
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lipid bilayer environments control exchange kinetics of deep cavitand hosts and enhance disfavored guest conformations†
Lizeth
Perez‡
,
Bethany G.
Caulkins‡
 ,
Magi
Mettry‡
,
Leonard J.
Mueller
and
Richard J.
Hooley
*
,
Magi
Mettry‡
,
Leonard J.
Mueller
and
Richard J.
Hooley
*
Department of Chemistry, University of California – Riverside, Riverside, CA 92521, USA. E-mail: richard.hooley@ucr.edu
First published on 11th January 2018
Abstract
The effects on the molecular recognition properties of water-soluble deep cavitand hosts upon embedding them in phosphocholine lipid bilayer environments have been studied by 2D NMR experiments. By employing suitable guests containing 19F or 13C nuclei that can be encapsulated inside the host, 2D EXSY NMR experiments can be used to analyze and compare the in/out guest exchange rates in aqueous solution, isotropically tumbling micelles, or magnetically ordered bicelles. These analyses show that embedding the deep cavitands in lipid bilayers slows the guest exchange rate, due to the lipids acting as a “compression sleeve” around the host, restricting guest egress. This effect also enhances guest conformations in the host that are not observed in free solution, such as axial cyclohexane conformers and ketone hydrates.
Introduction
When analyzing the conformation and motion of molecules confined in small spaces, the predominant tool is NMR spectroscopy.1 The sensitivity of 1H NMR experiments, along with the large changes in proton chemical shifts possible upon surrounding small hydrocarbons with aromatic π clouds, have opened a window into the physical behavior of molecules in enclosed environments. Quantitation of the thermodynamics2 and kinetics2a,3 of substrate binding is possible, as well as investigations into the orientation,4 conformation,5 motion2a,6 and unusual isomerism7 of bound small molecule substrates. Molecular confinement can lengthen the lifetime of reactive intermediates8 and unstable species:9 observing these phenomena often relies on 1H NMR spectroscopy. To maximize detection, these investigations are generally performed in controlled environments, in deuterated solvents and in the absence of NMR-visible additives and impurities.Synthetic host molecules are capable of selective molecular recognition in far more complex and challenging environments than pure solvent, however. Hosts such as deep cavitand 1 (Fig. 1)2a,10 have been shown to bind targets while embedded in supported phosphocholine (PC) lipid bilayers11 and even in living cells.12 The recognition capabilities of cavitand 1 in membrane bilayer systems have been shown via indirect methods, such as surface plasmon resonance (SPR) spectroscopy of cavitand:supported lipid bilayer (SLB) aggregates11 and capillary electrophoresis (CE) of liposome:cavitand systems.11d Other techniques can also be used, including fluorescence spectroscopy13 and isothermal calorimetry.2c,10a While these techniques have their advantages, none of them are as enticing as NMR spectroscopy, which allows sensitive interrogations of guest conformations, motions and dynamics, such as in/out exchange rates and the molecular motion of molecules bound in the host's interior. 1H NMR spectroscopy of these events has been limited to simple 1D experiments in fast tumbling micelles,14 although some examples of molecular recognition in more complex systems such as human serum or urine15 are known.
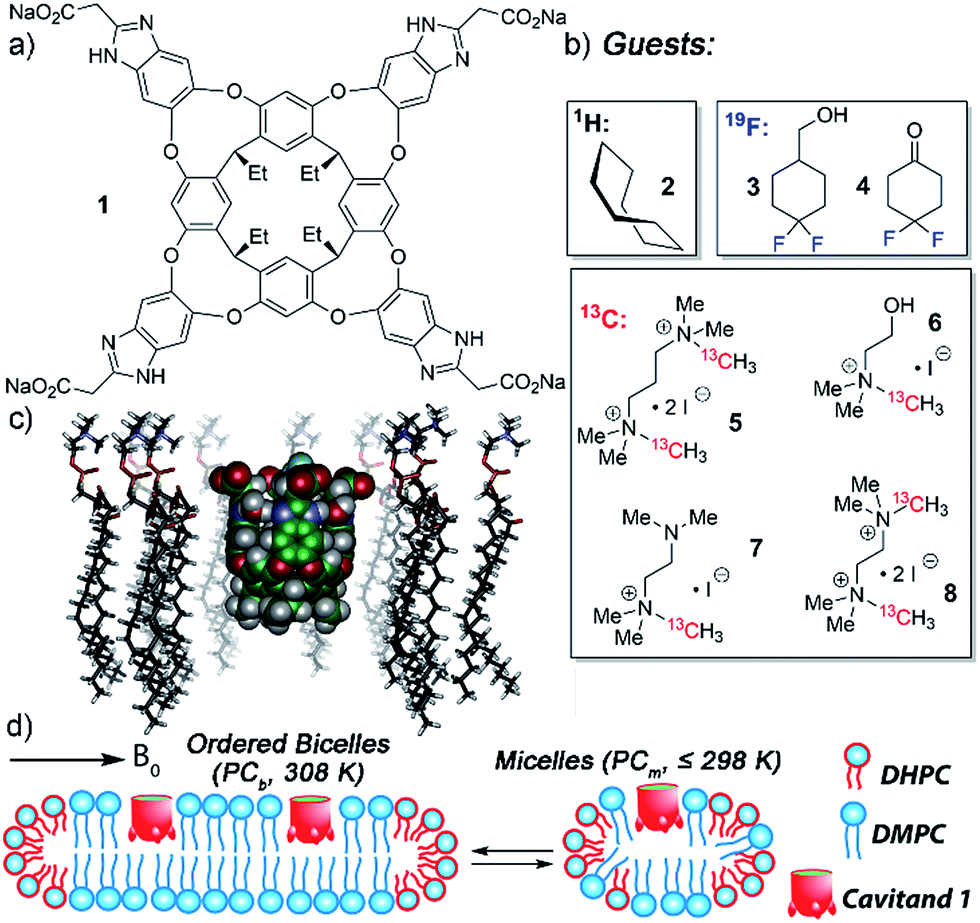 | ||
| Fig. 1 Structure of (a) water-soluble deep cavitand 1; (b) guests used in this study; (c) representation of 1 embedded in a DMPC lipid monolayer (SPARTAN, AMBER forcefield); (d) representation of possible DMPC/DHPC lipid structures, either magnetically ordered bicelles or isotropically disordered micelles. | ||
The tetracarboxylate cavitand 1 is soluble in water up to ∼20 mM, but is highly lipophilic, and is smoothly incorporated into a variety of lipid aggregates.11,12 It can bind a wide range of suitably sized guest molecules in aqueous solution, ranging from hydrocarbons2a to substituted trimethylammonium (R-NMe3+) salts such as choline.10,12 In pure D2O solution, the association constants of guests such as 1-adamantanemethanol (Ka = 2.9 × 105 M−1), cyclohexanone (Ka = 1.6 × 105 M−1), choline (Ka = 2.6 × 104 M−1) and acetylcholine (Ka = 1.2 × 105 M−1) are relatively consistent, within an order of magnitude or so.2a,10b The “upper limit” for guest association in free solution is on the order of 2 × 105 M−1. The in/out kinetics of bound guests, as well as their motion while inside the cavity, have been extensively investigated in pure D2O by 2D NMR techniques.2a,4a The mechanism of in/out exchange is a dissociative process, independent of guest concentration.2a The cavitand releases guest via an “SN1-like” mechanism, whereby the walls flex open to unfold the cavitand in the rate-determining step, followed by rapid guest exchange. In water, the energy barrier consists of three major components: the energy barrier to rotate around the C–O bonds in the cavitand walls16 (∼11 kcal mol−1), plus the energetic penalties of unfavorable solvation of the cavitand walls and the bound guest once unfolded. Depending on guest size, these barriers range from 16.0–17.2 kcal mol−1,2a,17 with observed rates ranging from 1.8 s−1 (adamantanol) to 14.6 s−1 (cyclohexane). All these investigations used 1H NMR for analysis, as the 1H peaks for bound guest are shifted strongly upfield, and are easily visible at negative ppm, unhindered by 1H peaks from the host or excess free guest.
