
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diastereo- and enantioselective additions of α-nitro esters to imines for anti-α,β-diamino acid synthesis with α-alkyl-substitution†
Daniel J.
Sprague
,
Anand
Singh
 and
Jeffrey N.
Johnston
*
and
Jeffrey N.
Johnston
*
Department of Chemistry, Vanderbilt Institute of Chemical Biology, Vanderbilt University, Nashville, Tennessee 37235, USA. E-mail: jeffrey.n.johnston@vanderbilt.edu
First published on 31st January 2018
Abstract
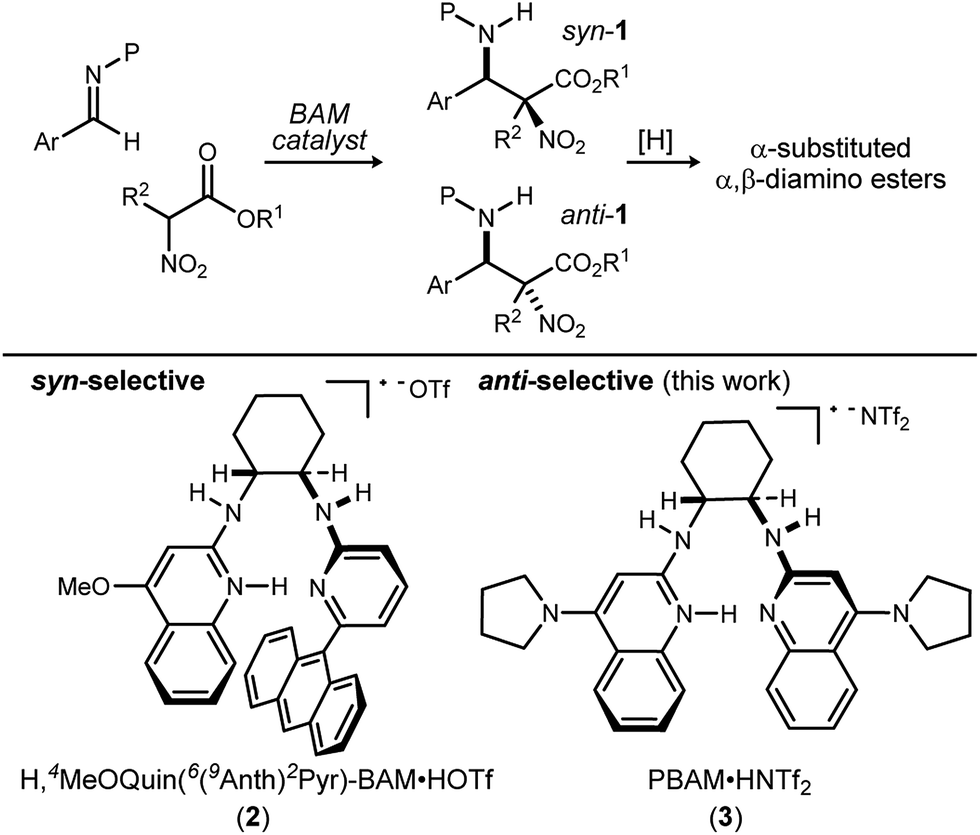
The discovery that a C2-symmetric bis(AMidine) [BAM] catalyst promotes an anti-selective addition of α-substituted α-nitro esters to imines is described, providing α-substituted α,β-diamino ester products with high diastereo- and enantioselectivity. When compared to the function of a BAM catalyst reported previously, the pair offer a rare example of diastereodivergence using a bifunctional Brønsted acid–base organocatalyst.
