
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceXBphos-Rh: a halogen-bond assembled supramolecular catalyst†
Lucas
Carreras
 a,
Marta
Serrano-Torné
a,
Piet W. N. M.
van Leeuwen
*b and
Anton
Vidal-Ferran
*ac
a,
Marta
Serrano-Torné
a,
Piet W. N. M.
van Leeuwen
*b and
Anton
Vidal-Ferran
*ac
aInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology, Av. Països Catalans 16, 43007 Tarragona, Spain. E-mail: avidal@iciq.cat
bLPCNO, INSA, 135 Avenue de Rangueil, F-31077 Toulouse, France
cICREA, Pg. Lluís Companys 23, 08010 Barcelona, Spain
First published on 19th February 2018
Abstract
The use of halogen bonding as a tool to construct a catalyst backbone is reported. Specifically, pyridyl- and iodotetrafluoroaryl-substituted phosphines were assembled in the presence of a rhodium(I) precursor to form the corresponding halogen-bonded complex XBphos-Rh. The presence of fluorine substituents at the iodo-containing supramolecular motif was not necessary for halogen bonding to occur due to the template effect exerted by the rhodium center during formation of the halogen-bonded complex. The halogen-bonded supramolecular complexes were successfully tested in the catalytic hydroboration of terminal alkynes.
Introduction
The evolution of metal-based homogeneous catalysis has run in parallel with the development of structurally diverse ligands.1 Ligand design allows the behaviour of the metal centre to be influenced and its catalytic activity and selectivity to be modified.Supramolecular chemistry has emerged as a more efficient tool for the construction of (enantiopure) ligand backbones of metal complexes when compared with standard covalent chemistry. Supramolecular strategies mainly rely on the self-assembly of building blocks that contain both the complementary motifs required for the assembly, and the binding groups necessary for the desired catalysis event. This approach has been successfully employed for preparing catalysts by assembling supramolecularly complementary building blocks through hydrogen-bonding, metal-ligand, or ionic interactions.2 However, the use of halogen bonding for constructing the backbone of a catalyst remains, to the best of our knowledge, unexplored.3
The directionality and strength of halogen bonding,4 together with a greater tolerance to solvent polarity changes,5 prompted us to design and develop new halogen-bonded metal catalysts (see Scheme 1). We speculated that the halogen bonding-mediated assembly of two appropriate building blocks, each bearing a ligating group, could lead to the formation of a metal complex due to the chelation effect that should be exhibited in the presence of the metal centre. With regard to the ligating groups, we focused our attention on phosphino groups due to their pre-eminence in homogeneous catalysis6 and their tolerance to functionalisation.7 With respect to the halogen bonding motifs, we envisaged that the use of a fluorinated iodoarene and a pyridine group could be suitable due to the reported complementarity of both supramolecular motifs.8 Concerning the metal centre, we focused on rhodium as its complexes are catalytically active in pivotal organic transformations. In the discussion that follows, we report our results in the preparation and full characterisation of the first examples of halogen-bonded rhodium(I) complexes and their catalytic performance in alkyne hydroborations.
