DOI:
10.1039/C7SC05328B
(Edge Article)
Chem. Sci., 2018,
9, 2831-2841
Insertion, protonolysis and photolysis reactivity of a thorium monoalkyl amidinate complex†
Received
16th December 2017
, Accepted 9th February 2018
First published on 16th February 2018
Abstract
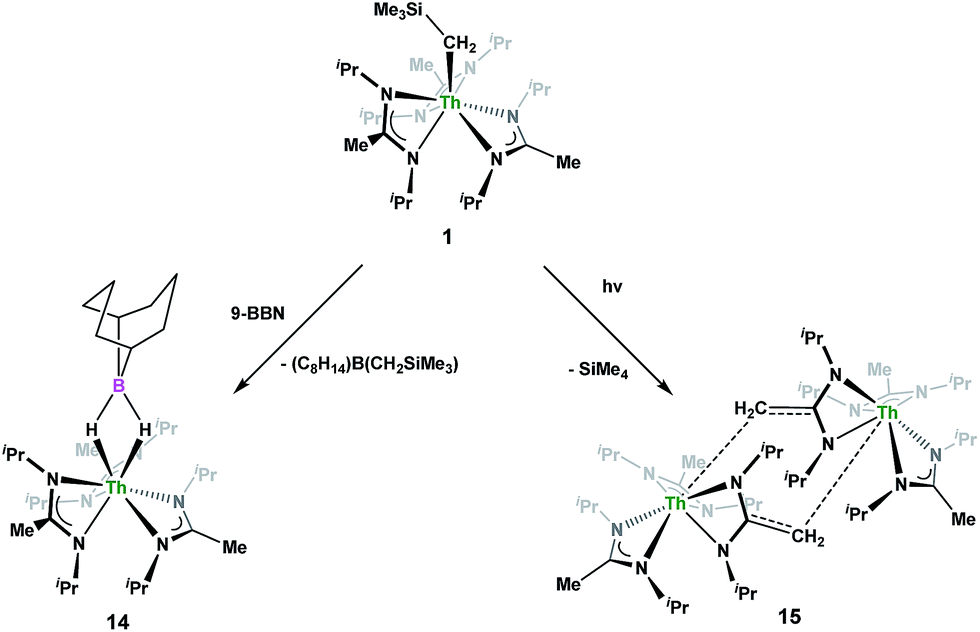
The reactivity of the thorium monoalkyl complex Th(CH2SiMe3)(BIMA)3 [1, BIMA = MeC(NiPr)2] with various small molecules is described. While steric congestion prohibits the insertion of N,N′-diisopropylcarbodiimide into the Th–C bond in 1, the first thorium tetrakis(amidinate) complex, Th(BIMA)4 (2), is synthesized via an alternative salt metathesis route. Insertion of p-tolyl azide leads to the triazenido complex Th[(p-tolyl)NNN(CH2SiMe3)-κ2N1,2](BIMA)3 (3), which then undergoes thermal decomposition to the amido species Th[(p-tolyl)N(SiMe3)](BIMA)3 (4). The reaction of 1 with 2,6-dimethylphenylisocyanide results in the thorium iminoacyl complex Th[η2-(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N)-2,6-Me2-C6H3(CH2SiMe3)](BIMA)3 (5), while the reaction with isoelectronic CO leads to the products Th[OC(CH2)SiMe3](BIMA)3 (6) and Th[OC(NiPr)C(CH2SiMe3)(C(Me)N(iPr))O-κ2O,O′](BIMA)2 (7), the latter being the result of CO coupling and insertion into an amidinate ligand. Protonolysis is achieved with several substrates, producing amido (9), aryloxide (10), phosphido (11a,b), acetylide (12), and cationic (13) complexes. Ligand exchange with 9-borabicyclo[3.3.1]nonane (9-BBN) results in formation of the thorium borohydride complex (BIMA)3Th(μ-H)2[B(C8H14)] (14). Complex 1 also reacts under photolytic conditions to eliminate SiMe4 and produce Th(BIMA)2(BIMA*) [15, BIMA* = (iPr)NC(CH2)N(iPr)], featuring a rare example of a dianionic amidinate ligand. Complexes 2, 3, 5, 6, 11a, and 12–15 were characterized by 1H and 13C{1H} NMR spectroscopy, FTIR, EA, melting point and X-ray crystallography. All other complexes were identified by one or more of these spectroscopic techniques.
N)-2,6-Me2-C6H3(CH2SiMe3)](BIMA)3 (5), while the reaction with isoelectronic CO leads to the products Th[OC(CH2)SiMe3](BIMA)3 (6) and Th[OC(NiPr)C(CH2SiMe3)(C(Me)N(iPr))O-κ2O,O′](BIMA)2 (7), the latter being the result of CO coupling and insertion into an amidinate ligand. Protonolysis is achieved with several substrates, producing amido (9), aryloxide (10), phosphido (11a,b), acetylide (12), and cationic (13) complexes. Ligand exchange with 9-borabicyclo[3.3.1]nonane (9-BBN) results in formation of the thorium borohydride complex (BIMA)3Th(μ-H)2[B(C8H14)] (14). Complex 1 also reacts under photolytic conditions to eliminate SiMe4 and produce Th(BIMA)2(BIMA*) [15, BIMA* = (iPr)NC(CH2)N(iPr)], featuring a rare example of a dianionic amidinate ligand. Complexes 2, 3, 5, 6, 11a, and 12–15 were characterized by 1H and 13C{1H} NMR spectroscopy, FTIR, EA, melting point and X-ray crystallography. All other complexes were identified by one or more of these spectroscopic techniques.
Introduction
The synthesis and reactivity of metal alkyl complexes have been a focus for organometallic chemists for decades due to their fundamental interest and relevance to catalytic and industrial processes. Studies of f-block alkyl complexes have been performed to a lesser extent, particularly those containing actinide metal centers. Actinide complexes display divergent coordination chemistry compared to the rest of the periodic table, and their large size combined with the accessibility of various oxidation states makes them particularly interesting for structural, electronic and reactivity studies.1–6 Much of the focus concerning organoactinide investigations has been devoted to uranium, due to its redox capabilities.4 Fewer studies have involved thorium, despite a fundamental question regarding whether thorium acts more as an actinide or group IV metal.7,8 Marks pioneered much of the work regarding thorium organometallic species bearing carbocyclic ancillary ligands (C5R5),9–15 and many groups have continued to explore the reactivity of these systems.16–20 Others have turned to non-carbocyclic ligands, in an attempt to investigate how complex stability and reactivity is affected by modifying the steric and electronic properties of the ancillary ligand framework.21–28 Having used a variety of non-carbocyclic ligand frameworks to stabilize and the explore the reactivity of both transition metal and actinide complexes,29–34 our group endeavored to expand these studies to thorium alkyl species. We recently reported on the synthesis of a thorium monoalkyl species utilizing a tris-amidinate ancillary framework, specifically Th(CH2SiMe3)(BIMA)3 (where BIMA = MeC(NiPr)2) (1), and its ability to insert chalcogen atoms to generate rare thorium chalcogenolate complexes.35 In addition to the chalcogen insertion reactivity, the increased electrophilicity of the metal center with respect to analogous Cp-based systems led to the rare C–H activation of trimethylamine N-oxide. Inspired by this result, we sought to investigate how the unique properties of this system would impact the reactivity of 1 with a variety of small molecules. Here we report on the reactivity of 1 with organic azides, isocyanide, CO, nitrile, 9-BBN, and various protic substrates, as well as the stability of 1 under photolytic conditions.
Results and discussion
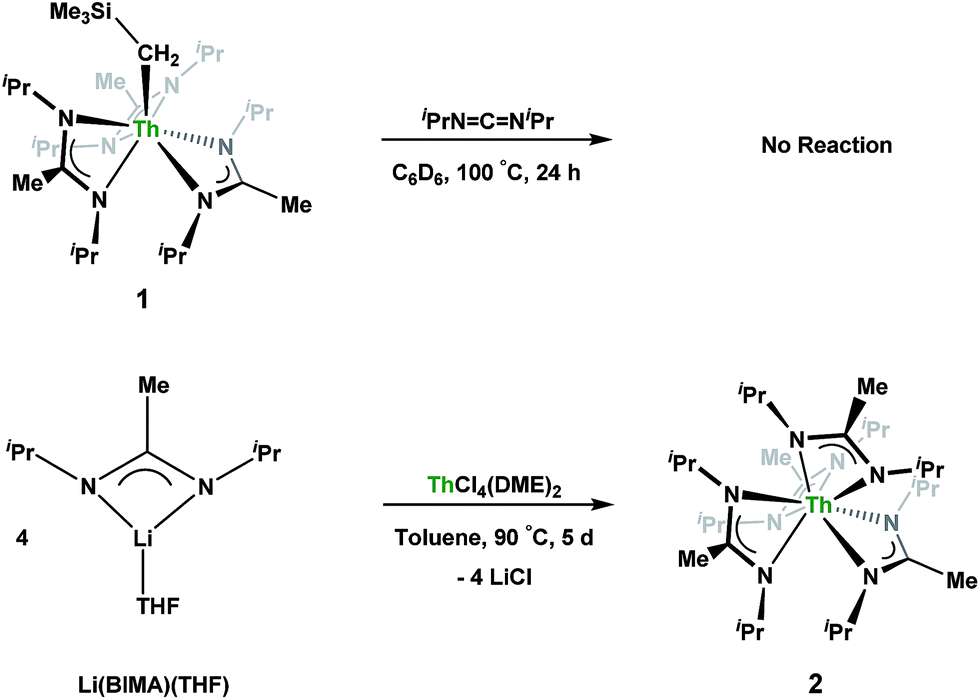
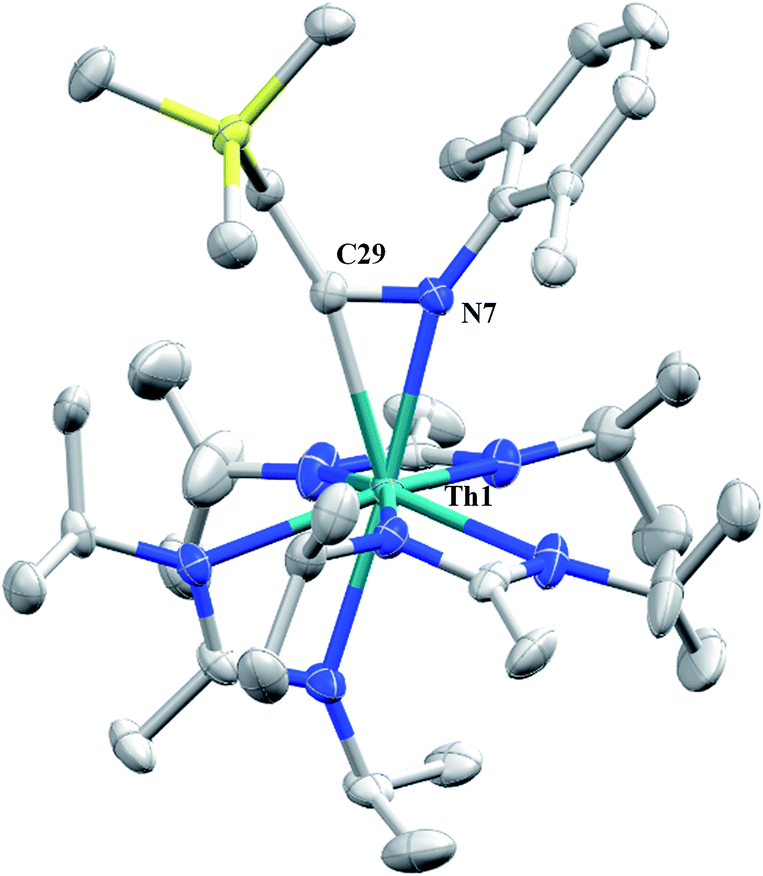
Having previously established the ability of 1 to undergo chalcogen atom insertion and generate unique thorium chalcogenolates,35 we next sought to examine potential insertion chemistry of this monoalkyl system with various small molecules. Evans has described the insertion of carbodiimides and organic azides into one alkyl moiety of 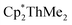 .16 With this in mind, we targeted the synthesis of a tetrakis(amidinate) complex through the reaction of 1 with N,N′-diisopropylcarbodiimide (Scheme 1). However, despite forcing conditions (100 °C), carbodiimide insertion into the Th–C bond was not observed, most likely a result of steric saturation around the metal center. Regardless, the alternative salt metathesis route using one equiv. of ThCl4(DME)2 (ref. 36) and 4.05 equiv. of Li(BIMA)(THF)37 and heating to 90 °C for 5 d afforded the desired homoleptic complex Th(BIMA)4 (2) as colourless crystals in 66% yield (Scheme 1). The 1H NMR spectrum of 2 reflects averaged C4-symmetry in solution, with one set of peaks observed for the equivalent amidinate ligands. The molecular structure of 2, determined by single-crystal X-ray diffraction studies, shows a pseudo-tetrahedral geometry of the amidinate ligands around the thorium center (Fig. 1). The Th–Namid bond lengths vary between 2.49 and 2.62 Å, a noticeably larger range than that seen in 1 (2.49–2.54 Å), indicating the significant steric congestion imposed by the isopropyl groups of the amidinate ligands. The structural parameters of 2 combined with the reaction conditions necessary to form 2 support the notion that insertion of carbodiimide into the Th–C bond was hindered by steric crowding, and indicate that related insertion reactions might be subject to this constraint. Due to the steric protection afforded by the tetrakis(amidinate) framework, and inspired by the work of Evans regarding low-valent thorium chemistry,38 we attempted the reduction of 2 with KC8 in the presence of 18-crown-6; nevertheless, no colour change was observed and only starting material was isolated.