What is not known is how embedding the cavitand in a lipid bilayer affects these molecular recognition properties. Other techniques have shown that host is capable of guest recognition in a bilayer, and that guest binding affinities are enhanced in some cases,11 but the effect on guest in/out kinetics and conformation is unknown. Cavitands are excellent mimics of proteins, and have shed light on many biomimetic recognition phenomena. Can they be used to illustrate the function of membrane-binding proteins, a far more elusive target?
NMR analysis of (bio)molecular structure and dynamics in membrane bilayers is well-studied,18 and exploits such biomimetic environments as isotropically tumbling19 and magnetically-oriented lipid bicelles, and unaligned and mechanically oriented phospholipid bilayers.18a,20 The aligned systems have the advantage of high resolution without the requirement of either fast isotropic reorientation (which restricts the dimensions of isotropic bicelles) or magic angle spinning (used for unaligned bilayers). These experiments often require isotopically enriched species for detection, and the nuclei of choice are generally 13C, 15N or 31P. Unfortunately, observing individual proton signals via1H NMR analysis of the binding processes in those cases is complicated by the presence of multiple hydrocarbon peaks from the lipids. More sensitive NMR experiments are rendered impractical by line broadening, and so dynamic NMR experiments that are essential for analysis of guest motion and binding kinetics in synthetic cavity-containing hosts are challenging. Here we employ a variety of guest molecules with different detectable nuclei for molecular recognition in a deep, water-soluble host, and investigate the effect of embedding the host in biomimetic membrane environments on the guest dynamics, conformation and reactivity.
Results and discussion
NMR analysis of the molecular recognition of small molecules by deep cavitands in lipid environments is complicated by a number of factors. The guest must bind in the cavitand, and be detectable when bound inside the host in multiple different lipid environments. In addition, study of the in/out kinetics requires guests that are sufficiently soluble to display peaks for both free and bound guest. Study of internal motion or conformational bias requires guests that display multiple conformations or orientations while bound inside the cavity: if 1H NMR analysis cannot be used, this introduces serious constraints on the nature of the guest. As such, we investigated a wide range of guest species (Fig. 1) for their suitability. The guest library consists of simple hydrocarbons such as cyclooctane 2, 19F-containing hydrophobic guests 3-4, and 13C-enriched R-NMe3+ guests 5–8. Each of these guests are either commercially available or accessible in one or two steps from commercial materials (see Experimental section for synthesis and characterization). Also, the host:guest library must be paired with a suitable membrane environment for analysis. Obviously, a natural cell membrane is challenging to use, but a number of surrogates are known and used for NMR analysis of membrane-bound biomolecules. One of the most effective mimics is a magnetically oriented bicelle, which maintains the bilayer sheet form of natural membranes while aligning in the magnetic field to allow analysis by ssNMR techniques.20 In addition, solution-phase NMR techniques can be employed with isotropically tumbling micelles. Each aggregate has benefits and drawbacks: bicelles are better mimics of natural membranes, but suffer from solid-state line broadening effects, whereas micelles are easier to analyze but are an imperfect membrane mimic. Fortunately, both bicelles and micelles can be accessed from the same lipid system, so we applied both types of lipid environment to the host:guest analysis.The lipid aggregates were formed from a 3.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mix of dimyristoylphosphocholine and diheptylphosphocholine lipids (DMPC/DHPC), which are well-known to allow formation of both magnetically oriented bicelles and smaller disoriented micelles.19 As illustrated in Fig. 1d, the type of lipid aggregate formed is dependent on temperature, with magnetically ordered bicelles dominant at 308 K, and disordered (isotropic) micelles favored at 298 K or lower. This can be seen by 31P NMR analysis of the lipid system. The disordered micelles display only one averaged 31P phosphate peak (see ESI, Fig. S-47 and S-48†), whereas the oriented bicelles display two peaks, due to the two different orientations of phosphate groups in the bicelle. The assembly and structure of these aggregates was not visibly affected by the presence of either 5 mM cavitand 1 or 5 mM 1 + 7 mM guest 6.
:1 mix of dimyristoylphosphocholine and diheptylphosphocholine lipids (DMPC/DHPC), which are well-known to allow formation of both magnetically oriented bicelles and smaller disoriented micelles.19 As illustrated in Fig. 1d, the type of lipid aggregate formed is dependent on temperature, with magnetically ordered bicelles dominant at 308 K, and disordered (isotropic) micelles favored at 298 K or lower. This can be seen by 31P NMR analysis of the lipid system. The disordered micelles display only one averaged 31P phosphate peak (see ESI, Fig. S-47 and S-48†), whereas the oriented bicelles display two peaks, due to the two different orientations of phosphate groups in the bicelle. The assembly and structure of these aggregates was not visibly affected by the presence of either 5 mM cavitand 1 or 5 mM 1 + 7 mM guest 6.
31P NMR analysis of the lipid phosphate groups is an invaluable tool to confirm the structure of the lipid aggregates, but does not allow investigation of the host:guest properties of 1. We initially approached the host:guest studies using 1H NMR spectroscopy with the simplest, most optimal guest possible. Cyclooctane 2 is easily extracted into the cavity of 1, has an affinity >104 M−1 by NMR,2a and tumbles rapidly on the NMR timescale, showing a single bound peak at −1.50 ppm corresponding to the averaged signal of all 16H in the guest. A premade sample of 1·2 in D2O was added to solutions of either DMPC:DHPC micelles (hereinafter denoted as PCm) or DMPC:DHPC bicelles (PCb) for a final [1] = [2] = 1.8 mM and the spectra acquired at 283 K and 308 K respectively (see ESI† for spectra). The spectrum in PCm shows that cavitand 1 binds 2 strongly in the hydrophobic lipid environment, as the expected sharp singlet for the 1·2 is retained. The cavitand is completely incorporated into the aggregates under the conditions used. T2-filtered spectra of the PCm·1·2 complex (see Fig. S-26†) show no peaks for either free, un-embedded cavitand or bound guest, indicating that all the detectable host is incorporated into the lipid aggregates under the conditions used. Unfortunately, no change in the conformational properties of 2 was observed: even at 10 °C, the guest tumbled rapidly in the cavity of 1. In addition, in the bicellar environment at 35 °C, only broad undefined peaks could be observed and no discrete peaks for 1 or bound 2 are visible, even when magic angle spinning (MAS) was applied.
As cyclooctane 2 was only partially useful, we turned to guests 3 and 4, containing 19F nuclei. As 19F peaks are broadened in the solid state in a similar manner to 1H, we focused on fast-tumbling micelles in the 1·3/4 analysis, with a lower lipid concentration of 60 mg mL−1 for ease of measurement. To allow analysis of conformation and in/out guest exchange, the guest must contain 19F nuclei that are bound inside the cavity of 1, and display the characteristic chemical shift variations caused by the magnetic anisotropy of the host. Fortunately, both guests 3 and 4 are suitable guests for 1 in aqueous solution, albeit displaying weaker binding than the equivalent hydrocarbons. 19F NMR spectra of the host:guest complexes shows that the bound 19F nuclei are upfield shifted, Δδ ∼ −2 ppm, indicating that the difluorocyclohexanyl group is oriented to the cavity interior. Presumably, the OH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups orient towards the external solvent to benefit from favorable H-bonding.
