
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phase-segregated NiPx@FePyOz core@shell nanoparticles: ready-to-use nanocatalysts for electro- and photo-catalytic water oxidation through in situ activation by structural transformation and spontaneous ligand removal†
Masaki
Saruyama
 *a,
Sunwon
Kim
b,
Toshio
Nishino
a,
Masanori
Sakamoto
a,
Mitsutaka
Haruta
a,
Hiroki
Kurata
a,
Seiji
Akiyama
cd,
Taro
Yamada
e,
Kazunari
Domen
e and
Toshiharu
Teranishi
*a
*a,
Sunwon
Kim
b,
Toshio
Nishino
a,
Masanori
Sakamoto
a,
Mitsutaka
Haruta
a,
Hiroki
Kurata
a,
Seiji
Akiyama
cd,
Taro
Yamada
e,
Kazunari
Domen
e and
Toshiharu
Teranishi
*a
aInstitute for Chemical Research, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan. E-mail: saruyama@scl.kyoto-u.ac.jp; teranisi@scl.kyoto-u.ac.jp
bDepartment of Chemistry, Graduate School of Science, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan
cMitsubishi Chemical Group Science and Technology Research Center, Inc., 1000 Kamoshida-cho, Aoba-ku, Yokohama 227-8502, Japan
dJapan Technological Research Association of Artificial Photosynthetic Chemical Process (ARPChem), 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
eDepartment of Chemical System Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
First published on 30th April 2018
Abstract
The high overpotential of the oxygen evolution reaction is a critical issue to be overcome to realize efficient overall water splitting and enable hydrogen generation powered by sunlight. Homogeneous and stable nanoparticles (NPs) dispersed in solvents are useful as both electrocatalysts and cocatalysts of photocatalysts for the electro- and photo-catalytic oxygen evolution reaction, respectively, through their adsorption on various electrode substrates. Here, phase-segregated NiPx@FePyOz core@shell NPs are selectively synthesized by the reaction of Fe(CO)5 with amorphous NiPx seed-NPs. The NiPx@FePyOz NPs on conductive substrates exhibit higher electrocatalytic activity in the oxygen evolution reaction than those of other metal phosphide-based catalysts. The NiPx@FePyOz NPs can also be used as a cocatalyst of an anodic BiVO4 photocatalyst to boost the photocatalytic water oxidation reaction. The excellent catalytic activity and high stability of the NiPx@FePyOz NPs without any post-treatments are derived from in situ activation through both the structural transformation of NiPx@FePyOz into mixed hydroxide species, (Ni, Fe)OxHy, and the spontaneous removal of the insulating organic ligands from NPs to form a smooth and robust (Ni, Fe)OxHy/substrate heterointerface during the oxygen evolution reaction.
Introduction
Hydrogen evolution by efficient and sustainable electrolysis of water is desirable for mass-production of hydrogen as a clean energy source.1 The water splitting reaction consists of two half reactions: the oxygen evolution reaction (OER) and the hydrogen evolution reaction. The OER is considered to be the bottle-neck in the water splitting reaction because the OER typically requires a multistep four-electron process for O–O bond formation, which is kinetically slow. A high overpotential for the OER is required in water electrolysis, even with the use of rare and expensive metal catalysts, such as Ir and Ru.2 These factors limit the mass-production of water electrolysis devices. Currently, extensive explorations have been made into earth-abundant, low cost, and highly active materials, with a focus on non-noble transition-metal-based materials.3 In particular, transition metal phosphides have attracted much attention as highly efficient earth-abundant electrocatalysts for the OER.4The most recently developed OER catalysts are micrometer-sized materials, in the form of powders,5 thin films,6 and microstructures grown on conductive substrates.7 Nano-sized OER catalysts have also been widely studied because they exhibit remarkable activities owing to their large specific surface areas.8 Well-dispersed OER catalyst nanoparticles (NPs) in solvents are considered to be particularly effective, because they can be used to modify various kinds of substrates, including conductive electrodes and photocatalyst semiconductors by simple deposition methods.8,9 Such catalyst NPs usually require a ligand removal process after deposition. Hence, OER catalyst NP “ink” systems, which do not require ligand removal processes, are attractive for developing both catalyst/electrode and cocatalyst/photocatalyst hybrid systems on a large scale.
In our investigations of such OER catalysts, we found that the phase-segregated NiPx@FePyOz core@shell NPs are colloidally stable and efficient OER active transition metal phosphide-based catalysts. The NiPx@FePyOz NPs could be adsorbed on various kinds of substrates by simple deposition methods. The resulting composites exhibited high and stable OER activity without the need for any post-treatments due to in situ activation of NiPx@FePyOz NPs. NiPx@FePyOz NP-loaded carbon substrates exhibited an OER overpotential of 0.25 V at 10 mA cm−2 in 0.1 M KOH. Adsorption of NiPx@FePyOz NPs also greatly enhanced the photocatalytic activity and durability of BiVO4, suggesting that the NiPx@FePyOz NPs can also be used to fabricate photocatalyst/cocatalyst hybrid systems on a large scale.
