
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the intrinsic dynamic nature of the rigid UiO-66 metal–organic framework†
Julianna
Hajek
,
Chiara
Caratelli
 ,
Ruben
Demuynck
,
Kristof
De Wispelaere
,
Louis
Vanduyfhuys
,
Michel
Waroquier
and
Veronique
Van Speybroeck
*
,
Ruben
Demuynck
,
Kristof
De Wispelaere
,
Louis
Vanduyfhuys
,
Michel
Waroquier
and
Veronique
Van Speybroeck
*
Center for Molecular Modeling (CMM), Ghent University, Technologiepark 903, B-9052 Zwijnaarde, Belgium. E-mail: veronique.vanspeybroeck@ugent.be
First published on 29th January 2018
Abstract
UiO-66 is a showcase example of an extremely stable metal–organic framework, which maintains its structural integrity during activation processes such as linker exchange and dehydration. The framework can even accommodate a substantial number of defects without compromising its stability. These observations point to an intrinsic dynamic flexibility of the framework, related to changes in the coordination number of the zirconium atoms. Herein we follow the dynamics of the framework in situ, by means of enhanced sampling molecular dynamics simulations such as umbrella sampling, during an activation process, where the coordination number of the bridging hydroxyl groups capped in the inorganic Zr6(μ3-O)4(μ3-OH)4 brick is reduced from three to one. Such a reduction in the coordination number occurs during the dehydration process and in other processes where defects are formed. We observe a remarkable fast response of the system upon structural changes of the hydroxyl group. Internal deformation modes are detected, which point to linker decoordination and recoordination. Detached linkers may be stabilized by hydrogen bonds with hydroxyl groups of the inorganic brick, which gives evidence for an intrinsic dynamic acidity even in the absence of protic guest molecules. Our observations yield a major step forward in the understanding on the molecular level of activation processes realized experimentally but that is hard to track on a purely experimental basis.
Introduction
Metal–organic frameworks (MOFs), among the most intriguing materials of current science, are hybrid materials constructed from metal ions or metal clusters linked together by multitopic organic linkers.1–3 The building block concept gives nearly infinite possibilities to vary the chemical composition of MOFs and make these materials very appealing for a broad set of applications such as gas storage and separation,4 detection and decomposition of warfare agents,5 drug delivery6 and catalysis.7–12 The main drawback of most MOFs is their weak stability under reaction conditions.13,14 However, to date, the amount of synthesized MOF structures has grown substantially, giving rise to chemically and thermally stable MOFs.14,15In this respect, UiO-66 has received considerable attention due to its exceptional thermal, mechanical and chemical stability, which is not typically found in other MOFs.16–19 Pristine UiO-66 is composed of inorganic Zr6(μ3-O)4(μ3-OH)4 (Fig. 1) bricks connected through ditopic organic ligands and was first synthesized by Lillerud and co-workers.20 In the inorganic brick, four three-fold bridging hydroxyl groups are symmetrically distributed around the building block such that the oxide and hydroxide anions are capping the faces of the Zr6 octahedron alternately to achieve minimum repulsion (Fig. 1, pristine brick).21,22 As will be shown here, the lability of the species that make up UiO-66 may play a critical role in processes where the coordination number of the Zr atoms changes, such as dehydration or exchange of either metal or linker ligands, and has a direct consequence in the formation of defects.
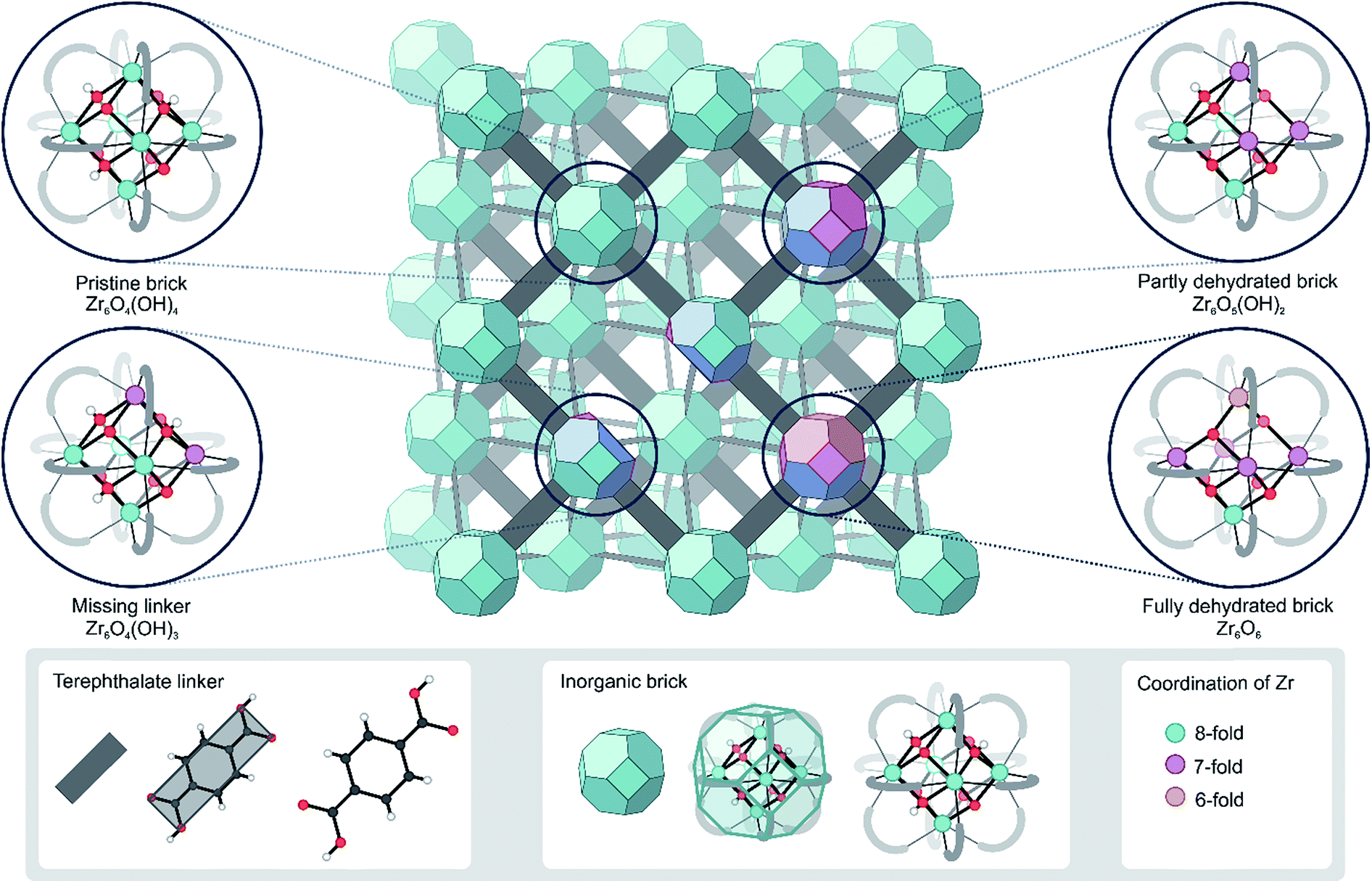 | ||
| Fig. 1 Schematic representation of the UiO-66 structure with possible configurations of the bricks that give rise to coordinatively unsaturated Zr atoms. The colors indicate the coordination of the Zr atoms. The representation of the UiO-66 unit cell has been adapted from ref. 23. | ||
The key to the exceptional stability can be traced back to the inherent composition of the framework, where each Zr atom is 8-fold coordinated by oxygen atoms and each inorganic brick is 12-fold coordinated by organic linkers in the defect-free material. Despite being exceptionally rigid, there are various indications that the structure is far more dynamic than originally believed.24 Dynamic behavior in this context should not be confused with the properties of flexible or dynamic frameworks, which have the ability to undergo large structural deformations while retaining their crystallinity upon exposure to external stimuli, such as light, temperature, pressure, and adsorption of guest molecules. Such materials are classified as third-generation soft porous crystals by Kitagawa and collaborators.25,26 The dynamic behavior explored here refers to the flexibility introduced by changing the coordination number of the metal, which affords to modify the framework by temporarily breaking the metal–ligand (M–L) bonds. The M–L bond is generally perceived as the weakest link in MOFs, both thermodynamically and kinetically.15 This has a negative effect on the thermal stability, but the ease of coordination change may also be used as an opportunity to modify the structure. In Zr-based MOFs, like UiO-66, M–L bonds are particularly stable as Zr is one of the most oxophilic metal elements,27 but such post-synthetic modifications have recently been realized experimentally in UiO-66.28–30