.16 With this in mind, we targeted the synthesis of a tetrakis(amidinate) complex through the reaction of 1 with N,N′-diisopropylcarbodiimide (Scheme 1). However, despite forcing conditions (100 °C), carbodiimide insertion into the Th–C bond was not observed, most likely a result of steric saturation around the metal center. Regardless, the alternative salt metathesis route using one equiv. of ThCl4(DME)2 (ref. 36) and 4.05 equiv. of Li(BIMA)(THF)37 and heating to 90 °C for 5 d afforded the desired homoleptic complex Th(BIMA)4 (2) as colourless crystals in 66% yield (Scheme 1). The 1H NMR spectrum of 2 reflects averaged C4-symmetry in solution, with one set of peaks observed for the equivalent amidinate ligands. The molecular structure of 2, determined by single-crystal X-ray diffraction studies, shows a pseudo-tetrahedral geometry of the amidinate ligands around the thorium center (Fig. 1). The Th–Namid bond lengths vary between 2.49 and 2.62 Å, a noticeably larger range than that seen in 1 (2.49–2.54 Å), indicating the significant steric congestion imposed by the isopropyl groups of the amidinate ligands. The structural parameters of 2 combined with the reaction conditions necessary to form 2 support the notion that insertion of carbodiimide into the Th–C bond was hindered by steric crowding, and indicate that related insertion reactions might be subject to this constraint. Due to the steric protection afforded by the tetrakis(amidinate) framework, and inspired by the work of Evans regarding low-valent thorium chemistry,38 we attempted the reduction of 2 with KC8 in the presence of 18-crown-6; nevertheless, no colour change was observed and only starting material was isolated.
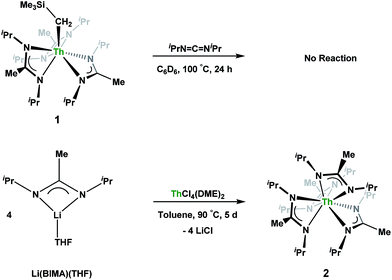 |
| | Scheme 1 Attempted insertion and salt metathesis routes to tetrakis(amidinate) species. | |
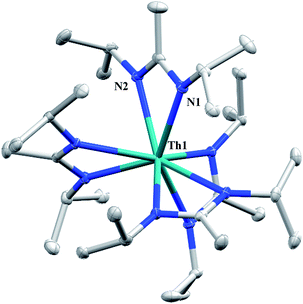 |
| | Fig. 1 Molecular structure of 2 (thermal ellipsoids drawn at the 50% probability level). Hydrogen atoms omitted for clarity. | |
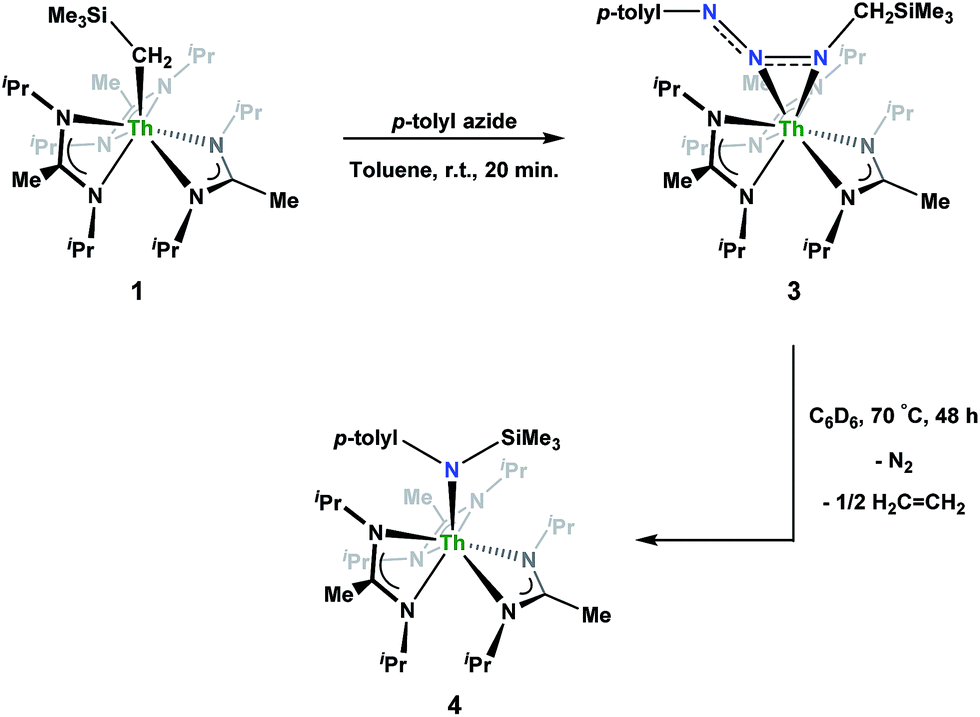
Reaction of 1 with an equivalent of p-tolyl azide resulted in insertion to form the triazenido complex Th[(p-tolyl)NNN(CH2SiMe3)-κ2N1,2](BIMA)3 (3) in 83% yield (Scheme 2). The 1H NMR spectrum displayed the diagnostic downfield shift of the methylene resonance which was also observed to result from chalcogen insertion.35 In the present case, we observed a shift from δ −0.08 in 1 to δ 3.99 in 3, alongside shifted amidinate resonances and the appearance of resonances attributable to the p-tolyl group. X-ray diffraction studies revealed a κ2N1,2 coordination mode of the triazenido moiety (Fig. 2), similar to that observed by Evans in the thorium metallocene system.16 The metrical parameters relating to the N3 fragment show the effect of delocalization, with bond lengths of 1.314(6) and 1.285(6) Å for N(7)–N(8) and N(8)–N(9), respectively, a difference of only ∼0.03 Å. In contrast, Evans' system that utilized adamantyl azide exhibited a more localized bonding for the analogous nitrogens, with bond lengths of 1.360(3) and 1.243(3) Å, a difference of ∼0.12 Å.16 The Th(1)–N(7) distance of 2.474(4) Å is ∼0.1 Å longer than that seen in the thorium metallocene system, while the Th(1)–N(8) distance of 2.597(4) Å is the same in both. This indicates that the Th(1)–N(7) interaction is best described as anionic, whereas Th(1)–N(8) is more dative. The difference between 3 and Evans' metallocene system is likely a combination of both steric and electronic effects. Insertion with tert-butyl azide was also achieved; however, a mixture of two species was always observed, with the major product slowly converting to the minor product in solution until a ratio of ∼4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was established. Heating did not alter this ratio, although elevated temperatures (100 °C) induced decomposition of the products. We postulate that the two products are both triazenido complexes that differ only in their coordination mode, with the κ2N1,2 and κ2N1,3 species present in solution. Exposing 3 to elevated temperatures in solution to see if a similar change in coordination mode would occur brought about a different result; 3 undergoes clean thermal decomposition to a new complex, with significantly shifted p-tolyl aromatic resonances and, most notably, no methylene resonance. Complete conversion was achieved within 48 h while heating at 70 °C. This new product was stable to further heating. Close inspection of the 1H NMR spectrum revealed a resonance attributable to ethylene. With this information we envisioned that 3 was losing diazomethane (N2CH2), which then decomposed to dinitrogen and ethylene,39 resulting in the thorium amido species Th[(p-tolyl)N(SiMe3)](BIMA)3 (4, Scheme 2). Ethylene formation may be the result of singlet methylene40 generation and coupling upon N2CH2 decomposition. In order to try and trap the transient singlet methylene, similar heating experiments were conducted in the presence of 2-butyne and 1,1-diphenylethylene and monitored by 1H NMR spectroscopy. Although it was difficult to unambiguously identify the trapped products (1,2-dimethylcyclopropene and 1,1-diphenylcyclopropane, respectively), the formation of ethylene was not seen with either trapping reagent, and a singlet at δ 1.13 was observed in the 1,1-diphenylethylene experiment, which we tentatively assign to 1,1-diphenylcyclopropane.41 We were able to confirm the identity of 4 as the thorium amido complex by X-ray diffraction studies (Fig. 2). To the best of our knowledge, this is the first example of clean thermal decomposition of an actinide triazenido complex to the corresponding amido species. Bart and co-workers have observed thermal instability in certain uranium triazenido species, which has led to intractable mixtures of products.42 However, the thermal decomposition of LtBuFe(η2-HNNNAd) (where LtBu = tert-butyl substituted-N,N′-diaryl-β-diketiminate, aryl = 2,6-iPr2-C6H3) to the corresponding primary amido species LtBuFeNHAd has been observed.43 This was rationalized based on the instability of free H2NNNR compounds with respect to loss of dinitrogen. The Th(1)–N(7) bond distance of 2.399(2) Å in 4 is very close to the Th–N bond distance of 2.389(2) Å observed by Walter and co-workers in [η5-1,2,4-(Me3C)3C5H2]2Th(Cl)-[N(p-tolyl)SiH2Ph], which has a similar silyl amide environment.44 The nitrogen atom of the amido exhibits a trigonal planar geometry (Σ∠ ≈ 360°), also consistent with Walter's complex. A series of NMR scale experiments revealed the thermal decomposition to be concentration dependent, with higher concentrations of 3 leading to the generation of the silyl amine (p-tolyl)NH(SiMe3) (as determined by 1H NMR spectroscopy) and a mixture of unknown species (see Fig. S7 in ESI†). The identity of these products, along with mechanistic studies regarding the formation of 4 from 3, is currently under investigation.
:1 was established. Heating did not alter this ratio, although elevated temperatures (100 °C) induced decomposition of the products. We postulate that the two products are both triazenido complexes that differ only in their coordination mode, with the κ2N1,2 and κ2N1,3 species present in solution. Exposing 3 to elevated temperatures in solution to see if a similar change in coordination mode would occur brought about a different result; 3 undergoes clean thermal decomposition to a new complex, with significantly shifted p-tolyl aromatic resonances and, most notably, no methylene resonance. Complete conversion was achieved within 48 h while heating at 70 °C. This new product was stable to further heating. Close inspection of the 1H NMR spectrum revealed a resonance attributable to ethylene. With this information we envisioned that 3 was losing diazomethane (N2CH2), which then decomposed to dinitrogen and ethylene,39 resulting in the thorium amido species Th[(p-tolyl)N(SiMe3)](BIMA)3 (4, Scheme 2). Ethylene formation may be the result of singlet methylene40 generation and coupling upon N2CH2 decomposition. In order to try and trap the transient singlet methylene, similar heating experiments were conducted in the presence of 2-butyne and 1,1-diphenylethylene and monitored by 1H NMR spectroscopy. Although it was difficult to unambiguously identify the trapped products (1,2-dimethylcyclopropene and 1,1-diphenylcyclopropane, respectively), the formation of ethylene was not seen with either trapping reagent, and a singlet at δ 1.13 was observed in the 1,1-diphenylethylene experiment, which we tentatively assign to 1,1-diphenylcyclopropane.41 We were able to confirm the identity of 4 as the thorium amido complex by X-ray diffraction studies (Fig. 2). To the best of our knowledge, this is the first example of clean thermal decomposition of an actinide triazenido complex to the corresponding amido species. Bart and co-workers have observed thermal instability in certain uranium triazenido species, which has led to intractable mixtures of products.42 However, the thermal decomposition of LtBuFe(η2-HNNNAd) (where LtBu = tert-butyl substituted-N,N′-diaryl-β-diketiminate, aryl = 2,6-iPr2-C6H3) to the corresponding primary amido species LtBuFeNHAd has been observed.43 This was rationalized based on the instability of free H2NNNR compounds with respect to loss of dinitrogen. The Th(1)–N(7) bond distance of 2.399(2) Å in 4 is very close to the Th–N bond distance of 2.389(2) Å observed by Walter and co-workers in [η5-1,2,4-(Me3C)3C5H2]2Th(Cl)-[N(p-tolyl)SiH2Ph], which has a similar silyl amide environment.44 The nitrogen atom of the amido exhibits a trigonal planar geometry (Σ∠ ≈ 360°), also consistent with Walter's complex. A series of NMR scale experiments revealed the thermal decomposition to be concentration dependent, with higher concentrations of 3 leading to the generation of the silyl amine (p-tolyl)NH(SiMe3) (as determined by 1H NMR spectroscopy) and a mixture of unknown species (see Fig. S7 in ESI†). The identity of these products, along with mechanistic studies regarding the formation of 4 from 3, is currently under investigation.