Results and discussion
NiPx@FePyOz core@shell NPs were synthesized through the reaction of a-NiPx NPs and Fe(CO)5 (see ESI† for details). The a-NiPx seed-NPs were 11.3 ± 0.7 nm in size (Fig. 1a) and their X-ray diffraction (XRD) pattern exhibited only one broad peak at 45°, indicating the amorphous structure of the NPs (Fig. 1c).10 After heating the a-NiPx NPs with Fe(CO)5 in a mixture of 1-octadecene, oleylamine, and tri-n-octylphosphine (TOP) at 270 °C for 1 h, the spherical a-NiPx NPs were transformed into a unique anisotropic structure, in which spherical particles (9.6 ± 0.7 nm) were connected with rod-shaped particles (10.8 ± 3.0 nm × 6.3 ± 1.2 nm) as shown in the transmission electron microscope (TEM) image (Fig. 1b). An XRD pattern of the resulting NPs featured diffraction peaks assigned to a mixture of the major Ni2P and minor Ni12P5 phases (Fig. 1c). Although no peaks from the Fe compounds were observed, we confirmed the presence of Fe [Ni/Fe = 78/22 (mol mol−1)] by X-ray fluorescence (XRF) analysis. High-resolution TEM (HRTEM) observations showed that both the spherical and rod-shaped phases were mainly composed of the Ni2P phase (Fig. 1d), and the Ni12P5 phase was rarely observed in our measurements (only one among eighteen NPs, Fig. S1†). These HRTEM images were consistent with the XRD results. The HRTEM images also show the presence of an amorphous shell layer surrounding the NiPx core. Scanning TEM-energy dispersive X-ray spectroscopy (STEM-EDS) mapping of a single NP revealed that the elements Ni and P were mainly located at the core, and the elements Fe, O, and P were located at the shell, and therefore we describe the resulting NPs as NiPx@FePyOz NPs (Fig. 1e).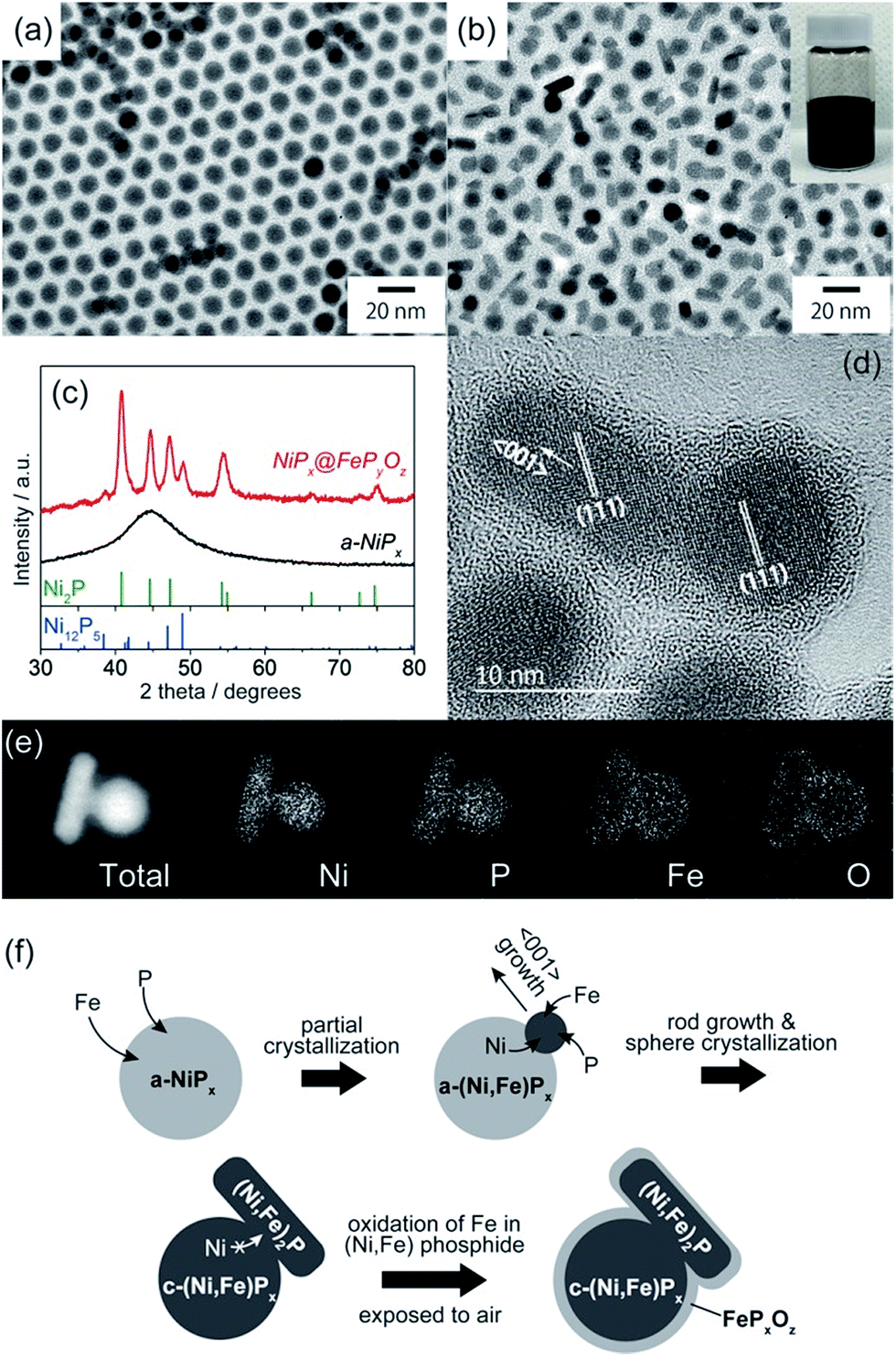 | ||
| Fig. 1 TEM images of (a) a-NiPx NPs and (b) NiPx@FePyOz NPs. The inset shows a hexane dispersion of the NiPx@FePyOz NPs stored for more than 6 months. (c) XRD patterns of a-NiPx NPs and NiPx@FePyOz NPs. (d) HRTEM image of NiPx@FePyOz NPs. (e) STEM-EDS mapping images of a NiPx@FePyOz NP. (f) Schematic of the formation mechanism of NiPx@FePyOz NPs. | ||
The structural evolution of the NiPx@FePyOz NPs was monitored during synthesis. At 10 min, large NiPx NPs with small spherical domains were observed (Fig. S2a†). As the reaction proceeded, these small domains grew larger. The XRD patterns indicate that a-NiPx started to change into crystalline NiPx (c-NiPx) phases, including Ni2P and Ni12P5 at 30 min. The peak intensities increased until 60 min (Fig. S2b†). The Fe/Ni molar ratios of the NiPx@FePyOz NPs increased as the reaction progressed, indicating that Fe atoms were gradually incorporated into a-NiPx seed-NPs (Fig. S3†).
