously, we have studied the isomerization of trans-cyclooctenes under various conditions, and have shown that isomerization of trans-cyclooctenes likely occurs via radical mechanism that can be promoted by high thiol concentrations. Consistent with the faster in cellulo deactivation of SiTCH-Halo relative to sTCO-Halo, we observe that SiTCH 7b (30 mM) completely isomerizes in 2 h at 22 °C in CD3OD with mercaptoethanol (30 mM), whereas as control sample without thiol was >98% stable under these conditions. At −17 °C, 7b isomerizes more slowly in the presence of 30 mM mercaptoethanol, with 47% isomerization after 24 h. By comparison, the trans-cyclooctenes d-TCO, s-TCO, and oxo-TCO are much more stable toward thiol promoted isomerization. In CD3OD with mercaptoethanol (30 mM) at room temperature, s-TCO (30 mM) isomerized only after an 8 hour induction period, with complete conversion to the cis-isomer after 4 additional hours.54 In a similar experiment with d-TCO (30 mM), the induction period was 10 hours. After the induction period, there was 42% isomerization after 4 hours, and 92% isomerization after 14 hours.56 5-OxoTCO (25 mM) in the presence of mercaptoethanol (25 mM) showed only 8% isomerization in CD3OD over a 22 hour period at room temperature.72
Together, the labeling experiments in bacteria and HEK293T cells show that SiTCH derivatives can serve as useful probe molecules in the cellular environment, as the unprecedented speed of the bioorthogonal reactions of SiTCH are much more rapid than competing deactivation pathways. However, the utility of SiTCH derivatives as protein tagging molecules, where extended incubation in the cellular environment takes place prior to bioorthogonal reactivity, appears much more limited in utility plausibly due to alkene isomerization in the cellular environment.
Conclusions
In conclusion, AgNO3 complexes of trans-cycloheptene and trans-1-sila-4-cycloheptene derivatives have been prepared via a flow photochemical synthesis, using a new low temperature flow photoreactor to enable the synthesis of carbocyclic TCH derivatives. TCH·AgNO3 complexes can be handled for brief periods at rt and stored for weeks in the freezer (−18 °C). Si-TCH·AgNO3 complexes are especially stable, and can be stored on the bench stable for >1 week at rt, and for months in the freezer. X-ray crystallography was used to characterize a Si-TCH·AgNO3 complex for the first time. With decomplexation of AgNO3in situ, metal-free TCO and Si-TCH derivatives can engage in a range of cycloaddition reactions as well as dihydroxylation reactions. Computation predicted that Si-TCH would display faster bioorthogonal reactions toward tetrazines than even the most reactive trans-cyclooctenes. Metal-free Si-TCH derivatives were shown to display good stability in solution, and to engage in the fastest bioorthogonal reaction reported to date (k2 1.14 × 107 M−1 s−1 in 9:1 H2O:MeOH). Utility in bioorthogonal protein labeling in live cells is described, including labeling of GFP with an unnatrual tetrazine-containing amino acid. The reactivity and specificity of the Si-TCH reagents with tetrazines in live mammalian cells was also evaluated using the HaloTag platform. The cell labeling experiments show that Si-TCH derivatives are suitable as highly reactive probe molecules in the cellular environment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NIH R01EB014354, R01DC014461, and NSF DMR-1506613. Spectra were obtained with instrumentation supported by NIH grants P20GM104316, P30GM110758, S10RR026962, S10OD016267 and NSF grants CHE-0840401, CHE-1229234, and CHE-1048367. We thank Joshua Judkins for preliminary cellular bioorthogonal labeling experiments, Dr Glenn Yap and Casey Rowland for X-ray crystallography, Shuyu Xu and He Zhang for IR(ATR) characterization and Prof. Dr Colin Thorpe for insightful discussions.Notes and references
- K. Ziegler and H. Wilms, Liebigs Ann. Chem., 1950, 567, 1 CrossRef CAS.
- A. C. Cope, R. A. Pike and C. F. Spencer, J. Am. Chem. Soc., 1953, 75, 3212 CrossRef CAS.