First of all, it has been found that the Zr6O4(OH)4 inorganic brick has the ability to be dehydrated reversibly, thereby changing the coordination number of the Zr metal centers between the hydrated and dehydrated forms.18,21,31,32 The dehydration and rehydration process of the inorganic Zr-brick in UiO-66 has been experimentally studied using in situ infrared (IR) spectroscopy and gravimetric techniques.31 The dehydration starts at 523 K and is completed at 573 K with the removal of two water molecules resulting in a cornerstone composition of Zr6O6 (Fig. 1, fully dehydrated brick). When starting from the defect-free material, the dehydration process is initiated by the first water removal and results in a Zr6O5(OH)2 brick, where the coordination number of the three Zr atoms is reduced from 8 to 7 (Fig. 1, partly dehydrated brick). A further reduction in the Zr coordination number takes place with the second removal of a water molecule. Fully dehydrated UiO-66 is composed of Zr6O6 distorted units and has variously coordinated Zr atoms, with coordination numbers ranging from 8 to 6 (Fig. 1). Despite these significant deformations in the inorganic brick, the original structure of UiO-66 is almost preserved and maintains the robustness of the brick, which was confirmed by PXRD patterns, in situ DRIFT spectra and gravimetric characterization techniques (TG-DSC).21,31 Moreover, a rehydration process can restore the material to its initial structure, with the hydrated Zr6O4(OH)4 inorganic cornerstone. This is a remarkable property of this MOF, giving an indication of the existence of mobile μ3-OH hydroxyl groups in the brick. By performing IR experiments on UiO-66, in which the temperature was gradually increased, Shearer et al.31 observed the isolated hydroxyl region by the presence of additional bands at 3600–3700 cm−1. Furthermore, recent experimental findings pointed towards a significant structural mobility in frameworks that are classified as rigid. By applying ultrafast 2D IR spectroscopy, structural fluctuations have been shown in functionalized Zr-based MOFs.33 These fast dynamics of the framework have a direct effect on an MOF's lability, flexibility and coordination changes. Furthermore, recent synthesis procedures have reached a higher level of perfection, allowing engineering of frameworks by concepts such as post-synthetic ligand exchange or solvent-assisted linker exchange.28 Within the UiO-66 framework, unfunctionalized linkers have been replaced by functionalized ones without compromising the MOF crystallinity or porosity.28
All these observed features in UiO-66 point towards an intrinsic dynamic flexibility of the material and are related to changes in the coordination number of the metal ions making up the inorganic bricks.26,34 A more general discussion on these aspects has been recently reviewed by Morris and Brammer.24 The ease of coordination change and ability to modify the structure without compromising its hydrothermal stability has shown to be extremely useful for adsorption and catalysis.12,35 However, despite this application-driven importance, the understanding of rearrangement of linkers, hydroxyl groups and other labile species on the molecular level is fairly limited. It poses a huge challenge for experimentalists to trace this intrinsic dynamic flexibility in situ during an activation process. In this sense, computational techniques offer an indispensable alternative to follow the dynamics of the system, as was suggested by Ling and Slater, who showed a dynamic acidity of the defective UiO-66 framework.36 Based on first-principles molecular dynamics simulations, proton shuttles have been observed between water molecules coordinated to the metals and charge-balancing hydroxide anions, which compensate for the missing linkers in defective UiO-66.36 Such proton shuttles may also be facilitated by the presence of other guest species in the pores, such as methanol.37
Herein, we take a major step forward in the structural understanding of the UiO-66 by using advanced or enhanced sampling molecular dynamics simulations, which allow to follow in situ the fast dynamics of the UiO-66 framework during activation processes. Typical molecular dynamics simulations only sample stable regions of the free energy surface, whereas enhanced sampling techniques allow visiting activated regions of the free energy surface, typically encountered during activation processes. More elaborate reviews on these advanced molecular dynamics simulations may be found in the work of Valsson and references therein.38 Such techniques have only recently found their way in the field of heterogeneous catalysis and have been successfully used to study chemical reactions at true operating conditions of temperature and pressure.39–45 A good overview of the recent advances in computational techniques for nanoporous materials is given in ref. 46 and 47 by the Coudert group. In this contribution, we study the UiO-66 framework along the activation path where one of the hydroxyl groups bridging the Zr atoms in the brick lowers its coordination number from 3 to 1, which is a crucial step in the dehydration process.32 Preliminary molecular insights are obtained from static calculations, neglecting the dynamic behavior of the system, and reveal the presence of transition states with loose hydroxyl groups and dangling linkers along the dehydration pathway (Reaction scheme depicted in Fig. S2 of the ESI†).48 The results here show a highly dynamic behavior of M–L bonding, whose lability changes the degree of network connectivity and coordination that leads to open metal sites. Linkers can easily decoordinate, translate, rotate and recoordinate, and hydroxyl groups show a high degree of mobility, even in the absence of external water or solvent molecules. All these rearrangements occur without deterioration of the structural integrity of the material. Our simulations show that even very stable MOFs like UiO-66 are much more dynamic than originally expected.