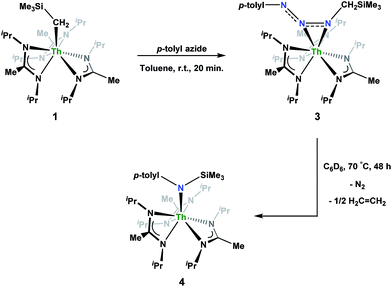 |
| | Scheme 2 Synthesis of 3 and thermal decomposition to 4. | |
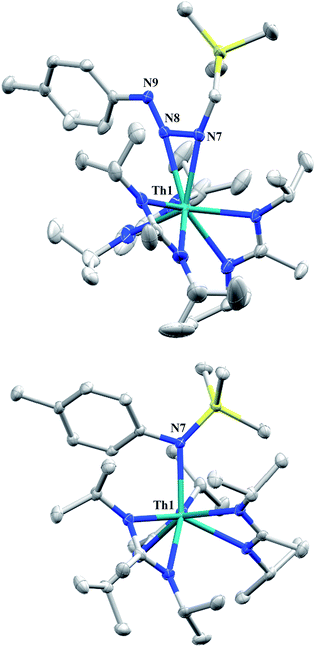 |
| | Fig. 2 Molecular structures of 3 (top) and 4 (bottom) (thermal ellipsoids drawn at the 50% probability level). Hydrogen atoms omitted for clarity. | |
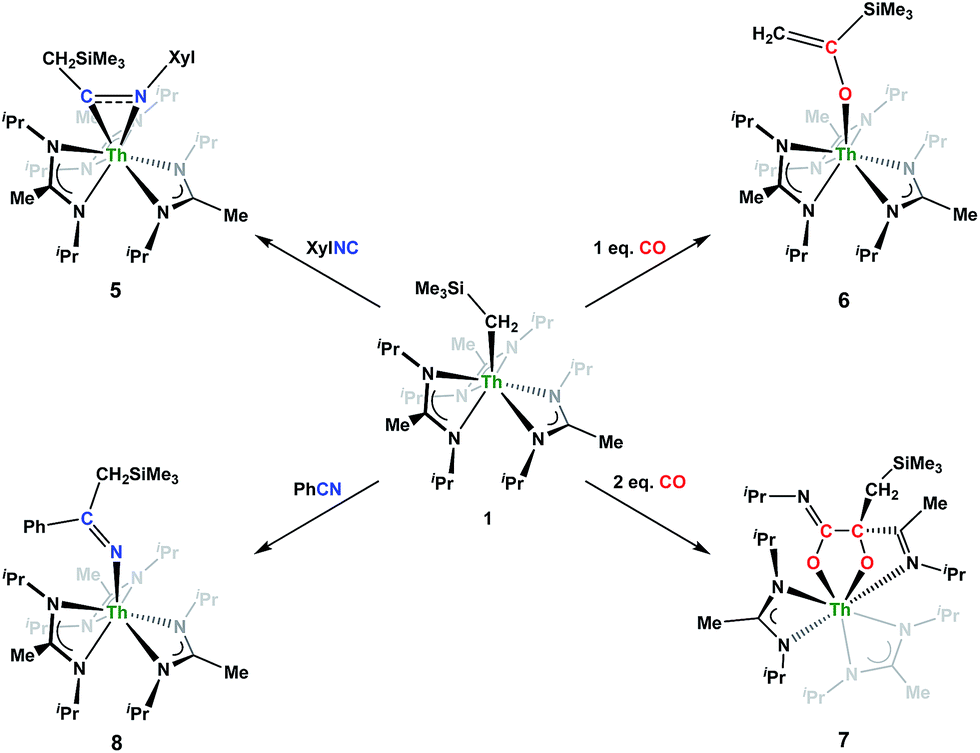
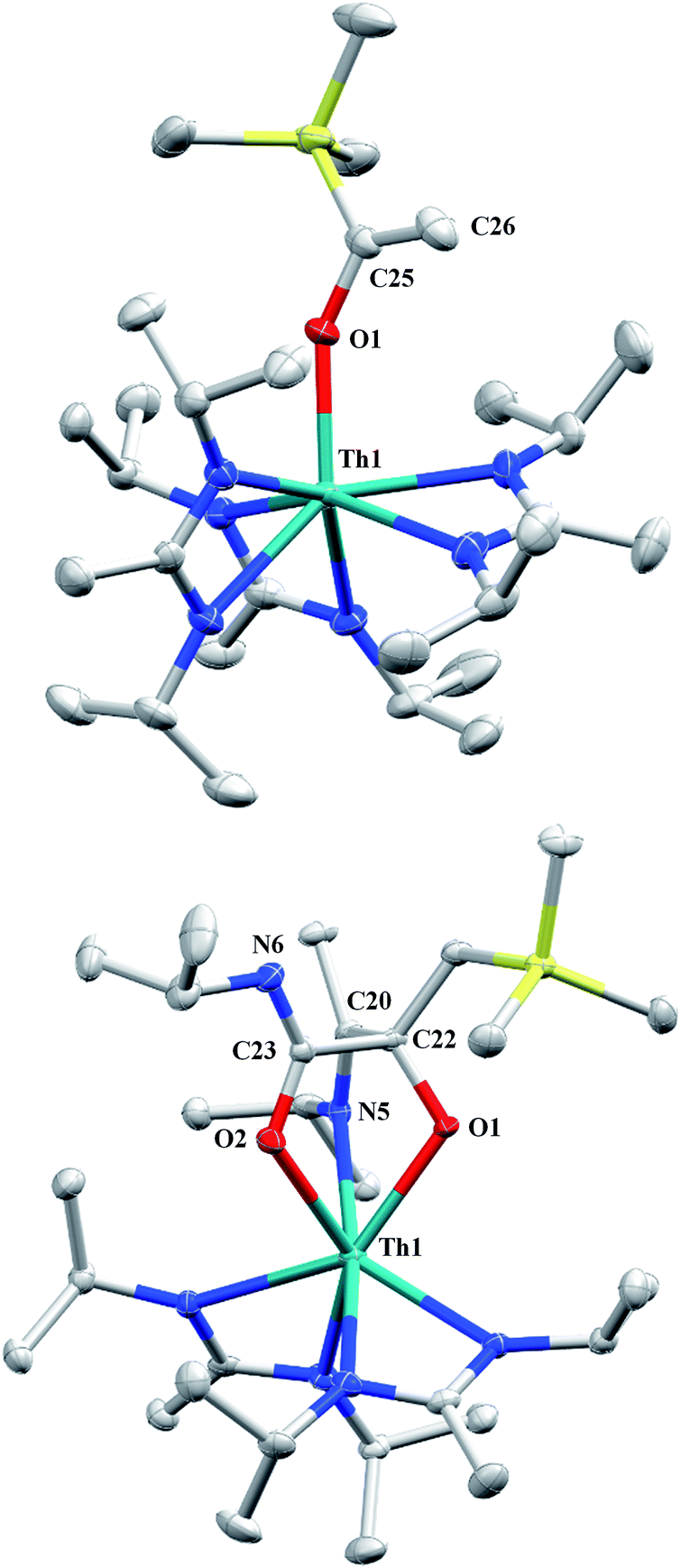
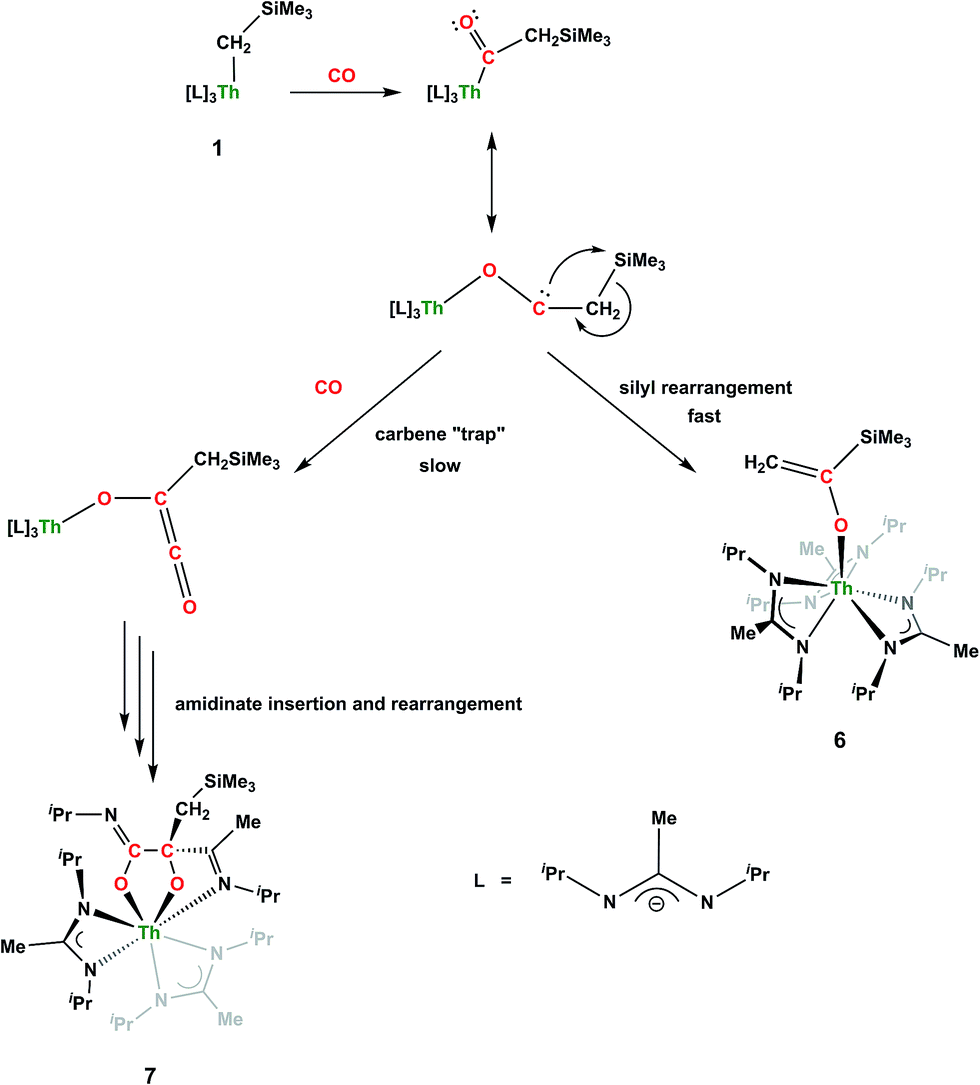
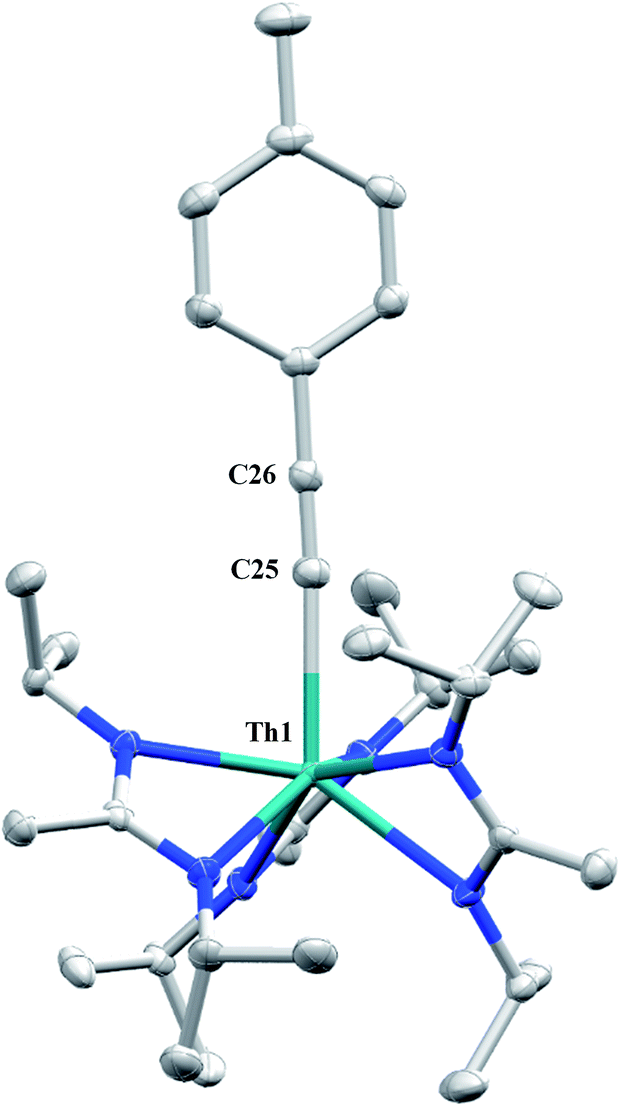
Although achieved with both transition metal45–48 and uranium49–52 species, isocyanide insertion into thorium alkyl bonds to form the corresponding η2-iminoacyl complexes has not yet been reported. While sterics precluded the insertion of N,N′-diisopropylcarbodiimide, isocyanide insertion was realized with one equivalent of 2,6-dimethylphenylisocyanide and moderate heating, resulting in Th[η2-(CN)-2,6-Me2-C6H3(CH2SiMe3)](BIMA)3 (5) as a colourless, crystalline solid in 81% yield (Scheme 3). To the best of our knowledge this is the first thorium η2-iminoacyl complex. Monitoring the reaction by 1H NMR spectroscopy confirms that this insertion proceeds slowly at room temperature (>95% conversion after 96 h), presumably due to the steric clash between the xylyl moiety and amidinate ligands. As expected, the diagnostic downfield shift of the methylene singlet to δ 2.85 indicated successful isocyanide insertion, along with new methyl and aromatic resonances corresponding to the xylyl group. X-ray diffraction studies revealed the η2-coordination mode of the imine moiety, with the N(7)–C(29) bond length of 1.299(4) Å falling in the range seen for other transition metal iminoacyl species (Fig. 3).45–52 The Th(1)–N(7) distance of 2.469(2) Å is noticeably shorter than that typically observed for a dative Th–N bond,53–56 while the Th(1)–C(29) bond length of 2.529(3) Å is in the range observed for other σ-bonded alkyl moieties.14,19,57 This insertion differs from that observed by Andersen and co-workers in their Th(CH2SiMe2NSiMe3)(NR2)2 (where R = SiMe3) complex, which undergoes insertion of tert-butyl isocyanide into the C–Si bond of the metallacycle to produce Th[(tBu)NC(CH2)SiMe2NSiMe3](NR2)2.58 Similar reactivity was observed with CO in Andersen's system, resulting in Th[(OC(CH2)SiMe2NSiMe3)](NR2)2. Exposing 1 to 1 atm of CO resulted in the formation of two products with similar solubilities in non-polar solvents, precluding their clean isolation and characterization, despite yields of ∼80% for the bulk mixture. The major species, as identified by 1H NMR spectroscopy, exhibited inequivalent methylene protons that are consistent with those observed in Th[(OC(CH2)SiMe2NSiMe3)](NR2)2 (see Fig. S10 in ESI†). This inequivalency would not be seen in the 1H NMR spectrum of the thorium acyl species generated by simple CO insertion into the Th–C bond; thus, we postulated that it was likely a similar insertion into the C–Si bond of 1 occurred (Scheme 3). This hypothesis was proven by X-ray diffraction studies, as a few X-ray quality crystals were isolated from a very concentrated pentane solution stored at −35 °C for 3 days, confirming the identity of the CO insertion as the enolate complex Th[OC(CH2)SiMe3](BIMA)3 (6) (Fig. 4). Complex 6 crystallized with two independent molecules in the asymmetric unit due to disorder in the enolate moiety; thus, the metrical parameters of only the nondisordered molecule will be discussed. The Th(1)–O(1) bond length of 2.216(2) Å is slightly longer than the Th–O bond length of 2.166(2) Å seen in Th(OCH2NMe2)(BIMA)3,35 while the C(25)–C(26) bond length of 1.338(5) Å is consistent with a carbon–carbon double bond, and the trigonal planar geometry of C25 (Σ∠ ≈ 360°) indicates sp2 hybridization. This type of reactivity has precedent in both uranium and thorium systems,52,58–61 and has been explained by initial CO insertion into the M–C bond to form the metal acyl, which then isomerizes to form a carbene-like intermediate that can then insert into the Si–C bond (Scheme 4). Alongside crystals of 6 were crystals of a different product, which we have tentatively assigned to the other resonances observed in the 1H NMR spectrum