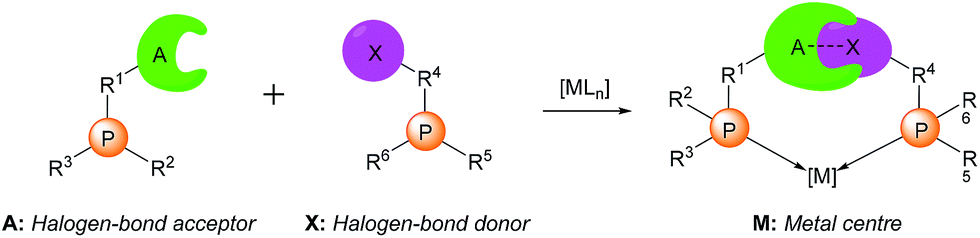 | ||
| Scheme 1 Supramolecular halogen-bonded catalysts (A: halogen bond acceptor; X: halogen bond donor; M: metal centre). | ||
Results and discussion
Examination of CPK models revealed that the relative arrangements of the P-ligating groups and the halogen bond acceptor and donor motifs in 2-pyridyldiphenylphosphine (1) and 2-iodo-3,4,5,6-tetrafluorophenyldiphenylphosphine (2), respectively, were adequate for the formation of the target halogen-bonded Rh(I) complex (see Scheme 2). Building blocks 1 and 2 were successfully prepared in good yields in a multigram scale by slight modification of well-established synthetic protocols.9 We envisaged that the use of [Rh(Cl)(CO)2]2 along with a halide scavenger would render a putative [Rh(CO)2]X intermediate that should react with phosphines 1 and 2. After some experimentation, halogen-bonded rhodium(I) complexes were obtained by first reacting [Rh(Cl)(CO)2]2 and NaBArF,10 and then adding ligands 1 and 2 as indicated in Scheme 2.11 The structure of the complex was confirmed by standard spectroscopic studies. The 31P{1H} NMR spectrum showed two complex multiplets, from which a high value 2JP–P coupling constant (i.e. 276 Hz; unequivocally assigned by 2D 31P{1H} J-resolved spectroscopy; Fig. SI 52 ESI†) was calculated. According to the literature reviewed, this high value 2JP–P coupling constant indicates a trans-spanning bisphosphine coordinated to the rhodium centre.12 We initially expected a dicarbonyl complex, but ESI-MS analysis identified complex [Rh(CO)(1)(2)]BArF (4), referred to as XBphos-Rh13 in the discussion that follows.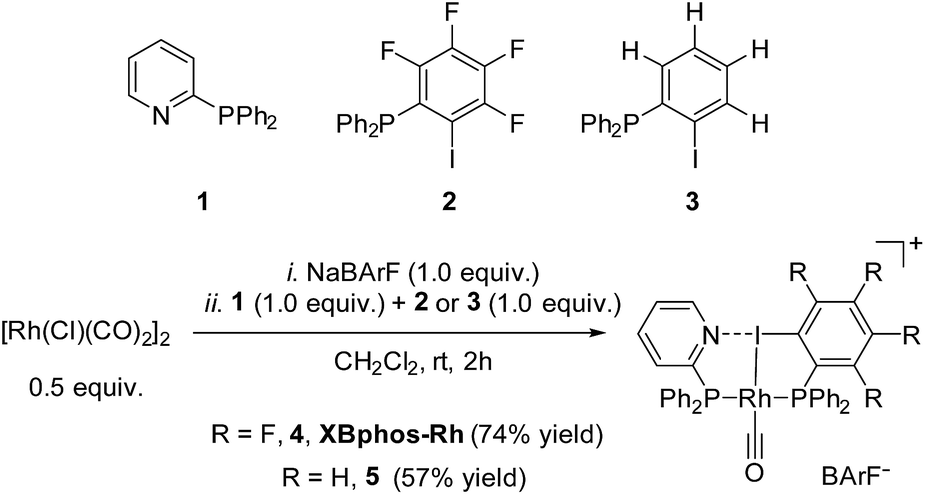 | ||
| Scheme 2 Synthesis of halogen-bonded rhodium(I) chelates. | ||
X-ray analysis confirmed the proposed halogen-bonded structure of the rhodium complex derived from 1 and 2 (see Scheme 2 for the structure of XBphos-Rh and Fig. 1A for the crystal structure). The nitrogen and iodine are aligned with a N–I distance of 2.757(8) Å and with a N⋯I–C bond angle of 169.4(5)°. These structural parameters are in agreement with those reported for halogen-bonded complexes involving pyridine and fluorinated iodoaryl moieties.14 Interestingly, the iodine appears to be coordinated to the rhodium centre (Rh–I distance = 2.5535(7) Å). Thus, the tridentate coordination of the halogen-bonded ligands in square planar XBphos-Rh resembles that observed in pincer-type complexes,15 and therefore this complex can be formally considered as the first reported P–I–P pincer-type complex.