Results and discussion
Herein, dynamical structural rearrangements of the UiO-66 framework are studied by lowering the coordination number of the hydroxyl groups bridging the Zr atoms from 3 to 1. This reshuffling is typically encountered during dehydration, which is a commonly applied activation procedure upon thermal treatment. Exploration of higher free energy regions requires usage of enhanced molecular dynamics simulations.49–55 Herein the umbrella sampling (US) methodology is used, whereby an external potential is added to the true Hamiltonian to enhance the sampling of low probability regions along certain coordinates of the system.55,56 These degrees of freedom are called collective variables and may be complicated functions of all internal degrees of freedom. An appropriate choice of these coordinates is far from trivial,57 but here we chose the coordination number of the hydroxyl oxygen to the three Zr atoms as a collective variable.58 This choice is inspired by the experimental observation of dangling, asymmetric hydroxyl groups in intermediate states during dehydration,31 and the theoretical reaction mechanism proposed in ref. 32. The coordination number is defined based on the distance between the hydroxyl group and the three surrounding Zr atoms, as schematically shown in Fig. 2.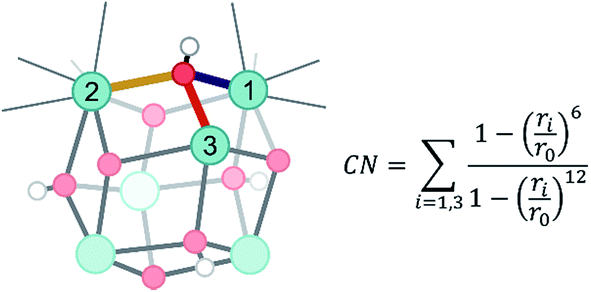 | ||
| Fig. 2 Schematic representation of the applied collective variable. Three critical Zr–O distances, represented with solid yellow, red and purple lines, are used as input in the formal definition of the collective variable. The index i labels each of the three Zr atoms surrounding the hydroxyl groups. | ||
With this choice, the coordination number at a value of about 2.17 represents the Zr brick in its equilibrium configuration, denoted as configuration 1 in Fig. 3, and reaches a value of about 0.9 when the hydroxyl group is covalently connected to only one Zr atom, giving rise to a bridging μ1-OH hydroxyl group (configuration 5). Partitioning the entire range of the reaction coordinate (1.7 ≤ CV ≤ 2.4) into 36 windows following the procedure of US as outlined in the Methodology section and in the ESI,† an umbrella sampling simulation has been performed in each window. The quality of the selection of the 36 windows along the reaction path and the magnitude of the spring constant characterizing the harmonic bias potential are assessed by sufficient overlap of the umbrellas covering the various windows (Fig. S1 of the ESI†), which is an indication that the sum of the three coordination numbers as CV is a well-suited choice in the reaction path towards the first dehydroxylation. Along the reaction path, five classes of configurations are distinguished, which differ in the degree of coordination of the hydroxyl group with the three neighboring Zr atoms going from a 3-fold to a 1-fold coordination (Fig. 3).
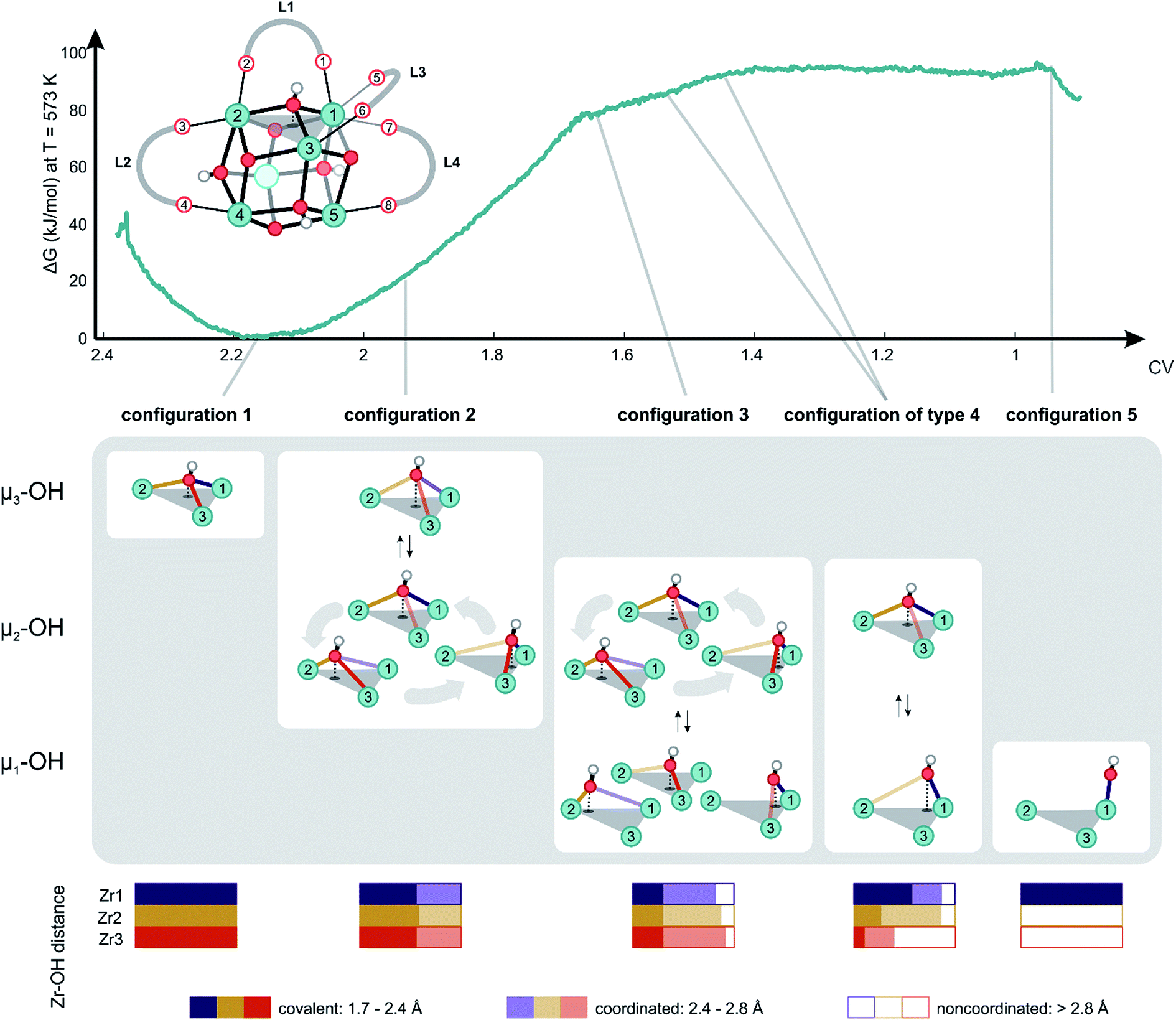 | ||
| Fig. 3 Free energy profile at 573 K along the reaction path of dehydration with the collective variable defined, as given by the equation in Fig. 2, as the reaction coordinate. Reference representation of the brick taken from configuration 1 is shown above the free energy profile, with labeling of all relevant linkers, carboxylate oxygens, hydroxyls and Zr atoms needed to describe the chemical transformation. Some essential turning points on the profile are indicated with a schematic display of their respective configurations. Probability distributions regarding the type of coordination of the hydroxyl group with respect to the Zr centers measured during the US simulation are reported in the bottom. | ||
Configuration 1 in Fig. 3 at 573 K shows the equilibrium structure, where the four μ3-OH groups are ordered symmetrically in a tetrahedral fashion and each Zr atom is 8-fold coordinated (Table S2 of the ESI†). When slightly activating the system towards configuration 2, which is characterized by a collective variable of 1.96, it becomes clear that the hydroxyl group is relatively mobile, changing its position equally between the three Zr atoms. The average position of the hydroxyl group remains centrally located between three Zr atoms in a μ3-OH configuration (Table S2 of the ESI†). Nevertheless, as the coordination number decreases, the mobility of the hydroxyl group becomes more distinct. Alternatively, the hydroxyl group makes a stronger connection to two out of three Zr atoms, breaking the trigonal symmetry (μ2-OH structure). The increase in free energy to reach this point is relatively modest with a value of about 20 kJ mol−1 from configuration 1. To obtain more information on the type of connection between the oxygen and the three Zr atoms, we also constructed probability distributions for each of the configurations (Fig. 3, bottom). Interestingly, at configuration 2, the probability of observing a looser OH-bond with one of the Zr atoms is the same for the three Zr atoms, which underlines the relatively mobile nature of the μ3-OH hydroxyl group in the pristine material. Additional inspection of the free energy profile shows that the dehydration process is initiated by configuration 2, as further progression towards lower coordination numbers does not reveal any more μ3-OH configurations.