of the bulk material (see Fig. S10 in ESI†). X-ray diffraction studies revealed this product to be Th[OC(NiPr)C(CH2SiMe3)(C(Me)N(iPr))O-κ2O,O′](BIMA)2 (7), the result of reductive CO coupling and insertion into an amidinate ligand (Fig. 4). The Th(1)–O(1) and Th(1)–O(2) bond lengths of 2.220(2) and 2.290(2) Å, respectively, are slightly longer than that seen in 6 and Th(OCH2NMe2)(BIMA)3,35 while the bond length of 2.764(3) Å for Th(1)–N(5) is indicative of a dative interaction.53–56 A single bond length of 1.558(5) Å is observed for C(22)–C(23), whereas imine bonds are seen for C(23)–N(6) and C(20)–N(5) (1.266(4) and 1.282(4) Å, respectively). This type of CO coupling mimics the enediolate formation observed with various actinide bis-alkyl systems.59,62 Regarding the formation of 7, it seems unlikely that this product is obtained from the interaction of a molecule of CO with 6, due to the intact nature of the trimethylsilylmethyl alkyl fragment. Instead, it is likely that 7 results from “trapping” of the carbene-like intermediate, which precedes formation of 6, by another molecule of CO to form a transient ketene,63 and subsequent insertion and rearrangement steps lead to 7 as the final product (Scheme 4). Kinetically this intermolecular process is slower than the intramolecular attack by the carbene intermediate on the C–Si bond, producing 6 as the major product. Exposing a mixture of 6 and 7 to additional CO did not change the ratio of products observed, confirming that 7 is not generated from 6. In an attempt to avoid the formation of 7, the slow addition of 1 eq. of CO to a stirred hexanes solution of 1 was conducted, resulting in clean formation of 6 in 64% yield. Development of a synthetic strategy to produce 7 is currently underway. Looking to other small molecules, insertion reactivity with CO2 and CS2 did not proceed cleanly, yielding intractable mixtures.
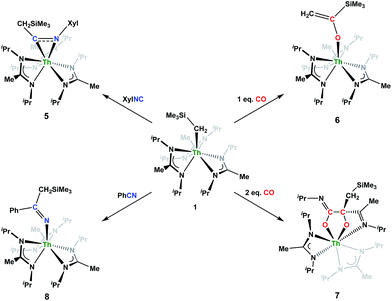 |
| | Scheme 3 Insertion reactivity of 1. | |
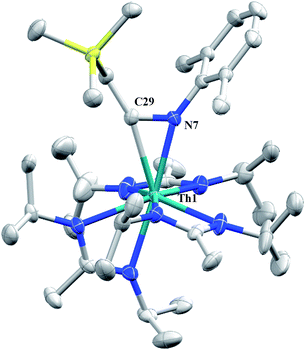 |
| | Fig. 3 Molecular structure of 5 (thermal ellipsoids drawn at the 50% probability level). Hydrogen atoms omitted for clarity. | |
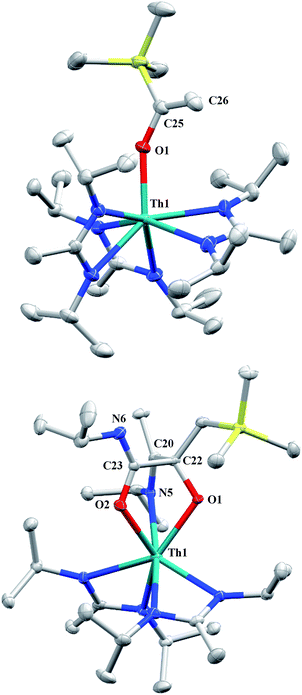 |
| | Fig. 4 Molecular structures of 6 (top) and 7 (bottom) (thermal ellipsoids drawn at the 50% probability level). Hydrogen atoms omitted for clarity. | |
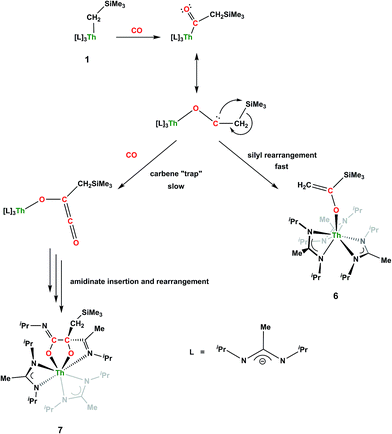 |
| | Scheme 4 Proposed pathways for the formation of 6 and 7. | |
Nitrile insertion was examined by the NMR-scale reaction of 1 and benzonitrile. The reaction was sufficiently complete (>95% conversion by 1H NMR spectroscopy) upon heating to 100 °C for 72 h, resulting in the ketimide complex Th[–NC(Ph)(CH2SiMe3)](BIMA)3 (8) (Scheme 3). The methylene singlet is seen downfield at δ 2.73, along with new resonances corresponding to the aromatic protons of the phenyl ring, as well as aromatic resonances corresponding to the cyclotrimerization product of benzonitrile, 2,4,6-triphenyl-1,3,5-triazine (see Fig. S13 in ESI†).64 The Lewis acid-catalysed cyclotrimerization of benzonitrile has been previously reported for lanthanide-imido species,64 although the active species in this transformation has not been identified.
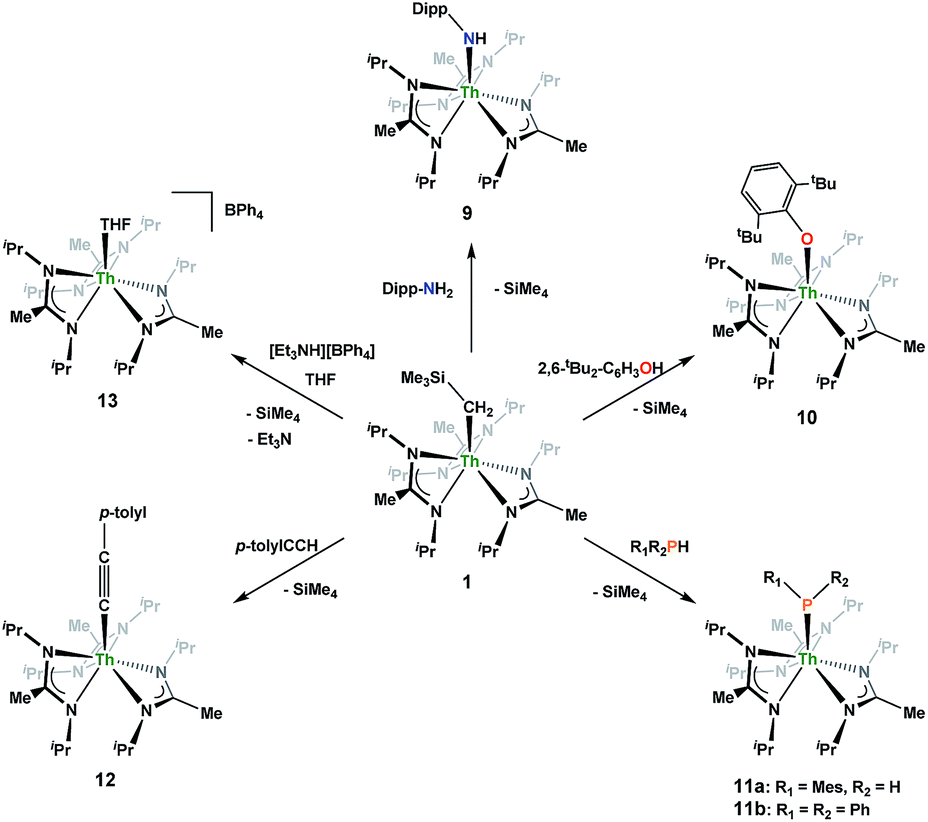
Looking to exploit the basic nature of the alkyl moiety of 1, protonolysis reactivity was explored with a variety of protic substrates, the results of which are summarized in Scheme 5. NMR-scale experiments were carried out with 1 and 2,6-diisopropylaniline, resulting in the primary amido species Th(NH-2,6-iPr2-C6H3)(BIMA)3 (9), as well as 1 and 2,6-di-tert-butylphenol, resulting in the aryloxide complex Th(O-2,6-tBu2-C6H3)(BIMA)3 (10), as determined by 1H NMR spectroscopy. While phenol addition and subsequent elimination of SiMe4 occurred within 12 h at room temperature, deprotonation of the aniline required extended reaction times at elevated temperatures before conversion to 9 was achieved. This can be rationalized on the basis of the higher pKa of the aniline (∼30 vs. 16.8 in DMSO),65,66 as the sterics imposed by the 2,6-di-tert-butylphenol are greater than that of 2,6-diisopropylaniline.