The effects of Fe(CO)5 on the transformation of the a-NiPx NPs were also studied. Without Fe(CO)5, the a-NiPx NPs crystallized in a spherical shape (Fig. S4†), indicating that the Fe atoms induced a partial transformation of the spherical a-NiPx into a rod-shaped phase.
In an XRD pattern of the NiPx@FePyOz NPs, the (111) peak slightly shifted from the position of the pure Ni2P phase owing to Fe incorporation into Ni2P.11 From the (111) peak position of NiPx@FePyOz NPs at 40.97°, the Fe content in the core of NiPx@FePyOz NPs was estimated to be ∼5 mol% (Fig. S5†).11 XRF analysis revealed the Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe molar ratio of the c-NiPx cores to be 96:4, through selective etching of the FePyOz shells by H2SO4. These results agreed with the XRD results (Fig. S6†). As previously reported, Ni2−xFexP NPs tend to form rods or wires, because the Ni2−xFexP phase preferentially grows along the 〈001〉 direction.12 HRTEM images of the NiPx@FePyOz NPs showed that the long axis of the rod domains also grew in the direction of the 〈001〉 plane for Ni2P (Fig. 1d); thus, both incorporation of Fe into NiPx and the crystallization contributed to the anisotropic growth of the NiPx NPs.
:Fe molar ratio of the c-NiPx cores to be 96:4, through selective etching of the FePyOz shells by H2SO4. These results agreed with the XRD results (Fig. S6†). As previously reported, Ni2−xFexP NPs tend to form rods or wires, because the Ni2−xFexP phase preferentially grows along the 〈001〉 direction.12 HRTEM images of the NiPx@FePyOz NPs showed that the long axis of the rod domains also grew in the direction of the 〈001〉 plane for Ni2P (Fig. 1d); thus, both incorporation of Fe into NiPx and the crystallization contributed to the anisotropic growth of the NiPx NPs.
From these results, we propose the following mechanism for the formation of NiPx@FePyOz NPs (Fig. 1f). Initially, Fe atoms become incorporated into the a-NiPx NPs. When the a-NiPx NPs partially crystallize into small Ni2−xFexP domains, they grow along the 〈001〉 direction to form rod structures. During rod growth, Ni atoms are supplied from spherical a-NiPx phases. When the a-NiPx phases are completely crystallized, the Ni migration and structural transformations terminate. As a result, anisotropic spherical and rod-shaped NPs are formed. Finally, the FePyOz shells are generated by surface oxidation during the purification step in air.
The NiPx@FePyOz NPs were stable in a hexane dispersion for more than half a year, and could be easily adsorbed on various kinds of substrates, including carbon powder, carbon paper, and FTO coated glass, by simple mixing or deposition methods (see ESI† for details). Their OER catalytic activities were examined without any post-treatments such as annealing or ligand exchange to remove the organic ligands. Cyclic voltammetry (CV) curves of the NiPx@FePyOz NPs, Ni2P NPs, FeOx NPs, and a mixture of Ni2P NPs and FeOx NPs [Ni2P + FeOx, Ni/Fe = 77/23 (mol mol−1)] loaded carbon powder in 0.1 M KOH (Fig. 2a, S7 and S8†) are shown in Fig. 2b. Interestingly, the simply mixed Ni2P + FeOx NPs showed a lower overpotential than those of Ni2P and FeOx NPs; however, the NiPx@FePyOz NPs exhibited a further lower overpotential of 0.28 V at 10 mA cm−2 (0.36 V without iR compensation, Fig. S9†). NiPx@FePyOz NPs with different Ni/Fe molar ratios (85/15 and 72/28) were synthesized by changing the reaction time and showed overpotentials of 0.29 and 0.30 V at 10 mA cm−2 (Fig. S10†). Thus, the NiPx@FePyOz NPs with a Ni/Fe molar ratio of 78/22 were found to be the best OER catalyst in this work. The overpotential of 0.28 V is lower than those of many other previously reported metal phosphide-based OER catalysts (Table S1†).8a,13 More importantly, the amount of loaded NPs (0.02 mg cm−2) was much smaller than those of other catalysts, further confirming the excellent OER activity of the NiPx@FePyOz NPs.8a,13 The Tafel slope of the NiPx@FePyOz NPs, 43 mV dec−1, was smaller than those of Ni2P (44 mV dec−1), FeOx (64 mV dec−1), and Ni2P + FeOx (48 mV dec−1) NPs (Fig. 2c), and was also better than those of most of previously reported phosphide-based OER catalysts.8a,13 These results suggest favorable OER kinetics for the NiPx@FePyOz NPs. We also checked the NP loading amount dependent OER activity of NiPx@FePyOz NPs/carbon paper composites (Fig. S11†). The densely loaded electrode (0.5 mg cm−2) showed an overpotential of 0.25 V at 10 mA cm−2, which is better than those of other transition metal phosphide OER catalysts reported recently (Table S1†). Additionally, long-term chronoamperometry (CA) testing of NiPx@FePyOz NP-loaded carbon powder and paper showed no major decrease of the current densities during the continuous OER for 10 h, indicating the high OER operational stability of the NiPx@FePyOz NPs (Fig. 2d and S11c†).