O groups orient towards the external solvent to benefit from favorable H-bonding.
Both guests 3 and 4 show interesting and unexpected behavior when bound to 1, and provide excellent examples of the effect of embedding the host in the PC lipid environment. The 1H NMR spectrum of the 1·3 complex (Fig. 2d) showed two sets of peaks for bound 3. The 1H chemical shifts for the guest CH peaks in each conformation were relatively similar, indicating that the two conformations in the host:guest complex are not up/down carceroisomers,4a,21 rather the axial/equatorial ring flip conformers of 3. This observation was unexpected, and provided an opportunity to investigate the effects of molecular recognition on conformations of bound guest. In the absence of host in either CDCl3 or D2O solution, only one conformation of 3 can be observed in the 1H or 19F NMR spectrum, presumably that of the lower energy equatorial conformer. At the concentrations used, this indicates that <0.5% of the axial conformer is present in solution. The 19F spectrum is most useful for this assignment: the peak for the axial F is a doublet of triplets (see Fig. S-2†), due to trans-diaxial coupling with the vicinal H atoms. The equatorial F is a doublet, and shows no visible peak splitting due to coupling to the protons. The highly different coupling patterns shown by the two fluorines indicate that no appreciable rapid interconversion between conformers is occurring. In contrast, when bound in the cavity of 1, 12% of the population of 1·3 corresponds to the axial conformer. 2D COSY analysis (Fig. 2e) clearly shows the two separate conformers, and molecular modeling (Fig. 2a) illustrates that the axial conformer of 3 easily fits inside the host cavity. The behavior of guest 3 in the PCm·1 system gives the first indication of the effect of embedding the host in lipid environment. Fig. 2c shows the upfield region of the 1H spectrum of PCm·1·3, and it is immediately obvious that the proportion of axial conformer is greatly increased when compared to that seen in the 1·3 complex. In this case, 28% of the bound 3 exists in the axial conformer, compared to only 12% in 1·3 and <0.5% in free solution.
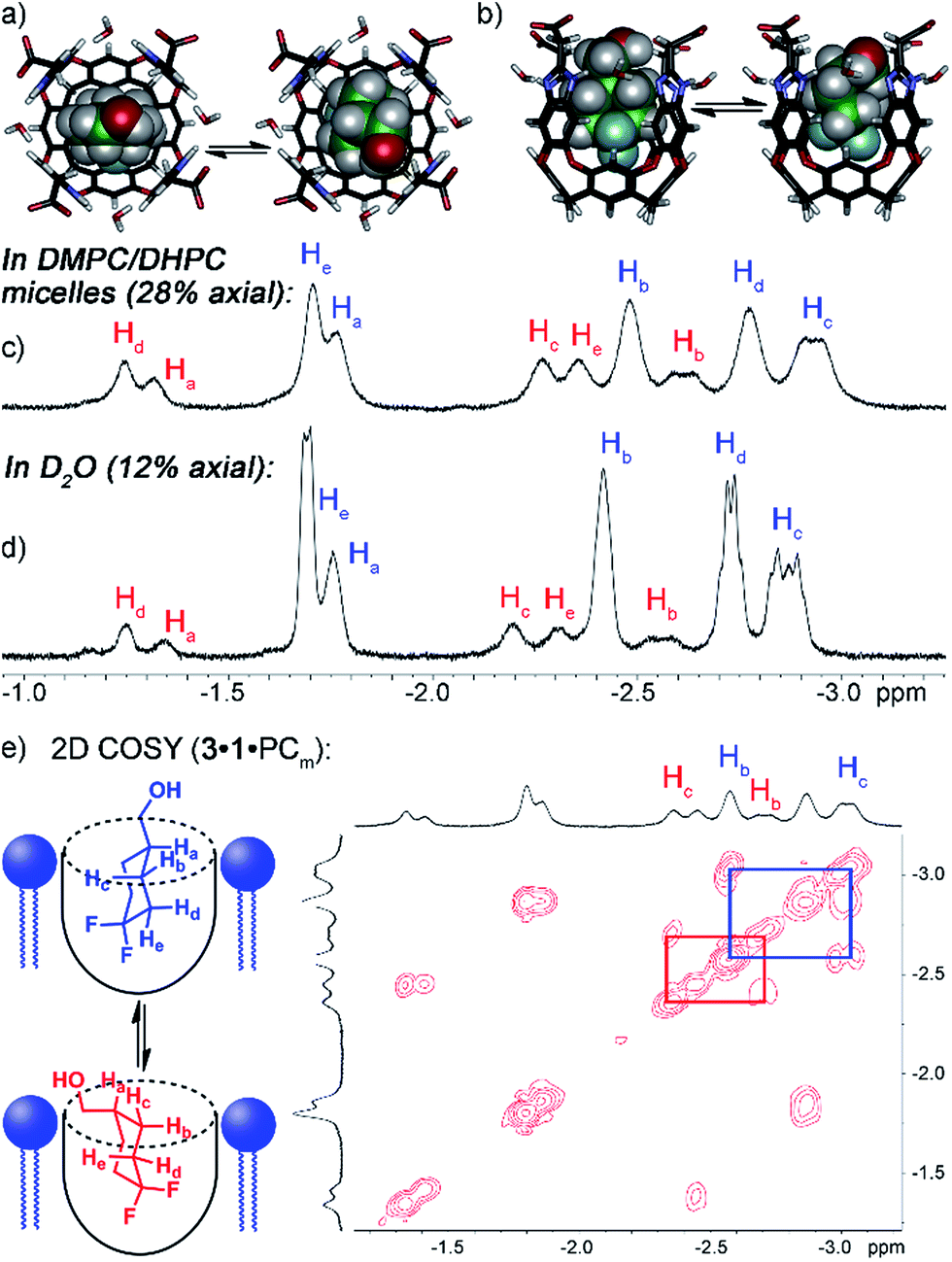 | ||
| Fig. 2 Enhanced axial conformation of bound guest 3. (a and b) Minimized structures of the 1·3eq and 1·3ax complexes (SPARTAN, AMBER forcefield). Upfield regions of the 1H NMR spectra of (c) PCm·1·3; (d) 1·3; (e) 2D COSY spectrum of PCm·1·3 (700 MHz, 298 K, [1] = 5.8 mM, [3] = 39.5 mM, ratio DMPC/DHPC = 3.2:1, 60 mg mL−1 total lipid concentration). | ||
Interestingly, the 1H and 19F NMR spectra of the 1·4 complex are reminiscent of those of 1·3, in that two different guests are bound, even though only one was seen in free solution. Again, the chemical shifts of the CH protons in the bound guests do not match the expected signals for up/down carceroisomers, but represent the recognition of 1·4 and 1·4hyd, namely the ketone and hydrated gem-diol form (Fig. 3). The 19F spectrum of hydrated 1·4hyd is highly reminiscent of 1·3: whereas the 19F peaks for cyclohexanone 1·4 are close in shift, the all-sp3 cyclohexane skeleton of 1·4hyd (similar to that of 1·3) separates the two fluorine peaks to reflect the distinct axial and equatorial positions (see Fig. 5 for full spectrum). The equilibrium between these two states strongly favors the ketone form in the case of unactivated ketones such as acetone, but the presence of the electron-withdrawing groups such as halogens increases the favorability of the hydrate (Khyd(acetone) = 1.4 × 10−3, Khyd(fluoroacetone) = 0.11).22 NMR analysis of guest 4 in D2O in the absence of cavitand showed no obvious hydrate present. At the concentrations used, this indicates that <0.5% hydrate is present in solution. In contrast, when bound inside the cavity of 1, 13% of bound 4 exists in the hydrated form at 298 K. Embedding the host in PC lipids also biases the hydration equilibrium of 4, similar to the conformational bias seen in the binding of 3. When 4 was added to the 1·PCm system, 23% of the bound 4 was present in the hydrated form, compared to only 13% in 1·4 and <0.5% in free solution.