Methods to prepare enantioenriched α-amino acids are in demand, and there are few direct solutions to those bearing α-alkyl substituents.1 Fewer still are methods that deliver α,β-diamino acids bearing α-alkyl substituents.2 These unnatural amino acids are desirable precursors to peptide sequences due to their effect on the conformation and activity of the peptide sequences into which they are incorporated.3–5 The enantioselective aza-Henry (nitro-Mannich) reaction6 serves as a convergent approach to α,β-diamino acid derivatives, but its adaptation to α-alkyl-α-nitroester substrates (Scheme 1) is more rare, owing to the congestion provided by the additional substituent. Within this realm are the highly diastereoselective examples of the anti-selective reaction by Jorgensen,7 Shibasaki,8 Wu,9 and Huang-Dong.10 In contrast, syn-selective reactions are the exception,11 with reports by us12 and Ooi.13 Diastereodivergence in enantioselective catalysis is a characteristic driving modern catalyst development,14–16 and it motivated us to develop an anti-selective variant using the same bifunctional Brønsted acid/base catalyst design (Scheme 1).12 We report the finding that a C2-symmetric ligand design, in combination with sterically hindered esters of α-nitro acids, can lead to highly anti-diastereoselective and enantioselective additions to N-Boc imine electrophiles. This creates a rare example in which a pair of organocatalyzed reactions with generally conserved design features, exhibit diastereodivergence and high selectivity.14,17,18
 | ||
| Scheme 1 Development of a diastereodivergent aza-Henry reaction of α-alkyl α,β-nitroesters: syn-selective (prior work) and anti-selective (this work) catalysts using a common bifunctional design. | ||
We previously reported the organocatalytic synthesis of α-substituted syn-α,β-diamino acid derivatives syn-1.12 Key to that success was the finding that unsymmetrical quinoline catalyst 219 was necessary to achieve adequate reactivity, wherein the methoxy substituent imparted a more Brønsted basic 2-aminoquinoline for efficient activation of the sterically demanding nitro ester pronucleophile.20 Additionally, hindered aryl esters found synergism with the crowded pocket of 2 to provide high syn-selectivity, good yield, and high enantioselection.21–23
A return to symmetrical catalyst 324 (Scheme 1) was made in order to examine the impact of a less congested binding pocket to selectivity. In doing so, retention of catalyst activation using a pyrrolidine at the quinoline 4-position was anticipated. In the event, the level of diastereoselection with a small alkyl ester was low, but again increased with ester size (as in Scheme 2) and with the distinction that the anti-diastereomer was favored. As before, the ester size works synergistically with the catalyst to achieve increasing levels of selectivity, particularly diastereoselectivity (10d → 11d, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, 94% ee). Finally, changing the solvent to toluene and the counteranion to triflimide afforded a combination producing optimal stereoselectivity (Table 1, entry 1) overall.25
:1 dr, 94% ee). Finally, changing the solvent to toluene and the counteranion to triflimide afforded a combination producing optimal stereoselectivity (Table 1, entry 1) overall.25
 | ||
| Scheme 2 Determinants of diastereoselection: synergism between catalyst 3 and ester size. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entrya | R | 13 | drb | eec | Yieldd |
|
a All reactions were 0.7 M in imine, used 1.1 equiv. of the α-nitro ester, and had a standard 48 h reaction time.
b Diastereomer ratios measured using 1H NMR.
c Enantiomeric ratios measured using HPLC and a chiral stationary phase.
d Yields are for isolated, analytically pure adduct.
e For comparison, use of the triflic acid salt of the catalyst provides this anti-product in 17:1 dr and 97% ee. rac-PBAM (free base) affords the adduct in 2:1 dr.
|
|||||
| 1 | Me | a | >20:1 |
99 | 70 |
| 2e | Et | b | >20:1 |
99 | 66 |
| 3 | n Pr | c | >20:1 |
96 | 72 |
| 4 | n Bu | d | 11:1 |
97 | 64 |
| 5 | Allyl | e | 9:1 |
97 | 71 |
| 6 | Bn | f | 4:1 |
83 | 65 |
| 7 | c Pr | g | 15:1 |
98 | 68 |
| 8 | i Pr | h | >20:1 |
93 | 66 |
| 9 | c Hex | i | >20:1 |
87 | 46 |
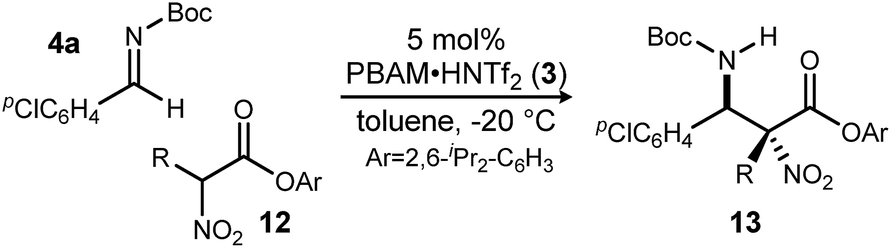
Having maximized the favored ester/catalyst combination to effect high anti-selectivity while maintaining high enantioselection, we turned to an evaluation of substrate scope. The effect of the size of the alkyl substituent presented by the hindered nitro ester was probed first by increasing chain length (Table 1, entries 1–4) using pCl-phenyl aldimine 4a as a standard electrophile. α-Nitro propionoate (12a), butanoate (10d/12b), pentanoate (12c), and hexanoate (12d) each afforded product in good yield with excellent diastereoselection (11:1 → 20:1 dr) and uniformly high enantioselection (96–99% ee). As this substituent is changed further, only those with sp2-hybridization resulted in lower diastereoselection (down to 4:1) (Table 1, entries 5–6).‡ Branching alkyl substituents, however, returned selectivity to >15:1 dr (Table 1, entries 7–9). α-Cyclopropyl nitroacetate 12g afforded product in 68% isolated yield with 15:1 dr and 98% ee (Table 1, entry 7), and α-isopropyl nitroacetate 12h afforded product in 66% isolated yield with >20:1 dr and 93% ee (Table 1, entry 8). α-Cyclohexyl nitroacetate 12i gave the desired diamine derivative in >20:1 dr, and 87% ee, albeit in a lower isolated yield (46%, Table 1, entry 9). The lower conversion, and consequently lower isolated yield, reflect the steric bulk surrounding the nucleophilic carbon. Nevertheless, synthetically useful amounts of stereoenriched product 13i can be obtained under the reaction conditions. An allyl group was incorporated at the α-position in good isolated yield, dr, and high ee (Table 1, entry 5). This installs a handle for further synthetic manipulations.