A critical point along the reaction path is encountered at configuration 3 corresponding to a collective variable of 1.64, where the hydroxyl group is covalently bonded to only one of the Zr atoms. Nevertheless, some coordination with another metal is maintained, but the coordination with the third Zr atom is no longer observed. Interestingly, even for such lower coordinated structures, where the hydroxyl group changes substantially its position, there is no clear preference for the coordination of the hydroxyl group to one of the three Zr atoms and there is still a complete symmetry in the average coordination behavior of the hydroxyl group with the three Zr atoms (Table S2 of the ESI†). Note that this region is already substantially highly activated with a value of about 80 kJ mol−1 that is typically reached during the activation processes. During the simulation, the hydroxyl group travels from one position to the other, systematically establishing a covalent bond with one of the three adjacent Zr atoms to create open metal sites that are 7-fold coordinated. For the first time along the trajectory, we observe a tendency of the linkers to elongate the M–L bond and deform the phenyl ring out-of-plane to make space for the traveling hydroxyl group. Such elongations and deformations indicate that the system enters the transition state region, explaining the increased free energy.
All structures encountered after configuration 3, are situated in a higher energy plateau. They are almost isoenergetic since the collective variable only reflects the degree of coordination of the hydroxyl group to the three Zr atoms to which it was originally attached. The broad set of structures encountered in this window of the collective variable show new internal deformation modes, which include decoordination, translation, rotation, and recoordination of the linkers and hydroxyl group bond cleavages. Each of the structures of configuration of type 4 has a clear preference to form μ1-OH configuration with one of the three Zr atoms. However, the hydroxyl group seeks stabilizing interactions with the other Zr atoms. For configurations of type 4, the probability distributions for the various Zr–O bonds show a clear asymmetry for the three Zr atoms. A strong covalent bond is observed between the hydroxyl group and a Zr atom from which a linker has been decoordinated (vide infra). Indeed, substantial mobility of linkers is observed in this region, with strong deformation modes of four linkers. This is explained in greater detail hereafter for configurations of type 4 and 5. For the labeling pattern of the various linkers and atoms, we refer to the reference structure as displayed in Fig. 3 above of the free energy profile.
For configurations of type 4, in which one Zr atom (Zr1) makes a strong, covalent bond with the hydroxyl group, two possible motions of the BDC linker are observed, which are further illustrated in Fig. 4. The first defined motion (Fig. 4, left column) corresponds to a translation of the linker along the axis of the two Zr atoms to which it is bonded (Movie S1 of the ESI†). In this case, the linker decoordination starts on the Zr with the μ1-OH configuration and is induced by the steric interactions, which are directly correlated to the position of the hydroxyl group. The linker (L1) decoordinates from Zr (O1 from Zr1) and further forms an intermediary chelated structure in which both carboxylic oxygens are connected to the same Zr atom (Zr2 and O1, O2). The chelated structure is a short-living metastable state from which one oxygen of the linker (O2) subsequently decoordinates from Zr2 (configuration 4t in Fig. 4), thereby changing the parent connectivity between the metal ion and the carboxylic oxygen from the linker (Zr2 is now connected to O1 instead of O2). During the simulation of this configuration, the hydroxyl group keeps its bond with Zr1 and systematically switches between μ2 and μ1 connectivity by establishing a coordination with the Zr atom (Zr2) to which the linker is η1 chemically bonded. Another significant aspect of configuration of type 4 that exhibits the translation mode is the progressive change in the coordination number of the two Zr atoms. Firstly, linker decoordination lowers the coordination number of Zr1 from 8 to 7. Secondly, in the starting configuration of type 4 and final configuration 4t, Zr2 is 7-fold coordinated, but during the process of linker translation, an intermediary coordination of 8 is present in the chelated structure. This evidence of the presence of an intermediary chelated structure has also been observed by Puchberger et al.59 in the case of an isolated Zr6 cluster. The change in coordination number of the various Zr atoms is schematically represented by a color code in Fig. 4.