- A. C. Cope, C. R. Ganellin, H. W. Johnson, T. V. Van Auken and H. J. S. Winkler, J. Am. Chem. Soc., 1963, 85, 3276 CrossRef CAS.
- F. Palacios, I. Perez de Heredia and G. Rubiales, Tetrahedron Lett., 1993, 34, 4377 CrossRef CAS.
- H. Jendralla, Tetrahedron, 1983, 39, 1359 CrossRef CAS.
- U. Thalhammer, U. Wallfahrer and J. Sauer, Tetrahedron Lett., 1990, 31, 6851 CrossRef.
- K.-J. Shea and J.-S. Kim, J. Am. Chem. Soc., 1992, 114, 4846 CrossRef CAS.
- W. Weyler Jr, L. R. Byrd, M. C. Caserio and H. W. Moore, J. Am. Chem. Soc., 1972, 94, 1027 CrossRef.
- H. Jendralla and K. Laumen, Chem. Ber., 1983, 116, 2136 CrossRef CAS.
- H. Jendralla, Synthesis, 1983, 111 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, ACS Chem. Biol., 2014, 9, 16 CrossRef CAS PubMed.
- C. S. McKay and M. G. Finn, Chem. Biol., 2014, 21, 1075 CrossRef CAS PubMed.
- C. P. Ramil and Q. Lin, Chem. Commun., 2013, 49, 11007 RSC.
- R. Rossin and M. S. Robillard, Curr. Opin. Chem. Biol., 2014, 21, 161 CrossRef CAS PubMed.
- J.-P. Meyer, P. Adumeau, J. S. Lewis and B. M. Zeglis, Bioconjugate Chem., 2016, 27, 2791 CrossRef CAS PubMed.
- M. Vrabel and T. Carell, Cycloadditions in Bioorthogonal Chemistry, 2016 Search PubMed.
- I. Nikic and E. A. Lemke, Curr. Opin. Chem. Biol., 2015, 28, 164 CrossRef CAS PubMed.
- R. Selvaraj and J. M. Fox, Curr. Opin. Chem. Biol., 2013, 17, 753 CrossRef CAS PubMed.
- A. Darko, S. Wallace, O. Dmitrenko, M. M. Machovina, R. A. Mehl, J. W. Chin and J. M. Fox, Chem. Sci., 2014, 5, 3770 RSC.
- H. Wu and N. K. Devaraj, Top. Curr. Chem., 2015, 374, 3 CrossRef PubMed.
- N. K. Devaraj and R. Weissleder, Acc. Chem. Res., 2011, 44, 816 CrossRef CAS PubMed.
- E. J. Corey, F. A. Carey and R. A. E. Winter, J. Am. Chem. Soc., 1965, 87, 934 CrossRef CAS.
- J. A. Marshall, Acc. Chem. Res., 1969, 2, 33 CrossRef CAS.
- P. J. Kropp, Mol. Photochem., 1978, 9, 39 CAS.
- J. Moran, P. H. Cebrowski and A. M. Beauchemin, J. Org. Chem., 2008, 73, 1004 CrossRef CAS PubMed.
- Y. Inoue, T. Ueoka, T. Kuroda and T. Hakushi, J. Chem. Soc., Chem. Commun., 1981, 1031 RSC.
- R. Hoffmann and Y. Inoue, J. Am. Chem. Soc., 1999, 121, 10702 CrossRef CAS.
- M. E. Squillacote, J. DeFellipis and Q. Shu, J. Am. Chem. Soc., 2005, 127, 15983 CrossRef CAS PubMed.
- G. M. Wallraff and J. Michl, J. Org. Chem., 1986, 51, 1794 CrossRef CAS.
- (a) R. Bonneau, P. Forner de Violet and J. Joussot-Dubien, Nouv. J. Chim., 1977, 1, 31 CAS; (b) R. Bonneau, J. Joussot-Dubien, J. Yarwood and J. Pereyre, Tetrahedron Lett., 1977, 235 CrossRef CAS.