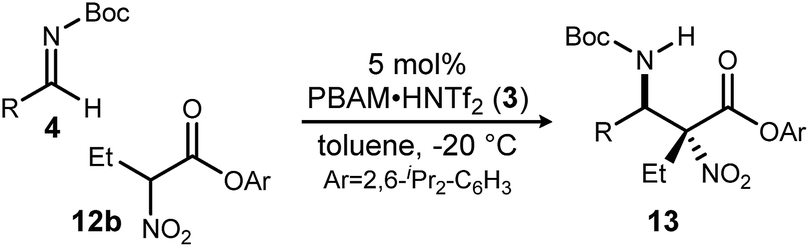
With these results in hand, α-nitro butanoate 10d/12b was employed as a standard pronucleophile to evaluate an electronically and sterically diverse group of aldimines in the reaction (Table 2). Electronically neutral aldimines (Table 2, entries 1, 4, 9, 10, and 13) resulted in good isolated yield (54–76%), high diastereoselection (12:1 → >20:1 dr) and high enantioselection (96–99% ee). Notably, sterically demanding 1-naphthyl (Table 2, entry 9) and para-phenyl benzaldimine (Table 2, entry 13) were tolerated well, with high stereoselection. Electron deficient aldimines were also competent electrophiles. Trifluoromethylphenyl-substituted imine 4t (Table 2, entry 12) afforded adduct 13t in 74% isolated yield with 15:1 dr and 97% ee. Both chloro- and bromo-substituted imines (Table 2, entries 2 and 3) afforded the corresponding adducts in good yield with excellent diastereoselection (>20:1 dr) and enantioselection (99% ee). Thiophenyl and pyridyl aldimines were equally amenable to addition (Table 2, entries 8 and 11). Electron-rich rings (Table 2, entries 5–7) afforded the aza-Henry adducts in good yields with notably lower diastereoselectivity, though enantioselectivity was generally maintained. The erosion of diastereoselection may be attributed to a less electrophilic azomethine, leading to a longer electrophile-nucleophile distance in the bond-forming step, or a diminished secondary interaction between the nitro and azomethine. Unfortunately, N-Boc ketimines exhibited their typical unreactive nature in this system, likely due to the severe steric congestion in the adducts, despite stirring at room temperature for 7 days. And while some product could be obtained using aliphatic N-Boc aldimines in exploratory experiments, selectivities were low.