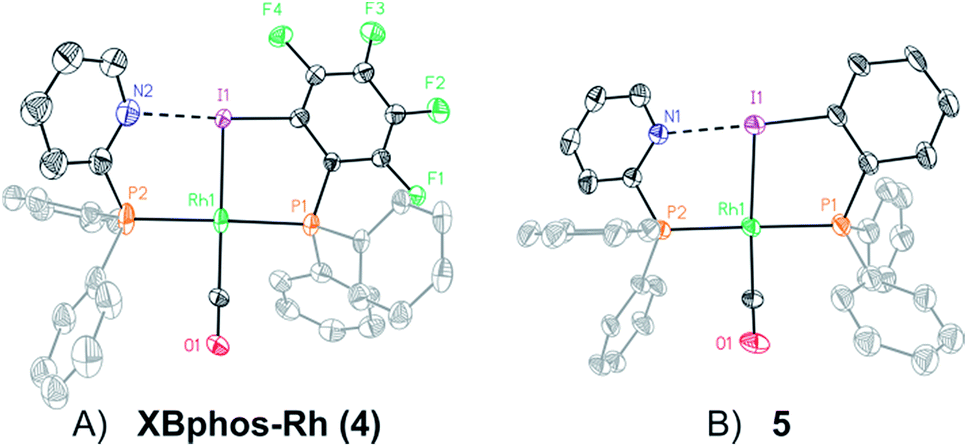 | ||
| Fig. 1 Crystal structures of XBphos-Rh (A) and 5 (B). Hydrogen atoms and the BArF unit have been omitted for the sake of clarity. Colour scheme: C: black, P: orange, Rh: green, F: green, N: blue, I: purple, O: red. Atomic displacement ellipsoids are drawn at a 50% probability. | ||
Given the favourable template effect exerted by the rhodium centre on the coordinated atoms in XBphos-Rh, we envisaged that fluorine-free compound 3 could also be used as the halogen bond donor. Thus, complex 5 could be prepared in good yields following a synthetic protocol analogous to that employed for XBphos-Rh (Scheme 2). Crystals from 5 suitable for X-ray analysis confirmed the proposed structure for the rhodium complex derived from 1 and 3 (Scheme 2 and Fig. 1B).
The analysis of the solid-state structures revealed a longer N–I distance for 5 than for XBphos-Rh (2.84(1) Å for 5 and 2.757(8) Å for 4), which indicates a stronger halogen bond interaction in the fluorine-containing complex.
The formation of a halogen bonding interaction between the pyridine and iodoarene motifs is driven by an enthalpy gain but penalised by an entropic unfavourable contribution arising from the geometry constraints for effective bonding (i.e. linear arrangement of the N⋯I–C atoms). Overall, the free energy associated with a single interaction is low.16 In order to gain a deeper insight into the halogen bonding event, full level DFT calculations were carried out. Geometries and stabilities of [Rh(CO)(1)(2)]+ and [Rh(CO)(1)(3)]+ and those of the starting building blocks were computed with Gaussian 09 (ref. 17) with the TPSS18-D3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 density functional employing a medium-sized triple-zeta (def2TZVP20) basis set, which is a good compromise between the computational cost and the reliability in describing halogen bonding.21 Selected geometrical parameters and computed binding free energies are summarised in Table 1. To aid comparison, the supramolecular interaction between 4MePy and IPFB (see footnote a in Table 1 for the abbreviations) was also computed and was found to be a slightly endergonic process with a calculated binding constant value being in agreement with that experimentally measured (i.e.; Kexp = 1 ± 1 M−1 in toluene in ref. 16 and Kcalc = 0.03 M−1 from the predicted ΔΔG value in entry 1 in Table 1).
19 density functional employing a medium-sized triple-zeta (def2TZVP20) basis set, which is a good compromise between the computational cost and the reliability in describing halogen bonding.21 Selected geometrical parameters and computed binding free energies are summarised in Table 1. To aid comparison, the supramolecular interaction between 4MePy and IPFB (see footnote a in Table 1 for the abbreviations) was also computed and was found to be a slightly endergonic process with a calculated binding constant value being in agreement with that experimentally measured (i.e.; Kexp = 1 ± 1 M−1 in toluene in ref. 16 and Kcalc = 0.03 M−1 from the predicted ΔΔG value in entry 1 in Table 1).