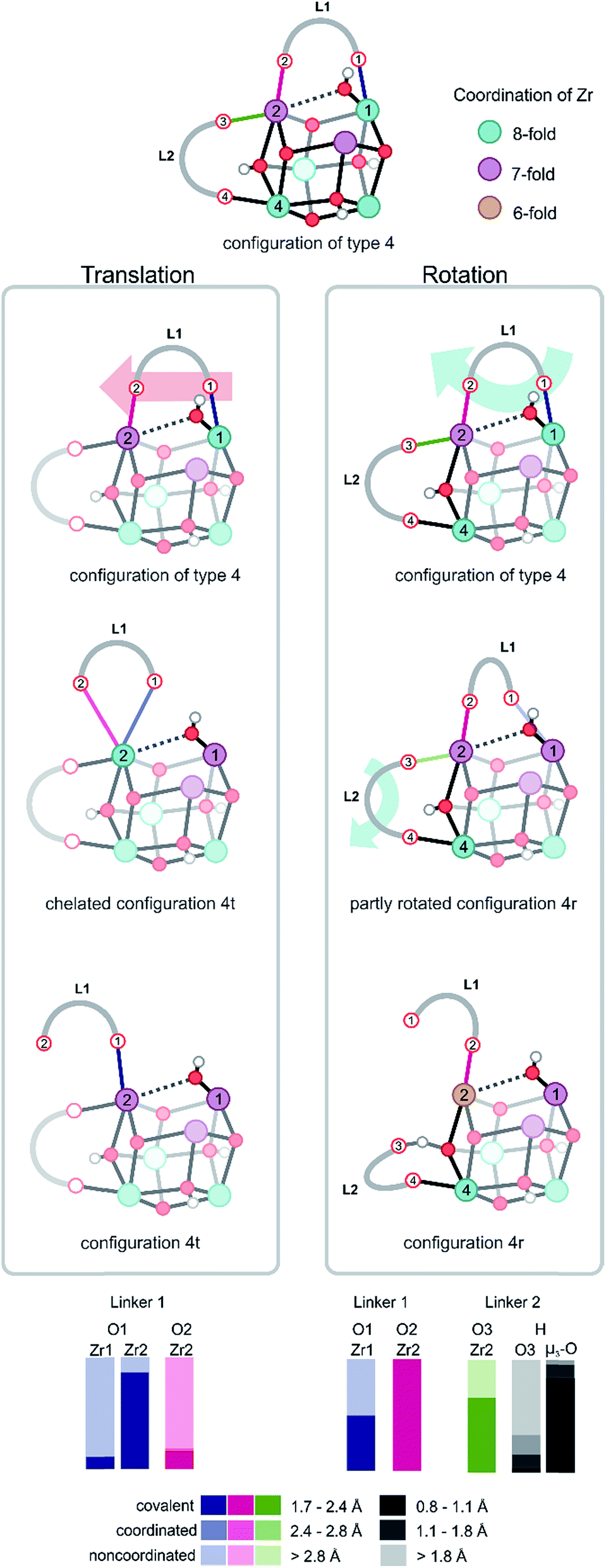 | ||
| Fig. 4 Umbrella sampling in two windows of CV = 1.45 and 1.51, showing two distinct motions of the linkers. On the left column, a translation of the linker L1 generates a chelated structure and a subsequent shift in the carboxylic oxygen connected to Zr2 (configuration 4t). On the right column, a rotation of the linker L1 and a partial decoordination of linker L2 forming a hydrogen bond with an μ3-OH hydroxyl group is shown (configuration 4r). A proton transfer between the carboxylic oxygen O3 and the bridging μ3-O is also observed and is evidenced by the measured probability distribution (bottom). It is an indication of the occurrence of an intrinsic dynamic acidity. Colors indicate the coordination number of Zr atoms. | ||
The second defined motion observed in configurations of type 4 corresponds to a rotation mode of the linker (Fig. 4, right column), which is again prompted by the μ1-OH hydroxyl group dislocation. At this case, the linker breaks a bond with a Zr atom (O1 with Zr1) and rotates along the Zr2–O2 bond (Movie S2 of the ESI†). The rearrangement of this linker (L1), with its steric hindrance, influences the coordination pattern of another linker connected to the same Zr (L2 to Zr2), which is pushed away by linker L1 and detaches from Zr2. This second linker (L2) immediately stabilizes itself by coordinating to the closest μ3-OH group (originally attached in between Zr2 and Zr4). This linker decoordination is clearly visible in the probability distribution of the O3 and Zr2 bond. In the remaining part of the simulation, the proton of the hydroxyl group travels from the μ3-O atom to the carboxylic oxygen O3 and is donated to the linker. The appearance of phenomena such as proton transfer processes between partly decoordinated carboxylic ligands and hydroxyl groups generates an intrinsic dynamic acidity. Proton mobility in UiO-66 was reported earlier by Ling and Slater,36 but in that work, the dynamic acidity was caused by shuttling protons between water molecules coordinated to the metals and charge balancing hydroxide anions, which compensate for the missing linkers in the defective material. This proton transfer process is reversible and elucidates the unusual simultaneous action of carboxylic ligands as hydrogen donors and hydrogen acceptors, which is an indication of the existence of an intrinsic dynamic acidity in defect-free UiO-66. During the rotational deformation mode observed for configuration of type 4, substantial rearrangements of the coordination numbers of the various Zr atoms are observed. The linker decoordination leaves an open metal site on Zr1, which changes the chemical bonding pattern of this atom from 8 to 7. Additionally, the rotation of the linker around Zr2 results in the second decoordination from this site, decreasing the Zr coordination number from 7 to 6, as shown in configuration 4r in Fig. 4.
The reversible proton shuttles observed in configuration 4r require some additional attention. First, a similar pattern has been noticed by Haigis et al.44 in metadynamics simulations performed at high temperatures on MIL-53(Ga), where the hydroxyl group of the inorganic chain is hydrogen bonded with the carboxylic oxygen of the BDC linker, forming an intermediate structure. Second, the ability to reversibly protonate the carboxylate groups of linker ligands in UiO-66 has been experimentally evidenced by DeCoste et al.18 by observing some specific peaks of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in the IR spectrum when adding hydrochloric acid to the material. The simulations performed here give evidence of an intrinsic dynamic acidity with rapid proton transfers between the hydroxyl groups and linkers, even in the absence of protic molecules in the pores of the material. This aspect gives evidence for a similar linker, and hydrogen bond stabilization during the reactions in which UiO-66 undergoes chemical transformations, like in the case of ligand exchange process and substantiates the structural stability.
O in the IR spectrum when adding hydrochloric acid to the material. The simulations performed here give evidence of an intrinsic dynamic acidity with rapid proton transfers between the hydroxyl groups and linkers, even in the absence of protic molecules in the pores of the material. This aspect gives evidence for a similar linker, and hydrogen bond stabilization during the reactions in which UiO-66 undergoes chemical transformations, like in the case of ligand exchange process and substantiates the structural stability.
Configurations of type 5, encountered in the window with collective variables smaller than 1.0, are typical for processes, where eventually water could also be formed. However, one should be careful; there is not one single water formation mode, but the system can assume a plethora of configurations of almost equal free energy, which are characterized by a large mobility of both linkers and protons originally connected to the inorganic brick. Herein, we demonstrate the presence of complex, fast structural fluctuations that can be rationalized as a combination of the two previously described motions of linker translation and rotation. In all these configurations located in the configuration 5 region (Fig. 5), which is characterized by an initial collective variable of 0.94, the hydroxyl group is bonded to a Zr atom in a stable μ1-OH position (Movie S3 of the ESI†). The starting configuration of the inorganic brick is composed of three 7-fold and three 8-fold coordinated Zr atoms, but the coordination pattern of the Zr atoms rapidly evolves, giving different types of open metal sites. During the simulation, two linkers (L1 and L3) undergo translational and rotational motions at the same time, modifying the chemical properties of four Zr sites by changing the coordination number between 8 and 6. In addition, it is important to notice that linker decoordination occurs from the same metal ion, which can lead to linker vacancies. Interestingly, linkers also recoordinate to their parent position, providing an explanation for the remarkable stability of UiO-66. The ability of the linkers to reversibly decoordinate and coordinate to the Zr atoms is intrinsically related to the oxophilic character of the metal. Upon linker recoordination, the hydroxyl group travels to the other side of the brick with spontaneous decoordination of another linker (L4) (Fig. 5). These intrinsic motions show the ability of the structure to easily respond to deformations of linkers and hydroxyl groups. Water formation can eventually occur by a process where a decoordinated hydroxyl group captures a proton from the μ3-OH group (Fig. 5, last configuration). All structural reorganizations observed so far give rise to the final configuration, which is made up of four 7-fold and two 8-fold coordinated Zr atoms.