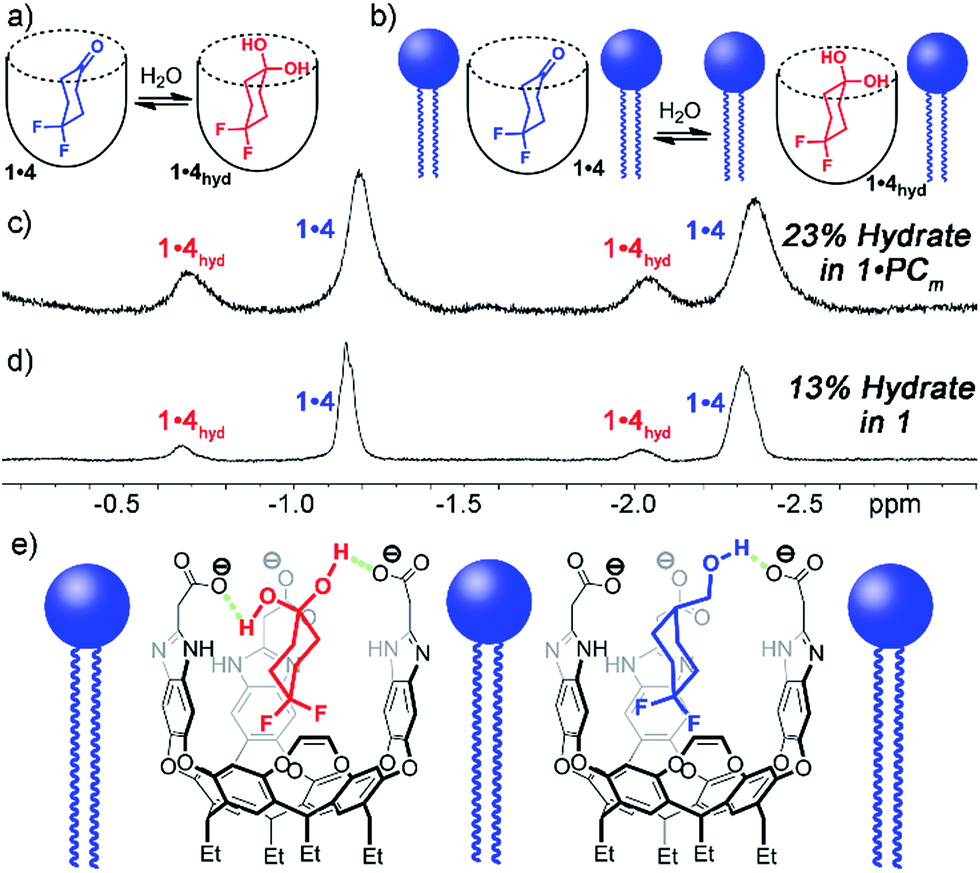 | ||
| Fig. 3 Enhanced hydration of bound guest 3. (a and b) Illustration of the equilibrium process. Upfield regions of the 1H NMR spectra of (c) PCm·1·4; (d) 1·4 (400 MHz, 298 K, [1] = 5.8 mM, [4] = 39.5 mM, ratio DMPC/DHPC = 3.2:1, 60 mg mL−1 total lipid concentration) (e) illustration of the favorable hydrogen bonding present in PCm·1·4hyd and PCm·1·3ax. | ||
These observations introduce the question of why binding in 1 stabilizes normally unfavorable guest structures, and why this effect is enhanced when 1 is embedded in lipid aggregates. Cavitand 1 is well-known to display dual-mode recognition, whereby both the defined cavity and the upper rim groups can affect guest binding.13a The upper rim carboxylates have been shown to accelerate solvolysis reactions of bound guests,4a and control binding selectivity for functionalized guests in different pH conditions.13b The presence of anionic carboxylate functions at the upper rim of the cavity confers favorable H-bonding to acidic groups in the guest positioned in close proximity, more so than the external bulk water. Guests containing properly positioned H-bond donors such as ammonium ions,10b,13a thioureas13 or even hydroxy groups2a,4a,10b,13a have been shown to have stronger affinity for 1 than those with esters, ethers or ketones.4a,10b,13a The axial conformer of 3 evidently positions the OH group in closer proximity to the rim carboxylates, increasing the favorability of that conformer when bound. The hydrated gem-diol of 4 is capable of H-bonding with the carboxylates, whereas the ketone is not, hence the increased favorability of the bound hydrate (Fig. 3e).
Why this effect is enhanced when 1 is bound inside lipid micelles is less clear, but two possibilities present themselves. The cavitand could be positioned in the bilayer such that a small “hydrophobic” pocket is created above the cavitand rim, hiding the bound guest somewhat from external water and increasing the effect of the H-bonding between guest and host by limiting competitive H-bonding with the external water. This theory was previously used to explain the enhanced binding of cationic proteins to a 1·POPC supported lipid bilayer interface.11d However, we have no concrete information about the position of the cavitand in the PCm aggregate, so there is little hard evidence for this theory. The other possibility is that the lipids act as a “compression sleeve”, forcing the cavitand walls closer to the guest than normally observed in pure water. Cavitand 1 is highly flexible, and the exact position of the walls varies with guest size. A restricted “breathing” motion of the host walls would strengthen intermolecular host:guest interactions, as has been seen for numerous other encapsulation complexes, in water and in organic solvents.3a,b,23
To shed light on this, as well as to gain valuable insight on the host:guest kinetics, we investigated the in/out exchange properties of 1 with guests 3 and 4 in solution and in the PCm aggregate. NMR experiments that take advantage of magnetization transfer are ideally suited to kinetic analysis of host:guest systems.24 2D 1H–1H EXSY experiments were previously used to show the exchange rates of small guests in and out of 1 in aqueous solution,2a but the presence of large interfering peaks from the lipids limits the effectiveness of these experiments. The presence of guest nuclei not present in the lipid aggregates allows exchange analysis, however, and 2D 19F–19F EXSY proved effective for kinetic analysis of guests 3 and 4. Fig. 4 shows partial 19F–19F EXSY spectra for the 1·3 and 1·4 complexes, obtained under the same conditions as the spectra in Fig. 2 and 3. The major diagonal peak corresponds to the signals from the free and bound axial F in each molecule. At a mixing time of τ = 100 ms, exchange crosspeaks are easily observed, illustrating the in/out exchange process. No crosspeaks are seen at τ = 3 ms. The spectra in Fig. 4 clearly illustrate the qualitative differences in exchange behavior of 1·3 and 1·4 in D2O and in PCm. Whereas exchange crosspeaks are clearly visible at τ = 100 ms for 1·3 (D2O), the same exchange conditions show only minimal crosspeaks for 1·3·PCm (Fig. 4a and b). Only at longer mixing times are crosspeaks observed, indicating a substantial slowing of the exchange rate of 3 when 1 is embedded in the PCm aggregate. A similar, although less obvious effect is seen for guest 4: exchange crosspeaks are smaller in the 1·4·PCm system than in 1·4 at 100 ms (Fig. 4c and d).