| Entry | Complex | d N–I (Å) | N⋯I–C bond angle (°) | ΔΔG (kcal· mol−1) |
|---|---|---|---|---|
| a 4MePy·IPFB = complex between 4-methylpyridine and iodopentafluorobenzene. b ΔΔG = ΔG4MePy·IPFB − ΔG4MePy − ΔGIPFB. c X-ray values in parentheses. d ΔΔG = ΔG[Rh(CO)(1)(2or3)]+ + ΔGCO − ΔG[Rh(CO)2]+ − ΔG1 − ΔG2or3. | ||||
| 1 | 4MePy·IPFBa | 2.714 | 179.4 | +2.03b |
| 2c | [Rh(CO)(1)(2)]+ | 2.709 (2.757(8)) | 175.2 (169.4(5)) | −112.83d |
| 3c | [Rh(CO)(1)(3)]+ | 2.827 (2.84(1)) | 176.2 (171.2(7)) | −111.97d |
The computed N–I distances were in agreement with the experimental values and their formations were calculated to be highly exergonic processes. Computational studies confirmed a slightly higher stability for [Rh(CO)(1)(2)]+ than for [Rh(CO)(1)(3)]+ (entries 2 and 3, Table 1). Electrostatic potential surfaces were calculated at the level of theory described above (see Fig. 2 for 2 and [Rh(CO)(1)(2)]+ and ESI† for all information). The maximum values of the σ-hole in 2 (Fig. 2) and fluorine-free derivative 3 (ESI†) are 25.1 and 12.5 kcal.mol−1, respectively. A lower σ-hole value indicates a lower donor character of the halogen atom to the supramolecular bond. This observation derived from electrostatic potentials is in agreement with previously discussed X-ray data (i.e. a longer N–I bond for 5 than for XBphos-Rh).
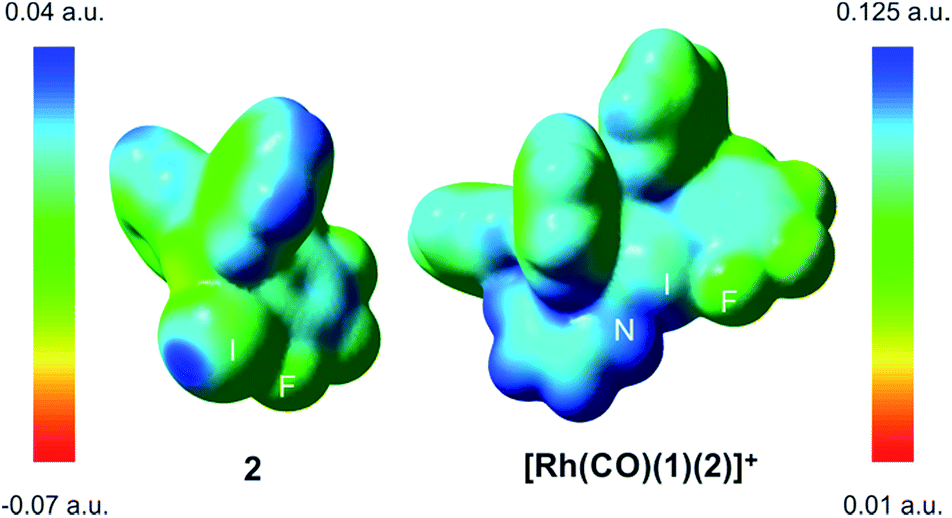 | ||
| Fig. 2 Electrostatic potential surfaces at an isovalue of 0.001 a.u. | ||
The interesting structural features of XBphos-Rh and 5 (Table 2),22 prompted us to investigate reactivity features as catalysts that could complement existing catalytic tools. These complexes were tested in the hydroboration of terminal alkynes towards vinylboronic acid derivatives, as the outcome of these reactions is heavily influenced by the electronic and steric properties of the ligand.23 Methods for synthesizing vinylboronic acid derivatives, which are important synthetic intermediates, by an atom-economical hydroboration reaction24 are scarce in the literature.23,25
To aid comparison of the results obtained with XBphos-Rh and 5, we included the monodentate [Rh(CO)(PPh3)3]BArF (6) complex in our catalytic studies.26 A summary of the results obtained is shown in Table 3. Both XBphos-Rh and 5 were active hydroboration catalysts, and E-isomers 8 were preferentially formed in all cases. Although XBphos-Rh is the most selective hydroboration catalyst for phenylacetylene 7a (88% overall yield for HBpin and 51% for HBcat; entries 1 and 4 in Table 3), hydroboration selectivities for 1-octyne 7b using XBphos-Rh are slightly lower than those with 6 (entries 7–12, Table 3). It is important to mention that in all cases selectivity towards branched derivatives is higher with the halogen-bonded catalysts than with the background catalyst 6.26 For example, a ten-fold increase in yield was obtained for the branched product 10a,pin when XBphos-Rh was used (entries 1 and 3, Table 3). It is also noteworthy that, as far as we are aware, XBphos-Rh provides the highest reported yield for the branched derivative 10b,cat (44% branched product with respect to all hydroboration products, entry 10, Table 3). Regarding electronic effects of the product distribution in the hydroboration of aryl-substituted acetylenes, higher ratios of the branched product were obtained for electron-deficient arene 7c than for electron rich derivative 7d (entries 13 and 14, Table 3).