|
|
|||||
|---|---|---|---|---|---|
| Entrya | R | 13 | drb | eec | Yieldd |
| a All reactions were 0.7 M in imine, used 1.1 equiv. of the α-nitro ester, and had a standard 48 h reaction time. b Diastereomer ratios measured using 1H NMR. c Enantiomeric ratios measured using HPLC and a chiral stationary phase. d Yields are those of isolated, analytically pure adduct. | |||||
| 1 | C6H5 | j | >20:1 |
96 | 76 |
| 2 | 4Cl–C6H4 | b | >20:1 |
99 | 66 |
| 3 | 4Br–C6H4 | k | >20:1 |
99 | 71 |
| 4 | 3Me–C6H4 | l | 12:1 |
97 | 71 |
| 5 | 3MeO–C6H4 | m | 5:1 |
96 | 71 |
| 6 | 4MeO–C6H4 | n | 5:1 |
78 | 68 |
| 7 | 2Furyl | o | 4:1 |
91 | 63 |
| 8 | 2Thiophene | p | >20:1 |
97 | 63 |
| 9 | 1Naphthyl | q | 15:1 |
99 | 54 |
| 10 | 2Naphthyl | r | >20:1 |
96 | 70 |
| 11 | 3Pyridyl | s | 9:1 |
96 | 48 |
| 12 | 4CF3–C6H4 | t | 15:1 |
97 | 74 |
| 13 | 4Ph–C6H4 | u | >20:1 |
99 | 73 |
In addition to absolute and relative stereochemical assignment by X-ray for 11d,12 the absolute stereochemistry of adduct 1426 was assigned via chemical correlation to known compound 15. (S,S)-15 was reported to have a rotation of +44. Synthetic 15 using catalyst 3 exhibited a measured rotation of −39. Therefore, the adducts produced by (R,R)-PBAM·HNTf2 have the configuration of (R,R) as depicted in Scheme 3.10
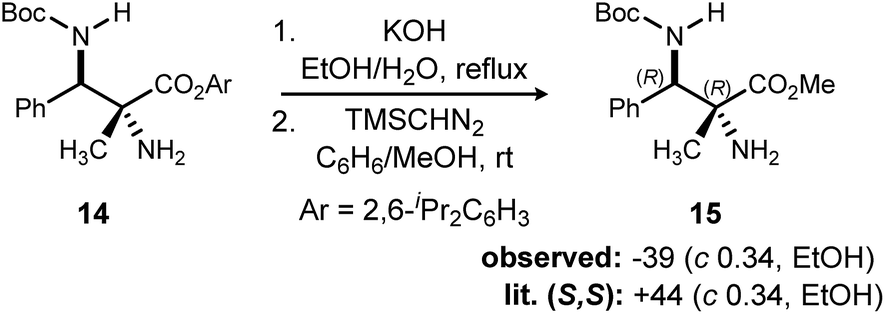 | ||
| Scheme 3 Determination of absolute and relative configuration by chemical correlation. | ||
Conclusions
In conclusion, we have developed the second enantioselective addition of α-alkyl α-nitro esters to imines using chiral proton catalysis, but with anti-diastereoselection. Taken together, these reactions are among the first highly selective hydrogen bond-catalyzed reactions exhibiting diastereodivergence. More remarkable is the use of a common catalyst design to reverse diastereoselection without compromise to enantioselection. We hypothesize that the key difference between 2 and 3 is the level of steric congestion in the binding pocket of the catalyst. Future studies will interrogate this hypothesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DJS is grateful for support from the D. Stanley and Ann T. Tarbell Endowment Fund and the Vanderbilt Institute of Chemical Biology. We are grateful to the National Institute of General Medical Sciences (NIH GM 084333) for financial support, and Dr Maren Pink (Indiana University Molecular Structure Center) for X-ray analysis of the original adducts.Notes and references
- A. E. Metz and M. C. Kozlowski, J. Org. Chem., 2015, 80, 1–7 CrossRef CAS PubMed.
- (a) K. Bera and I. N. N. Namboothiri, Asian J. Org. Chem., 2014, 3, 1234–1260 CrossRef CAS; (b) C. K. De, N. Mittal and D. Seidel, J. Am. Chem. Soc., 2011, 133, 16802–16805 CrossRef CAS PubMed.
- M. Goodman and S. Ro, in Burger's Medicinal Chemistry and Drug Discovery, ed. M. E. Wolff, Wiley, 1995, pp. 803–861 Search PubMed.
- (a) W. S. Saari, M. B. Freedman, R. D. Hartman, S. W. King, A. W. Raab, W. C. Randall, E. L. Engelhardt, R. Hirschmann and A. Rosegay, J. Med. Chem., 1978, 21, 746–753 CrossRef CAS PubMed; (b) W. S. Saari, W. Halczenko, D. W. Cochran, M. R. Dobrinska, W. C. Vincek, D. C. Titus, S. L. Gaul and C. S. Sweet, J. Med. Chem., 1984, 27, 713–717 CrossRef CAS PubMed; (c) J. J. Walsh, D. E. Metzler, D. Powell and R. A. Jacobson, J. Am. Chem. Soc., 1980, 102, 7136–7138 CrossRef CAS.
- T. Degenkolb, A. Berg, W. Gams, B. Schlegel and U. Grafe, J. Pept. Sci., 2003, 9, 666–678 CrossRef CAS PubMed.
- (a) A. Noble and J. C. Anderson, Chem. Rev., 2013, 113, 2887–2939 CrossRef CAS PubMed; (b) R. G. Arrayas and J. C. Carretero, Chem. Soc. Rev., 2009, 38, 1940–1948 RSC.
- K. R. Knudsen and K. A. Jorgensen, Org. Biomol. Chem., 2005, 3, 1362–1364 CAS.