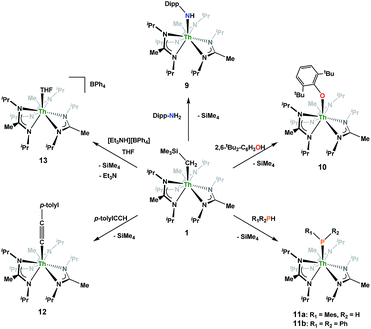 |
| | Scheme 5 Protonolysis reactivity of 1. | |
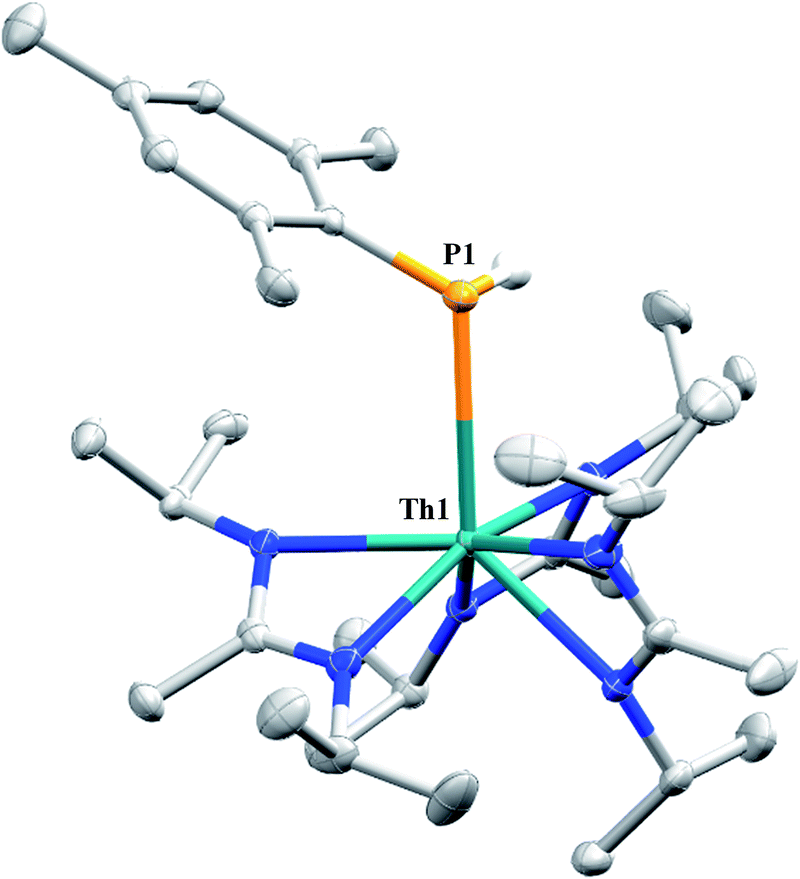
Similar NMR experiments were also performed with primary and secondary phosphines, namely mesitylphosphine and diphenylphosphine, resulting in the thorium phosphido complexes Th(PHMes)(BIMA)3 (11a) (Mes = 2,4,6-trimethylphenyl) and Th(PPh2)(BIMA)3 (11b), respectively, as determined by 1H and 31P NMR spectroscopy. Complex 11a exhibits a doublet at δ −45.4 in the 31P NMR spectrum with a 1JP,H coupling constant of 195 Hz; the corresponding doublet in the 1H NMR is observed at δ 3.41. In the 31P NMR spectrum of 11b a singlet is observed at δ 89.5 corresponding to the thorium phosphido, alongside peaks consistent with the dehydrocoupled product Ph2P–PPh2 (δ −14.9)67 and an unidentified phosphorus-containing species (δ 106). These 31P chemical shifts are in the range reported for other primary and secondary thorium phosphido species.68–70 The dehydrocoupling of phosphines has been reported with zirconium phosphido complexes under similar reaction conditions.71 Both complexes 11a and 11b required elevated temperatures and prolonged reaction times to reach completion. We sought a more scalable strategy to synthesize 11a without the need for harsh reaction conditions, as complex 11a has the potential to form a thorium phosphinidene via deprotonation of the phosphide ligand. With few examples of thorium phosphinidenes available, this would provide valuable information regarding metal–ligand multiple bonding between thorium and the heavier pnictogens.72–74 Salt metathesis between the previously reported ThCl(BIMA)3 (ref. 35) and KPHMes70 provided a more direct route to 11a as bright yellow crystals in 58% yield. The 13C{1H} NMR spectrum of 11a exhibits rare through-space coupling of the phosphorus atom and the isopropyl methyl carbons of the BIMA ligand (TSJP,C = 2.1 Hz), as well as the ortho-methyls of the mesitylene ring (TSJP,C = 9.6 Hz).75–78 This coupling is supported by the X-ray crystal structure of 11a, which displays a close proximity of the phosphorus atom to one of the BIMA isopropyl methyls (3.746(4) and 3.779(4) Å) and mesityl methyls (3.062(3) and 3.070(3) Å), along with significant pyramidalization at P (Σ∠ ≈ 311°) in the two independent molecules found in the asymmetric unit (Fig. 5). The orientation of the phosphorus lone pair toward these carbon atoms facilitates this spin–spin interaction. This is the first crystallographically characterized example of a thorium monophosphido species bearing a primary phosphide ligand; to date, only a handful of primary bis(phosphido)-thorium species have been isolated and characterized.70,72,79 The Th–P bond lengths seen in the two molecules in the asymmetric unit (3.0497(8) and 3.0404(8) Å) are ∼0.15 Å longer than those observed in the previously reported bis(phosphido)-thorium complexes. Attempts to deprotonate 11a to form the corresponding thorium phosphinidene have thus far proven unsuccessful.
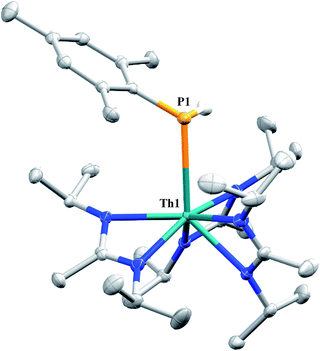 |
| | Fig. 5 Molecular structure of 11a (thermal ellipsoids drawn at the 50% probability level). Non-phosphorus-bound hydrogen atoms omitted for clarity. | |
The reaction of 1 with p-tolylacetylene proceeds cleanly, providing the thorium acetylide complex Th(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C-p-tolyl)(BIMA)3 (12) as colourless crystals in 95% yield. This new alkynyl species may serve as a useful starting material for future chemistry, as other thorium acetylide complexes have been shown to be active catalysts for the linear oligomerization of terminal alkynes.80 The 1H NMR spectrum displays a C3-symmetric amidinate environment along with resonances attributable to the p-tolyl group, while the 13C{1H} NMR spectrum features a downfield resonance of δ 189.2 corresponding to the thorium-bound carbon atom of the alkyne, consistent with other thorium and group IV acetylides.81–84 The IR spectrum exhibits a characteristic signal at 2061 cm−1 assigned to the CC stretch. X-ray diffraction studies revealed a near-linear Th–CC bond angle of 175.7(2)° and bond lengths of 2.542(2) and 1.219(3) Å for Th(1)–C(25) and C(25)–C(26), respectively (Fig. 6). The Th–C bond length in 12 is ∼0.05 Å longer than that of the few other thorium acetylide species to have been characterized crystallographically, namely [(L)Th(CCSiMe3)2] and [(L)Th(CCSiiPr3)2] (where L = trans-calix[2]benzene[2]pyrrolide), and Th(BcMes)2(CC-p-tolyl)2 (where BcMes = mesityl-substituted bis(NHC)borate, NHC = N-heterocyclic carbene), which were only recently reported.27,75
C-p-tolyl)(BIMA)3 (12) as colourless crystals in 95% yield. This new alkynyl species may serve as a useful starting material for future chemistry, as other thorium acetylide complexes have been shown to be active catalysts for the linear oligomerization of terminal alkynes.80 The 1H NMR spectrum displays a C3-symmetric amidinate environment along with resonances attributable to the p-tolyl group, while the 13C{1H} NMR spectrum features a downfield resonance of δ 189.2 corresponding to the thorium-bound carbon atom of the alkyne, consistent with other thorium and group IV acetylides.81–84 The IR spectrum exhibits a characteristic signal at 2061 cm−1 assigned to the CC stretch. X-ray diffraction studies revealed a near-linear Th–CC bond angle of 175.7(2)° and bond lengths of 2.542(2) and 1.219(3) Å for Th(1)–C(25) and C(25)–C(26), respectively (Fig. 6). The Th–C bond length in 12 is ∼0.05 Å longer than that of the few other thorium acetylide species to have been characterized crystallographically, namely [(L)Th(CCSiMe3)2] and [(L)Th(CCSiiPr3)2] (where L = trans-calix[2]benzene[2]pyrrolide), and Th(BcMes)2(CC-p-tolyl)2 (where BcMes = mesityl-substituted bis(NHC)borate, NHC = N-heterocyclic carbene), which were only recently reported.27,75
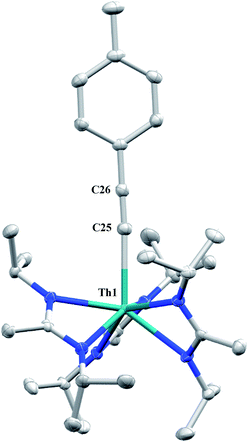 |
| | Fig. 6 Molecular structure of 12 (thermal ellipsoids drawn at the 50% probability level). Hydrogen atoms omitted for clarity. | |
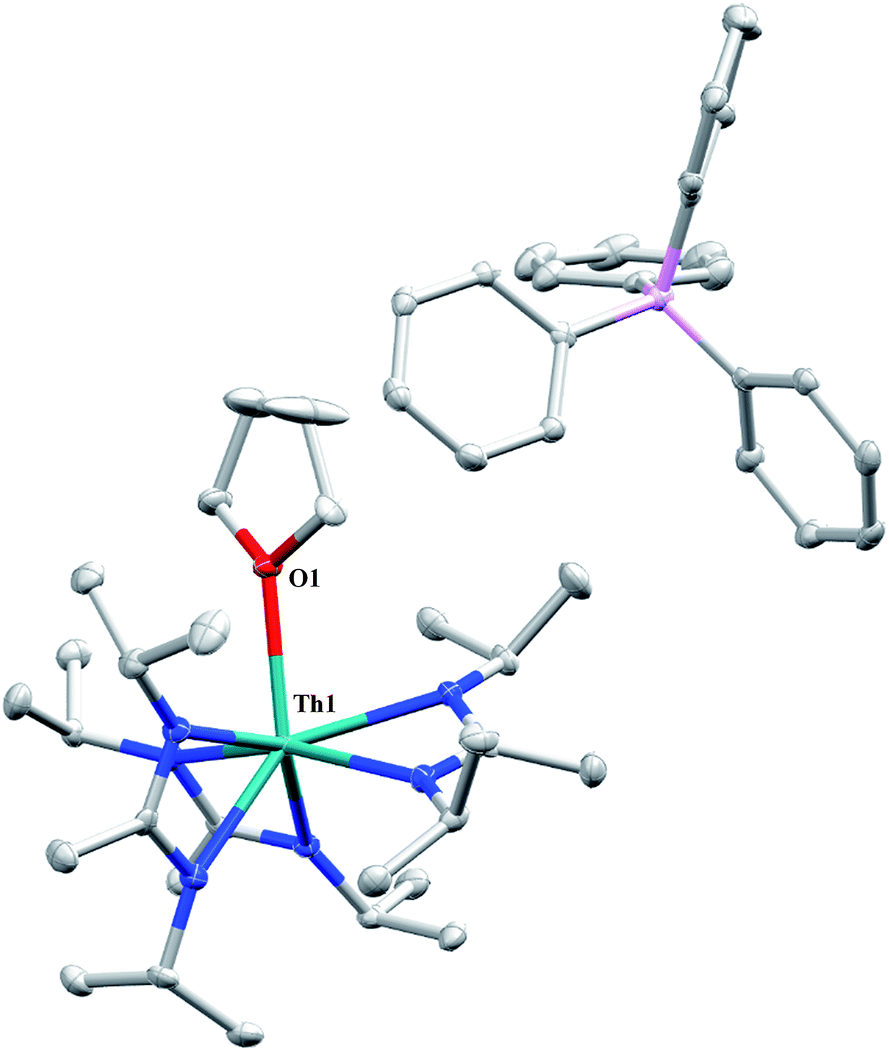
A cationic species was targeted as a potential precursor to a Th(III) amidinate complex, as Evans has shown that reduction of a mixed cyclopentadienyl amidinate thorium cation, namely {(C5Me5)2[iPrNC(Me)NiPr]Th}{BPh3Me}, can be achieved with KC8.85 Treatment of 1 with [Et3NH][BPh4]86 in THF led to the isolation of [Th(THF)(BIMA)3][BPh4] (13) as colourless crystals in 81% yield. The NH3 and SiMe4 byproducts were easily removed under vacuum upon workup, and X-ray diffraction studies show a well separated ion pair with one THF molecule coordinated to the thorium center and another co-crystallized in the lattice (Fig. 7). The Th–Namid bond lengths are noticeably shorter (2.45–2.48 Å) than that of 1 or 2, likely due to reduced steric congestion and higher electrophilicity of the metal center. The Th(1)–O(1) distance of 2.504(2) Å is in the range observed for other THF-bonded thorium complexes.87–90 The second equivalent of THF can be removed under high vacuum. The 1H NMR spectrum of dried 13 in CDCl3 displays equivalent amidinate resonances, the aromatic peaks of the BPh4 anion, and one set of resonances corresponding to coordinated THF. The complex displays appreciable stability in CDCl3, but begins to decompose after ∼24 h at room temperature. Attempted reduction of 13 with KC8 in THF led to the isolation of 2. No colour change was observed throughout the reaction. Increasing the sterics of the R groups on the amidinate nitrogens may help stabilize a tris-amidinate Th(III) complex, and work is currently ongoing to test this hypothesis. Attempts to utilize H2 as a protic substrate and form a thorium hydride complex were not successful.