- X.-N. Wang, E. H. Krenske, R. C. Johnston, K. N. Houk and R. P. Hsung, J. Am. Chem. Soc., 2015, 137, 5596 CrossRef CAS PubMed.
- E. J. Corey, M. Tada, R. LaMahieu and L. Libit, J. Am. Chem. Soc., 1965, 87, 2051 CrossRef CAS.
- P. E. Eaton and K. Lin, J. Am. Chem. Soc., 1965, 87, 2052 CrossRef CAS.
- J. Moran, P. Dornan and A. M. Beauchemin, Org. Lett., 2007, 9, 3893 CrossRef CAS PubMed.
- H. Dorr and V. H. Rawal, J. Am. Chem. Soc., 1999, 121, 10229 CrossRef CAS.
- H. M. L. Davies, Ø. Loe and D. G. Stafford, Org. Lett., 2005, 7, 5561 CrossRef CAS PubMed.
- R. G. Salomon, K. Folting, W. E. Streib and J. K. Kochi, J. Am. Chem. Soc., 1974, 96, 1145 CrossRef CAS.
- T. Spee and A. Mackor, J. Am. Chem. Soc., 1981, 103, 6901 CrossRef CAS.
- J. T. M. Evers and A. Mackor, Recl. Trav. Chim. Pays-Bas, 1979, 98, 423 CrossRef CAS.
- H. Nishiyama, T. Naitoh, Y. Motoyama and K. Aoki, Chem.–Eur. J., 1999, 5, 3509 CrossRef CAS.
- H. Jendralla, Angew. Chem., Int. Ed., 1980, 19, 1032 CrossRef.
- H. Jendralla, Chem. Ber., 1980, 113, 3557 CrossRef CAS.
- T. Shimizu, K. Shimizu and W. Ando, J. Am. Chem. Soc., 1991, 113, 354 CrossRef CAS.
- A. W. Krebs, K.-I. Pforr, W. Raffay, B. Thölke, W. A. König, I. Hardt and R. Boese, Angew. Chem., Int. Ed., 1997, 36, 159 CrossRef CAS.
- A. W. Krebs, B. Thölke, K.-I. Pforr, W. A. König, K. Scharwächter, S. Grimme, F. Vögtle, A. Sobanski, J. Schramm and J. Hormes, Tetrahedron: Asymmetry, 1999, 10, 3483 CrossRef CAS.
- J. Santucci, J. R. Sanzone and K. A. Woerpel, Eur. J. Org. Chem., 2016, 2016, 2933 CrossRef CAS.
- M. A. Greene, M. Prévost, J. Tolopilo and K. A. Woerpel, J. Am. Chem. Soc., 2012, 134, 12482 CrossRef CAS PubMed.
- B. Hurlocker, C. Hu and K. A. Woerpel, Angew. Chem., Int. Ed., 2015, 54, 4295 CrossRef CAS PubMed.
- (a) J. R. Sanzone, C. T. Hu and K. A. Woerpel, J. Am. Chem. Soc., 2017, 139, 8404 CrossRef CAS PubMed; (b) J. R. Sanzone and K. A. Woerpel, Synlett, 2017, 28, 2478–2482 CrossRef CAS.
- K. Tomooka, S. Miyasaka, S. Motomura and K. Igawa, Chem.–Eur. J., 2014, 20, 7598 CrossRef CAS PubMed.
- M. Royzen, G. P. A. Yap and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 3760 CrossRef CAS PubMed.
- M. T. Taylor, M. L. Blackman, O. Dmitrenko and J. M. Fox, J. Am. Chem. Soc., 2011, 133, 9646 CrossRef CAS PubMed.
- M. Royzen, M. T. Taylor, A. DeAngelis and J. M. Fox, Chem. Sci., 2011, 2, 2162 RSC.
- H. E. Murrey, J. C. Judkins, C. W. Am Ende, T. E. Ballard, Y. Fang, K. Riccardi, L. Di, E. R. Guilmette, J. W. Schwartz, J. M. Fox and D. S. Johnson, J. Am. Chem. Soc., 2015, 137, 11461 CrossRef CAS PubMed.