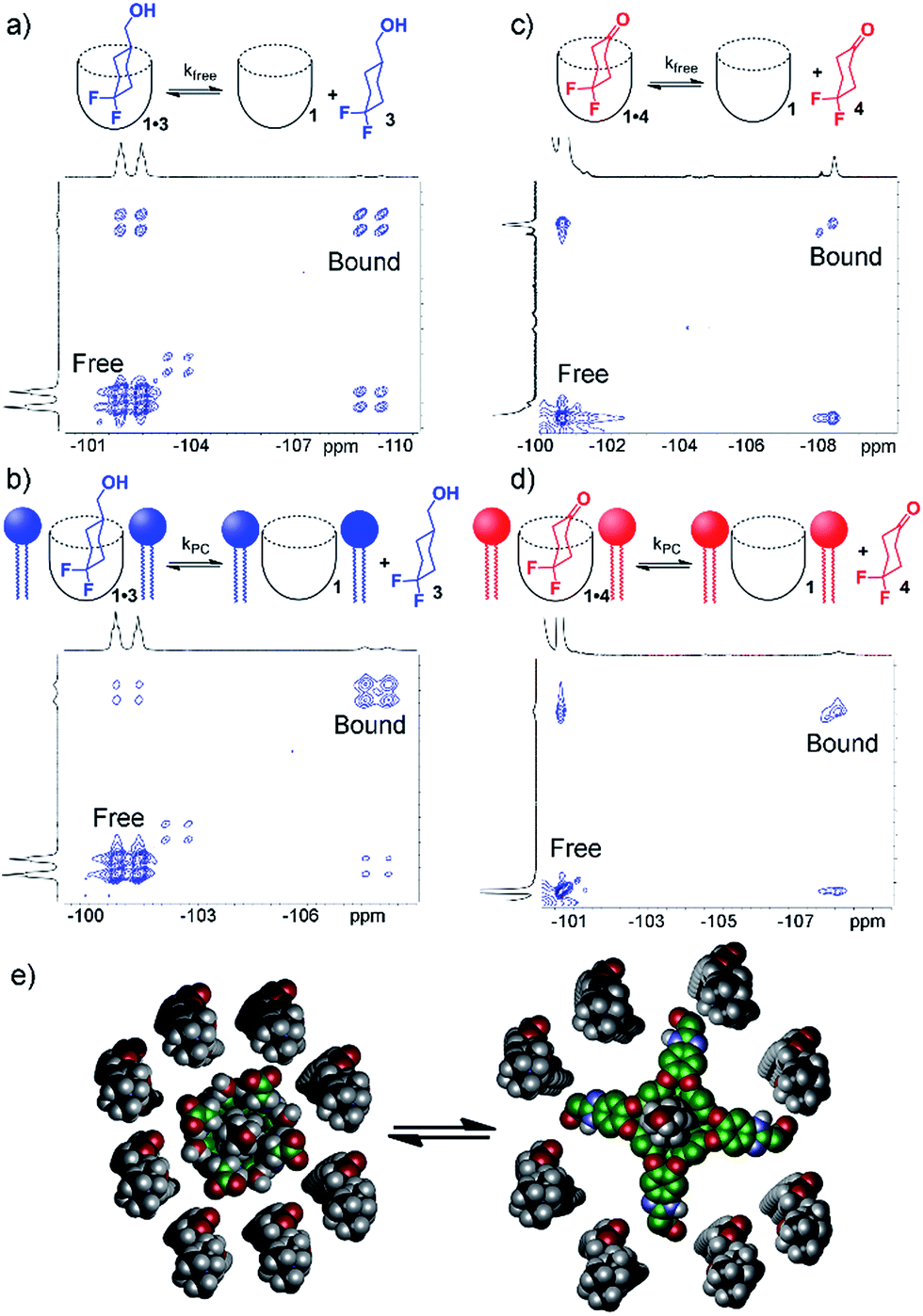 | ||
| Fig. 4 In/out exchange of guests 3 and 4 in 1 and 1·PCm. 19F–19F EXSY NMR spectra at mixing time τ = 100 ms of (a) 1·3 in D2O solution; (b) 1·3·PCm; (c) 1·4 in D2O solution; (d) 1·4·PCm (376.50 MHz, 298 K, [1] = 5.8 mM, [3,4] = 39.5 mM, ratio DMPC/DHPC = 3.2:1, 60 mg mL−1 total lipid concentration); (e) representation of the exchange dynamics in cavitand 1 in a DMPC/DHPC lipid bilayer environment. | ||
By taking the exchange spectra at multiple different mixing times, quantitation of the exchange rates was possible. The EXSY spectra of 1·3 and 1·4 were surprisingly complex. Fig. 4 shows the relevant sections of the 19F–19F EXSY spectra (at mixing time τ = 100 ms) used to determine exchange rates; the full spectra are shown in the ESI.† Multiple peaks are observed in the full spectra: the axial and equatorial F atoms both show free and bound peaks, which show chemical exchange with each other. In addition, the geminal fluorines show NOE crosspeaks to each other, and small peaks are present from the other conformer with CH2OH axial. As such, the in/out exchange rates were determined by fitting the intensity of the four exchange peaks shown in Fig. 4 (obtained by extracting 1D slices from the 2D EXSY plots) against mixing time. At higher mixing times (τ ≥ 300 ms), the multiple methods of magnetization transfer in the system caused inaccuracies in the fitting, so the initial rate regions of the plot were used. For a detailed description of the fitting method, please see ESI.†
Table 1 shows the results of the exchange analysis. The fitting process gives the rate k−1 (or “koff”) for each guest, obtained at identical concentrations and temperatures for each guest in either aqueous solution (kfree) or in the micelle environment (kPC). Eyring analysis25 of the rate constants gives the exchange barriers ΔG‡. As expected, the rate is dependent on the nature of the guest, but most interestingly, it is also dependent on the external environment. The larger guest 3 shows a kfree = 4.2 s−1, comparable to that previously obtained for cyclooctanol.2a In the presence of lipids, however, the exchange rate drops by over a factor of two, with kPC = 1.8 s−1, corresponding to an additional 0.5 kcal mol−1 additional barrier conferred by the external environment surrounding the cavitand host. The same “compression sleeve” effect that enhanced the axial conformation of bound 3 slows the in/out exchange rate as well.