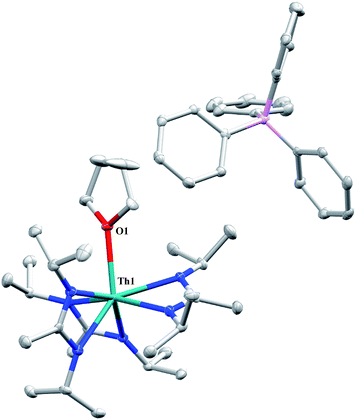 |
| | Fig. 7 Molecular structure of 13 (thermal ellipsoids drawn at the 50% probability level). Hydrogen atoms and THF solvent molecule omitted for clarity. | |
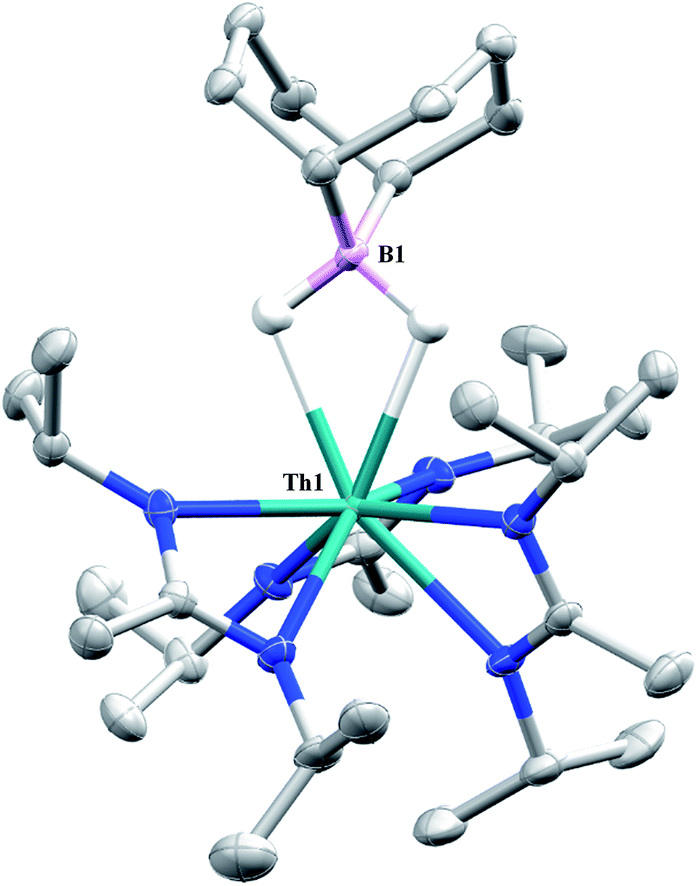
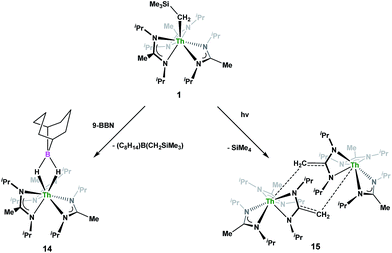
Complex 1 undergoes ligand exchange with one equivalent of 9-borabicyclo[3.3.1]nonane (9-BBN), affording (BIMA)3Th(μ-H)2[B(C8H14)] (14) alongside one equivalent of (C8H14)B(CH2SiMe3), as determined by 11B{1H} NMR spectroscopy (resonance observed at δ 84.3 in C6D6).91 Complex 14 was isolated in 66% crystalline yield after workup (Scheme 6). The thorium borohydride complex exhibits equivalent amidinate ligands in solution according to 1H NMR spectroscopy. Additionally, broad μ-H resonances and several multiplets corresponding to the C8H14 fragment are observed. The 11B{1H} NMR spectrum exhibits a single resonance at δ 4.90, which is in the range typically observed for boron hydrides.92 FTIR spectroscopy reveals a broad B–H stretch centered at 2021 cm−1. X-ray diffraction studies show an eight-coordinate thorium center bearing bridging hydrides bound to the 9-BBN moiety (Fig. 8). The hydrides were located in the Fourier difference map and refined isotropically. The Th(1)–B(1) distance of 2.952(9) Å is significantly longer than typically observed with other thorium complexes containing bridging borohydrides (2.49(6)–2.670(2) Å),93–97 but is within the range observed by Girolami and co-workers in the complexes [Th(H3BNMe2BH3)4] and [Th(H3BNMe2BH3)2(BH4)2], which exhibit Th–B distances between 2.848(9) to 3.193(5) Å.97 Complex 14 is surprisingly stable both under photolytic and elevated temperature conditions, with no decomposition or elimination of H2 observed.
 |
| | Scheme 6 Ligand exchange and photolytic reactivity of 1. | |
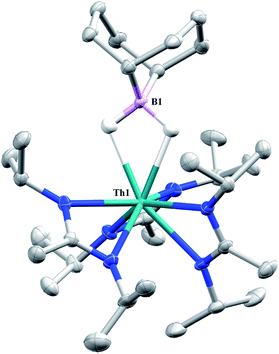 |
| | Fig. 8 Molecular structure of 14 (thermal ellipsoids drawn at the 50% probability level). Non-hydride hydrogen atoms omitted for clarity. | |
Storage of 1 at room temperature under dry nitrogen for several weeks led to slight discolouration of the crystalline solid. While the 1H NMR spectrum of this material showed very little change from that of pure 1, we decided to investigate the stability of 1 under photolytic conditions. The UV-Vis spectrum of 1 features an absorption with λmax = 295 nm, likely a ligand-based π–π* transition (see Fig. S31 in ESI†). Irradiation of 1 with UV-light centered at 253 nm in C6D6 and monitoring by 1H NMR spectroscopy showed elimination of SiMe4 alongside the production of a single new product, which displayed inequivalent amidinate ligands. Reaction times ranged from 24 h to 10 days, depending on the concentration of 1 in the sample (∼0.02–0.45 M), with higher concentrations taking longer. Optimization of the reaction conditions, by use of a quartz reaction vessel, cyclohexane-d12 and a xenon arc lamp, resulted in significantly reduced reaction times (∼2 h for ∼0.03 M solution). Removal of SiMe4 under vacuum and crystallization from toluene afforded Th(BIMA)2(BIMA*) (15) in 53% yield [BIMA* = (iPr)NC(CH2)N(iPr)]. Heating a solution of 1 at 100 °C for 24 h and monitoring by 1H NMR spectroscopy showed no decomposition of 1 or production of 15, eliminating the possibility that conversion of 1 to 15 was thermally-induced.
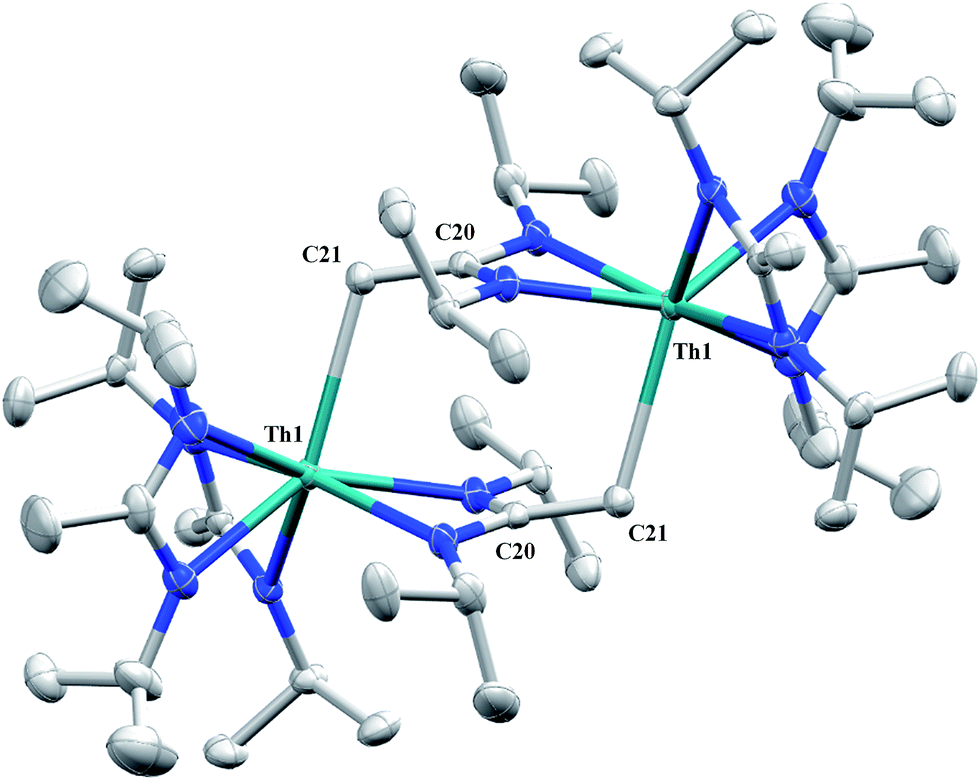
Irradiation of 1 results in C–H activation of a methyl group on an amidinate ligand by the –CH2SiMe3 moiety, eliminating SiMe4 and reducing the activated amidinate to a dianionic ligand (Scheme 6). Heterolytic bond cleavage of the Th–C bond resulting in a –CH2SiMe3 anion, which then attacks the methyl backbone of an amidinate ligand, is a possible explanation for the C–H activation and subsequent ligand reduction observed. This is a rare example of an amidinate dianion,64 and the first generated under photolytic conditions. The 1H NMR spectrum of 15 exhibits a set of equivalent amidinate resonances, alongside the resonances attributable to the amidinate dianion, specifically a 2H septet at δ 4.24, a 2H singlet at δ 3.44, and a 12H doublet at δ 1.53. The 6H singlet corresponding to the methyl groups of the monoanionic amidinates also appears at δ 1.53 (see Fig. S29 in ESI†). In the 13C{1H} NMR spectrum the terminal methylene carbon resonance is observed noticeably downfield at δ 53.3, shifted by ∼40 ppm from the amidinate methyls seen at δ 12.2, but further upfield than that typically seen with alkenes. The 1JC–H coupling constant of 157.6 Hz (as measured from the 13C satellites observed in the 1H NMR spectrum) is consistent with sp2 hybridization and similar to that observed for ethylene.98 The ipso-carbon of the amidinate dianion is shifted upfield to δ 154.2 from δ 172.8 as observed for the monoanionic amidinates. X-ray diffraction studies revealed a dimeric structure where the thorium centers are bridged by the methylene carbon of the dianionic amidinate ligand (Fig. 9). Complex 15 crystallizes in P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with the asymmetric unit containing only the monomer unit; the dimer is generated through inversion symmetry. The Th(1)–C(21) bond distance of 2.749(3) Å is significantly longer than the Th–C bond observed in 1 (2.557(3) Å), but shorter than the long Th–σ-alkyl bond distance of 2.875(9) Å observed by Liddle and co-workers in [Th{N(CH2CH2NSiMe2tBu)2(CH2CH2NSiMetBu-μ-CH2)}]2.90 The C(20)–C(21) bond distance of 1.438(4) is longer by ∼0.1 Å than typically observed for C–C double bonds, but this lengthening may be a result of delocalized electron density involved in the Th–C contact, reminiscent of a three-center two-electron bond. This is also manifested in the lack of planarity seen in the –N(CH2)CN– unit of the amidinate dianion.