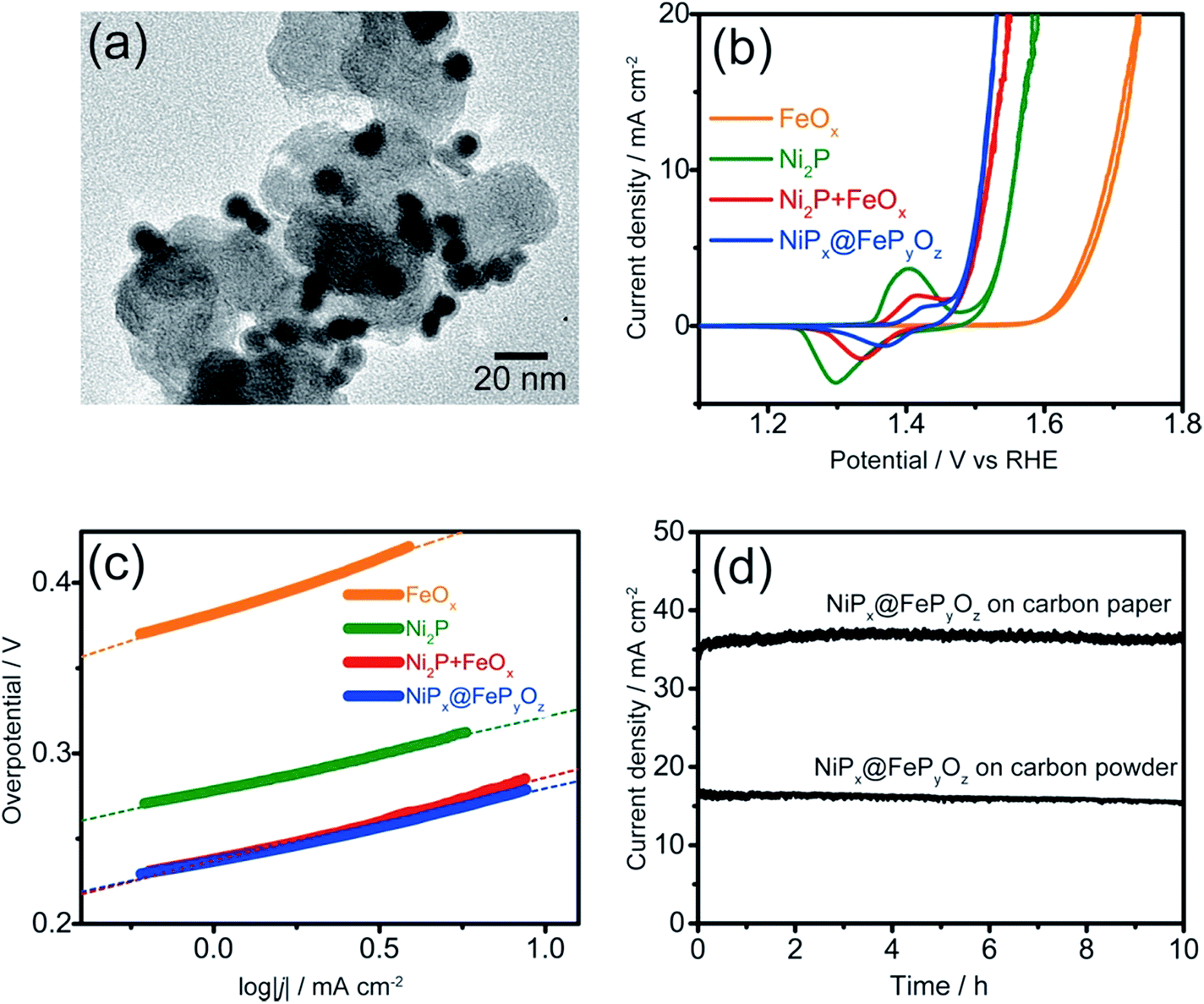 | ||
| Fig. 2 (a) TEM image of NiPx@FePyOz NP-loaded carbon powder. (b) Cyclic voltammograms and (c) Tafel plots of NiPx@FePyOz, FeOx, Ni2P, and FeOx + Ni2P NPs supported on carbon powder in 0.1 M KOH at 10 mV s−1. (d) CA curves of the NiPx@FePyOz NPs (0.075 mg cm−2) loaded on carbon paper and carbon powder at an overpotential of 0.35 V and 0.30 V in 0.1 M KOH, respectively. | ||
To understand the origin of the high OER activity of the NiPx@FePyOz NPs, we performed XRF and X-ray photoelectron spectroscopy (XPS) on the NiPx@FePyOz NP-loaded carbon paper before and after the OER (100 cycles of CV in 0.1 M KOH). The XRF results of the NiPx@FePyOz NPs after the OER revealed a considerable decrease of the element P, while the Ni:Fe molar ratio was maintained. Thus, P was selectively eliminated during the OER (Fig. S12†). Core level XPS spectra of the NiPx@FePyOz NPs before and after the OER are shown in Fig. S13.† Before the OER, the Ni 2p peak intensity was small because of coverage of the NiPx cores by FePyOz shells (Fig. S13a†). After the OER, the Ni 2p peak at 857 eV clearly emerged, which was attributed to the Ni 2p3/2 peak of Ni oxide or hydroxide.14 This result also indicates that Ni2+ ions were exposed to the surface of catalysts during the OER. For the case of P, before the OER, P 2p peaks appeared at 133 and 130 eV corresponding to PO43− species in the FePyOz shells and P0 in the partially exposed NiPx cores, respectively (Fig. S13b†).15 After the OER, these P 2p peaks completely disappeared owing to the elimination of P, which is consistent with the XRF results. The Fe 2p peak at 711 eV before the OER could be assigned to Fe oxide or phosphate in FePyOz shells. This peak markedly shifted to 714 eV, corresponding to Fe (oxy)hydroxide, after the OER (Fig. S13c†).16 In the case of O, before the OER, the O 1s peak at 531 eV, attributed to metal oxide or phosphate, shifted to 532 eV, which could be assigned to metal (oxy)hydroxide after the OER (Fig. S13d†).17 We conclude from the XPS results that the NiPx@FePyOz NPs were transformed into (Ni, Fe)OxHy during the OER. After the transformation, the elements Ni and Fe were homogeneously distributed over the entire catalyst surface, and P was dissolved. XPS spectra measured after Ar bombardment also indicated this transformation occurred (see Fig. S13† for details). Fe-doped NiOxHy has been reported to have much higher activity than pure NiOxHy, because the Fe ions surrounded by the Ni ions behave as active centres for the OER.18 Because the active Ni species in the NiPx@FePyOz NPs are covered with the FePyOz shell, the catalytic activity for the OER should be low, as shown in the case of the FeOx NPs. However, the elimination of element P and the subsequent structural transformation of NiPx@FePyOz into (Ni, Fe)OxHy create the OER active sites.