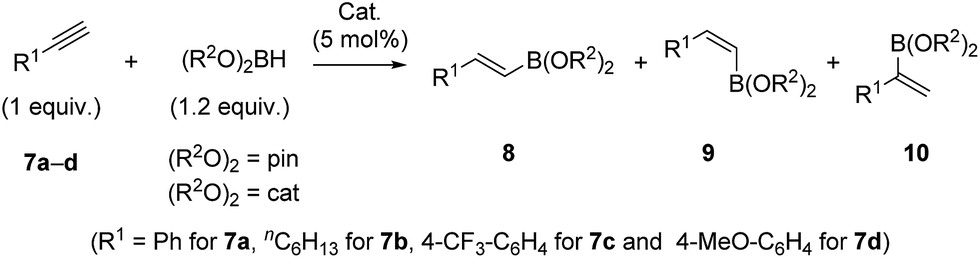
| Entry | Alkyne, (R2O)2BHb | Cat. | 8 + 9 + 10 yield% | Ratio 8:9:10 |
|---|---|---|---|---|
|
a Reactions were performed in CD2Cl2 (0.5 M) under N2 and reacted for 24 h at room temperature. Yields were determined by 1H NMR using 1,2,4,5-tetramethylbenzene as the internal standard. See ESI† for details.
b (R2O)2BH: HBpin = pinacolborane, HBcat = catecholborane.
c 2 equiv. of (R2O)2BH.
d Results obtained with bidentate complex [Rh(CO)(Xantphos)]BArF as catalyst in the reaction of 7a and HBpin are as follows: 69% overall yield (ratio 8:9:10 = 97:2:1).
|
||||
| 1c | 7a, HBpin | XBphos-Rh | 88 | 78:2:20d |
| 2 | 7a, HBpin | 5 | 36 | 72:5:23 |
| 3 | 7a, HBpin | 6 | 54 | 94:3:3 |
| 4 | 7a, HBcat | XBphos-Rh | 51 | 82:2:16 |
| 5 | 7a, HBcat | 5 | 49 | 82:2:16 |
| 6 | 7a, HBcat | 6 | 50 | 86:9:5 |
| 7 | 7b, HBpin | XBphos-Rh | 49 | 79:4:17 |
| 8 | 7b, HBpin | 5 | 50 | 79:4:17 |
| 9 | 7b, HBpin | 6 | 59 | 93:2:5 |
| 10 | 7b, HBcat | XBphos-Rh | 50 | 52:4:44 |
| 11 | 7b, HBcat | 5 | 51 | 66:4:30 |
| 12 | 7b, HBcat | 6 | 58 | 86:4:10 |
| 13c | 7c, HBpin | XBphos-Rh | 58 | 71:2:27 |
| 14c | 7d, HBpin | XBphos-Rh | 74 | 83:3:14 |
Conclusions
In summary, we have successfully prepared and fully characterised the first halogen-bonded supramolecular rhodium(I) complexes. To the best of our knowledge, these are the first examples where halogen bonding is the driving force for the assembly of the catalyst backbone. Furthermore, we have demonstrated that the presence of fluorine groups at the iodo-containing supramolecular motif is not necessary for efficient halogen bonding due to the favourable template effect exerted by the rhodium centre. The two new catalysts are active in the hydroboration of terminal alkynes, with the catalytic activity of XBphos-Rh rivalling that of the highest performing catalysts. Moreover, this catalyst favours enhanced ratios of the branched alkenyl boronic acid products. Ongoing investigations in our group include mechanistic studies on the outcome of the reaction,27 application of this system to other catalytically relevant transformations, extension of the application of this halogen bonding approach to other transition metals, and studies into alternative ligand designs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank MINECO (CTQ2014-60256-P, CTQ2017-89814-P and Severo Ochoa Excellence Accreditation 2014–2018 SEV-2013-0319) and the ICIQ Foundation for the financial support. L. C. thanks MINECO for a FPI-SO pre-doctoral fellowship (BES-2015-071872). The Université Fédérale Toulouse et Midi-Pyrénées is thanked for an IDEX Chaire d'attractivité. We also thank Dr E. Martin and J. Benet-Buchholz for X-ray crystallographic data, Dr N. Bampos for NMR 31P{1H} J-resolved data, Dr J. L. Núñez-Rico for his support with graphic design and Ms R. Somerville for proof reading the manuscript.Notes and references
- Homogeneous Catalysis: Understanding the Art, ed. P. W. N. M. van Leeuwen, Kluwer Academic Publishers, Dordrecht, 2004 Search PubMed.