with the asymmetric unit containing only the monomer unit; the dimer is generated through inversion symmetry. The Th(1)–C(21) bond distance of 2.749(3) Å is significantly longer than the Th–C bond observed in 1 (2.557(3) Å), but shorter than the long Th–σ-alkyl bond distance of 2.875(9) Å observed by Liddle and co-workers in [Th{N(CH2CH2NSiMe2tBu)2(CH2CH2NSiMetBu-μ-CH2)}]2.90 The C(20)–C(21) bond distance of 1.438(4) is longer by ∼0.1 Å than typically observed for C–C double bonds, but this lengthening may be a result of delocalized electron density involved in the Th–C contact, reminiscent of a three-center two-electron bond. This is also manifested in the lack of planarity seen in the –N(CH2)CN– unit of the amidinate dianion.
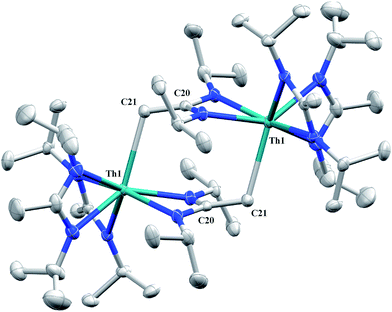 |
| | Fig. 9 Molecular structure of 15 (thermal ellipsoids drawn at the 50% probability level). | |
Conclusions
The amidinate-supported thorium monoalkyl complex 1 exhibits a variety of reactivity with small molecules, including insertion, protonolysis and photolysis. The insertion of p-tolyl azide lead to the thorium triazenido complex 3 which undergoes clean thermal decomposition at low concentrations to the corresponding amido complex 4, through the loss of the unstable “N2CH2” fragment. This is the first example of an actinide complex undergoing this rare transformation. Insertion of xylyl isocyanide results in the first crystallographically characterized thorium iminoacyl complex. This insertion reactivity differs from that observed with CO, which instead results in the corresponding enolate species 6 upon CO insertion and rearrangement, as well as the unique double CO insertion and amidinate cleavage product 7. The utility of the alkyl moiety as an internal base was demonstrated with a variety of protic substrates, with the thorium phosphido complex 11a generated via both protonolysis and salt metathesis routes, the latter providing a more scalable option for the synthesis of 11a. The photolytic elimination of SiMe4 concomitant with the reduction of an amidinate ligand to form complex 15 is unprecedented reactivity with amidinate-supported metal complexes, and a rare example of a complex bearing a dianionic amidinate ligand. Mechanistic and reactivity investigations of several of the complexes reported are currently ongoing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Director, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences Heavy Element Chemistry Program of the U.S. Department of Energy (DOE) at LBNL under Contract No. DE-AC02-05CH11231. We would like to acknowledge the NIH shared instrumentation grant S10-RR027172 for use in X-ray diffraction studies. N. S. S. acknowledges the Department of Energy Nuclear Energy University Programs for a graduate research fellowship. N. S. S. gratefully acknowledges Ms M. Garner, Mr M. Boreen and Mr T. Lohrey for their valuable discussions, and Mr P. Smith for assistance with the xenon arc lamp experiment.
References
-
C. J. Burns and M. S. Eisen, in The Chemistry of the Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein and J. Fuge, Springer, Berlin, Heidelberg, 2006, vol. 5 Search PubMed.
- T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC.
- M. B. Jones and A. J. Gaunt, Chem. Rev., 2013, 113, 1137–1198 CrossRef CAS PubMed.
- S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 2–40 CrossRef PubMed.
- M. Ephritikhine, Organometallics, 2013, 32, 2464–2488 CrossRef CAS.
- A. R. Fox, S. C. Bart, K. Meyer and C. C. Cummins, Nature, 2008, 455, 341–349 CrossRef CAS PubMed.
- K. C. Jantunen, B. L. Scott and J. L. Kiplinger, J. Alloys Compd., 2007, 444, 363–368 CrossRef.
- A. Yahia and L. Maron, Organometallics, 2009, 28, 672–679 CrossRef CAS.
- T. J. Marks and W. A. Wachter, J. Am. Chem. Soc., 1976, 98, 703–710 CrossRef CAS.
- D. G. Kalina, T. J. Marks and W. A. Wachter, J. Am. Chem. Soc., 1977, 99, 3877–3879 CrossRef CAS.
- J. W. Bruno, D. G. Kalina, E. A. Mintz and T. J. Marks, J. Am. Chem. Soc., 1982, 104, 1860–1869 CrossRef CAS.
- J. W. Bruno, T. J. Marks and V. W. Day, J. Am. Chem. Soc., 1982, 104, 7357–7360 CrossRef CAS.
- K. G. Moloy and T. J. Marks, J. Am. Chem. Soc., 1984, 106, 7051–7064 CrossRef CAS.
- J. W. Bruno, G. M. Smith, T. J. Marks, C. K. Fair, A. J. Schultz and J. M. Williams, J. Am. Chem. Soc., 1986, 108, 40–56 CrossRef CAS.
- K. G. Moloy, P. J. Fagan, J. M. Manriquez and T. J. Marks, J. Am. Chem. Soc., 1986, 108, 56–67 CrossRef CAS.
- W. J. Evans, J. R. Walensky, J. W. Ziller and A. L. Rheingold, Organometallics, 2009, 28, 3350–3357 CrossRef CAS.
- K. C. Jantunen, C. J. Burns, I. Castro-Rodriguez, R. E. Da Re, J. T. Golden, D. E. Morris, B. L. Scott, F. L. Taw and J. L. Kiplinger, Organometallics, 2004, 23, 4682–4692 CrossRef CAS.
- J. A. Pool, B. L. Scott and J. L. Kiplinger, J. Am. Chem. Soc., 2005, 127, 1338–1339 CrossRef CAS PubMed.
- R. J. Butcher, D. L. Clark, S. K. Grumbine, B. L. Scott and J. G. Watkin, Organometallics, 1996, 15, 1488–1496 CrossRef CAS.
- E. J. Schelter, P. Yang, B. L. Scott, R. E. Da Re, K. C. Jantunen, R. L. Martin, P. J. Hay, D. E. Morris and J. L. Kiplinger, J. Am. Chem. Soc., 2007, 129, 5139–5152 CrossRef CAS PubMed.
- C. A. Cruz, D. J. H. Emslie, L. E. Harrington, J. F. Britten and C. M. Robertson, Organometallics, 2007, 26, 692–701 CrossRef CAS.
- C. A. Cruz, D. J. H. Emslie, L. E. Harrington and J. F. Britten, Organometallics, 2008, 27, 15–17 CrossRef CAS.
- C. A. Cruz, D. J. H. Emslie, C. M. Robertson, L. E. Harrington, H. A. Jenkins and J. F. Britten, Organometallics, 2009, 28, 1891–1899 CrossRef CAS.
- B. M. Gardner, P. A. Cleaves, C. E. Kefalidis, J. Fang, L. Maron, W. Lewis, A. J. Blake and S. T. Liddle, Chem. Sci., 2014, 5, 2489–2497 RSC.
- L. A. Seaman, J. R. Walensky, G. Wu and T. W. Hayton, Inorg. Chem., 2013, 52, 3556–3564 CrossRef CAS PubMed.
- A. C. Behrle, A. J. Myers, P. Rungthanaphatsophon, W. W. Lukens, C. L. Barnes and J. R. Walensky, Chem. Commun., 2016, 52, 14373–14375 RSC.
- M. Suvova, K. T. P. O'Brien, J. H. Farnaby, J. B. Love, N. Kaltsoyannis and P. L. Arnold, Organometallics, 2017, 36, 4669–4681 CrossRef CAS.
- E. Mora, L. Maria, B. Biswas, C. Camp, I. C. Santos, J. Pécaut, A. Cruz, J. M. Carretas, J. Marçalo and M. Mazzanti, Organometallics, 2013, 32, 1409–1422 CrossRef CAS.
- T. D. Lohrey, R. G. Bergman and J. Arnold, Angew. Chem., Int. Ed., 2017, 56, 14241–14245 CrossRef CAS PubMed.
- S. Hohloch, M. E. Garner, B. F. Parker and J. Arnold, Dalton Trans., 2017, 46, 13768–13782 RSC.
- J. A. Ziegler, H. L. Buckley and J. Arnold, Dalton Trans., 2017, 46, 780–785 RSC.
- M. E. Garner, S. Hohloch, L. Maron and J. Arnold, Organometallics, 2016, 35, 2915–2922 CrossRef CAS.
- C. Camp, N. Settineri, J. Lefevre, A. R. Jupp, J. M. Goicoechea, L. Maron and J. Arnold, Chem. Sci., 2015, 6, 6379–6384 RSC.
- M. A. Boreen, B. F. Parker, T. D. Lohrey and J. Arnold, J. Am. Chem. Soc., 2016, 138, 15865–15868 CrossRef CAS PubMed.
- N. S. Settineri, M. E. Garner and J. Arnold, J. Am. Chem. Soc., 2017, 139, 6261–6269 CrossRef PubMed.
- T. Cantat, B. L. Scott and J. L. Kiplinger, Chem. Commun., 2010, 46, 919–921 RSC.
- S. Hao, S. Gambarotta, C. Bensimon and J. J. H. Edema, Inorg. Chim. Acta, 1993, 213, 65–74 CrossRef CAS.
- R. R. Langeslay, M. E. Fieser, J. W. Ziller, F. Furcher and W. J. Evans, Chem. Sci., 2015, 6, 517–521 RSC.
- T. Kurogi, M. V. Mane, S. Zheng, P. J. Carroll, M. Baik and D. J. Mindiola, Angew. Chem., Int. Ed., 2018, 57, 1978–1981 CrossRef CAS PubMed.
- R. Hoffmann, R. Gleiter and F. B. Mallory, J. Am. Chem. Soc., 1970, 92, 1460–1466 CrossRef CAS.
- R. Waterman and G. L. Hillhouse, J. Am. Chem. Soc., 2003, 125, 13350–13351 CrossRef CAS PubMed.
- S. J. Kraft, P. E. Fanwick and S. C. Bart, Organometallics, 2013, 32, 3279–3285 CrossRef CAS.
- Y. Yu, A. R. Sadique, J. M. Smith, T. R. Dugan, R. E. Cowley, W. W. Brennessel, C. J. Flaschenriem, E. Bill, T. R. Cundari and P. L. Holland, J. Am. Chem. Soc., 2008, 130, 6624–6638 CrossRef CAS PubMed.