The formation of (Ni, Fe)OxHy was further supported by the CV results. The CV of NiPx@FePyOz NPs showed a smaller redox peak area at 1.48 V vs. RHE than that of Ni2P (Fig. 2b). This result implies that Fe diffused into the NiOxHy, because the Fe cations doped into NiOxHy suppressed oxidation of Ni2+.19 Although, Ni2P + FeOx NPs also showed a smaller redox peak area of Ni2+ than that of Ni2P, and the peak was larger than that of the NiPx@FePyOz NPs. This suggests that the Fe diffusion was incomplete for the case of Ni2P + FeOx NPs because the Ni2P and FeOx NPs were spatially separated. Thus, the direct contact of Ni- and Fe-containing phases in NiPx@FePyOz NPs was advantageous for fabricating homogeneously mixed metal compound catalysts.
Upon chemical transformation, the morphology of the NiPx@FePyOz NPs on the substrates changed to a film-like structure owing to fusion of the NiPx@FePyOz NPs (Fig. 3a, b and S14†), which led to the drastic change of their absorption spectrum. The transmittance of the NiPx@FePyOz NPs film on the FTO-coated glass became higher after 30 CV cycles in 0.1 M KOH owing to the formation of hydroxide species with a low absorption coefficient (Fig. 3c and S15†).20 Highly transparent catalysts in the visible region are particularly beneficial as cocatalysts for photocatalysts, because they do not obstruct incident light from reaching the photocatalysts. Furthermore, Fourier transform infrared (FT-IR) spectroscopy of the NiPx@FePyOz NPs on FTO before and after CV revealed that the C–H stretching vibration peaks at 2849 and 2918 cm−1 disappeared after the CV scans, indicating that the organic ligands (oleylamine and TOP) were completely removed during CV (Fig. 3d). This spontaneous removal of insulating ligands is a major advantage of the NiPx@FePyOz NPs as both a ready-to-use electrocatalyst and as a cocatalyst for photocatalysts, because post-treatment processes can be omitted to form NP/substrate heterointerfaces directly (Fig. 3e).
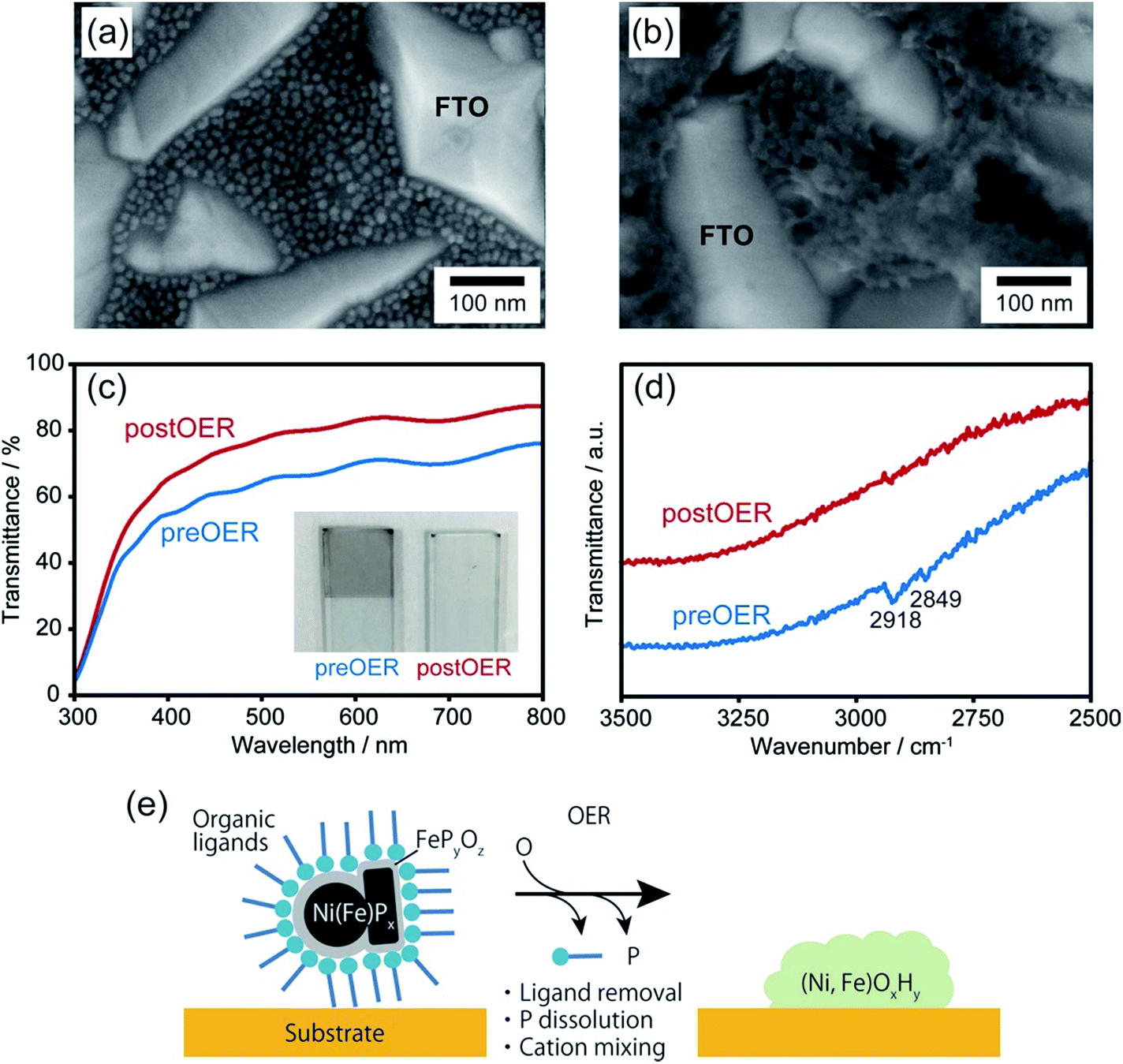 | ||
| Fig. 3 SEM images of NiPx@FePyOz NP coated FTO glass (a) before and (b) after 30 CV cycles in 0.1 M KOH. (c) Transmittance and (d) FT-IR spectra of NiPx@FePyOz NP coated FTO glass (blue) before and (red) after 30 CV cycles in 0.1 M KOH. The inset in (c) shows the photograph of NiPx@FePyOz NP coated FTO glass. (e) Schematic illustration of the transformation of NiPx@FePyOz NPs into (Ni, Fe)OxHy during the OER. | ||
To prove the versatile application of NiPx@FePyOz NPs, we applied the NiPx@FePyOz NPs as an OER cocatalyst with an anodic semiconductor photocatalyst to boost photocatalytic water oxidation (light source: 300 W Xe lamp with a 385 nm short-cut filter). A hexane solution of FeOx, Ni2P, Ni2P + FeOx, or NiPx@FePyOz NPs was deposited on porous BiVO4 film electrodes21 and spin-dried, followed by washing with ethanol (Fig. S16†). Note that no post-treatment processes were performed in the following measurements. Linear sweep voltammetry (LSV) measurements with chopped light in 0.125 M K2B4O7, in Fig. 4a, showed that the loading of the NPs enhanced the photocurrents and that the NiPx@FePyOz NP-loaded BiVO4 film electrode exhibited the largest photocurrent among the NPs used in this work. CA measurements with continuous light irradiation (@1.23 V vs. RHE) showed that the NiPx@FePyOz NPs loaded