| Guest | k free, s−1 | k PC, s−1 | ΔG‡free, kcal mol−1 | ΔG‡PC, kcal mol−1 |
|---|---|---|---|---|
|
a Exchange rates determined by fitting 2D EXSY crosspeaks (see ESI for fit plots and model). kfree = “off” exchange rate k−1 of guest from 1 in D2O. kfree = “off” exchange rate k−1 of guest from 1·PCm in 1 mM HEPES/D2O, ratio DMPC/DHPC = 3.2:1, 60 mg mL−1 total lipid concentration. [1] = 5.8 mM, [3,4] = 39.5 mM, [6,7] = 16 mM. Exchange barriers determined via the Eyring equation.25
|
||||
| 3 | 4.2 ± 0.9 | 1.8 ± 0.8 | 16.6 | 17.1 |
| 4 | 8.7 ± 1.3 | 5.2 ± 1.6 | 16.2 | 16.5 |
| 4hyd | 5.7 ± 0.8 | N/A | 16.4 | N/A |
| 6 | N/A | 3.0 ± 0.2 | N/A | 16.7 |
| 7 | N/A | 5.7 ± 0.5 | N/A | 16.4 |
EXSY analysis of guest 4 showed that kfree = 8.7 s−1, similar to that to that previously obtained for cyclohexanone, as expected.2a In a lipid environment, the exchange rate slowed again, with kPC = 5.2 s−1. The retardation of exchange rate is slightly less in the case of the smaller guest 4, with a 0.3 kcal mol−1 additional barrier. Surprisingly, the EXSY spectrum of 1·4 allowed analysis of the in/out exchange of the hydrated gem-diol form of 4hyd, as the crosspeaks were large enough to observe (Fig. 5). The additional hydrogen bonding present in 4hyd slows the exchange rate when compared to the ketone form, and kfree (4hyd) = 5.7 s−1. Unfortunately, the equivalent crosspeaks in the PCm system were too small to accurately fit, so determination of kPC was unsuccessful in that case.
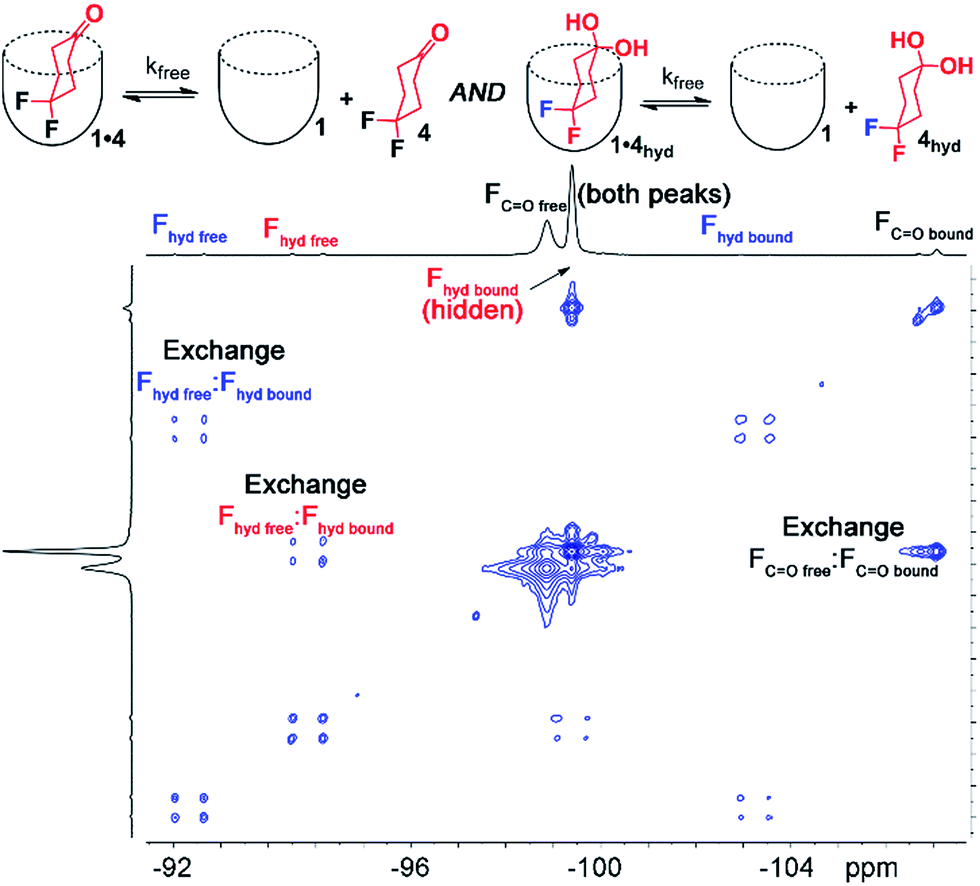 | ||
| Fig. 5 Full 19F EXSY spectrum of the cavitand 1· guest 4 complex in pure D2O with peak assignments (D2O, 150.84 MHz, 298 K, mixing time = 150 ms, [1] = 5.8 mM, [4] = 39.5 mM). | ||
The use of guest nuclei other than 1H to analyze the in/out exchange allows analysis of other, more biorelevant guests such as choline. While association constants for R-NMe3+ guests are easily determinable by ITC10b or indicator displacement assays,13 NMR analysis of the exchange kinetics are complicated by self-aggregation. Cavitand 1 is susceptible to aggregation in free solution in the presence of lipophilic salts such as choline. Stable 1:1 complexes can be observed by NMR when substoichiometric amounts of guest are used,10b but addition of excess RNMe3+ salt causes aggregation and peak broadening, limiting analysis of the in/out rate by exchange NMR.13 This does not occur in lipid environments, rendering this system ideal for analyzing exchange of hydrophilic, yet strongly binding salts 5–8. 13C-enriched R-NMe3+ guests were easily accessed via reaction of the corresponding dimethylamino precursor with enriched 13CH3I. As cavitand 1 is capable of binding numerous R-NMe3+ salts in lipid environment, we investigated a series of guests 5–8 with variable upper rim functionality. The bola-type bis-NMe3+ guests 5 and 8 were initially targeted to investigate the possibility of slowed tumbling in the cavity of 1, as opposed to in/out guest exchange. Encapsulation in 1 slows the up/down interconversion rate of hydrocarbons such as trans-decalin, but quantitation is challenging due to peak broadening. As 5 and 8 are symmetrical, it was envisaged that exchange would be observed between the two conformers in the 1·5·PCm system via13C–13C EXSY NMR analysis. Unfortunately, neither guest 5 nor 8 are bound by 1 in either free aqueous solution or in the PCm environment. Evidently, the nature of the upper rim has a large effect on guest recognition, so we turned to guests that can display favorable H-bonds with the carboxylate groups, choline 6 and the dimethylamino-variant 7.
Analysis of the unsymmetrical guests 6 and 7 was far more successful, and representative examples of the 13C–13C EXSY spectra are shown in Fig. 6. The peak for free R-NMe3+ guest (6 or 7) overlaps with peaks from the R-NMe3+ group in the phosphocholine lipids, as would be expected. Even though 6/7 are 13C-enriched, a significant proportion of 13C-PC is present due to the excess of lipids in the sample. Despite the interfering peaks for free guest, 13C peaks for both bound 1·6 and 1·7 are easily observable, with the characteristic upfield shift observed. The relative change in 13C δ upon binding is proportionally smaller than that for 1H, with Δδ ∼ −2 ppm, but this is easily enough to allow exchange analysis via13C–13C EXSY.
 | ||
| Fig. 6 In/out exchange of guests 6 and 7 in 1 and 1·PCm. 13C–13C EXSY NMR spectra at mixing time τ = 100 ms of (a) 1·7·PCm; (b) 1·6·PCm (2.5 mM HEPES/D2O, 150.84 MHz, 298 K, [1] = 5.8 mM, [6,7] = 16 mM, ratio DMPC/DHPC = 3.2:1, 60 mg mL−1 total lipid concentration). | ||
13C–13C EXSY spectra with varying mixing times were obtained for samples of 1·6·PCm and 1·7·PCm at 298 K with 5.8 mM 1, 16 mM guest and 60 mg mL−1 lipid, as usual. The exchange rates were acquired via fitting the crosspeak intensities extracted as slices from the 2D spectra. In this case, the diagonal peak corresponding to free guest overlapped with peaks from the NMe3+ groups in the lipids. As the concentration of the lipids was constant, the signal from the lipids remained constant in the low mixing time experiments, and the accuracy of the fit for the off-rate was not compromised. The rates are shown in Table 1. Interestingly, the rates are broadly similar to those observed with the difluorocyclohexanyl guests, with kPC (6) = 3.0 s−1 and kPC (7) = 5.7 s−1. The bulkier 7 exchanges more rapidly than choline 6, in contrast to the results for hydrophobic guests, where larger guests showed slowed exchange. It is likely that positioning the extra steric bulk at the upper rim lowers the affinity of 7 for the cavitand, as has been observed for other R-NMe3+ species, and a more rapid exchange rate is seen. The results from Table 1 also allow an estimate of the kfree for 6 and 7: if the “compression sleeve” effect of the micelle environment is assumed to be constant, then kfree for 6 and 7 would be on the order of ∼6 s−1 and ∼10 s−1, respectively.