- Z. Chen, H. Morimoto, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2008, 130, 2170–2171 CrossRef CAS PubMed.
- anti-Selective: X. X. Yan, Q. Peng, Q. Li, K. Zhang, J. Yao, X. L. Hou and Y. D. Wu, J. Am. Chem. Soc., 2008, 130, 14362–14363 CrossRef CAS PubMed.
- B. Han, W. Huang, Z. R. Xu and X. P. Dong, Chin. Chem. Lett., 2011, 22, 923–926 CrossRef CAS.
- syn-Diastereoselectivity reported by Wang was later corrected to anti (see p. 2902, ref. 6): X. X. Jiang, Y. F. Zhang, L. P. Wu, G. Zhang, X. Liu, H. L. Zhang, D. Fu and R. Wang, Adv. Synth. Catal., 2009, 351, 2096–2100 CrossRef CAS.
- A. Singh and J. N. Johnston, J. Am. Chem. Soc., 2008, 130, 5866–5867 CrossRef CAS PubMed.
- D. Uraguchi, K. Koshimoto and T. Ooi, J. Am. Chem. Soc., 2008, 130, 10878–10879 CrossRef CAS PubMed.
- S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 5627–5639 CrossRef CAS PubMed.
- L. Lin and X. Feng, Chem.–Eur. J., 2017, 23, 6464–6482 CrossRef CAS PubMed.
- M. Bihani and J. C. G. Zhao, Adv. Synth. Catal., 2017, 359, 534–575 CrossRef CAS.
- Diastereodivergence driven by the electronic character of a ligand: X. X. Yan, Q. Peng, Q. Li, K. Zhang, J. Yao, X. L. Hou and Y. D. Wu, J. Am. Chem. Soc., 2008, 130, 14362–14363 CrossRef CAS PubMed.
- Substrate control: J. Hernández-Toribio, R. G. Arrayás and J. C. Carretero, J. Am. Chem. Soc., 2008, 130, 16150–16151 CrossRef PubMed . (anti-selective), compare to ref. 23c (syn-selective).
- (a) B. Shen, D. M. Makley and J. N. Johnston, Nature, 2010, 465, 1027–1032 CrossRef CAS PubMed; (b) B. Shen and J. N. Johnston, Org. Lett., 2008, 10, 4397–4400 CrossRef CAS PubMed.
- T. A. Davis, J. C. Wilt and J. N. Johnston, J. Am. Chem. Soc., 2010, 132, 2880–2882 CrossRef CAS PubMed.
- syn-Selective α-iminyl glycine additions have been developed: (a) Z. L. Tao, A. Adele, X. Wu and L. Z. Gong, Chin. J. Chem., 2014, 32, 969–973 CrossRef CAS; (b) A. Cayuelas, L. Serrano, C. Nájera and J. M. Sansano, Tetrahedron: Asymmetry, 2014, 25, 1647–1653 CrossRef CAS . (without simultaneous high dr/ee).
- For syn-selectivity (but not enantioselective): L. Qiu, D. Wang, F. Lv, X. Guo, W. Hu, L. Yang and S. Liu, Tetrahedron, 2014, 70, 1471–1477 CrossRef CAS.
- syn-Selective Mannich reaction using glycine Schiff base: (a) T. Arai, A. Mishiro, E. Matsumura, A. Awata and M. Shirasugi, Chemistry, 2012, 18, 11219–11222 CrossRef CAS PubMed; (b) L. Bernardi, A. S. Gothelf, R. G. Hazell and K. A. Jorgensen, J. Org. Chem., 2003, 68, 2583–2591 CrossRef CAS PubMed; (c) J. Hernández-Toribio, R. Gómez Arrayás and J. C. Carretero, Chem.–Eur. J., 2010, 16, 1153–1157 CrossRef PubMed; (d) K. Imae, K. Shimizu, K. Ogata and S. Fukuzawa, J. Org. Chem., 2011, 76, 3604–3608 CrossRef CAS PubMed.
- (a) T. A. Davis, M. C. Dobish, K. E. Schwieter, A. C. Chun and J. N. Johnston, Org. Synth., 2012, 89, 380–393 CrossRef CAS PubMed; (b) T. A. Davis, M. W. Danneman and J. N. Johnston, Chem. Commun., 2012, 48, 5578–5580 RSC.
- The triflate salt afforded slightly inferior results; see Table 1, footnote e.
- See ESI† for preparative details.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc05176j |
| ‡ These diastereomers are separable using silica gel chromatography. |
| This journal is © The Royal Society of Chemistry 2018 |