on BiVO4 possessed the highest durability to continuous water photo-oxidation and maintained 92% of their photocurrent after 1000 s (Fig. 4b and S17†). However, the current densities of the bare, Ni2P, FeOx, and Ni2P + FeOx NP-loaded BiVO4 decreased to 30, 49, 43, and 61% of their initial current densities, respectively. Interestingly, we found that the photocurrent of the NiPx@FePyOz NP-loaded BiVO4 was further enhanced after the CA measurement (Fig. 3c, d and S18e†), because the CA measurement promoted the transformation of the NiPx@FePyOz NPs into (Ni, Fe)OxHy. We also confirmed the transformation of NiPx@FePyOz NPs and enhanced OER activity in 0.125 M K2B4O7 (Fig. S19†). Conversely, the photocurrents of bare and other NP-loaded BiVO4 electrodes decreased after CA measurements (Fig. 3c, d and S18a–d†). This effect was likely caused by degradation of BiVO4 owing to accumulation of photogenerated holes in BiVO4.22 Namely, slow water oxidation kinetics at the surface of BiVO4 led to photo-corrosion under continuous light irradiation. By loading efficient OER cocatalysts onto the photoanode, holes were immediately consumed to oxidize water, preventing photo-corrosion and improving stability. These results also indicate the excellent catalytic OER activity of NiPx@FePyOz NPs as a cocatalyst.
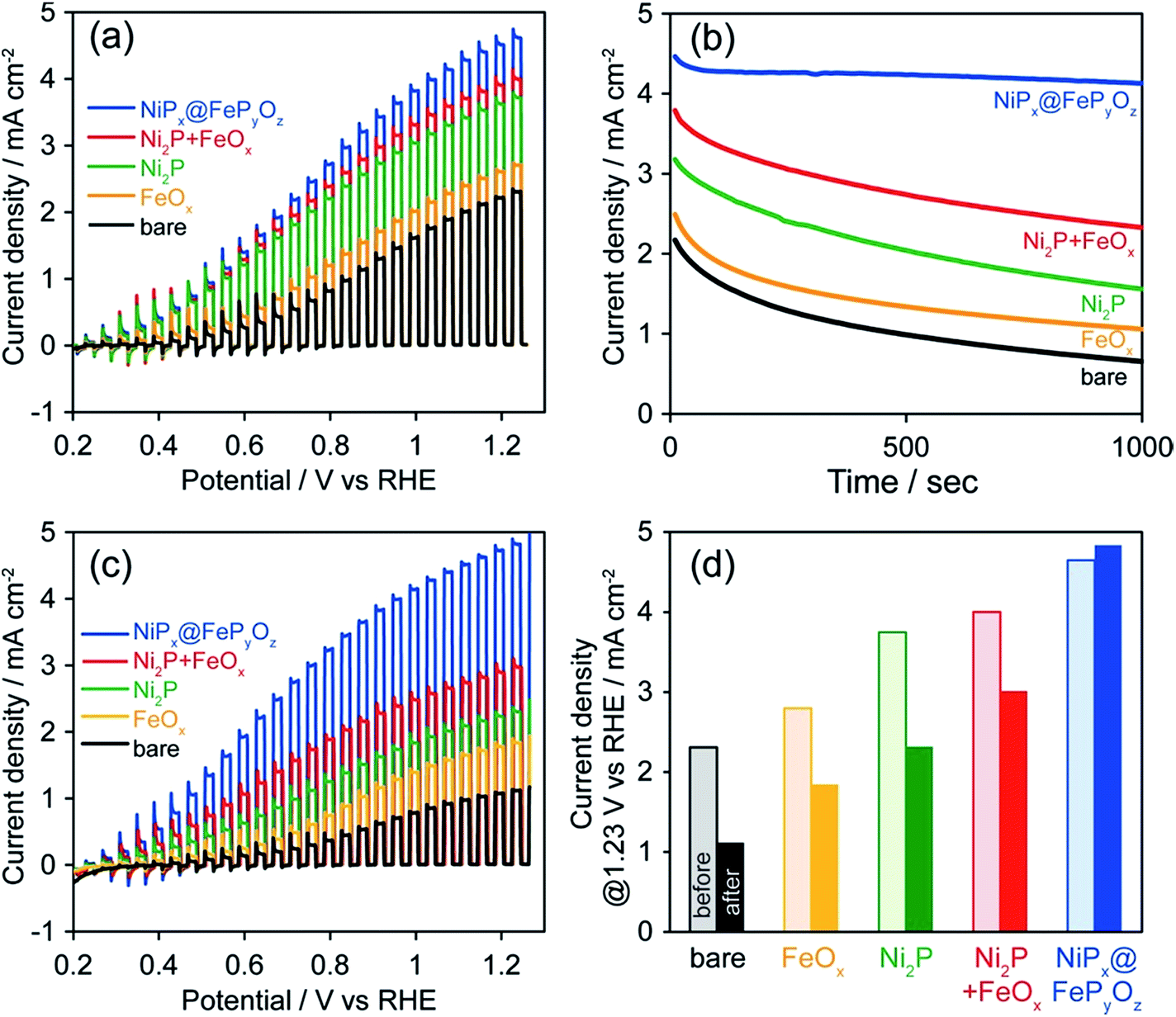 | ||
| Fig. 4 (a) Photocurrent density curves before CA, (b) CA curves, and (c) photocurrent density curves after CA, and (d) photocurrent densities at 1.23 V vs. RHE before and after CA of bare and NP-loaded BiVO4 in 0.125 M K2B4O7 at 1.23 V vs. RHE (300 W Xe lamp with a <385 nm cut filter). | ||
For practical use, the photoelectrochemical measurements of NiPx@FePyOz NPs/BiVO4 were also conducted under simulated sunlight (Fig. S20†). By loading NPs, the photocurrent density of NiPx@FePyOz NPs/BiVO4 at 1.23 V vs. RHE reached 2.3 mA cm−2, which is more than double that of bare BiVO4 (1.1 mA cm−2). Loading NiPx@FePyOz NPs increased the surface charge transfer efficiency (ηsurface) from 40% to 73% at 1.23 V vs. RHE (Fig. S20b†). Especially, the ηsurface at lower potential, 0.6 V vs. RHE, was greatly improved from 9% to 58%, also proving the fast OER kinetics of NiPx@FePyOz NP cocatalyst. A long-term stability test was also conducted for each electrode, and the highest durability was 62% photocurrent retention in 3 h at 1.23 V vs. RHE (Fig. S20d†).
Generally, a robust cocatalyst layer on BiVO4 drastically improves both activity and durability (Table S2†).21 On the other hand, partial coverage of BiVO4 with nanosized particles or molecules tends to show limited improvement of BiVO4 stability (Table S2†).22 Because, in our case, the NiPx@FePyOz NPs partially attach to BiVO4, it seems to be insufficient to fully boost the activity of BiVO4. However, ηsurface = 58% at 0.6 V vs. RHE is relatively high and the best durability (62% in 3 h) is better than that of partially covered BiVO4, despite the use of an ultimately simple and fast method (completed within ∼10 s under ambient conditions). However, there is plenty of room for further improvement of the photocatalyst performance. In addition to the OER kinetics on the surface of the cocatalyst, the hole transfer from the photocatalyst to the cocatalyst should be considered. Tuning the band structure of NiFe(OH)x must be effective and may be realized by incorporating a foreign metal into NiPx@FePyOz NPs.23