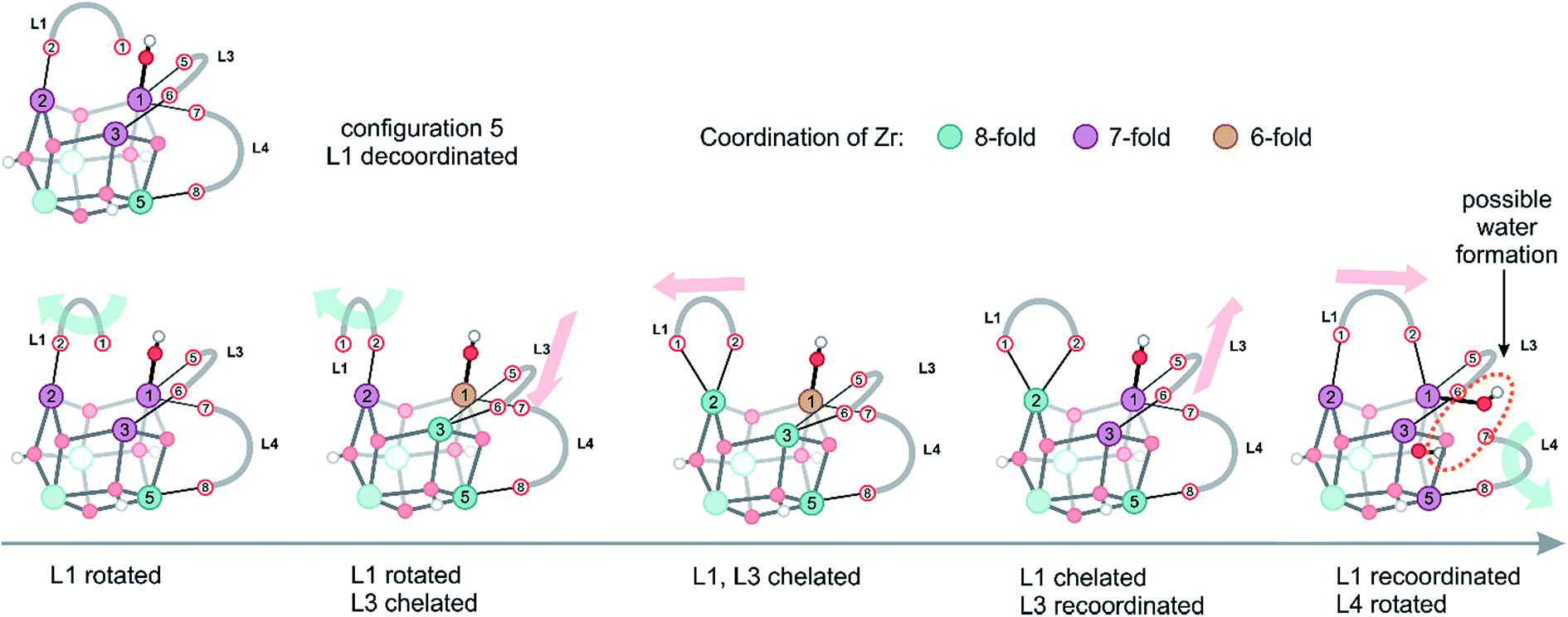 | ||
| Fig. 5 Different structures observed in the simulation window of configuration 5, which are rationalized by the two motions presented in Fig. 4. The colors of the Zr atoms indicate their coordination number. | ||
At this point, it is also interesting to note the differences between the chemical features shown in this study and the earlier reaction profile determined with static NEB calculations. The earlier static calculations allow two transition states to be located with barriers of 121.3 and 178.7 kJ mol−1 at 593 K.48 The processes observed here consist translation and rotation of a linker attached originally to two Zr atoms with the hydroxyl group in a μ1-OH configuration and situated on a multidimensional free energy plateau at about 90 kJ mol−1. The simulations shown here reveal that deformations in one linker induces the mobility of various nearby linkers and another hydroxyl group in the inorganic brick. Such metastable configurations are stabilized by the presence of hydrogen bonds and are maintained for a consistent part of the simulation. This has been further validated by an additional set of regular MD simulations of 50 ps without the presence of activated umbrellas starting from the final configuration obtained in the last window. This regular MD simulation confirms the mobility of the hydroxyl groups and supports the hypothesis of a strongly varying intrinsic dynamic proton acidity (Fig. S3 of the ESI†).
Overall, our simulations show that linkers are much more mobile than originally anticipated. During an activation process, they can easily change their position via decoordinations, translations, rotations and recoordinations, while the inorganic brick shows no major distortions. The latter is shown in the distribution of the Zr–Zr and Zr–O distances in the brick (Fig. S4 of the ESI†). Such effects point towards a remarkable intrinsic flexibility of the framework, where the coordination numbers can easily vary between 8 and 6. Defects on the structure are crucial for the catalytic activity and the dehydration mechanism may have a decisive effect on certain catalytic reactions, where next to the Lewis acid site also, the neighboring Brønsted base or acid sites may take an interactive role in the reaction mechanism.60,61 Examples of such reactions are jasminaldehyde condensation,62 in which first the C–C coupling product gets protonated by the hydrogen attached to the μ3-oxygen atom and this leads to the aldol product. Also, in the case of esterification reaction, the hydrogen bond with the μ3-oxygen atom plays a crucial role in the proposed reaction mechanism.60 For the Oppenauer oxidation studied in ref. 61, Lewis and dynamic Brønsted sites are required. For this reaction, a conversion of 68% was found to the desired product for an almost ideal UiO-66 material (11.6 linker per brick). The mobility of the linkers observed in this study could explain these findings, as linkers might temporarily decoordinate from the Zr atoms and be stabilized by protons from the framework, giving more accessible Lewis acid sites.61 The unraveled dynamic behavior of UiO-66 can serve as a platform to rationalize the dynamic coordination change and accessibility of active sites in other ‘inert’ MOFs characterized by exceptional stability and higher surface-to-volume ratio.