- For selected reviews, see: (a) B. Breit, Angew. Chem., Int. Ed., 2005, 44, 6816–6825 CrossRef CAS PubMed; (b) A. J. Sandee and J. N. H. Reek, Dalton Trans., 2006, 3385–3391 RSC; (c) B. Breit, Pure Appl. Chem., 2008, 80, 855–860 CrossRef CAS; (d) S. Carboni, C. Gennari, L. Pignataro and U. Piarulli, Dalton Trans., 2011, 40, 4355–4373 RSC; (e) R. Bellini, J. I. van der Vlugt and J. N. H. Reek, Isr. J. Chem., 2012, 52, 613–629 CrossRef CAS; (f) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC; (g) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC. For a selected example on hydroborations employing supramolecular catalysts, see: (h) S. A. Moteki and J. M. Takacs, Angew. Chem., Int. Ed., 2008, 47, 894–897 CrossRef CAS PubMed.
- For the application of halogen bonding as a secondary interaction for substrate recognition in catalysis, see for example: (a) T. Caronna, R. Liantonio, T. A. Logothetis, P. Metrangolo, T. Pilati and G. Resnati, J. Am. Chem. Soc., 2004, 126, 4500–4501 CrossRef CAS PubMed. For the use of halogen bonding in the stabilisation of the ligand conformation in a metal catalysed transformation, see for example: (b) V. N. G. Lindsay, W. Lin and A. B. Charette, J. Am. Chem. Soc., 2009, 131, 16383–16385 CrossRef CAS PubMed. For the application of halogen bonding in organocatalysis, see for example: (c) D. Bulfield and S. M. Huber, Chem.–Eur. J., 2016, 22, 14434–14450 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- M. G. Sarwar, B. Dragisic, L. J. Salsberg, C. Gouliaras and M. S. Taylor, J. Am. Chem. Soc., 2010, 132, 1646–1653 CrossRef CAS PubMed.
- Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, ed. P. C. J. Kamer and P. W. N. M. van Leeuwen, John Wiley & Sons Ltd., United Kingdom, 2012 Search PubMed.
- For examples on the design and use of functionalised phosphorus-based catalysts, see for example: H. Fernández-Pérez, P. Etayo, A. Panossian and A. Vidal-Ferran, Chem. Rev., 2011, 111, 2119–2176 CrossRef PubMed.
- S. Tsuzuki, A. Wakisaka, T. Ono and T. Sonoda, Chem.–Eur. J., 2012, 18, 951–960 CrossRef CAS PubMed.
- For the preparation of 1, see: (a) P. A. A. Klusener, J. C. L. Suykerbuyk and P. A. Verbrugge, Shell Internationale Research Maatschappij B. V., Eur. Pat. Appl., EP499328A2, 1992. For the preparation of 2 and 3, see: (b) P. G. Eller and D. W. Meek, J. Organomet. Chem., 1970, 22, 631–636 CrossRef CAS.
- The tetrakis[3,5-bis(trifluoromethyl)phenyl]borate counter-ion enhances the solubility and crystallinity of the resulting derivatives, which we deemed as useful for further catalytic studies. See: L. Carreras, L. Rovira, M. Vaquero, I. Mon, E. Martin, J. Benet-Buchholz and A. Vidal-Ferran, RSC Adv., 2017, 7, 32833–32841 RSC.