Recent studies on efficient Ni–Fe oxide, hydroxide, or oxyhydroxide based OER electrocatalysts showed significantly small overpotentials less than 0.3 V at 10 mA cm−2.24 Most of these catalysts are in bulk form, such as micrometer scale NiFe layered double hydroxides,24a,b composites with carbon,24c and Ni foam.24d Such bulk electrocatalysts, however, are difficult to directly hybridize with semiconductor photocatalysts, because of the small interfacial contact area and the lack of a robust bond between electrocatalysts and photocatalysts by simple mixing. This would be the reason why excellent electrocatalyst/photocatalyst hybrid system combinations have been rarely reported. Our NiPx@FePyOz NPs allow us to readily form a number of durable NPs/substrate heterointerfaces through an in situ activation and provide excellent OER activity with various kinds of conductive and semiconductive substrates. This feature is a considerable advantage of our catalytic NPs in the fabrication of large scale electro- and photo-catalyst systems.
Conclusions
In conclusion, we developed a selective synthesis of monodisperse, colloidally stable, and phase-segregated NiPx@FePyOz core@shell NPs with high OER activity. Using our NiPx@FePyOz NP ink, we loaded the NPs onto various conductive and semiconductor substrates and found excellent OER activity. We discovered that migration of Ni and Fe occurred between the phase separated NiPx and FePyOz phases, which served as efficient OER active sites for the OER. This process also induced spontaneous removal of ligands and in situ formation of the NP/substrate heterointerfaces, which provided ready-to-use OER hybrid catalysts without the need for any post-treatments. We demonstrated that even phase-segregated structures could be transformed into homogeneous active phases, suggesting a new way to design efficient nanostructured catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by the Artificial Photosynthesis Project of the New Energy and Industrial Technology Development Organization (NEDO) of Japan and JSPS KAKENHI for Scientific Research B (Grant No. JP16H03826), Scientific Research on Innovative Areas (Grant No. JP16H06520 (Coordination Asymmetry)) (T. T.), and Young Scientists B (Grant No. 17K14081) (M. S.).Notes and references
- (a) N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 43, 15729 CrossRef PubMed; (b) Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511 CrossRef CAS.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347 CrossRef CAS PubMed.
- (a) R. D. L. Smith, M. S. Prévot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60 CrossRef CAS PubMed; (b) M. S. Burke, L. J. Enman, A. S. Batchellor, S. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549 CrossRef CAS.
- A. Dutta and N. Pradhan, J. Phys. Chem. Lett., 2017, 8, 144 CrossRef CAS PubMed.
- (a) B. Zhang, X. Zhang, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. G. de Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. De Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333 CrossRef CAS PubMed; (b) J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. S. Horn, Science, 2011, 334, 1383 CrossRef CAS PubMed.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253 CrossRef CAS PubMed.