The energy required to induce such internal rearrangements are easily accessible with common experimental activation procedures such as a temperature increase and can be accommodated with this exceptionally stable material. Furthermore, hydrogen bonding appears to be an important part of the interaction between hydroxyl group and the decoordinated linkers. The hydrogen-bonding pattern observed during dehydration is remarkable and can stabilize the structure in processes that involve decoordination of the linkers. One may anticipate that further stabilization patterns might occur when a solvent such as methanol would be present. The observed structural dynamic flexibility of robust UiO-66 is unique and our results confirmed that the chemistry of M–L bond is reversible, as was indicated by Kim et al.28 in the linker exchange reaction. This demonstrates that in all chemical processes where the coordination number of the metal ion changes, there is fast structural evolution in which the linkers play a major role, constantly switching their position. Experimentally, the characteristics of structural dynamic motions on the picosecond timescale have been seen in the case of functionalized MOFs by applying ultrafast 2D IR spectroscopy33 and can be expanded to track changes during reversible dehydration process of UiO-66.
Conclusions
The UiO-66 framework is archetypal for its structural stability, but also allows structural deformations by processes such as dehydration, linker exchange, and defect formation while maintaining its structural integrity. Herein, we tried by means of enhanced sampling molecular dynamics simulations such as umbrella sampling techniques to give a better insight in the factors based on the observed intrinsic dynamic flexibility of the framework, whereby the coordination number of the Zr atoms can easily change between 8 and 6. By following the system on the fly at 573 K, during an activated process where the coordination number of the hydroxyl group bridging the Zr atoms of the inorganic brick varies from 3 to 1, a remarkable fast dynamics of the system is observed. Depending on the free energy window explored, major rearrangements are observed for the hydroxyl group, which induce a strong mobile behavior of various connecting linkers. In lower activated regions, the hydroxyl group can easily travel among the three Zr atoms to which it is originally connected in a μ3-OH configuration. Once higher activated plateau is reached at about 90 kJ mol−1, reversible mobility of the linkers is observed, while the rigidity of the inorganic brick is retained. We observe the processes of proton shuttling even in defect-free UiO-66 at elevated temperature, where a proton transfer occurs between a μ3-OH and carboxylic oxygen of a partly decoordinated linker. These results expand the earlier concept of dynamic acidity introduced by Ling and Slater36 to defect-free UiO-66, even in the absence of protic species. General deformation modes are detected for the linkers during activation, which correspond to translational and rotational modes. The system has a remarkable intrinsic dynamic ability to respond to deformations, where closely connected linkers decoordinate and are eventually stabilized by hydrogen bonding interactions with adjacent hydroxyl groups. We can safely assume that partly decoordinated linkers would also be stabilized by hydrogen bonding interactions with other solvent species during processes such as solvent assisted ligand exchange. The change in coordination of the hydroxyl group simulated here is an essential step during the dehydroxylation process. Furthermore, our observations are based on simulations in defect-free UiO-66, but these rearrangements may be expected to take place in defective UiO-66, as we have already established similar dehydration pathways in defective materials. The understanding of rearrangements of linker ligands or bridging hydroxyl groups in the inorganic node during activation has so far been insufficient, as it poses a huge challenge to follow the system in situ during activation based on a purely experimental basis. Herein, we took a major step forward for the structural understanding in the intrinsic dynamic behavior of UiO-66.Methodology
The ab initio molecular dynamics (AIMD) simulations were performed within the DFT level of theory in a full periodic defect-free UiO-66 in the NPT ensemble using the CP2K simulation package interfaced with the advanced simulation library PLUMED.63,64 The revPBE functional65 with inclusion of Grimme D3 dispersion corrections66 was chosen using the DZVP-GTH basis sets for C, O and H atoms, which is a combination of Gaussian basis functions and plane waves (GPW)67 with a cut-off energy of 350 Ry. For Zr, the DZVP-MOLOPT-SR basis set has been applied. Along the reaction path, we performed advanced molecular dynamic simulations by means of umbrella sampling (US). The crucial step in the US methodology was the selection of the various windows on the reaction path along the collective variable (CV) and determination of the initial structures in each of these windows. We applied a protocol in the form of a moving umbrella, which during the duration of the molecular dynamics crosses the entire range of CV with a constant velocity. This constrained MD concept was introduced by Grubmüller et al.68 but it was not applied in this context. 36 snapshots were extracted from this moving umbrella simulation, and in each of those, an MD simulation with a static harmonic bias was initiated. Those MD simulations were run for 25 ps and the frequently visited configurations in each umbrella sampling were stored and their specific features were discussed. The free energy profile was constructed by application of the WHAM methodology.69,70 More details of the calculations were included in the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is supported by the Research Foundation Flanders (FWO), the Research Board of Ghent University (BOF) and BELSPO in the frame of IAP/7/05. This project received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No. 641887 (project acronym: DEFNET). Funding was also received from the European Union's Horizon 2020 research and innovation programme [consolidator ERC grant agreement no. 647755-DYNPOR (2015–2020)]. The computational resources and services used in this work were provided by the VSC (Flemish Supercomputer Center), funded by the FWO. Molecular visualizations were produced with VMD software support. VMD is developed with NIH support by the Theoretical and Computational Biophysics group at the Beckman Institute, University of Illinois, at Urbana-Champaign.References
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- H. C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- J. E. Mondloch, M. J. Katz, W. C. Isley Iii, P. Ghosh, P. Liao, W. Bury, G. W. Wagner, M. G. Hall, J. B. DeCoste, G. W. Peterson, R. Q. Snurr, C. J. Cramer, J. T. Hupp and O. K. Farha, Nat. Mater., 2015, 14, 512–516 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- M. Ranocchiari and J. A. v. Bokhoven, Phys. Chem. Chem. Phys., 2011, 13, 6388–6396 RSC.
- P. Valvekens, F. Vermoortele and D. De Vos, Catal. Sci. Technol., 2013, 3, 1435–1445 CAS.
- A. Corma, H. Garcia and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- S. M. J. Rogge, A. Bavykina, J. Hajek, H. Garcia, A. I. Olivos-Suarez, A. Sepúlveda-Escribano, A. Vimont, G. Clet, P. Bazin, F. Kapteijn, M. Daturi, E. V. Ramos-Fernandez, F. X. Llabrés i Xamena, V. Van Speybroeck and J. Gascon, Chem. Soc. Rev., 2017, 46, 3134–3184 RSC.
- K. Leus, T. Bogaerts, J. De Decker, H. Depauw, K. Hendrickx, H. Vrielinck, V. Van Speybroeck and P. Van Der Voort, Microporous Mesoporous Mater., 2016, 226, 110–116 CrossRef CAS.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS PubMed.
- S. M. J. Rogge, J. Wieme, L. Vanduyfhuys, S. Vandenbrande, G. Maurin, T. Verstraelen, M. Waroquier and V. Van Speybroeck, Chem. Mater., 2016, 28, 5721–5732 CrossRef CAS PubMed.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- J. B. DeCoste, G. W. Peterson, H. Jasuja, T. G. Glover, Y.-g. Huang and K. S. Walton, J. Mater. Chem. A, 2013, 1, 5642–5650 CAS.
- A. De Vos, K. Hendrickx, P. Van Der Voort, V. Van Speybroeck and K. Lejaeghere, Chem. Mater., 2017, 29, 3006–3019 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS PubMed.