- Attempts to prepare homocomplexes [Rh(CO)(1)2]BArF or [Rh(CO)(2)2]BArF following an analogous strategy to that used for XBphos-Rh failed, as complex mixtures of rhodium derivatives were obtained in all cases. Experimental reaction conditions for the selective preparation of rhodium complexes arising from C–I oxidative addition processes in 2 have been also developed (see p. SI-12 in ESI†). It is interesting to note that 31P NMR signals that could correspond to the above mentioned homocomplexes, or complexes arising from C–I oxidative additions in 2, were not found in any spectra of the crude reaction mixtures.
- Z. Freixa and P. W. N. M. van Leeuwen, Coord. Chem. Rev., 2008, 252, 1755–1786 CrossRef CAS.
- XB in XBphos-Rh stands for halogen (X) bonding (B).
- V. Vasylyeva, L. Catalano, C. Nervi, R. Gobetto, P. Metrangolo and G. Resnati, CrystEngComm, 2016, 18, 2247–2250 RSC.
- For representative literature reports on pincer-type metal complexes, see: Pincer and Pincer-Type Complexes: Applications in Organic Synthesis and Catalysis, ed. K. J. Szabó and O. F. Wendt, Wiley-VCH, Weinheim, 2014 Search PubMed.
- C. C. Robertson, J. S. Wright, E. J. Carrington, R. N. Perutz, C. A. Hunter and L. Brammer, Chem. Sci., 2017, 8, 5392–5398 RSC.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- (a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (b) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- R. Sure and S. Grimme, Chem. Commun., 2016, 52, 9893–9896 RSC.
- Examples of ligands with a bite angle close to 180° are scarce in the literature. See, for example: (a) A. J. Sandee, L. A. Van der Veen, J. N. H. Reek, P. C. J. Kamer, M. Lutz, A. L. Spek and P. W. N. M. Van Leeuwen, Angew. Chem., Int. Ed., 1999, 38, 3231–3235 CrossRef CAS; (b) P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, Acc. Chem. Res., 2001, 34, 895–904 CrossRef CAS PubMed.
- See, for example: (a) T. Ohmura, Y. Yamamoto and N. Miyaura, J. Am. Chem. Soc., 2000, 122, 4990–4991 CrossRef CAS; (b) J. Cid, J. J. Carbó and E. Fernández, Chem.–Eur. J., 2012, 18, 1512–1521 CrossRef CAS PubMed.
- These compounds can also be synthesised by hydroboration of alkynes with diboron derivatives. For selected examples, see the following references and those cited therein: (a) H. Jang, A. R. Zhugralin, Y. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 7859–7871 CrossRef CAS PubMed; (b) D. P. Ojha and K. R. Prabhu, Org. Lett., 2016, 18, 432–435 CrossRef CAS PubMed; (c) B. M. Trost, J. J. Cregg and N. Quach, J. Am. Chem. Soc., 2017, 139, 5133–5139 CrossRef CAS PubMed.
- For selected examples, see the following references and those cited therein: (a) I. D. Gridnev, N. Miyaura and A. Suzuki, Organometallics, 1993, 12, 589–592 CrossRef CAS; (b) S. Pereira and M. Srebnik, Tetrahedron Lett., 1996, 37, 3283–3286 CrossRef CAS; (c) D. M. Khramov, E. L. Rosen, J. A. V. Er, P. D. Vu, V. M. Lynch and C. W. Bielawski, Tetrahedron, 2008, 64, 6853–6862 CrossRef CAS; (d) N. Iwadate and M. Suginome, Org. Lett., 2009, 11, 1899–1902 CrossRef CAS PubMed.
- The bidentate rhodium complex [Rh(CO)(Xantphos)]BArF was also considered a background catalyst in this transformation and incorporated to catalytic hydroboration studies of alkyne 7a (see footnote d in Table 3). [Rh(CO)(Xantphos)]BArF displayed a lower performance than XBphos-Rh in terms of overall yield of hydroboration products of 7a with HBpin.
- As a detailed mechanism on Rh-catalysed hydroboration of terminal alkynes has still to be established (see: B. M. Trost and Z. T. Ball, Synthesis, 2005, 853–887 CrossRef CAS ), elementary mechanistic steps involving XBphos-Rh cannot be unequivocally proposed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic and crystallographic data. CCDC 1815870 and 1815871. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc00233a |
| This journal is © The Royal Society of Chemistry 2018 |