As the 13C-labeled guests 6 and 7 were amenable to EXSY analysis in isotropically tumbling micelles, we next employed these guests towards detection of in/out exchange of host 1 in the more challenging, yet more relevant magnetically ordered bicelles. The bicelles were formed as described above (also see Experimental), loaded into a 4 mm Bruker solid state rotor, and solutions of cavitand 1 and guests 6 and 7 were added. As might be expected, the larger concentration of lipids (and their overlapping phosphocholine groups) made analysis via 1D NMR challenging, even with 13C-enriched guests. Fortunately, the magnetic alignment at 308 K was good, and the presence of exchanging guest 7 was observable by 13C–13C EXSY analysis (see Fig. 7a). The signal:noise ratio was poor for 13C choline 6, so we focused on guest 7 for bicellar analysis. At mixing time τ = 20 ms, exchanging crosspeaks corresponding to the 13CH3 signal from bound and free guest 7 can be seen. At the elevated temperatures required for magnetically ordered bicelle formation, the in/out rate occurs more rapidly, and a shorter mixing time was needed to see exchange. The nature of the crosspeaks was corroborated by the EXSY spectrum taken with mixing time τ = 0 ms (Fig. 7b), where no crosspeaks could be seen. Unfortunately, accurate quantitation of the exchange rate in the bicellar system proved challenging. The smaller sample volume necessitated a greater amount of signal averaging to obtain good signal, and required long acquisition times (∼48 h per spectrum). In addition, the bicelles decomposed after ∼1 week at 308 K. The experiments were performed on the same sample to avoid differences in peak intensity due to any slight differences in concentration between samples, and to obtain spectra in manageable timeframes, the resolution in the F2 dimension was reduced. As a result, the spectra were suitable for only qualitative analysis rather than quantitation of the exchange rate. However, the exchange could be clearly seen for guest 7, and illustrates the power of the system: 2D NMR analysis of the host:guest properties of cavitand 1 is possible in different types of complex lipid aggregates, and the guest kinetics can be analyzed for guests containing suitable nuclei for detection.
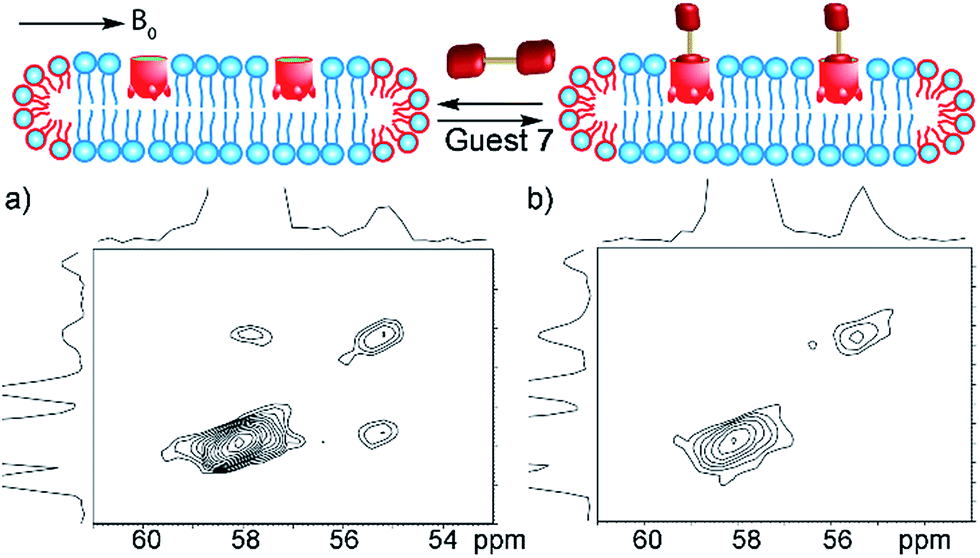 | ||
| Fig. 7 In/out exchange of guest 7 in the magnetically ordered bicelle system PCb. 13C–13C EXSY NMR spectra at mixing time (a) τ = 20 ms; (b) τ = 0 ms of 1·7·PCb; (1 mM HEPES/D2O, 100.69 MHz, 298 K, [1] = 20 mM, [7] = 36 mM, ratio DMPC/DHPC = 3.2:1, 150 mg mL−1 total lipid concentration). | ||
Conclusions
By employing guests with detectable nuclei, NMR analysis of how the external environment affects the recognition properties of a water-soluble deep cavitand is possible. Nuclei such as 13C or 19F are usually not used to analyze conformation and motion of small molecules in confined environments, as the chemical shift changes are small relative to 1H and lone-pair containing groups can lower affinity, especially for aromatic hosts. The systems studied here illustrate a wide variety of effects that can be conferred on a small molecule guest from molecules outside the host. Embedding the deep cavitand in lipids compresses the flexible walls of the host, enhancing its recognition properties and providing an additional barrier to wall-opening. By forcing the walls of the closer to the guest, unfavorable conformational or reaction equilibria can be enhanced: favorable H-bonding with the upper rim carboxylates enhances the population of an axial cyclohexyl conformer, as well as favoring ketone hydration. These unusual conformations are present for the host:guest complexes in water, but are enhanced in the lipid environment. In addition, 2D EXSY NMR spectroscopy using either 13C or 19F as detectable nucleus allows analysis of the in/out exchange properties of bound guests in phosphocholine lipid micelles and magnetically ordered bicelles. Embedding the host in a lipid aggregate slows the exchange rate of small molecule guests by over a factor of two, due to the energetic penalty conferred on the opening of the host walls by the external lipid aggregate. Both solution- and solid-state NMR techniques were employed to show this exchange process, providing the first detailed view of the exchange process of flexible supramolecular host molecules in biomimetic lipid membranes. Further studies of molecular recognition in complex systems is underway in our laboratory.Experimental
General information
1D NMR experiments (1H, 13C, 19F) were performed on a Bruker Avance NEO 400 9.4 T spectrometer with a 5 mm Prodigy CPP BBO BB-H&F z-gradient cryo-probe or a Bruker 14.1 T (600.01 MHz 1H) Avance I spectrometer equipped with a 5 mm BBO Z-grad probe. Micelle (PCm) experiments (1H–1H COSY, 2H–2H EXSY, 13C–13C EXSY, 19F–19F EXSY) were performed on a Bruker Avance NEO 400 9.4 T spectrometer with a 5 mm Prodigy CPP BBO BB-H&F z-gradient cryo-probe, a Bruker 14.1 T (600.01 MHz 1H) Avance I spectrometer equipped with a 5 mm BBO Z-grad probe or a Bruker Avance III 700 16.44 T spectrometer with a 5 mm CP TCI H–C/N-D z-gradient cryo-probe. Magnetically ordered bicelle (PCb) experiments (13C, 13C–13C EXSY) were performed at 9.4 T (400.37 MHz 1H, 100.69 MHz 13C, 162.07 MHz 31P) on a Bruker AVIII spectrometer equipped with a double resonance, 4 mm MAS probe. Proton (1H) and