- M. J. Cliffe, W. Wan, X. D. Zou, P. A. Chater, A. K. Kleppe, M. G. Tucker, H. Wilhelm, N. P. Funnell, F. X. Coudert and A. L. Goodwin, Nat. Commun., 2014, 5, 4176 CAS.
- R. E. Morris and L. Brammer, Chem. Soc. Rev., 2017, 46, 5444–5462 RSC.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- M. Kim, J. F. Cahill, Y. Su, K. A. Prather and S. M. Cohen, Chem. Sci., 2012, 3, 126–130 RSC.
- K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498–519 RSC.
- S. M. Cohen, J. Am. Chem. Soc., 2017, 139, 2855–2863 CrossRef CAS PubMed.
- G. C. Shearer, S. Forselv, S. Chavan, S. Bordiga, K. Mathisen, M. Bjorgen, S. Svelle and K. P. Lillerud, Top. Catal., 2013, 56, 770–782 CrossRef CAS.
- M. Vandichel, J. Hajek, F. Vermoortele, M. Waroquier, D. E. De Vos and V. Van Speybroeck, CrystEngComm, 2015, 17, 395–406 RSC.
- J. Nishida, A. Tamimi, H. Fei, S. Pullen, S. Ott, S. M. Cohen and M. D. Fayer, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 18442–18447 CrossRef CAS PubMed.
- T. D. Bennett, A. K. Cheetham, A. H. Fuchs and F.-X. Coudert, Nat. Chem., 2017, 9, 11–16 CrossRef CAS PubMed.
- A. D. Wiersum, E. Soubeyrand-Lenoir, Q. Yang, B. Moulin, V. Guillerm, M. B. Yahia, S. Bourrelly, A. Vimont, S. Miller, C. Vagner, M. Daturi, G. Clet, C. Serre, G. Maurin and P. L. Llewellyn, Chem.–Asian J., 2011, 6, 3270–3280 CrossRef CAS PubMed.
- S. Ling and B. Slater, Chem. Sci., 2016, 7, 4706–4712 RSC.
- C. Caratelli, J. Hajek, S. M. J. Rogge, S. Vandenbrande, E. J. Meijer, M. Waroquier and V. Van Speybroeck, ChemPhysChem DOI:10.1002/cphc.201701109.
- O. Valsson, P. Tiwary and M. Parrinello, in Annual Review of Physical Chemistry, Vol 67, ed. M. A. Johnson and T. J. Martinez, 2016, vol. 67, pp. 159–184 Search PubMed.
- K. De Wispelaere, S. Bailleul and V. Van Speybroeck, Catal. Sci. Technol., 2016, 6, 2686–2705 CAS.
- K. De Wispelaere, B. Ensing, A. Ghysels, E. J. Meijer and V. Van Speybroeck, Chem.–Eur. J., 2015, 21, 9385–9396 CrossRef CAS PubMed.
- P. Cnudde, K. De Wispelaere, J. Van der Mynsbrugge, M. Waroquier and V. Van Speybroeck, J. Catal., 2017, 345, 53–69 CrossRef CAS.
- J. Hajek, J. Van der Mynsbrugge, K. De Wispelaere, P. Cnudde, L. Vanduyfhuys, M. Waroquier and V. Van Speybroeck, J. Catal., 2016, 340, 227–235 CrossRef CAS.
- V. Van Speybroeck, K. De Wispelaere, J. Van der Mynsbrugge, M. Vandichel, K. Hemelsoet and M. Waroquier, Chem. Soc. Rev., 2014, 43, 7326–7357 RSC.
- V. Haigis, F.-X. Coudert, R. Vuilleumier, A. Boutin and A. H. Fuchs, J. Phys. Chem. Lett., 2015, 6, 4365–4370 CrossRef CAS PubMed.
- T. Bučko, L. Benco, J. Hafner and J. G. Angyan, J. Catal., 2011, 279, 220–228 CrossRef.
- J. D. Evans, G. Fraux, R. Gaillac, D. Kohen, F. Trousselet, J. M. Vanson and F. X. Coudert, Chem. Mater., 2017, 29, 199–212 CrossRef CAS.
- G. Fraux and F. X. Coudert, Chem. Commun., 2017, 53, 7211–7221 RSC.
- M. Vandichel, J. Hajek, A. Ghysels, A. De Vos, M. Waroquier and V. Van Speybroeck, CrystEngComm, 2016, 18, 7056–7069 RSC.
- Y. Sugita and Y. Okamoto, Chem. Phys. Lett., 1999, 314, 141–151 CrossRef CAS.
- U. H. E. Hansmann, Chem. Phys. Lett., 1997, 281, 140–150 CrossRef CAS.
- L. Maragliano and E. Vanden-Eijnden, Chem. Phys. Lett., 2006, 426, 168–175 CrossRef CAS.
- R. W. Zwanzig, J. Chem. Phys., 1954, 22, 1420–1426 CrossRef CAS.
- J. G. Kirkwood, J. Chem. Phys., 1935, 3, 300–313 CrossRef CAS.
- A. Laio and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12562–12566 CrossRef CAS PubMed.
- G. N. Patey and J. P. Valleau, J. Chem. Phys., 1975, 63, 2334–2339 CrossRef CAS.
- G. M. Torrie and J. P. Valleau, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- M. A. Rohrdanz, W. Zheng and C. Clementi, Annu. Rev. Phys. Chem., 2013, 64, 295–316 CrossRef CAS PubMed.
- R. Demuynck, S. M. J. Rogge, L. Vanduyfhuys, J. Wieme, M. Waroquier and V. Van Speybroeck, J. Chem. Theory Comput., 2017, 13, 5861–5873 CrossRef CAS PubMed.
- M. Puchberger, F. R. Kogler, M. Jupa, S. Gross, H. Fric, G. Kickelbick and U. Schubert, Eur. J. Inorg. Chem., 2006, 2006, 3283–3293 CrossRef.
- C. Caratelli, J. Hajek, F. G. Cirujano, M. Waroquier, F. X. Llabrés i Xamena and V. Van Speybroeck, J. Catal., 2017, 352, 401–414 CrossRef CAS.
- J. Hajek, B. Bueken, M. Waroquier, D. De Vos and V. Van Speybroeck, ChemCatChem, 2017, 9, 2203–2210 CrossRef CAS PubMed.
- J. Hajek, M. Vandichel, B. Van de Voorde, B. Bueken, D. De Vos, M. Waroquier and V. Van Speybroeck, J. Catal., 2015, 331, 1–12 CrossRef CAS.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS.
- K. Yang, J. J. Zheng, Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2010, 132, 10 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- G. Lippert, J. Hutter and M. Parrinello, Theor. Chem. Acc., 1999, 103, 124–140 CrossRef CAS.
- H. Grubmüller, B. Heymann and P. Tavan, Science, 1996, 271, 997–999 Search PubMed.
- A. Grossfield, http://membrane.urmc.rochester.edu/content/wham.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04947a |
| This journal is © The Royal Society of Chemistry 2018 |