- (a) C. Tang, N. Cheng, Z. Pu, W. Xing and X. Sun, Angew. Chem., Int. Ed., 2015, 54, 9351 CrossRef CAS PubMed; (b) J. X. Feng, H. Xu, Y. T. Dong, S. H. Xe, Y. X. Tong and G. R. Li, Angew. Chem., Int. Ed., 2016, 55, 3694 CrossRef PubMed; (c) X. Lu and C. Zhao, Nat. Commun., 2015, 6, 6616 CrossRef CAS PubMed.
- (a) D. Li, H. Baydoun, C. N. Verani and S. L. Brock, J. Am. Chem. Soc., 2016, 138, 4006 CrossRef CAS PubMed; (b) Z. Zhuang, W. Sheng and Y. Yan, Adv. Mater., 2014, 26, 3950 CrossRef CAS PubMed; (c) L. Wu, Q. Li, C. H. Wu, H. Zhu, A. M. Garcia, B. Shen, J. Guo and S. Sun, J. Am. Chem. Soc., 2015, 13, 7071 CrossRef PubMed.
- (a) K. Maeda, A. Xiong, T. Yoshinaga, T. Ikeda, N. Sakamoto, T. Hisatomi, M. Takashima, D. Lu, M. Kanehara, T. Setoyama, T. Teranishi and K. Domen, Angew. Chem., Int. Ed., 2010, 49, 4096 CrossRef CAS PubMed; (b) T. Ikeda, A. Xiong, T. Yoshinaga, K. Maeda, K. Domen and T. Teranishi, J. Phys. Chem. C, 2013, 117, 2467 CrossRef CAS; (c) X. Chang, T. Wang, J. Zhang, A. Li and J. Gong, J. Am. Chem. Soc., 2015, 137, 8356 CrossRef CAS PubMed.
- E. Muthuswamy, G. H. L. Savithra and S. L. Brock, ACS Nano, 2011, 5, 2402 CrossRef CAS PubMed.
- A. H. Mudiyanselage, M. P. Arachchige, T. Seda, G. Lawes and S. L. Brock, Chem. Mater., 2015, 27, 6592 CrossRef.
- (a) K. Y. Yoon, Y. Jang, J. Park, Y. Hwang, B. Koo, J. G. Park and T. Hyeon, J. Solid State Chem., 2008, 181, 1609 CrossRef CAS; (b) J. Park, B. Koo, Y. Hwang, C. Bae, K. An, J. G. Park, H. M. Park and T. Hyeon, Angew. Chem., Int. Ed., 2004, 43, 2282 CrossRef CAS PubMed.
- (a) A. M. Garcia, H. Zhu, Y. Yu, Q. Li, L. Zhou, D. Su, M. J. Kramer and S. Sun, Angew. Chem., Int. Ed., 2015, 54, 9642 CrossRef PubMed; (b) J. Ryu, N. Jung, J. H. Jang, H. J. Kim and S. J. Yoo, ACS Catal., 2015, 5, 4066 CrossRef CAS; (c) N. Jiang, B. You, M. Sheng and Y. Sun, Angew. Chem., Int. Ed., 2015, 54, 6251 CrossRef CAS PubMed; (d) C. G. Read, J. F. Callejas, C. F. Holder and R. E. Schaak, ACS Appl. Mater. Interfaces, 2016, 8, 12798 CrossRef CAS PubMed; (e) J. Li, J. Li, X. Zhou, Z. Xia, W. Gao, Y. Ma and Y. Qu, ACS Appl. Mater. Interfaces, 2016, 8, 10826 CrossRef CAS PubMed; (f) M. Ledendecker, S. K. Calderýn, C. Papp, H. P. Steinrîck, M. Antonietti and M. Shalom, Angew. Chem., Int. Ed., 2015, 54, 12361 CrossRef CAS PubMed; (g) L. A. Stern, L. Feng, F. Song and X. Hu, Energy Environ. Sci., 2015, 8, 2347 RSC.
- A. Han, H. Chen, Z. Sun, J. Xu and P. Du, Chem. Commun., 2015, 51, 11626 RSC.
- D. R. Liyanage, S. J. Danforth, Y. Liu, M. E. Bussell and S. L. Brock, Chem. Mater., 2015, 27, 4349 CrossRef CAS.
- G. Zou, K. Xiong, C. Jiang, H. Li, T. Li, J. Du and Y. Qian, J. Phys. Chem. B, 2005, 109, 18356 CrossRef CAS PubMed.
- H. Ali-Löytty, M. W. Louie, M. R. Singh, L. Li, H. G. S. Casalongue, H. Ogasawara, E. J. Crumlin, Z. Liu, A. T. Bell, A. Nilsson and D. Friebel, J. Phys. Chem. C, 2016, 120, 2247 Search PubMed.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M. J. Cheng, D. Sokaras, T. C. Weng, R. A. Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305 CrossRef CAS PubMed.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329 CrossRef CAS PubMed.
- M. Liu, T. Wang, H. Ma, Y. Fu, K. Hu and C. Guan, Sci. Rep., 2014, 4, 7147 CrossRef CAS PubMed.
- T. W. Kim and K. S. Choi, Science, 2014, 343, 990 CrossRef CAS PubMed.
- T. W. Kim and K. S. Choi, J. Phys. Chem. Lett., 2016, 7, 447 CrossRef CAS PubMed.
- Y. Zhao, X. Jia, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung, D. O'Hare and T. Zhang, Adv. Energy Mater., 2016, 6, 1501974 CrossRef.
- (a) L. Yu, J. F. Yang, B. Y. Guan, Y. Lu and X. W. D. Lou, Angew. Chem., Int. Ed., 2018, 57, 172 CrossRef CAS PubMed; (b) W. Ma, R. Ma, C. Wang, J. Liang, X. Liu, K. Zhou and T. Sasaki, ACS Nano, 2015, 9, 1977 CrossRef CAS PubMed; (c) M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452 CrossRef CAS PubMed; (d) X. Lu and C. Zhao, Nat. Commun., 2015, 6616 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional characterization and results. See DOI: 10.1039/c8sc00420j |
| This journal is © The Royal Society of Chemistry 2018 |