- E. Zhou, W. Ren, G. Hou, G. Zi, D.-C. Fang and M. D. Walter, Organometallics, 2015, 34, 3637–3647 CrossRef CAS.
- M. G. Thorn, J. Lee, P. E. Fanwick and I. P. Rothwell, J. Chem. Soc., Dalton Trans., 2002, 17, 3398–3405 RSC.
- M. Nechayev, T. L. Gianetti, R. G. Bergman and J. Arnold, Dalton Trans., 2015, 44, 19494–19500 RSC.
- F. Amor, A. Butt, K. E. du Plooy, T. P. Spaniol and J. Okuda, Organometallics, 1998, 17, 5836–5849 CrossRef CAS.
- R. Fandos, A. Otero, A. M. Rodriguez and S. Suizo, Organometallics, 2012, 31, 1849–1856 CrossRef CAS.
- W. J. Evans, M. K. Takase, J. W. Ziller and A. L. Rheingold, Organometallics, 2009, 28, 5802–5808 CrossRef CAS.
- N. A. Siladke, J. W. Ziller and W. J. Evans, J. Am. Chem. Soc., 2011, 133, 3507–3516 CrossRef CAS PubMed.
- W. J. Evans, J. R. Walensky and J. W. Ziller, Organometallics, 2010, 29, 945–950 CrossRef CAS.
- W. J. Evans, N. A. Siladke and J. W. Ziller, Chem.–Eur. J., 2010, 16, 796–800 CrossRef CAS PubMed.
- W. Ren, G. Zi, D.-C. Fang and M. D. Walter, J. Am. Chem. Soc., 2011, 133, 13183–13196 CrossRef CAS PubMed.
- D. M. Barnhart, D. L. Clark, J. C. Gordon, J. C. Huffman and J. G. Watkin, Inorg. Chem., 1994, 33, 3939–3944 CrossRef CAS.
- J. L. Kiplinger, B. L. Scott, E. J. Schelter and J. A. P. D. Tournear, J. Alloys Compd., 2007, 444, 477–482 CrossRef.
- D. L. Clark and J. G. Watkin, Inorg. Chem., 1993, 32, 1766–1772 CrossRef CAS.
- C. M. Fendrick, E. A. Mintz, L. D. Schertz and T. J. Marks, Organometallics, 1984, 3, 819–821 CrossRef CAS.
- S. J. Simpson and R. A. Andersen, J. Am. Chem. Soc., 1981, 103, 4063–4066 CrossRef CAS.
- J. M. Manriquez, P. J. Fagan, T. J. Marks, C. S. Day and V. W. Day, J. Am. Chem. Soc., 1978, 100, 7112–7114 CrossRef CAS.
- O. Bénaud, J.-C. Berthet, P. Thuéry and M. Ephritikhine, Inorg. Chem., 2010, 49, 8117–8130 CrossRef PubMed.
- P. L. Arnold, Z. R. Turner, A. I. Germeroth, I. J. Casely, G. S. Nichol, R. Bellabarba and R. P. Tooze, Dalton Trans., 2013, 42, 1333–1337 RSC.
- K. Tatsumi, A. Nakamura, P. Hofmann, R. Hoffmann, K. G. Moloy and T. J. Marks, J. Am. Chem. Soc., 1986, 108, 4467–4476 CrossRef CAS.
- N. S. Radu, M. P. Engeler, C. P. Gerlach, T. D. Tilley and A. L. Rheingold, J. Am. Chem. Soc., 1995, 117, 3621–3622 CrossRef CAS.
- D. Cui, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2005, 44, 959–962 CrossRef CAS PubMed.
- F. G. Bordwell and D. J. Algrim, J. Am. Chem. Soc., 1988, 110, 2964–2968 CrossRef CAS.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- O. T. Beachley Jr, D. J. MacRae and A. Y. Kovalevsky, Organometallics, 2003, 22, 1690–1695 CrossRef.
- D. A. Wrobleski, R. R. Ryan, H. J. Wasserman, K. V. Salazar, R. T. Paine and D. C. Moody, Organometallics, 1986, 5, 90–94 CrossRef CAS.
- S. W. Hall, J. C. Huffman, M. M. Miller, L. R. Avens, C. J. Burns, D. S. J. Arney, A. F. England and A. P. Sattelberger, Organometallics, 1993, 12, 752–758 CrossRef CAS.
- M. E. Garner and J. Arnold, Organometallics, 2017, 36, 4511–4514 CrossRef CAS.
- R. Waterman, Organometallics, 2007, 26, 2492–2494 CrossRef CAS.
- A. C. Behrle, L. Castro, L. Maron and J. R. Walensky, J. Am. Chem. Soc., 2015, 137, 14846–14849 CrossRef CAS PubMed.
- E. P. Wildman, G. Balázs, A. J. Wooles, M. Scheer and S. T. Liddle, Nat. Commun., 2016, 7, 12884–12895 CrossRef CAS PubMed.
- M. R. Duttera, V. W. Day and T. J. Marks, J. Am. Chem. Soc., 1984, 106, 2907–2912 CrossRef CAS.
- M. E. Garner, B. F. Parker, S. Hohloch, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2017, 139, 12935–12938 CrossRef CAS PubMed.
- J.-C. Hierso, V. V. Ivanov, R. Amardeil, P. Richard and P. Meunier, Chem. Lett., 2004, 33, 1296–1297 CrossRef CAS.
- A. Marchenko, A. Hurieva, H. Koidan, V. Rampazzi, H. Cattey, N. Pirio, A. N. Kostyuk and J.-C. Hierso, Organometallics, 2012, 31, 5986–5989 CrossRef CAS.
- J.-C. Hierso, Chem. Rev., 2014, 114, 4838–4867 CrossRef CAS PubMed.
- A. C. Behrle and J. R. Walensky, Dalton Trans., 2016, 45, 10042–10049 RSC.
- A. Haskel, T. Straub, A. K. Dash and M. S. Eisen, J. Am. Chem. Soc., 1999, 121, 3014–3024 CrossRef CAS.
- A. K. Dash, J. Q. Wang and M. S. Eisen, Organometallics, 1999, 18, 4724–4741 CrossRef CAS.
- I. Pappas and P. J. Chirik, Angew. Chem., Int. Ed., 2014, 53, 6241–6244 CrossRef CAS PubMed.
- S. K. Podiyanachari, R. Fröhlich, C. G. Daniliuc, J. L. Petersen, C. Mück-Lichtenfeld, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2012, 51, 8830–8833 CrossRef CAS PubMed.
- T. P. Vaid, A. S. Veige, E. B. Lobkovsky, W. V. Glassey, P. T. Wolczanski, L. M. Liable-Sands, A. L. Rheingold and T. R. Cundari, J. Am. Chem. Soc., 1998, 120, 10067–10079 CrossRef CAS.
- J. R. Walensky, R. L. Martin, J. W. Ziller and W. J. Evans, Inorg. Chem., 2010, 49, 10007–10012 CrossRef CAS PubMed.
- J.-C. Berthet, C. Villiers, J.-F. Le Maréchal, B. Delavaux-Nicot, M. Lance, M. Nierlich, J. Vigner and M. Ephritikhine, J. Organomet. Chem., 1992, 440, 53–65 CrossRef CAS.
- N. E. Travia, M. J. Monreal, B. L. Scott and J. L. Kiplinger, Dalton Trans., 2012, 41, 14514–14523 RSC.
- A. C. Behrle, J. R. Levin, J.-E. Kim, J. M. Drewett, C. L. Barnes, E. J. Schelter and J. R. Walensky, Dalton Trans., 2015, 44, 2693–2702 RSC.
- A. Zalkin, D. H. Templeton, C. Le Vanda and A. Streitwieser Jr, Inorg. Chem., 1980, 19, 2560–2563 CrossRef CAS.
- B. M. Gardner, W. Lewis, A. J. Blake and S. T. Liddle, Organometallics, 2015, 34, 2386–2394 CrossRef CAS.
- J. A. Soderquist and M. R. Najafi, J. Org. Chem., 1986, 51, 1330–1336 CrossRef CAS.
- G. R. Eaton, J. Chem. Educ., 1969, 46, 547–556 CrossRef CAS.
- R. Shinomoto, E. Gamp, N. M. Edelstein, D. H. Templeton and A. Zalkin, Inorg. Chem., 1983, 22, 2351–2355 CrossRef CAS.
- H. W. Turner, R. A. Andersen, A. Zalkin and D. H. Templeton, Inorg. Chem., 1979, 18, 1221–1224 CrossRef CAS.
- J. McKinven, G. S. Nichol and P. L. Arnold, Dalton Trans., 2014, 43, 17416–17421 RSC.
- T. M. Trnka, J. B. Bonanno, B. M. Bridgewater and G. Parkin, Organometallics, 2001, 20, 3255–3264 CrossRef CAS.
- S. R. Daly, P. M. B. Piccoli, A. J. Schultz, T. K. Todorova, L. Gagliardi and G. S. Girolami, Angew. Chem., Int. Ed., 2010, 49, 3379–3381 CrossRef CAS PubMed.
- J. Kaski, P. Lantto, J. Vaara and J. Jokisaari, J. Am. Chem. Soc., 1998, 120, 3993–4005 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, and X-ray crystallographic tables. CCDC 1811106–1811116. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc05328b |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab and
John
Arnold
ab and
John
Arnold
 .16 With this in mind, we targeted the synthesis of a tetrakis(amidinate) complex through the reaction of 1 with N,N′-diisopropylcarbodiimide (Scheme 1). However, despite forcing conditions (100 °C), carbodiimide insertion into the Th–C bond was not observed, most likely a result of steric saturation around the metal center. Regardless, the alternative salt metathesis route using one equiv. of ThCl4(DME)2 (ref. 36) and 4.05 equiv. of Li(BIMA)(THF)37 and heating to 90 °C for 5 d afforded the desired homoleptic complex Th(BIMA)4 (2) as colourless crystals in 66% yield (Scheme 1). The 1H NMR spectrum of 2 reflects averaged C4-symmetry in solution, with one set of peaks observed for the equivalent amidinate ligands. The molecular structure of 2, determined by single-crystal X-ray diffraction studies, shows a pseudo-tetrahedral geometry of the amidinate ligands around the thorium center (Fig. 1). The Th–Namid bond lengths vary between 2.49 and 2.62 Å, a noticeably larger range than that seen in 1 (2.49–2.54 Å), indicating the significant steric congestion imposed by the isopropyl groups of the amidinate ligands. The structural parameters of 2 combined with the reaction conditions necessary to form 2 support the notion that insertion of carbodiimide into the Th–C bond was hindered by steric crowding, and indicate that related insertion reactions might be subject to this constraint. Due to the steric protection afforded by the tetrakis(amidinate) framework, and inspired by the work of Evans regarding low-valent thorium chemistry,38 we attempted the reduction of 2 with KC8 in the presence of 18-crown-6; nevertheless, no colour change was observed and only starting material was isolated.
.16 With this in mind, we targeted the synthesis of a tetrakis(amidinate) complex through the reaction of 1 with N,N′-diisopropylcarbodiimide (Scheme 1). However, despite forcing conditions (100 °C), carbodiimide insertion into the Th–C bond was not observed, most likely a result of steric saturation around the metal center. Regardless, the alternative salt metathesis route using one equiv. of ThCl4(DME)2 (ref. 36) and 4.05 equiv. of Li(BIMA)(THF)37 and heating to 90 °C for 5 d afforded the desired homoleptic complex Th(BIMA)4 (2) as colourless crystals in 66% yield (Scheme 1). The 1H NMR spectrum of 2 reflects averaged C4-symmetry in solution, with one set of peaks observed for the equivalent amidinate ligands. The molecular structure of 2, determined by single-crystal X-ray diffraction studies, shows a pseudo-tetrahedral geometry of the amidinate ligands around the thorium center (Fig. 1). The Th–Namid bond lengths vary between 2.49 and 2.62 Å, a noticeably larger range than that seen in 1 (2.49–2.54 Å), indicating the significant steric congestion imposed by the isopropyl groups of the amidinate ligands. The structural parameters of 2 combined with the reaction conditions necessary to form 2 support the notion that insertion of carbodiimide into the Th–C bond was hindered by steric crowding, and indicate that related insertion reactions might be subject to this constraint. Due to the steric protection afforded by the tetrakis(amidinate) framework, and inspired by the work of Evans regarding low-valent thorium chemistry,38 we attempted the reduction of 2 with KC8 in the presence of 18-crown-6; nevertheless, no colour change was observed and only starting material was isolated.