- B. D. A. Hook, W. Dohle, P. R. Hirst, M. Pickworth, M. B. Berry and K. I. Booker-Milburn, J. Org. Chem., 2005, 79, 7558 CrossRef PubMed.
- G. V. Los, L. P. Encell, M. G. McDougall, D. D. Hartzell, N. Karassina, C. Zimprich, M. G. Wood, R. Learish, R. F. Ohana, M. Urh, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris, J. Zhu, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, ACS Chem. Biol., 2008, 3, 373 CrossRef CAS PubMed.
- D. B. Janssen, Curr. Opin. Chem. Biol., 2004, 8, 150 CrossRef CAS PubMed.
- I. Rencken, J. C. A. Boeyens and S. W. Orchard, J. Crystallogr. Spectrosc. Res., 1988, 18, 293 CrossRef CAS.
- M. Tsutsui, M. N. Levy and A. Nakamura, Introduction to Metal π-complex chemistry, Springer, New York, 1970, p. 40 Search PubMed.
- A. A. Vasil'ev, N. A. Donskaya, G. V. Cherkaev and N. M. Yur'eva, J. Org. Chem., 1991, 27, 273 Search PubMed.
- J. H. Smitrovich and K. A. Woerpel, J. Org. Chem., 1996, 61, 6044 CrossRef CAS.
- R. Rossin, S. M. J. Van Duijnhoven, W. Ten Hoeve, H. M. Janssen, L. H. J. Kleijn, F. J. M. Hoeben, R. M. Versteegen and M. S. Robillard, Bioconjugate Chem., 2016, 27, 1697 CrossRef CAS PubMed.
- R. M. Versteegen, R. Rossin, W. Ten Hoeve, H. M. Janssen and M. S. Robillard, Angew. Chem., Int. Ed., 2013, 52, 14112 CrossRef CAS PubMed.
- J. Li, S. Jia and P. R. Chen, Nat. Chem. Biol., 2014, 10, 1003 CrossRef CAS PubMed.
- X. Fan, Y. Ge, F. Lin, Y. Yang, G. Zhang, W. S. C. Ngai, Z. Lin, S. Zheng, J. Wang, J. Zhao, J. Li and P. R. Chen, Angew. Chem., Int. Ed., 2016, 55, 14046 CrossRef CAS PubMed.
- (a) G. de Almeida, E. M. Sletten, H. Nakamura, K. K. Palaniappan and C. R. Bertozzi, Angew. Chem., Int. Ed., 2012, 51, 2443 CrossRef CAS PubMed; (b) G. de Almeida, L. C. Townsend and C. R. Bertozzi, Org. Lett., 2013, 15, 3038 CrossRef CAS PubMed; (c) M. Martinek, L. Filipova, J. Galeta, L. Ludvikova and P. Klan, Org. Lett., 2016, 18, 4892 CrossRef CAS PubMed, For earlier work on heteroatom containing cycloheptynes: (d) H. Kimling and A. Krebs, Liebigs Ann. Chem., 1974, 2074 Search PubMed; (e) A. Krebs and H. Kimling, Tetrahedron Lett., 1970, 11, 761 CrossRef.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518 CrossRef CAS PubMed.
- N. K. Devaraj, S. Hilderbrand, R. Upadhyay, R. Mazitschek and R. Weissleder, Angew. Chem., Int. Ed., 2010, 49, 2869 CrossRef CAS PubMed.
- R. J. Blizzard, D. R. Backus, W. Brown, C. G. Bazewicz, Y. Li and R. A. Mehl, J. Am. Chem. Soc., 2015, 137, 10044 CrossRef CAS PubMed.
- W. D. Lambert, S. L. Scinto, O. Dmitrenko, S. J. Boyd, R. Magboo, R. A. Mehl, J. W. Chin, J. M. Fox and S. Wallace, Org. Biomol. Chem., 2017, 15, 6640 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full procedures, computational details and characterization data. CCDC 1583975 and 1583976. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04773h |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2018 |