carbon (13C) chemical shifts are reported in parts per million (δ) with respect to tetramethylsilane (TMS, δ = 0). Phosphorus (31P) chemical shifts are reported in parts per million (δ), and referenced internally with respect to 85% H3PO4. Fluorine (19F) chemical shifts are reported in parts per million (δ), and referenced internally with respect to CF3COOH. Deuterated NMR solvents were obtained from Cambridge Isotope Laboratories, Inc., Andover, MA, and used without further purification. Mass spectra were recorded on an Agilent 6210 LC TOF mass spectrometer using electrospray ionization with fragmentation voltage set at 115 V and processed with an Agilent MassHunter Operating System. All other materials (including guests 2–4, synthetic precursors for guests 5–8, dimyristoylphosphocholine (DMPC), and diheptylphosphocholine (DHPC)) were obtained from Aldrich Chemical Company, St. Louis, MO, or TCI, Tokyo, Japan and were used as received. Solvents were dried through a commercial solvent purification system (Pure Process Technologies, Inc.). Molecular modeling (molecular mechanics calculations) was carried out using the AMBER force field26 with the solvation (dielectric) setting for water as implemented by SPARTAN. Cavitand 1 was synthesized according to published procedures:10b also see this paper for the NMR spectra of the 1· choline complex.Experimental procedures
Synthesis of new molecules
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the National Science Foundation (CHE-1748063 to R. J. H.; CHE-1710671 to L. J. M.) for support, and Dr Dan Borchardt and Alvicler Magalhaes for initial NMR spectra and testing. Funding for NMR spectrometers was provided by the National Science Foundation (CHE-1626673) and the Army Research Office (W911NF-16-1-0523).Notes and references
-
(a) J. Rebek Jr, Acc. Chem. Res., 2009, 42, 1660–1668 CrossRef PubMed
; (b) D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 351–360 CrossRef PubMed
-
(a) R. J. Hooley, H. J. Van Anda and J. Rebek Jr, J. Am. Chem. Soc., 2007, 129, 13464–13473 CrossRef CAS PubMed
-
(a) A. V. Davis, D. Fiedler, G. Seeber, A. Zahl, R. van Eldik and K. N. Raymond, J. Am. Chem. Soc., 2006, 128, 1324–1333 CrossRef CAS PubMed
-
(a) R. J. Hooley, J. V. Gavette, M. Mettry, D. Ajami and J. Rebek Jr, Chem. Sci., 2014, 5, 4382–4387 RSC
-
(a) A. Asadi, D. Ajami and J. Rebek Jr, J. Am. Chem. Soc., 2011, 133, 10682–10684 CrossRef CAS PubMed
-
(a) R. J. Hooley, S. R. Shenoy and J. Rebek Jr, Org. Lett., 2008, 10, 5397–5400 CrossRef CAS PubMed
- A. Shivanyuk and J. Rebek Jr, J. Am. Chem. Soc., 2002, 124, 12074–12075 CrossRef CAS PubMed
-
(a) R. J. Hooley, T. Iwasawa and J. Rebek Jr, J. Am. Chem. Soc., 2007, 129, 15330–15339 CrossRef CAS PubMed
-
(a) S. Horiuchi, T. Murase and M. Fujita, Angew. Chem., Int. Ed., 2012, 51, 12029–12031 CrossRef CAS PubMed
-
(a) F. Hof, L. Trembleau, E. C. Ullrich and J. Rebek Jr, Angew. Chem., Int. Ed., 2003, 42, 3150–3153 CrossRef CAS PubMed
-
(a) Y. Liu, P. Liao, Q. Cheng and R. J. Hooley, J. Am. Chem. Soc., 2010, 132, 10383–10390 CrossRef CAS PubMed
- Y.-J. Ghang, M. P. Schramm, F. Zhang, R. A. Acey, C. N. David, E. H. Wilson, Y. Wang, Q. Cheng and R. J. Hooley, J. Am. Chem. Soc., 2013, 135, 7090–7093 CrossRef CAS PubMed
-
(a) Y. Liu, L. Perez, M. Mettry, A. D. Gill, S. R. Byers, C. J. Easley, C. J. Bardeen, W. Zhong and R. J. Hooley, Chem. Sci., 2017, 8, 3960–3970 RSC
-
(a) M. P. Schramm, R. J. Hooley and J. Rebek Jr, J. Am. Chem. Soc., 2007, 129, 9773–9779 CrossRef CAS PubMed
-
(a) D. A. Ryan and J. Rebek Jr, J. Am. Chem. Soc., 2011, 133, 19653–19655 CrossRef CAS PubMed
- D. J. Cram, H. J. Choi, J. A. Bryant and C. B. Knobler, J. Am. Chem. Soc., 1992, 114, 7748–7765 CrossRef CAS
- S. L. Craig, S. Lin, J. Chen and J. Rebek Jr, J. Am. Chem. Soc., 2002, 124, 8780–8781 CrossRef CAS PubMed
-
(a) S. H. Park, B. B. Das, F. Casagrande, Y. Tian, H. J. Nothnagel, M. Chu, H. Kiefer, K. Maier, A. A. De Angelis, F. M. Marassi and S. J. Opella, Nature, 2012, 491, 779–783 CrossRef CAS PubMed
-
(a) U. H. N. Dürr, M. Gildenberg and A. Ramamoorthy, Chem. Rev., 2012, 112, 6054–6074 CrossRef PubMed
-
(a) S. J. Opella, Acc. Chem. Res., 2013, 46, 2145–2153 CrossRef CAS PubMed
- P. Timmerman, W. Verboom, F. C. J. M. Vanveggel, J. P. M. Vanduynhoven and D. N. Reinhoudt, Angew. Chem., Int. Ed., 1994, 33, 2345–2348 CrossRef
-
F. A. Carey and R. J. Sundberg, in Advanced Organic Chemistry Part A: Structure and Mechanisms, Springer, 2008 Search PubMed
-
(a) D. M. Rudkevich, G. Hilmersson and J. Rebek Jr, J. Am. Chem. Soc., 1998, 120, 12216–12225 CrossRef CAS
-
(a) C. L. Perrin and T. J. Dwyer, Chem. Rev., 1990, 90, 935–967 CrossRef CAS
-
E. V. Anslyn and D. A. Dougherty, in Modern Physical Organic Chemistry, University Science Books, Sausalito, California, 2006 Search PubMed
- F. Mohamadi, N. G. J. Richards, W. C. Guida, R. Liskamp, M. Lipton, C. Caufield, G. Chang, T. Hendrickson and W. C. Still, J. Comput. Chem., 1990, 11, 440–467 CrossRef CAS
- K. P. Howard and S. J. Opella, J. Magn. Reson., Ser. B, 1996, 112, 91–94 CrossRef CAS PubMed
- A. A. De Angelis and S. J. Opella, Nat. Protoc., 2007, 2, 2332–2338 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Spectral data not included in the text, detailed description of the exchange fitting process. See DOI: 10.1039/c7sc05155g |
| ‡ These authors all contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2018 |